

Abstract
Kaposi sarcoma-associated herpesvirus (KSHV) is a human γ-herpesvirus associated with several human malignancies. The replication and transcription activator (RTA) is necessary and sufficient for the switch from KSHV latency to lytic replication. Toll-interleukin-1 receptor (TIR) domain-containing adaptor-inducing β-interferon (TRIF, also called TIR-domain-containing adaptor molecule-1 (TICAM-1)) is a signaling adaptor molecule that is critically involved in the Toll-like receptor 3 (TLR-3) and TLR-4 signaling pathways for type I interferon (IFN) production, a key component of innate immunity against microbial infection. In this report, we find a new mechanism by which RTA blocks innate immunity by targeting cellular TRIF. RTA specifically degrades TRIF by shortening the half-life of TRIF protein. This RTA-mediated degradation is at least partially mediated through the ubiquitin-proteasome pathway because proteasome inhibitors as well as knockdown of cellular ubiquitin expression alleviate the degradation. RTA may not directly interact with TRIF and may activate TRIF degradation indirectly through an unknown mediator(s). RTA targets multiple regions of TRIF and may use its ubiquitin ligase domain for the degradation. In addition, physiological levels of TRIF protein are down-regulated during KSHV lytic replication when RTA is expressed. Finally, RTA down-regulates double-stranded RNA-initiated activation of TLR-3 pathway, in the absence of degradation of IFN regulatory factor 7 (IRF-7). Taken together, these data suggest that KSHV employs a novel mechanism to block the innate immunity by degrading TRIF protein. This work may contribute to our understandings on how KSHV evades host immunity for its survival in vivo.
Keywords: Innate Immunity, Interferon, Toll IL-1 Receptor (TIR) Domain, Toll-like Receptors (TLR), TRIF, Ubiquitin, KSHV, RTA
Introduction
Kaposi sarcoma (KS)2-associated herpesvirus (KSHV), also known as human herpesvirus 8 (HHV-8), is a γ-herpesvirus. It is believed to be the etiological agent of KS (1–3). KSHV is also implicated in the pathogenesis of AIDS-associated primary effusion lymphoma (also called body cavity-based lymphoma (BCBL)) and a lymphoproliferative disorder known as multicentric Castleman disease (1, 4, 5).
As with other herpesviruses, KSHV goes through both latency and lytic replication cycles. The expression of the KSHV replication and transcription activator (RTA) is necessary and sufficient for the switch from latency to lytic replication (4, 5). RTA is an immediate early gene (6–8) and a sequence-specific DNA-binding protein (7–13). RTA also interacts with other factors to modulate its transcription potential, such as with cellular recombination signal sequence-binding protein-Jκ (RBP-Jκ) (14–16).
RTA also modulates cellular activities through protein degradation. The proteasome is a multicomponent macromolecule that is ubiquitous in eukaryotic cells and works as a cellular machinery for degradation of proteins (17). The proteasome degradation is usually ubiquitin-dependent involving ubiquitin-activating enzyme E1, ubiquitin-conjugating enzyme E2, and ubiquitin ligase E3 (17–19). RTA was first documented as an E3 ligase for degradation of a cellular interferon regulatory factor 7 (IRF-7) (20). Two more cellular proteins, K-RBP and Hey, both are repressors of KSHV lytic replication and have been identified as targets for degradation by RTA with the same pathway (21, 22).
The host innate immune system is essential for the initial detection of invading viruses and subsequent activation of adaptive immunity. Toll-like receptors (TLRs) are a family of evolutionarily conserved receptors. TLRs are able to recognize molecular patterns unique to pathogens and activate host innate immunity against the pathogen (23, 24). One of the major products from TLR activation is the production of type I interferons (IFN). IFN is a key component to mount a proper and robust immune response to a viral infection (25, 26).
Toll-IL-1 receptor (TIR) domain-containing adaptor-inducing IFN-β (TRIF), also called TIR domain-containing adaptor molecule-1(TICAM-1), is an adaptor protein that is involved in the signaling transduction during the activation of some TLRs, leading to activation of NF-κB and type I IFN (27–29). TLR-3 and TLR-4 seem to be dependent on TRIF for their downstream cascades (27). Because TRIF is critically involved in TLR and IFN production, it would be a natural strategy for viruses to nullify TRIF for their own benefit. Vaccinia virus encodes two genes to differentially affect TRIF signaling (30, 31). Also, hepatitis C virus encodes a viral protease that cleaves TRIF (32). Interestingly, the same viral protease also digests another gene critically involved in IFN production (33).
IFN inhibits the KSHV replication in vitro (34–36). TLR-4 has been identified as an important molecule against KSHV infection, and KSHV has developed a mechanism to rapid suppression of TLR-4 expression (37). Also, murine γ-herpesvirus 68 (MHV68) is another herpesvirus with significant similarities to KSHV. Activation of the TLR-3/TLR-4 pathway potently inhibits the replication of MHV68 in vivo (38). At the same time, KSHVs have a segment genomic DNA and protein product (K8.1) that induce the production of IFNs (39, 40). KSHV infects many cell types, including B lymphocytes and dendritic cells (41–44). These cells express multiple TLRs, and dendritic cells are potent IFN producers. Thus, a successful counteraction of IFN activation may be a necessity for the survival of KSHV in vivo.
