

Abstract
Prion diseases are characterized by deposits of abnormal conformers of the PrP protein. Although large aggregates of proteinase K-resistant PrP (PrPres) are infectious, the precise relationships between aggregation state and infectivity remain to be established. In this study, we have fractionated detergent lysates from prion-infected cultured cells by differential ultracentrifugation and ultrafiltration and have characterized a previously unnoticed PrP species. This abnormal form is resistant to proteinase K digestion but, in contrast to typical aggregated PrPres, remains in the soluble fraction at intermediate centrifugal forces and is not retained by filters of 300-kDa cutoff. Cell-based assay and inoculation to animals demonstrate that these entities are infectious. The finding that cell-derived small infectious PrPres aggregates can be recovered in the absence of strong in vitro denaturating treatments now gives a biological basis for investigating the role of small PrP aggregates in the pathogenicity and/or the multiplication cycle of prions.
Keywords: Neurological Diseases, Prions, Protein Conformation, Transgenic, Ultracentrifugation, Aggregates, Cell Model, Soluble Prions
Introduction
Prion diseases are caused by the misfolding of the endogenously expressed PrP protein, and the most widely accepted model of prion multiplication is that abnormally folded PrP recruits and converts the host PrP into new abnormal forms (1–3). Historically, abnormal PrP was operationally defined as insoluble and resistant to protease digestion (4) and was subsequently termed PrPres.4 Although normal PrP protein is soluble in detergents and sensitive to proteinase K (PK) digestion, part of PrP in infected tissues is PK-resistant and recovered in the pellets of centrifuged samples. These large PrPres aggregates are infectious (5), but it is unclear whether infectivity is entirely congruent with these species (6, 7) or whether they are the more efficient species to promote infection. Efficient transmission in the absence of detectable typical PrPres has been observed in some models of prion diseases (8, 9), and in other situations, infectivity and the bulk of large PrPres aggregates have distinct sedimentation profiles in density gradients (10). In addition, large aggregates of PrPres are not the only abnormal forms in diseased tissues, as exemplified by the characterization of PK-sensitive abnormal forms of PrP (11–15). Collectively, these and other findings (16) indicated that abnormal PrP is a set of diverse populations, displaying various biochemical properties and different morphological deposits in the brain, and their biological properties have yet to be established.
Here, we investigated the diversity of infectious abnormal PrP species in a cell line and in neuronal primary cultures infected with a sheep scrapie agent. Although we obtained no evidence for the presence of PK-sensitive infectivity, we identified a previously unnoticed abnormal PrP species. Solubility and ultrafiltration assays indicate that these PK-resistant PrP entities are small aggregates. These species are infectious and may explain the perplexing presence of infectivity in brain samples devoid of detectable large aggregates of PrPres.
EXPERIMENTAL PROCEDURES
Cells and Transgenic Mice
Rov cells (the Rov9 clone) were maintained at 37 °C in 5% CO2 in Opti-MEM (Invitrogen) supplemented with 10% fetal bovine serum, 100 units/ml penicillin, and 10 μg/ml streptomycin (17). To induce the expression of ovine PrP, 1 μg/ml doxycycline (Sigma-Aldrich) was added to the culture medium.
Primary neuronal cultures were established from transgenic mice expressing the ovine PrP (tg338) or the human PrP on a mouse PrP0/0 background, as described (18). Cerebellar granule neurons (CGNs) were extracted from 5- to 7-day-old mice by enzymatic and mechanical dissociation and were plated at a density of 2,500 cells/mm2 in plastic culture wells coated with 10 μg/ml poly-d-lysine (Sigma-Aldrich) at 37 °C and 5% CO2. Cells were cultured in DMEM-GlutaMAX (Invitrogen) containing 10% heat-inactivated fetal bovine serum, 25 mm KCl, penicillin and streptomycin, N2 and B27 supplements (Invitrogen). The medium was complemented weekly with 1 mg/ml glucose and 10 μm antimitotics uridine and fluorodeoxyuridine (Sigma-Aldrich). For immunofluorescence analysis, CGNs were plated on poly-d-lysine-coated glass coverslips and analyzed 13–20 days later. Cultures were prefixed by adding 1 volume of paraformaldehyde (4% w/v in PBS) to the culture medium. Ten minutes later, the cells were subjected to a second round of fixation by adding 4% paraformaldehyde for 10 min. The cells were permeabilized with 0.2% Triton X-100 in PBS and stained for microtubule-associated protein 2 with mAb HM-2 (Sigma-Aldrich) and for β-tubulin III with affinity-purified polyclonal antibodies (Sigma-Aldrich). Bound antibodies were visualized with Alexa Fluor-conjugated secondary antibodies (Molecular Probe), and coverslips were mounted on slides using ProLong Gold antifade reagent (Invitrogen) containing DAPI.
Ovine transgenic mice (tg338 line) are transgenic for the ovine PrP on a mouse PrP0/0 background (19). The mouse line transgenic for the human PrP will be described elsewhere.
Prion Agent and Infection of Cultured Cells
The sheep isolate (PG127) (20) propagated once in sheep was used to infect tg338 mice. At the terminal stage, brains were homogenized at 10% (w/v) in 5% sterile glucose using a high speed homogenizer (TeSeE Precess 48 system), and infectious inoculum was heated at 70 °C for 20 min. Persistently infected Rov cultures were obtained by exposing uninfected cells to 0.5% infectious brain homogenate for a few days. The resulting cultures were grown for 1–2 months and used to prepare cell lysates. Mock-infected Rov cultures were grown and processed in parallel. CGNs (at day 3 after plating) were infected by diluting the 10% infectious homogenate 1,000-fold in cell culture medium.