KSHV encodes several genes to counteract the innate IFN response. IRF-7 was first cloned in the context of Epstein-Barr virus (EBV) latency (45) and is a master gene for IFN production (46). KSHV employs two mechanisms to nullify the function of IRF-7; RTA mediates IRF-7 degradation in certain cell lines (20), and ORF45 blocks the activation of IRF-7 to impair IFN production (47). In addition, KSHV latency-associated nuclear antigen (LANA) is able to block the activation of TBK1-mediated activation of IRF-3 (48). KSHV encodes several viral homologs of the IRF family of transcription factors (vIRF). vIRF1, vIRF2, and vIRF3 are all able to block the production of IFNs by functioning as dominant negative versions of cellular IRFs (4, 5). Furthermore, KSHV ORF10 is also able to block the IFN signaling in KSHV-infected cells (49).
We have found a cell line in which IRF-7 could not be degraded by RTA. However, IFN production was still inhibited by RTA as reported previously (20). We have examined the mechanism of this IRF-7 degradation-independent IFN inhibition and found that RTA degrades cellular TRIF protein, an upstream adaptor protein involved in IFN production. This report provides another mechanism by which KSHV nullifies host innate immunity.
MATERIALS AND METHODS
Plasmids, Antibodies, and siRNA
Expression plasmids of KSHV RTA and its mutant (RTA-K152E), EBV RTA, IRF-7, pcDNA3.1-Myc-TRIF, and p56-luc (561-pGL3 luciferase reporter construct) are as described previously (45, 50–54). FLAG-TLR3 was a gift from Dr. Katherine Fitzgerald (55). Ubiquitin-expressing plasmid was a gift from Dr. Clinton Jones. The mutagenesis to generate a mutant with deletion (amino acids 318–514; TRIF-Del) was done by the use of PCR site-directed mutagenesis kit (Invitrogen). The TRIF-N (amino acids 1–514) and TRIF-C (amino acids 318–712) were done with the use of PCR and cloned into 3×-FLAG vector (Sigma). The expression clones were all sequenced to verify the validity of the clones. IRF-7 antibody was as described previously (45). Tubulin (T6557) and FLAG antibody (F1804) were obtained from Sigma. The antibodies for Myc (SC-40), IRF-1 (SC-497), ubiquitin (SC-9133), and GAPDH (SC-47724) were from Santa Cruz Biotechnology. TRIF antibody was from Cell Signaling (catalog number 4596). siRNA to ubiquitin (siUb) was from Santa Cruz Biotechnology (SC-29513), and siLuc was from Thermo Scientific Dharmacon (D-002050-01-05).
Cell Culture
293T is a human fibroblast line. WT11(clone 9) is a TLR-3-expressing cell line (56) (gift from Dr. Ganes Sen). Both cells were grown in Dulbecco's modified Eagle's medium (DMEM, Invitrogen) supplemented with 10% fetal bovine serum (FBS; Invitrogen) and 1% penicillin-streptomycin at 37 °C in 5% CO2 incubation. 400 μg/ml G418 (Invitrogen) was used to maintain the TLR-3 expression in WT11 cells. IB4 is a KSHV-negative but EBV-positive cell line. BCBL1 is a KSHV-positive primary effusion lymphoma line. These cells were maintained in RPMI 1640 plus 10% FBS.
Transient Transfection, Drug Treatment, and Reporter Assays
Effectene (Qiagen, Valencia, CA) was used for the transfection of 293T or WT11-9 cell lines following the manufacturer's recommendation. Proteasome inhibitors were as follows; lactacystin (80052-806) was from VWR International and used at the 1–10 μm range, and MG132 (HPK-116) was purchased from Assay Designs and used at 1–10 mm. For lactacystin and MG132 treatment, cells were transfected, and medium were removed 4–6 h after transfection. Fresh medium plus proper concentrations of drugs were added. Cells were collected 12–24 h later. Poly(I-C) (double-stranded RNA) was purchased from InvivoGen (tlrl-pic) and used at 10 μg/ml. Luciferase assays were performed using the assay kit from Promega according to the manufacturer's recommendation. β-Galactosidase assays were also performed for transfection efficiency. Data were averaged from the triplicates.
Protein Stability Assays
Protein biosynthesis inhibitor, cycloheximide, was purchased from Sigma (C4859) and used at 50–100 μg/ml. Cells were transfected in 10-cm dishes, and transfected cells were split 4–6 h after transfection into a 6-well plate. On the next day, the cells were treated with cycloheximide for the indicated period, and cell lysates were made and used for Western blot analysis.
Western Blot Analysis, RNA Extraction, and RNase Protection Assays (RPAs)
Standard Western blot analysis was performed as described (57–59). Total RNA was isolated from cells using the RNeasy total RNA isolation kit (Qiagen) or TRIzol extraction. RPA was performed with 10 μg of total RNA using the RNase protection assay kit II (Ambion, Houston, TX) at 55 °C. Sometimes, gradient temperatures were performed for RPA when difficulties in RPA were encountered (60). The GAPDH probe was from United States Biochemicals, Inc. The probe for IFN-β was a gift from Dr. Ganes Sen.
RESULTS
RTA Has an Additional Mechanism(s) to Block the IFN Production
RTA has been shown to degrade cellular IRF-7 for the blockage of IFN production (20). We have examined whether RTA could degrade IRF-7 extensively and found that RTA was unable to degrade IRF-7 in the cell line we used (293T) (Fig. 1A). Because RTA blocks the production of IFN, this particular line is therefore very useful to examine the role of IRF-7 in RTA-mediated IFN production. IRF-7 was transfected with or without RTA, and the cells were infected by Sendai virus. As shown in Fig. 1B, endogenous IFN-β production was greatly enhanced with IRF-7 expression. However, with the expression of KSHV RTA, the IFN production was drastically reduced. The use of IFN-β as a marker for type I IFN production is well established and appreciated in the field. Because in our experimental system there was no apparent IRF-7 degradation, even after the 6-h Sendai infection (Fig. 1A and data not shown), the data suggest that KSHV RTA has an additional mechanism(s), other than degradation of IRF-7, for the blockage of IFN production.