PK Digestion of Cell Lysates and Brain Extracts
Cell cultures were rinsed with cold PBS and solubilized for 10 min at 4 °C in Triton-DOC lysis buffer (50 mm Tris-HCl (pH 7.4), 0.5% Triton X-100, 0.5% sodium deoxycholate). The lysates were clarified by low speed centrifugation (425 × g, 1 min), and cellular proteins in the postnuclear supernatants were quantified by bicinchoninic acid (Pierce). Digestion with PK (recombinant grade; Roche Applied Science) was performed for 2 h at 37 °C with a mass ratio of 4 μg of PK/mg of cellular proteins, and the reaction was stopped by the addition of Pefabloc (Sigma-Aldrich) to 4 mm. Small volumes of 10% brain homogenates were solubilized in Triton-DOC lysis buffer and processed as cell lysates for detection of PrPres. PK-digested samples were centrifuged for 1 h at 10,000–20,000 × g, and pellets were analyzed by Western blotting.
Search for PK-sensitive Infectivity in Cell Lysates
Lysates (the equivalent of 250 μg of proteins) from infected Rov cultures were incubated for 2 h at 37 °C with 1 μg of PK, and digestion was stopped with Pefabloc. Mock-treated lysates were incubated with Pefabloc and no PK. Proteins were precipitated with 4 volumes of methanol, resuspended in PBS, and used to inoculate Rov cells.
Isolation of Soluble and Insoluble Fractions
Preparative isolation of the soluble and insoluble fractions was carried out with three different infected Rov cultures with similar results. Typically, 35 ml of PK-digested cell lysates (1 mg/ml) were centrifuged at 100,000 × g (gav) for 1 h at 4 °C in polyallomer tubes in a SW32 rotor (Beckman). The supernatant was removed, and 4 volumes of methanol were added to precipitate proteins. The insoluble pellet was dried at room temperature, transferred with 400 μl of PBS into a microtube, and stored at −20 °C. The methanol-precipitated supernatant fraction was centrifuged for 30 min at 10,000 × g in 50-ml tubes, and pelleted material was air-died and resuspended in 400 μl of PBS and transferred into a microtube. Pellets from both fractions were sonicated for a total of 6 min at power 9 with a Cup-Horn apparatus to help dissolution and stored at −20 °C until used for immunoblotting, guanidine denaturation assays, cell-based assay, and bioassay in mice. In some experiments, the supernatant obtained after a 1-h centrifugation at 100,000 × g was transferred to a new clean tube and subjected to a second centrifugation at 100,000 × g.
In some experiments, analytical PK-digested samples were centrifuged in 1.5-ml microtubes either in a TLA-110 rotor (Beckman) or in a microcentrifuge. Rotors with different sedimentation pathlengths have distinct efficiencies to pellet a given particle, and these can be compared using their k factors. At 100,000 × g, k factors of SW32 and TLA-110 rotors are 262 and 50, respectively, indicating that the TLA-110 is about 5-fold more efficient than SW32 to pellet a given particle. Therefore, centrifugation at 20,000 × g or 100,000 × g with TLA-110 corresponds to 100,000 × g or 500,000 × g, respectively, with the SW32 rotor. Pellets and methanol-precipitated supernatants were resuspended in small volumes of PBS without sonication and analyzed by immunoblotting and in the cell-based assay.
Guanidine Denaturation Assays
Insoluble and soluble fractions in PBS were mixed with a noninfected cell lysate to achieve a final protein concentration of 870 μg/ml. Aliquots (30 μl) were mixed with the same volume of H2O or GdnHCl to achieve GdnHCl final concentrations of 1, 1.5, and 2 m and were incubated for 1 h at room temperature. After methanol precipitation, the samples were digested with 0.1 μg of PK for 2 h at 37 °C and analyzed by immunoblotting.
Immunoblotting
Samples were separated by 12% SDS-PAGE before transfer to PVDF membranes (Bio-Rad). The Western blots were stained for PrP with Sha31 mAb (21). Filters were developed using an ECL Plus reagent kit (Amersham Biosciences-GE Healthcare) and visualized with a Bio-Rad VersaDoc imaging system. The signals were quantified with Quantity One software (Bio-Rad).
Ultrafiltration of Cell Lysates
The total and the soluble fractions of PK-digested infected Rov cell lysates were ultrafiltrated as follows. We used filters with cutoffs of 0.2 μm, 1,000 kDa, 300 kDa (Vivaspin 500; Sartorius), and 100-kDa (Microcon; Millipore). Before use, the filters were rinsed with the lysis buffer to remove the preservatives. For each filter, 350 μg of PK-digested lysates (1 mg/ml) were loaded. The ultrafiltration was performed at 5,000 rpm, at room temperature for 15 min in a microcentrifuge. The volumes of the collected filtrates were the same as the loaded volumes. The retentates were recovered by the addition of 350 μl of the lysis buffer. The filtrates and retentates were either methanol-precipitated or subjected to centrifugation (10,000 × g, 1 h) to generate soluble and insoluble fractions.
Immunoprecipitation of PrPres
Purified βS36 antibody (22) and BSA were coupled to Dynabeads M-270 (Invitrogen) following the manufacturer's protocol. Lysate from infected Rov cells (1 mg/ml) was PK-digested and centrifuged for 1 h at 100,000 × g in the SW32 rotor. Supernatant (1.1 ml) was incubated with 5.5 mg of βS36- or BSA-coupled Dynabeads for 2 h, at room temperature. A fraction of the supernatant (100 μl) and the equivalent amount of magnetic beads were analyzed by Western blotting. The remainder (1 ml) was methanol-precipitated, resuspended with sonication in 300 μl of PBS, and used in the cell-based assay.
Cell-based Assay and Bioassay of the Soluble and Insoluble Fractions
Uninfected Rov cells were seeded in 6-well plates in the presence of doxycycline. When confluent, the cultures were exposed to 3 ml of culture medium containing 2, 8, or 32 μl of the insoluble or soluble preparative fractions. In parallel, 32 μl of each fraction was used to inoculate Rov cells in the absence of doxycycline. For cell-based assay of immunodepleted supernatants, from 30 to 300 μl were tested. Infection was allowed to proceed for 3–4 weeks at the end of which the cultures were solubilized. In each assay, 3 ml of culture medium containing serial 10-fold dilutions of the PG127 10% infectious brain homogenate was inoculated in parallel to Rov cells. Cell lysates (500 μg of proteins) were PK-digested and centrifuged at 10,000 × g for 1 h, and pellets were analyzed for insoluble PrPres by Western blotting.
Insoluble and soluble preparative fractions (4 μl each, corresponding to 350 μg of cellular proteins digested with PK) were diluted to 200 μl with 10% brain homogenate from a healthy sheep, and 20 μl was inoculated intracerebrally into six tg338 mice. Mice were then clinically monitored until the occurrence of scrapie clinical signs, at which time they were euthanized.
RESULTS
Detection of Infectious PrPres in the 100,000 × g Supernatants of PK-digested Cell Lysates
The sheep prion agent used here was selected because it multiplies stably, efficiently, and apparently faithfully in previously described Rov and MovS cell lines (23) and in cultured primary neurons (18). In addition, infectivity levels can be quantified either through bioassay in tg338 transgenic mice expressing the ovine PrP protein (20) or through a cell-based assay.
Previous studies reported the presence of abnormal, yet PK-sensitive, forms of PrP in infected tissues and cultured cells from various species (11–15). However, it is not known whether these particular abnormal PrPs are infectious. To clarify this issue in our model, chronically infected Rov cultures were solubilized in nondenaturing detergents. Cell lysates were subjected to PK digestion (2 h at 37 °C, with 4 μg of PK/mg of cellular proteins) or left untreated, and the samples were precipitated with methanol. Infectivity in the resuspended materials was quantified through a Rov cell-based assay. In this assay, Rov cultures inoculated with increasing infectivity accumulate dose-dependent amounts of PrPres (Fig. 1A). Uninfected Rov cells were exposed to mock- and PK-digested infected Rov extracts, and the inoculated cultures were analyzed 3 weeks later for the presence of PrPres. We found that Rov cells inoculated with mock- or PK-digested samples had accumulated similar amounts of PrPres, indicating that infectivity in the two inocula was similar (PrPres amounts in cultures inoculated with mock-treated lysates was 19.5% ± 22% (mean ± S.E., n = 6) above those in cultures inoculated with PK-treated lysates; see a representative Western blot analysis in Fig. 1B). We thus concluded that the vast majority of infectivity propagated in Rov cells is resistant to PK digestion and that PK-sensitive infectivity, if any, is marginal.
FIGURE 1.
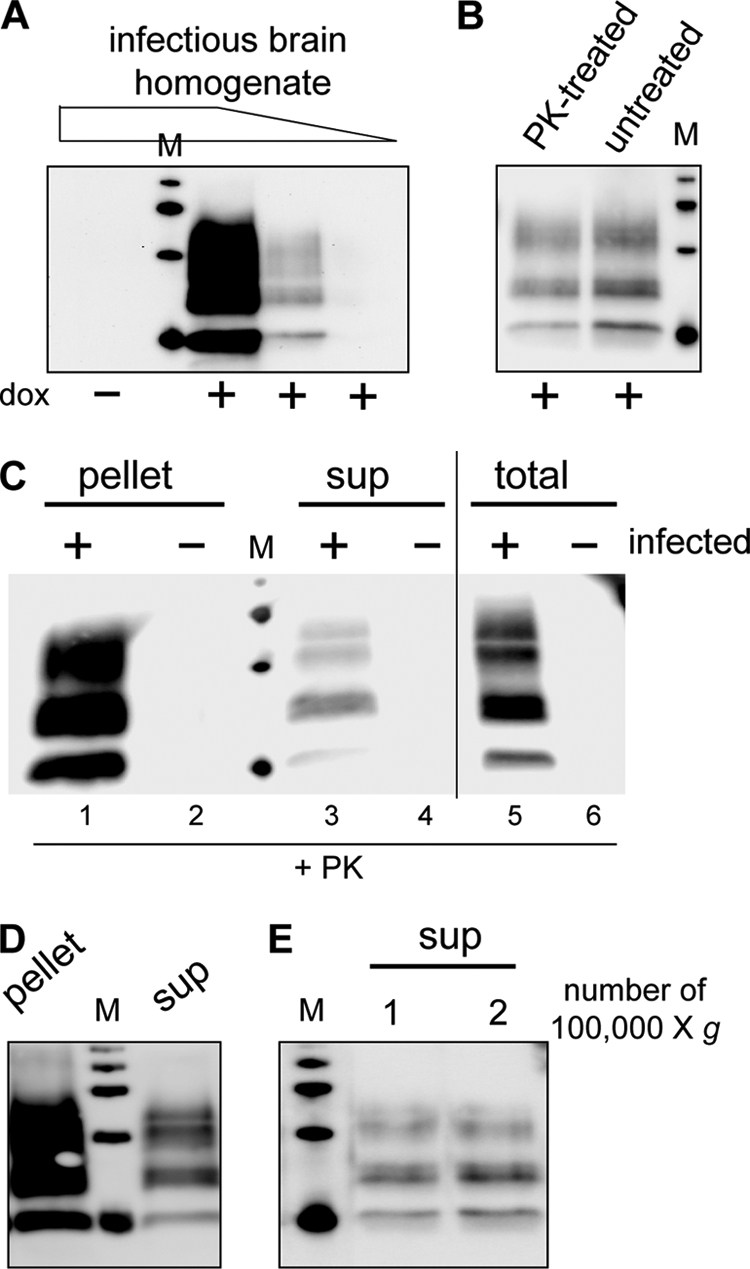
PrPres species is present in the soluble 100,000 × g fraction of infected Rov cells. A, Rov cell-based assay is shown. Uninfected Rov cells were inoculated with three 10-fold dilutions of an infectious brain homogenate. Three weeks later, the inoculated cultures were detergent-extracted and digested with PK, and PrPres was analyzed by immunoblotting. Please note that no PrPres was detected when inoculated Rov cultures did not express ovine PrP (dox−) and that the amounts of accumulated PrPres are dependent on the amount of infectivity used to inoculate the cells. M are standard molecular mass marker proteins (20, 30, 40, and 50 kDa). B, cell-based assay indicates that infectivity propagated in Rov cells is resistant to PK digestion. Infectious Rov cell extracts either PK-treated or left untreated were used to inoculate uninfected Rov cells. Three weeks later, accumulated PrPres in the inoculated cultures was analyzed by immunoblotting. C, lysates (250 μg of proteins) from mock-infected (−) or infected (+) Rov cultures were digested with PK and either methanol-precipitated (lanes 5 and 6) or centrifuged at 10,000 × g for 1 h to obtain pellet and supernatant (sup) fractions (lanes 1-4). All the samples were analyzed for the presence of PrPres by immunoblotting. D, PK-digested lysates from infected Rov cells were centrifuged for 1 h at 100,000 × g (SW32 rotor). Equivalent amounts of the pellet or the supernatant (sup) fractions were analyzed for PrPres. E, PrPres in the supernatant fractions after one and two successive centrifugations at 100,000 × g was analyzed. 71% of PrPres remained in the supernatant after a second round of ultracentrifugation (n = 2).