FIGURE 1.

KSHV RTA blocks IFN production without IRF-7 degradation. A, RTA could not degrade IRF-7 in the 293T cells. IRF-7 expression plasmid (0.1 μg) plus various amounts of RTA (0.2 μg) were transfected into 293T cells in a 6-well plate. Total DNA for transfection was maintained the same with the use of vector DNA (pcDNA3). Lysates were used for Western blot analysis. The identity of proteins is as shown. B, RTA inhibits IFN production. IRF-7 expression plasmid (0.4 μg) plus RTA (0.8 μg) were transfected into 293T cells in a 10-cm dish. 1 day later, cells were infected with Sendai virus (20 HA units/ml) for 6 h. Total RNA was isolated, and RPA was performed with IFN-β and GAPDH probes. The identity of RNA is as shown.
RTA Reduces the Expression of TRIF Protein
The data in Fig. 1 suggested that RTA blocks transcription of IFN by targeting a different protein involved in the innate immune response. We thus examined several intermediates involved in IFN induction for putative RTA targets, such as TRIF and RIG-I. 293T cells were co-transfected with target gene and RTA expression plasmids, and 1 day later, the cell lysates were used to examine the target gene expression. As shown in Fig. 2A, we found that TRIF protein was reduced in the presence of RTA. This reduction seems to be specific for KSHV RTA as EBV RTA (E-RTA) could not reduce the expression of TRIF at the level expressed (Fig. 2B). EBV RTA and KSHV RTA have significant homologies, and both viruses are γ-herpesvirus. We further examined whether TRIF RNA levels were reduced when RTA was present. The semiquantitative RT-PCR results indicated that TRIF RNA was not obviously affected by RTA expression (data not shown). Thus, the data suggest that RTA may reduce TRIF protein expression to block IFN production.
FIGURE 2.

RTA reduces the expression of TRIF protein. A, RTA reduces TRIF protein expression. 293T cells in 6-well plates were transfected with pcDNA3, RTA (0.2 μg), and TRIF expression plasmid (0.1 μg) in various combinations as shown on the top. Total DNA for transfection was maintained the same with the use of vector DNA. The cell lysates were obtained 1 day later for Western blot analysis. The identity of proteins is as shown. B, EBV RTA could not reduce the expression of TRIF. 293T cells were transfected with pcDNA3, RTA (0.2 μg), E-RTA (0.2 μg), and TRIF expression plasmid (0.1 μg) in various combinations as shown on the top. The cell lysates were obtained 1 day later. The membrane was stripped and probed with another antibody. The images in the same box indicate that they are derived from the same membranes. The identity of proteins is as shown.
RTA Modulates Protein Stability of TRIF
To test whether RTA can alter TRIF protein stability, 293T cells were transfected with TRIF, with or without RTA expression plasmid. 1 day later, cells were treated with cycloheximide for blockage of protein synthesis. Cell lysates were made at various time points after the drug treatment. As shown in Fig. 3, the TRIF protein was quite stable at 7 h after treatment. However, the half-life of TRIF was reduced in the presence of RTA (Fig. 3B). The half-life of IRF-1 protein was also examined and was short as expected (61). These results indicated that RTA reduced TRIF expression by altering protein stability.
FIGURE 3.

RTA alters the TRIF protein stability. 293T cells in a 10-cm dish were transfected with TRIF (0.4 μg) (A) or TRIF plus RTA (0.8 μg) expression plasmid (B). Total DNA for transfection was maintained the same with the use of vector DNA. 6 h after transfection, cells were split into a 6-well plate. Cycloheximide (100 μg/ml) was added after a 12-h incubation. Cell lysates were made at various time intervals (in hours) as shown on the top, and Western blot analysis was performed. The membrane was stripped and probed with another antibody. The images in the same box indicate that they are derived from the same membranes. The identity of proteins is as shown.
An RTA Mutant Does Not Significantly Degrade the Expression of TRIF
To determine the region of RTA to promote the degradation of TRIF, an RTA mutant (RTA-K152E) was used. This mutant cannot induce the lytic replication of KSHV (50, 62) and fails to induce the degradation of K-RBP (22). As shown in Fig. 4, the expression of TRIF was significantly reduced in cells transfected with RTA; however, TRIF expression was partially restored in the cells transfected with RTA-K152E. The region (amino acids 118–207; Cys/His-rich domain) was shown to be the putative domain as E3 ligase for IRF-7 and other proteins (20, 22). The mutation (K152E) is within the putative E3 ligase domain.
FIGURE 4.
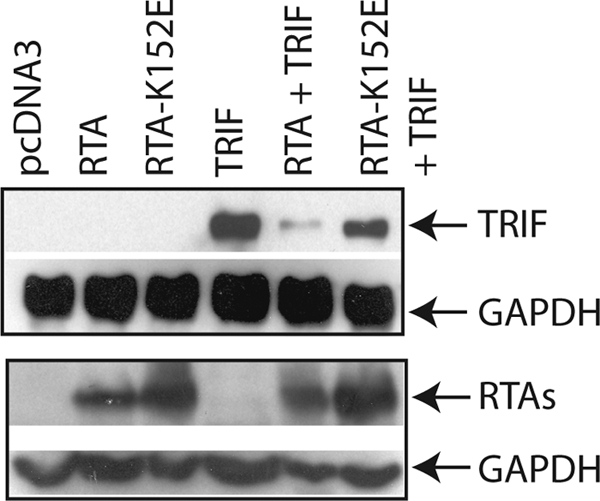
An RTA mutant failed to reduce the protein expression of TRIF. 293T cells were transfected with vector pcDNA3, TRIF (0.1 μg), RTA (0.2 μg), and RTA-K152E (0.1 μg) expression plasmid in various combinations as shown on the top. Total DNA for transfection was maintained the same with the use of vector DNA. Cell lysates were made 1 day later, and Western blot analysis was performed. The membrane was stripped and probed with another antibody. The images in the same box indicate that they are derived from the same membranes. The identity of proteins is as shown.
Multiple Regions of TRIF Are Targeted by RTA
To narrow down the region(s) of TRIF for RTA-mediated degradation, we have made several mutants as shown in Fig. 5A. The TRIF-Del was made to remove the majority of the TIR domain in TRIF. The TIR domain is a signature and used for interaction with other proteins in TLR signaling. The deletion mutant was transfected into 293T cells, and whether RTA degrades the TRIF mutant was examined. As shown in Fig. 5B, the TIR deletion TRIF was degraded by RTA. Next, we made mutants containing the N or C terminus of TRIF and the TIR domain. As shown in Fig. 5C, both TRIF-N and TRIF-C were degraded in the presence of RTA. However, the RTA-K152E mutant marginally degraded the proteins. The RTA mutant also failed to degrade TRIF-Del (data not shown). These data suggest that the RTA targets multiple regions of TRIF for degradation.
FIGURE 5.

RTA targets multiple regions of TRIF for degradation. A, schematic diagram of TRIF mutant constructs. The numbers denote the amino acid positions. The drawing is not to scale. B, TRIF-Del is degraded by RTA. 293T cells were transfected with vector pcDNA3, TRIF (0.1 μg), RTA (0.2 μg), and TRIF-Del mutant (0.1 μg) expression plasmids in various combinations as shown on the top. Total DNA for transfection was maintained the same with the use of vector DNA. Cell lysates were made 1 day later, and Western blot analysis was performed. Myc and GAPDH antibodies were used. The identity of proteins is as shown. C, both TRIF-N and TRIF-C are degraded by RTA. The two TRIF mutants, RTA, and RTA-K152E, were transfected into 293T cells with various combinations. FLAG, Myc, GAPDH, and RTA antibodies were used for the Western blot. The images in the same box indicate that they are derived from the same membranes. The identity of proteins is as shown.
RTA-mediated TRIF Degradation Is Mediated through Proteasome
RTA has been shown to degrade several proteins through the proteasome pathway (20, 22). To address whether TRIF is also degraded through the proteasome, we first used the proteasome inhibitor lactacystin. As shown in Fig. 6A, lactacystin partially restored the expression of TRIF. Another proteasome inhibitor, MG132, also partially restored the TRIF protein expression (data not shown). Second, we used siUb to knock down the endogenous ubiquitin. As shown in Fig. 6B, siUb could partially restore the expression of TRIF, but siRNA for luciferase (siLuc) failed to do so. The expression of ubiquitin was reduced in the presence of siUb (Fig. 6B). Finally, whether ubiquitin could be added to the TRIF molecule and increase its molecular weight was examined. However, we could not detect any slowly migrating, high molecular weight TRIF present under our experimental conditions (data not shown). It is possible that the TRIF molecule might be too large, and the addition of limited ubiquitin molecule might not be as obvious for observation. We then tested whether the ubiquitination could be observed in the TRIF-C as it is relatively small in molecular weight. As shown in Fig. 6C, in the presence of RTA, a slowly migrating, high molecular weight smear of TRIF-C, was observed (Fig. 6C, lanes 1 and 2). Furthermore, increased levels of TRIF-C smears were observable when ubiquitin-expressing plasmid was co-transfected (lanes 3 and 4). The expression of ubiquitin was confirmed (data not shown). These data provided evidence that RTA, at least partially, promoted TRIF degradation via the proteasome pathway.
FIGURE 6.

RTA degrades TRIF through proteasome pathway. A, proteasome inhibitor alleviates TRIF degradation. 293T cells were transfected with pcDNA3, RTA (0.2 μg), and TRIF expression plasmid (0.1 μg) in various combinations as shown on the top. Total DNA for transfection was maintained the same with the use of vector DNA. 6 h after transfection, cells were washed and treated with lactacystin. The cell lysates were obtained 1 day later for Western blot analysis. The identity of proteins is as shown. B, knockdown of ubiquitin alleviates TRIF degradation. 293T cells were transfected with pcDNA3, RTA (0.1 μg), and TRIF (0.05 μg) expression plasmids in various combinations as shown on the top. In addition, siUb or siLuc (5 pmol of each) were also transfected. Cell lysates were made 1 day later, and Western blot analysis with TRIF antibody was performed. The membrane was stripped and probed with another antibody. The images in the same box indicate that they are derived from the same membranes. The identity of proteins is as shown. C, ubiquitin was added to TRIF-C. TRIF-C or RTA+TRIF-C were transfected with or without HA-Ub expression plasmid. The cell lysates were obtained 1 day later for Western blot analysis. More cell lysates were used for RTA+TRIF-C transfections. Two different exposures of the same membrane are shown. The identity of proteins is as shown.
Expressions of RTA and TRIF Are Inversely Correlated at Physiological Concentrations
To examine whether RTA could degrade TRIF under physiological conditions, we induced the lytic replication process in the KSHV latently infected BCBL cell line. RTA is not expressed during the viral latency, but it is a key mediator for the lytic replication. When KSHV-positive BCBL1 cells were treated with sodium butyrate, the expression of RTA was significantly increased (Fig. 7A). In addition, a simultaneous down-regulation of endogenous TRIF expression was observed. The same lysates were also used for the detection of IRF-7, and the expression of IRF-7 was increased with the sodium butyrate treatment, which is in a agreement with a previous report (63, 64). Furthermore, we tested whether TRIF was degraded in KSHV-negative cell lines. IB4 is an EBV-transformed cell line with no KSHV. With the sodium butyrate treatment, no degradation of TRIF was observed (Fig. 7B). The induction of lytic replication by sodium butyrate is not 100%. However, endogenous TRIF is apparently low as we had to concentrate the cell lysates for the detection of endogenous TRIF. A slight induction of RTA might be sufficient to degrade endogenous TRIF. All these results suggest that the reduction of TRIF is specifically associated with the induction of KSHV RTA under physiological conditions.