Fig. 1C illustrates the PrPres species recovered in nondenaturing detergent lysates of chronically infected Rov cells. Immunoblot analysis of methanol-precipitated, PK-digested samples reveals the presence of PrPres in lysates from infected cultures (lane 5). We reasoned that small PrPres aggregates, if present, might remain in the supernatant after centrifugation of PK-digested lysates. PK-digested lysates were subjected to a standard centrifugation step (e.g. 10,000–20,000 × g for 1 h in a microcentrifuge). Western blot analysis of pellets revealed the presence of PrPres (lane 1), consistent with the known insolubility of large PrPres aggregates. Strikingly, a PrPres signal was consistently observed when material from methanol-precipitated supernatant was analyzed (lane 3). No PrPres was observed in either fraction when uninfected cultures were analyzed (lanes 2, 4, and 6).
To characterize the PrPres species in the supernatant and pellet fractions, large volumes of infected cell lysates were digested with PK and ultracentrifuged at 100,000 × g for 1 h in a SW32 rotor to generate soluble (supernatant) and insoluble (pellet) fractions. Soluble material was recovered by methanol precipitation and resuspended in PBS by sonication along with the original insoluble pellet. Immunoblot analysis of the supernatant and pellet fractions showed that PrPres was still present in 100,000 × g supernatants (Fig. 1D). Importantly, this PrPres species quantitatively remained in the soluble fraction when the supernatant was submitted to a second 100,000 × g centrifugation step (Fig. 1E). Quantification through Western blot analysis of various amounts of the two fractions indicated that PrPres in the supernatant represents 12% ± 8% of total PrPres (mean ± S.D., four determinations from three different infected cultures). To test for the presence of infectivity in the soluble fraction, we first used the Rov cell-based assay. Uninfected Rov cells were exposed to increasing equivalent amounts of the soluble and insoluble fractions and analyzed 3 weeks later for the presence of PrPres. Although no PrPres was observed in control cells not expressing ovine PrP, the dose-dependent presence of PrPres in Rov cells challenged with the 100,000 × g supernatant demonstrated that the supernatant was infectious (Fig. 2A). After quantification of PrPres levels in the cultures exposed to the different amounts of the soluble and insoluble fractions, we estimated that 18.5% ± 5% of infectivity was soluble (mean ± S.D., four determinations from three experiments). To investigate the physical support for infectivity, we sought to deplete PrPres from the infectious supernatants by immunoprecipitation with βS36, a monoclonal antibody that specifically immunoprecipitates PrPres aggregates (22). βS36- but not BSA-coupled magnetic beads removed most (85%, n = 2) of the PrPres in the 100,000 × g supernatants (Fig. 2B). Strikingly, ∼90% (n = 2) of infectivity was also removed from the immunodepleted supernatants, as assessed by Rov cell-based assay (Fig. 2C). Soluble and insoluble fractions were also inoculated into tg338 transgenic mice highly susceptible to the ovine prion strain used in this study (19). All of the inoculated mice died with typical neurological symptoms and accumulated abnormal PrP in their brains (data not shown). Mice inoculated with the pellet and supernatant fractions died 74 days ± 4 days (n = 5) and 73 days ± 4 days (n = 4) after inoculation, respectively. Mice inoculated with fractions from uninfected cultures remained clinically healthy and were killed at 110 days. These similar incubation periods correspond to infectious titers within the same order of magnitude and confirm that the 100,000 × g supernatants are infectious. Altogether, our data show that PK-resistant PrP recovered in a detergent-soluble fraction is infectious.