FIGURE 7.
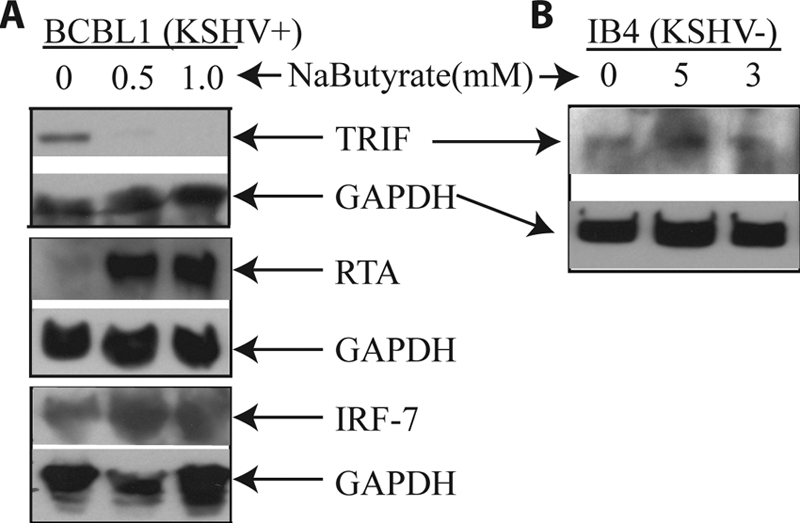
TRIF is down-regulated during KSHV lytic replication. A, the inverse correlation between the expression of RTA and TRIF. BCBL1 cells were treated with sodium butyrate (NaButyrate) for 24 h. Cell lysates were made, and the expression of endogenous proteins was analyzed by Western blot analysis. The membrane was stripped and probed with another antibody. The same cell lysates were used, and images in the same box indicate that they are derived from the same membranes. The identity of proteins is as shown. B, sodium butyrate did not affect TRIF in KSHV-negative cell line (KSHV−). IB4 cells (KSHV−) were treated with sodium butyrate for 24 h, and the cell lysates were made. The expression of TRIF was examined.
RTA Down-regulates the Activation of TLR-3 Pathway
Recently, it has been shown that Sendai infection activates the RIG-I pathway for IFN induction (65). Thus, it is possible that the IFN blockage observed in Fig. 1 may not be related to TRIF degradation completely. We thus examined whether RTA could block a signaling pathway that absolutely requires TRIF. TRIF is absolutely required for TLR-3 signaling (27). It is well established that p56 is a good target and a marker of TLR-3 activation (56, 66, 67). We thus tested whether the activation of p56 promoter by TLR-3 is affected by RTA expression. 293T cells were transfected with RTA- and TLR-3-expressing plasmids, and transfected cells were treated with or without synthetic double-stranded RNA (dsRNA). As shown in Fig. 8A, p56 promoter reporter was activated in response to TLR-3 and dsRNA (columns 2 and 3). However, in the presence of RTA, the activation was drastically reduced (columns 5 and 6). In addition to 293T cells, we also used the TLR-3-expressing cells (WT11) (56). As shown in Fig. 8B, RTA reduced the dsRNA-mediated activation of the p56 promoter reporter. All these data suggested that RTA reduced TLR-3 signaling activities. Of note, in our 293T cells, IRF-7 was not degraded by RTA (Fig. 1A). Therefore, the blockage of TLR-3 signaling may be related to potential TRIF degradation.
FIGURE 8.

RTA blocks TLR-3 signaling. A, RTA blocks TLR-3 signaling in 293T cells. Various plasmids were transfected into 293T cells as shown on the top. 0.1 or 0.2 μg of TLR-3 (columns 2, 3, 5, and 6) was used, respectively. RTA (0.2 μg) was used for transfection. Total DNA for transfection was maintained the same with the use of vector DNA. After 4–6 h of transfection, the cells were then treated (+) or not treated (−) with dsRNA (10 μg/ml). 1 day later, the cells were collected, and luciferase and β-gal assays were used for detection of reporter activation. The relative activation of p56 reporter (+dsRNA/−dsRNA) is as shown. Error bars indicate S.D. B, RTA blocks TLR-3 signaling in TLR-expressing cells. Various plasmids were transfected into TLR-3-expressing cells (WT11-9) as shown on the top. 0.2 or 0.4 μg of RTA was used, respectively. The cells were then treated (+) or not treated (−) with dsRNA (10 μg/ml). The relative activation of p56 reporter (+dsRNA/−dsRNA) is shown. One representative result is shown. Error bars indicate S.D.
DISCUSSION
IFN is important to control KSHV infection, and a successful counteraction of IFN signaling may be a necessity for the survival of KSHV in vivo. It is thus not surprising that KSHV encodes several genes to counteract the innate IFN system. IRF-7 is targeted by at least two KSHV proteins (20, 47). Latency-associated nuclear antigen is able to block the activation of IFN through IRF-3 (48). KSHV IRF homologs (vIRF1, vIRF2, and vIRF3) are able to block the production of IFNs by functioning as dominant negative versions of cellular IRFs. ORF10 blocks the IFN signaling in KSHV-infected cells (49).