FIGURE 2.

PrPres from the soluble fraction is infectious in the Rov cell-based assay. A, pellet and supernatant (sup) fractions were recovered after 100,000 × g centrifugation of PK-digested lysates from infected Rov cells. Equivalent portions (from 1 to 16, 1 corresponds to 175 μg of proteins in the original lysate) of each fraction were used to infect target Rov cultures. Three weeks later the cultures were solubilized and analyzed for the presence of PrPres. No PrPres was detected when inoculated Rov cultures did not express ovine PrP (dox−). B, PrPres was depleted from the supernatant fraction. PrPres remaining in the supernatant fraction (sup) after immunoprecipitation with βS36 mAb- or BSA-coupled magnetic beads was analyzed. Immunoprecipitated PrPres was also analyzed in parallel (beads). The anti-PrP mAb used to reveal the blot was coupled with peroxidase. C, Rov cells were infected with immunodepleted supernatants. Various amounts (from 1/10 to 1/1) of either βS36-depleted supernatants (supβS36) or control BSA-depleted supernatants (supBSA) were used to infect target Rov cells. Four weeks later, the cultures were solubilized and analyzed for the presence of PrPres. No PrPres was detected when inoculated Rov cultures did not express ovine PrP (dox−).
PrPres Is Recovered in the Detergent-soluble Fraction of Infected Neurons and Prior to PK Digestion
We then asked whether PrPres in the soluble fraction could be a by-product generated during PK digestion of insoluble PrPres. To explore this possibility, analytical volumes (<1 ml) of lysates were centrifuged before PK digestion. The soluble fractions were then PK-digested and analyzed by Western blotting. Fig. 3A shows that the PrPres species can also be detected in the supernatants of non-PK-digested cell lysates, indicating that PK digestion is not required to generate this PrPres species. To examine whether PrPres species in the soluble and insoluble fractions differ in their PK resistance, cell lysates from infected cells were digested with increasing amounts of PK before isolation of the soluble and insoluble fractions. Immunoblot analysis (Fig. 3B) revealed that PrPres in the supernatant is as PK-resistant as insoluble PrPres. To study the structural stability of PrPres in the soluble and insoluble fractions, we performed guanidine denaturation assays. Both fractions were incubated with increasing amounts of GdnHCl and then digested with PK. Immunoblot analysis indicated that PrPres species in the soluble and insoluble fractions have similar stabilities to denaturation by GdnHCl (Fig. 3C).
FIGURE 3.

PrPres from supernatant is not generated through PK digestion of insoluble PrPres, and both species have similar resistance to PK digestion and GdnHCl denaturation. A, supernatant fractions were recovered after a 1-h centrifugation at 10,000 × g before (lane 1) or after (lane 2) PK digestion of cell lysates. Supernatant from the undigested lysate was PK-treated, and both samples were analyzed for PrPres by Western blotting. B, cell lysates were digested with increasing amounts of PK (1 stands for 1 μg of PK/250 μg of cellular proteins) and centrifuged at 10,000 × g for 1 h. Pellets and supernatants (sup) were tested for PrPres by Western blotting. C, pellet and supernatant (sup) fractions were incubated in the presence of the indicated concentrations of GdnHCl for 1 h. The samples were then digested with PK and analyzed for PrPres by Western blotting.
The ovine prion agent used here can also be propagated in tg338-derived neuronal primary cultures (18). Cultured CGNs from either tg338 (CGNov) (Fig. 4A) or transgenic mice expressing a heterologous PrP (human PrP, CGNhu, used as nonpermissive, negative controls) were infected 3 days after plating. The cells were solubilized and analyzed 7, 14, or 21 days after infection. A marked, time-dependent accumulation of PrPres was observed in CGNov, demonstrating active prion multiplication in the primary neuronal cultures derived from tg338 mice (Fig. 4B). Analysis of supernatant and pellet fractions from PK-digested CGNov lysates revealed that 12.5% ± 4% of the total PrPres was in the soluble fraction (mean ± S.D. of five determinations from two different infected CGNov cultures) (Fig. 4C). Lastly, brain homogenates from tg338 mice, either healthy or at the terminal stage, were solubilized in the Triton-DOC lysis buffer, subjected to PK digestion, and centrifuged. Analysis of the supernatant and pellet fractions showed that PrPres was also present in the soluble fraction of infected brain tissue (Fig. 4D).
FIGURE 4.

PrPres is present in detergent-soluble fractions from infected cultures of CGNs and from infected brain tissue. A, fluorescence analysis of tg338-derived cerebellar granule neurons (CGNov) stained for microtubule-associated protein 2 (green, right) and β-tubulin III (red, left) after 13 and 20 days in vitro, respectively. Nuclei are in blue. B, kinetics of PrPres accumulation in infected CGNov cultures. Proteins (75 μg) from cell lysates obtained at 7, 14, and 21 days postinfection (pi) were PK-digested and centrifuged at 10,000 × g for 1 h. The pellets were analyzed to detect PrPres. C, detection of PrPres in the soluble fraction of PK-digested CGNov lysates. Proteins (75 μg) from infected CGNov cultures (21 days postinfection) were PK-digested and centrifuged at 10,000 × g for 1 h. PrPres in the supernatant (sup) and in various portions of the pellet were analyzed by Western blotting. D, brain tissues from terminally sick or healthy tg338 mice. Tissues were solubilized, and 50 μg of brain proteins were PK-digested and centrifuged at 10,000 × g for 1 h. PrPres in the pellet and the supernatant (sup) fractions was analyzed.