Protein degradation has been shown to be important for antiviral responses (for a review, see Ref. 68). In this report, we have shown that RTA could block the IFN production without degradation of IRF-7 (Fig. 1). To address the possible mechanism behind this IRF-7 degradation-independent phenomenon, we find that RTA targets TRIF for the blockage of IFN production. First, we show that RTA reduces the expression of TRIF, and the reduction is likely to be specific to KSHV RTA as EBV RTA failed to reduce the expression of TRIF (Fig. 2). Second, RTA affects the stability of TRIF as its half-life is getting shorter in the presence of RTA (Fig. 3). In addition, RT-PCR results indicated that TRIF RNA was not affected by RTA (data not shown). Third, this RTA-mediated degradation of TRIF seemed to be present under physiological conditions as induction of lytic replication, and thus of the RTA expression, reduces the expression of endogenous TRIF protein (Fig. 7). Fourth and finally, the reduction of TRIF may be responsible for the blockage of TLR-3-mediated activation of p56 promoter in an IRF-7 degradation-independent manner (Figs. 1 and 8).
This RTA-mediated degradation of TRIF is apparently through the ubiquitin-mediated proteasome pathway. First, RTA has been shown to be an E3 ligase for the degradation of IRF-7, Hey1, and K-RBP. We have found that RTA-K152E mutant lost some activity in degrading TRIF (Figs. 4 and 5). The same mutant failed to degrade K-RBP (22). Also, the mutation is located in the Cys/His-rich region (amino acids 118–207), the putative E3 ligase domain (20, 22). Second, proteasome inhibitors (lactacystin and MG132) partially restore the TRIF protein expression (Fig. 6A and data not shown). Knockdown of ubiquitin increases the TRIF expression (Fig. 6B). RTA enhances high molecular weight smear formation in TRIF-C protein, and the smear is likely to be ubiquitin as the overexpression of ubiquitin enhances the formation (Fig. 6C). All these data suggest that RTA degrades TRIF, at least partially, through ubiquitin-mediated proteasome pathway.
TRIF is a cytoplasmic protein and mainly localized to the cell membrane (69), and KSHV RTA is a nuclear protein. We have failed to show that RTA is directly interacting with TRIF in co-immunoprecipitation assays (data not shown). In addition, we have done the immunostaining of the two proteins together in a cell and found that the two proteins were localized to the cytoplasm and nucleus, respectively, and no obvious co-localization was observed (data not shown). Thus, under our experimental conditions, we failed to obtain convincing evidence that the two proteins physically interact with each other directly. However, we cannot exclude the possibility that the two proteins may interact under some specific conditions/times. Based on our data, we propose that the RTA-mediated degradation of TRIF is through an indirect mechanism in which RTA activates an unknown cellular mediator(s) that facilitates the degradation of TRIF.
Our results here suggest another novel pathway for KSHV to evade the host innate immunity. Specifically, TLR-4 has been identified as an important molecule against KSHV infection recently, and KSHV has developed a mechanism for rapid suppression of TLR-4 expression (37). TLR-4 activation would lead to activation of TRIF, which further leads to the induction of type I IFNs. Therefore, this RTA-mediated degradation of TRIF may be used by KSHV to escape TLR-4-mediated innate immunity. Finally, the degradation of TRIF was observed in KSHV-infected cells under physiological concentrations (Fig. 7). Therefore, this phenomenon might naturally occur in individuals who have KSHV infection in vivo. The suppression of TLR-4 expression as well as TRIF degradation collectively suggest that the TLR-4 pathway is a critical one to control KSHV infection, and the virus employs two mechanisms to block TLR-4 activation.
It is known that KSHV infects B cells, where the majority of TLRs are present (70). In addition, TRIF is an unexpectedly multifunctional adaptor protein, mediating activation of several transcription factors including NF-κB, a key mediator for pro-inflammatory cytokines (71). Additionally, TRIF may trigger apoptosis, and thus, degradation of TRIF may alleviate the potential apoptosis process. It is tempting to speculate that the RTA-mediated TRIF degradation may not be limited to the blockage of IFNs, but also other cellular activities for the benefits of KSHV in vivo.
Acknowledgments
We thank Drs. Ganes Sen, Katherine Fitzgerald, and Clinton Jones for providing reagents for the study and the University of Nebraska – Lincoln Microscopy Facility for technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grants CA138213 and CA108951 and COBRA Grant RR15635 from the National Center for Research Resources (NCRR) (to L. Z.).
- KS
- Kaposi sarcoma
- KSHV
- Kaposi sarcoma-associated herpesvirus
- RTA
- replication and transcription activator
- E-RTA
- EBV RTA
- EBV
- Epstein-Barr virus
- TIR
- Toll-interleukin-1 receptor
- TRIF
- TIR domain-containing adaptor-inducing β-interferon
- TLR
- Toll-like receptor
- IRF
- IFN regulatory factor
- vIRF
- viral IRF
- BCBL
- body cavity-based lymphoma
- RPA
- RNase protection assay
- Ub
- ubiquitin.
REFERENCES
- 1. Moore P., Chang Y. (2001) in Virology (Knipe D. M. ed) pp. 2803–2833, Lippincott Williams and Wilkins, Philadelphia, PA [Google Scholar]
- 2. Chang Y., Cesarman E., Pessin M. S., Lee F., Culpepper J., Knowles D. M., Moore P. S. (1994) Science 266, 1865–1869 [DOI] [PubMed] [Google Scholar]
- 3. Chang Y., Moore P. S. (1996) Infect. Agents Dis. 5, 215–222 [PubMed] [Google Scholar]
- 4. West J. T., Wood C. (2003) Oncogene 22, 5150–5163 [DOI] [PubMed] [Google Scholar]
- 5. Dourmishev L. A., Dourmishev A. L., Palmeri D., Schwartz R. A., Lukac D. M. (2003) Microbiol. Mol. Biol. Rev. 67, 175–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sarid R., Flore O., Bohenzky R. A., Chang Y., Moore P. S. (1998) J. Virol. 72, 1005–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sun R., Lin S. F., Staskus K., Gradoville L., Grogan E., Haase A., Miller G. (1999) J. Virol. 73, 2232–2242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhu F. X., Cusano T., Yuan Y. (1999) J. Virol. 73, 5556–5567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sun R., Lin S. F., Gradoville L., Yuan Y., Zhu F., Miller G. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 10866–10871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lukac D. M., Kirshner J. R., Ganem D. (1999) J. Virol. 73, 9348–9361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sakakibara S., Ueda K., Chen J., Okuno T., Yamanishi K. (2001) J. Virol. 75, 6894–6900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gradoville L., Gerlach J., Grogan E., Shedd D., Nikiforow S., Metroka C., Miller G. (2000) J. Virol. 74, 6207–6212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Saveliev A., Zhu F., Yuan Y. (2002) Virology 299, 301–314 [DOI] [PubMed] [Google Scholar]
- 14. Liang Y., Chang J., Lynch S. J., Lukac D. M., Ganem D. (2002) Genes Dev. 16, 1977–1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liang Y., Ganem D. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 8490–8495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Carroll K. D., Bu W., Palmeri D., Spadavecchia S., Lynch S. J., Marras S. A., Tyagi S., Lukac D. M. (2006) J. Virol. 80, 9697–9709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bochtler M., Ditzel L., Groll M., Hartmann C., Huber R. (1999) Annu Rev Biophys. Biomol. Struct. 28, 295–317 [DOI] [PubMed] [Google Scholar]
- 18. Ciechanover A. (1998) EMBO J. 17, 7151–7160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hershko A., Ciechanover A. (1998) Annu. Rev. Biochem. 67, 425–479 [DOI] [PubMed] [Google Scholar]
- 20. Yu Y., Wang S. E., Hayward G. S. (2005) Immunity 22, 59–70 [DOI] [PubMed] [Google Scholar]
- 21. Gould F., Harrison S. M., Hewitt E. W., Whitehouse A. (2009) J. Virol. 83, 6727–6738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yang Z., Yan Z., Wood C. (2008) J. Virol. 82, 3590–3603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kawai T., Akira S. (2007) Semin. Immunol. 19, 24–32 [DOI] [PubMed] [Google Scholar]
- 24. Takeda K., Akira S. (2007) in Current Protocols in Immunology (Coligan J. E. ed) chapter 14 (unit 14):12, John Wiley & Sons, Inc., New York [Google Scholar]
- 25. Samuel C. E. (2001) Clin. Microbiol. Rev. 14, 778–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sen G. C. (2001) Annu. Rev. Microbiol. 55, 255–281 [DOI] [PubMed] [Google Scholar]
- 27. Yamamoto M., Sato S., Hemmi H., Hoshino K., Kaisho T., Sanjo H., Takeuchi O., Sugiyama M., Okabe M., Takeda K., Akira S. (2003) Science 301, 640–643 [DOI] [PubMed] [Google Scholar]
- 28. Yamamoto M., Sato S., Hemmi H., Uematsu S., Hoshino K., Kaisho T., Takeuchi O., Takeda K., Akira S. (2003) Nat. Immunol. 4, 1144–1150 [DOI] [PubMed] [Google Scholar]
- 29. Hoebe K., Du X., Georgel P., Janssen E., Tabeta K., Kim S. O., Goode J., Lin P., Mann N., Mudd S., Crozat K., Sovath S., Han J., Beutler B. (2003) Nature 424, 743–748 [DOI] [PubMed] [Google Scholar]
- 30. Stack J., Haga I. R., Schröder M., Bartlett N. W., Maloney G., Reading P. C., Fitzgerald K. A., Smith G. L., Bowie A. G. (2005) J. Exp. Med. 201, 1007–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Harte M. T., Haga I. R., Maloney G., Gray P., Reading P. C., Bartlett N. W., Smith G. L., Bowie A., O'Neill L. A. (2003) J. Exp. Med. 197, 343–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li K., Foy E., Ferreon J. C., Nakamura M., Ferreon A. C., Ikeda M., Ray S. C., Gale M., Jr., Lemon S. M. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 2992–2997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li X. D., Sun L., Seth R. B., Pineda G., Chen Z. J. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 17717–17722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Krug L. T., Pozharskaya V. P., Yu Y., Inoue N., Offermann M. K. (2004) J. Virol. 78, 8359–8371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Monini P., Carlini F., Stürzl M., Rimessi P., Superti F., Franco M., Melucci-Vigo G., Cafaro A., Goletti D., Sgadari C., Butto' S., Leone P., Chiozzini C., Barresi C., Tinari A., Bonaccorsi A., Capobianchi M. R., Giuliani M., di Carlo A., Andreoni M., Rezza G., Ensoli B. (1999) J. Virol. 73, 4029–4041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pozharskaya V. P., Weakland L. L., Offermann M. K. (2004) J. Gen. Virol. 85, 2779–2787 [DOI] [PubMed] [Google Scholar]