Infectious PrPres in the Detergent-soluble Fraction Are Small Aggregates
As a first step to investigate the reasons for apparent solubility of PrPres (small size or low density), we tested whether PrPres in the soluble fraction would be pelleted if centrifuged at a higher speed. To apply higher centrifugation forces, we used a TLA-110 rotor (Beckmann). Because its k factor is about 5-fold less than that of SW32, TLA-110 is about 5-fold more efficient to pellet a given particle (see “Experimental Procedures”), e.g. 20,000 × g supernatants in the TLA-110 are equivalent to 100,000 × g supernatants in SW32. Although normal PrP from uninfected cells remained in the supernatants (Fig. 5A, right panel), data in Fig. 5A, left panel, show that the vast majority (∼95%, n = 2) of the PrPres in the soluble fraction is pelleted when g force was increased to 100,000 × g in the TLA-110 rotor (equivalent to 500,000 × g with SW32). We also performed ultracentrifugation of the soluble fraction through a 10% sucrose cushion. As expected for small aggregates, PrPres did not float in the sucrose and were found in the pellet (Fig. 5B). Cell-based assays showed that infectivity in the 100,000 × g supernatants (Sup100K) was 10-fold lower to that in Sup20K (data not shown). Collectively, these data indicate that infectious PrPres species in the SW32 100,000 × g supernatants are not fully soluble.
FIGURE 5.

PrPres from supernatants is not fully soluble. A, left, PK-digested lysates from infected Rov cells were centrifuged in a TLA-110 rotor at 20,000 or 100,000 × g for 1 h. Various portions of the corresponding supernatants (Sup20K or Sup100K, respectively) were analyzed for PrPres by Western blotting. Right, undigested lysates from uninfected Rov cells were analyzed for normal PrP before (−) or after (+) 100,000 × g centrifugation. Please note that these ultracentrifugation conditions correspond to 100,000 × g or 500,000 × g with the SW32 rotor used previously (see “Experimental Procedures”). B, soluble fraction of a PK-digested cell lysate was ultracentrifuged for 1 h at 200,000 × g in a TLA-110 rotor either directly (−) or through a 10% (w/v) sucrose cushion (+). The pellets were analyzed for PrPres by Western blotting.
We then subjected PK-digested cell lysates to ultrafiltration, a convenient alternative to sucrose gradient for separating large PrPres aggregates from other filterable molecules (24, 25). PrPres species in the retained (retentates) and nonretained (filtrates) fractions were analyzed by immunoblotting. Data in Fig. 6A show that although most of PrPres was retained, approximately 10% of total PrPres passed through 300-kDa but not through 100-kDa filters. Before ultrafiltration, insoluble PrPres is the major (∼90%, Fig. 1) PK-resistant species. This was dramatically different in the filtrates from 1,000-kDa and 300-kDa membranes (Fig. 6B), indicating that PrPres in the soluble fraction passed much more readily through these filters than did insoluble PrPres. To confirm that the former quantitatively crosses filters of low molecular mass cutoff, PrPres from the soluble fraction was isolated by centrifugation and then submitted to ultrafiltration through a range of different filters. As expected, PrPres was not retained by 1,000- and 300-kDa filters as it was exclusively detected in the corresponding filtrates (Fig. 6C). In contrast, 100-kDa pore size fully retained PrPres. The above data indicate that the infectious PrPres recovered in the detergent-soluble fraction are small aggregates.
FIGURE 6.

PrPres from soluble fractions passes through 300-kDa filters. A, analysis of PK-digested lysates before (total) or after ultrafiltration through 300-kDa or 100-kDa filters. Retentates (R; 1/10 and 9/10 were loaded) and the whole filtrates (F) were analyzed for PrPres by immunoblotting. B, filtrate fractions obtained after ultrafiltration through 1,000-kDa and 300-kDa filters of PK-digested lysates. Fractions were centrifuged to separate the insoluble (pellet) and soluble (sup) materials. Insoluble (1/10) and the whole soluble fractions from the filtrates were analyzed for PrPres. C, supernatants of centrifuged PK-digested lysates ultrafiltrated through the indicated filters. PrPres in the retentates (R) and the filtrates (F) are shown.
DISCUSSION
After longstanding controversies, there is now a wide agreement that prion infectivity is associated with abnormally folded PrP. Historically, abnormal PrP has been operationally defined as PK-resistant and insoluble (26), and a wealth of studies indicates that fractions containing large insoluble PrPres aggregates are infectious. However, it does not imply that infectivity is exclusively associated with large aggregates of PrPres or that they are the more efficient species to promote infection. A number of studies reported successful disaggregation of at least part of prion infectivity by chemical and/or physical treatments. Early experiments showed that sonication of large PrPres aggregates (known as prion rods) in detergent and phospholipids can solubilize PrPres and infectivity (27). Denaturation by chaotropic salts and subsequent renaturation also led to infectivity recovery in low density fractions after sucrose gradients (28), and disruption of prion rods by sonication in detergents liberated infectivity in 100,000 × g supernatants (25). More recently, small PrP aggregates, isolated after physical disruption of detergent-extracted aggregates of PrPres, were shown to be more infectious than large ones (29), presumably by providing an increased number of multiplication-competent seeds. Although this study added strong support for small PrP aggregates as more active species to initiate infection, firm evidence for the presence of small PrPres aggregates in infected tissues or cells was lacking. Thus, the biological significance of the aforementioned experimental treatments remained an open question. An important outcome of our study is to show that small infectious PrPres aggregates can be recovered from infected cells after a standard nondenaturing solubilization step in the absence of further treatments. Importantly, these small infectious PrPres aggregates were also observed in the absence of PK digestion, indicating that they were not derived from PK proteolysis of the large aggregates. They were conveniently separated from large PrPres aggregates by a solubility assay and/or by ultrafiltration through low molecular weight cutoff filters. Two lines of evidence indicate that the PrPres species in the detergent-soluble 100,000 × g supernatants are small PrP aggregates. First, and in contrast to normal PrP, these small PrPres species are eventually pelleted after ultracentrifugation at higher centrifugal forces. Second, they are immunoprecipitated by βS36, a mAb directed against aggregated PrP from infected tissue and not recognizing normal PrP (22). However, further purification will be required to determine their molecular composition.