- 37. Lagos D., Vart R. J., Gratrix F., Westrop S. J., Emuss V., Wong P. P., Robey R., Imami N., Bower M., Gotch F., Boshoff C. (2008) Cell Host Microbe 4, 470–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Doyle S., Vaidya S., O'Connell R., Dadgostar H., Dempsey P., Wu T., Rao G., Sun R., Haberland M., Modlin R., Cheng G. (2002) Immunity 17, 251–263 [DOI] [PubMed] [Google Scholar]
- 39. Sanchez D. J., Miranda D., Jr., Arumugaswami V., Hwang S., Singer A. E., Senaati A., Shahangian A., Song M. J., Sun R., Cheng G. (2008) J. Virol. 82, 2208–2217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Perry S. T., Compton T. (2006) J. Virol. 80, 11105–11114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rappocciolo G., Hensler H. R., Jais M., Reinhart T. A., Pegu A., Jenkins F. J., Rinaldo C. R. (2008) J. Virol. 82, 4793–4806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rappocciolo G., Jenkins F. J., Hensler H. R., Piazza P., Jais M., Borowski L., Watkins S. C., Rinaldo C. R., Jr. (2006) J. Immunol. 176, 1741–1749 [DOI] [PubMed] [Google Scholar]
- 43. Wu W., Vieira J., Fiore N., Banerjee P., Sieburg M., Rochford R., Harrington W., Jr., Feuer G. (2006) Blood 108, 141–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cirone M., Lucania G., Bergamo P., Trivedi P., Frati L., Faggioni A. (2007) Immunol. Lett. 113, 40–46 [DOI] [PubMed] [Google Scholar]
- 45. Zhang L., Pagano J. S. (1997) Mol. Cell. Biol. 17, 5748–5757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Honda K., Yanai H., Negishi H., Asagiri M., Sato M., Mizutani T., Shimada N., Ohba Y., Takaoka A., Yoshida N., Taniguchi T. (2005) Nature 434, 772–777 [DOI] [PubMed] [Google Scholar]
- 47. Zhu F. X., King S. M., Smith E. J., Levy D. E., Yuan Y. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 5573–5578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cloutier N., Flamand L. (2010) J. Biol. Chem. 285, 7208–7221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bisson S. A., Page A. L., Ganem D. (2009) J. Virol. 83, 5056–5066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhang J., Wang J., Wood C., Xu D., Zhang L. (2005) J. Virol. 79, 5640–5652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lin R., Yang L., Nakhaei P., Sun Q., Sharif-Askari E., Julkunen I., Hiscott J. (2006) J. Biol. Chem. 281, 2095–2103 [DOI] [PubMed] [Google Scholar]
- 52. Jiang Y., Xu D., Zhao Y., Zhang L. (2008) PLoS ONE 3, e1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhao T., Yang L., Sun Q., Arguello M., Ballard D. W., Hiscott J., Lin R. (2007) Nat. Immunol. 8, 592–600 [DOI] [PubMed] [Google Scholar]
- 54. Grandvaux N., Servant M. J., tenOever B., Sen G. C., Balachandran S., Barber G. N., Lin R., Hiscott J. (2002) J. Virol. 76, 5532–5539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Schoenemeyer A., Barnes B. J., Mancl M. E., Latz E., Goutagny N., Pitha P. M., Fitzgerald K. A., Golenbock D. T. (2005) J. Biol. Chem. 280, 17005–17012 [DOI] [PubMed] [Google Scholar]
- 56. Sarkar S. N., Smith H. L., Rowe T. M., Sen G. C. (2003) J. Biol. Chem. 278, 4393–4396 [DOI] [PubMed] [Google Scholar]
- 57. Zhang L., Pagano J. S. (1999) Mol. Cell. Biol. 19, 3216–3223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhang L., Pagano J. S. (2000) J. Virol. 74, 1061–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhang L., Zhang J., Lambert Q., Der C. J., Del Valle L., Miklossy J., Khalili K., Zhou Y., Pagano J. S. (2004) J. Virol. 78, 12987–12995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhang L. (2000) Mol. Biotechnol. 14, 73–75 [DOI] [PubMed] [Google Scholar]
- 61. Harada H., Kitagawa M., Tanaka N., Yamamoto H., Harada K., Ishihara M., Taniguchi T. (1993) Science 259, 971–974 [DOI] [PubMed] [Google Scholar]
- 62. Papugani A., Coleman T., Jones C., Zhang L. (2008) Virus Res. 131, 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Poole L. J., Yu Y., Kim P. S., Zheng Q. Z., Pevsner J., Hayward G. S. (2002) J. Virol. 76, 3395–3420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhang L., Wu L., Hong K., Pagano J. S. (2001) J. Virol. 75, 12393–12401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Rehwinkel J., Tan C. P., Goubau D., Schulz O., Pichlmair A., Bier K., Robb N., Vreede F., Barclay W., Fodor E., Reis e Sousa C. (2010) Cell 140, 397–408 [DOI] [PubMed] [Google Scholar]
- 66. Sarkar S. N., Peters K. L., Elco C. P., Sakamoto S., Pal S., Sen G. C. (2004) Nat. Struct. Mol. Biol. 11, 1060–1067 [DOI] [PubMed] [Google Scholar]
- 67. Sen G. C., Sarkar S. N. (2005) Cytokine Growth Factor Rev. 16, 1–14 [DOI] [PubMed] [Google Scholar]
- 68. Viswanathan K., Früh K., DeFilippis V. (2010) Curr. Opin. Microbiol. 13, 517–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. McGettrick A. F., O'Neill L. A. (2010) Curr. Opin. Immunol. 22, 20–27 [DOI] [PubMed] [Google Scholar]
- 70. Gregory S. M., West J. A., Dillon P. J., Hilscher C., Dittmer D. P., Damania B. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 11725–11730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. O'Neill L. A., Bowie A. G. (2007) Nat. Rev. Immunol. 7, 353–364 [DOI] [PubMed] [Google Scholar]