The population of small PrPres aggregates, which represents roughly 10% of the total PrPres in infected Rov cells, is also observed in infected primary cultures of neurons and in brain tissue of infected mice, suggesting that it is produced by a wide variety of infected cells. An important issue is whether these small PrPres aggregates are more infectious than the large ones, as shown for small aggregates generated in vitro (29). Studies are now ongoing to address precisely this question. Another interesting point will be to determine whether the formation of small and large PrPres aggregates is due to different metabolic pathways. Alternatively, small aggregates could be metabolic intermediates in the formation of larger deposits. They could also be a cell-mediated catabolic product of large aggregates, contributing to an efficient transmission of infection during cell divisions (2). So far, and in contrast to yeast in which hsp104-dependent fragmentation is required for prion maintenance during successive cell divisions (30), no such activity has been identified for mammalian prions.
The discovery of small infectious PrPres aggregates further documents the diversity of abnormal PrP extracted from infected cells and tissues. Other small PrP aggregates have been detected and isolated in infected cells and tissues (11–15). In contrast to the small infectious PrPres aggregates described here, the previously described small PrP aggregates are sensitive to PK digestion, and no evidence indicates that they can transmit disease (31, 32). Small infectious PrPres aggregates, such as those described here, might give a molecular basis for the perplexing presence of infectivity in soluble preparations of infected hamster brain (33). Also, in a very recent study (10), density gradients were used to fractionate detergent-solubilized brain homogenate from tg338 mice infected with the ovine prion agent (PG127) used here. Strikingly, fractions of low density, well separated from the bulk of large PrPres aggregates in heavier fractions, contained high levels of infectivity. The molecular basis of infectivity in the light fractions, not addressed in Tixador's study, may involve the small PrPres aggregates characterized in our work.
Several neurodegenerative disorders, including Parkinson, Alzheimer, Huntington, and prion diseases, are characterized by the misfolding and the aggregation of specific proteins (34). Although early hypotheses considered the large insoluble aggregates as the only toxic entities, an important emerging view is that small soluble prefibrillar assemblies may be more efficient biological entities to promote cell death (35, 36). The toxic entities responsible for prion-mediated neuronal cell death and the underlying mechanisms are not yet clear (37, 38). The identification of naturally produced small PrPres aggregates in cultured neurons that will eventually undergo infection-dependent apoptosis (18, 39) may be useful to determine the potential neurotoxicity of these entities.
Acknowledgments
We thank J. Grassi (CEA, Saclay, France) for βS36 and Sha31 mAbs, H. Laude and J. L. Vilotte (INRA, Jouy-en-Josas, France) for tg338 mice, J. M. Torres (CISA-INIA, Madrid, Spain) for transgenic mice expressing the human PrP, M. C. Miquel and A. Bertholet for sharing expertise on primary neuronal cultures, L. Goulème for immunofluorescence analysis of CGN cultures, R. Melki (CNRS, Gif-sur-Yvette, France) for helpful initial discussions on ultrafiltration, and V. Beringue and V. Setola for helpful comments on the manuscript.
This work was supported in part by Agence Nationale de la Recherche Grant ANR-08-MIEN-010 (France) and the Department for Environment, Food and Rural Affairs (United Kingdom).
- PrPres
- proteinase-resistant PrP protein
- CGN
- cerebellar granule neuron
- PK
- proteinase K.
REFERENCES
- 1. Prusiner S. B. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 13363–13383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Weissmann C. (2004) Nat. Rev. Microbiol. 2, 861–871 [DOI] [PubMed] [Google Scholar]
- 3. Collinge J., Clarke A. R. (2007) Science 318, 930–936 [DOI] [PubMed] [Google Scholar]
- 4. Bolton D. C., McKinley M. P., Prusiner S. B. (1982) Science 218, 1309–1311 [DOI] [PubMed] [Google Scholar]
- 5. Prusiner S. B. (1992) Biochemistry 31, 12277–12288 [DOI] [PubMed] [Google Scholar]
- 6. Aguzzi A., Sigurdson C., Heikenwaelder M. (2008) Annu. Rev. Pathol. 3, 11–40 [DOI] [PubMed] [Google Scholar]
- 7. Soto C., Castilla J. (2004) Nat. Med. 10, S63–S67 [DOI] [PubMed] [Google Scholar]
- 8. Barron R. M., Campbell S. L., King D., Bellon A., Chapman K. E., Williamson R. A., Manson J. C. (2007) J. Biol. Chem. 282, 35878–35886 [DOI] [PubMed] [Google Scholar]
- 9. Lasmézas C. I., Deslys J. P., Robain O., Jaegly A., Beringue V., Peyrin J. M., Fournier J. G., Hauw J. J., Rossier J., Dormont D. (1997) Science 275, 402–405 [DOI] [PubMed] [Google Scholar]
- 10. Tixador P., Herzog L., Reine F., Jaumain E., Chapuis J., Le Dur A., Laude H., Béringue V. (2010) PLoS Pathog. 6, e1000859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pastrana M. A., Sajnani G., Onisko B., Castilla J., Morales R., Soto C., Requena J. R. (2006) Biochemistry 45, 15710–15717 [DOI] [PubMed] [Google Scholar]
- 12. Safar J., Wille H., Itri V., Groth D., Serban H., Torchia M., Cohen F. E., Prusiner S. B. (1998) Nat. Med. 4, 1157–1165 [DOI] [PubMed] [Google Scholar]
- 13. Safar J. G., Geschwind M. D., Deering C., Didorenko S., Sattavat M., Sanchez H., Serban A., Vey M., Baron H., Giles K., Miller B. L., Dearmond S. J., Prusiner S. B. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 3501–3506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Thackray A. M., Hopkins L., Bujdoso R. (2007) Biochem. J. 401, 475–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tzaban S., Friedlander G., Schonberger O., Horonchik L., Yedidia Y., Shaked G., Gabizon R., Taraboulos A. (2002) Biochemistry 41, 12868–12875 [DOI] [PubMed] [Google Scholar]
- 16. Caughey B., Baron G. S., Chesebro B., Jeffrey M. (2009) Annu. Rev. Biochem. 78, 177–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vilette D., Andreoletti O., Archer F., Madelaine M. F., Vilotte J. L., Lehmann S., Laude H. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 4055–4059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cronier S., Laude H., Peyrin J. M. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 12271–12276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Le Dur A., Béringue V., Andréoletti O., Reine F., Laï T. L., Baron T., Bratberg B., Vilotte J. L., Sarradin P., Benestad S. L., Laude H. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 16031–16036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vilotte J. L., Soulier S., Essalmani R., Stinnakre M. G., Vaiman D., Lepourry L., Da Silva J. C., Besnard N., Dawson M., Buschmann A., Groschup M., Petit S., Madelaine M. F., Rakatobe S., Le Dur A., Vilette D., Laude H. (2001) J. Virol. 75, 5977–5984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Féraudet C., Morel N., Simon S., Volland H., Frobert Y., Créminon C., Vilette D., Lehmann S., Grassi J. (2005) J. Biol. Chem. 280, 11247–11258 [DOI] [PubMed] [Google Scholar]
- 22. Morel N., Simon S., Frobert Y., Volland H., Mourton-Gilles C., Negro A., Sorgato M. C., Créminon C., Grassi J. (2004) J. Biol. Chem. 279, 30143–30149 [DOI] [PubMed] [Google Scholar]
- 23. Vilette D. (2008) Vet. Res. 39, 10. [DOI] [PubMed] [Google Scholar]
- 24. Hecker R., Taraboulos A., Scott M., Pan K. M., Yang S. L., Torchia M., Jendroska K., DeArmond S. J., Prusiner S. B. (1992) Genes Dev. 6, 1213–1228 [DOI] [PubMed] [Google Scholar]
- 25. Riesner D., Kellings K., Post K., Wille H., Serban H., Groth D., Baldwin M. A., Prusiner S. B. (1996) J. Virol. 70, 1714–1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Prusiner S. B. (1982) Science 216, 136–144 [DOI] [PubMed] [Google Scholar]
- 27. Gabizon R., McKinley M. P., Prusiner S. B. (1987) Proc. Natl. Acad. Sci. U.S.A. 84, 4017–4021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Manousis T., Verghese-Nikolakaki S., Keyes P., Sachsamanoglou M., Dawson M., Papadopoulos O., Sklaviadis T. K. (2000) J. Gen. Virol. 81, 1615–1620 [DOI] [PubMed] [Google Scholar]
- 29. Silveira J. R., Raymond G. J., Hughson A. G., Race R. E., Sim V. L., Hayes S. F., Caughey B. (2005) Nature 437, 257–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Romanova N. V., Chernoff Y. O. (2009) Protein Pept. Lett. 16, 598–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cronier S., Gros N., Tattum M. H., Jackson G. S., Clarke A. R., Collinge J., Wadsworth J. D. (2008) Biochem. J. 416, 297–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Deleault A. M., Deleault N. R., Harris B. T., Rees J. R., Supattapone S. (2008) J. Gen. Virol. 89, 2642–2650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Berardi V. A., Cardone F., Valanzano A., Lu M., Pocchiari M. (2006) Transfusion 46, 652–658 [DOI] [PubMed] [Google Scholar]
- 34. Selkoe D. J. (2003) Nature 426, 900–904 [DOI] [PubMed] [Google Scholar]
- 35. Caughey B., Lansbury P. T. (2003) Annu. Rev. Neurosci. 26, 267–298 [DOI] [PubMed] [Google Scholar]
- 36. Haass C., Selkoe D. J. (2007) Nat. Rev. Mol. Cell Biol. 8, 101–112 [DOI] [PubMed] [Google Scholar]
- 37. Harrison C. F., Barnham K. J., Hill A. F. (2007) J. Neurochem. 103, 1709–1720 [DOI] [PubMed] [Google Scholar]
- 38. Soto C. (2008) Dev. Cell 15, 339–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Carimalo J., Cronier S., Petit G., Peyrin J. M., Boukhtouche F., Arbez N., Lemaigre-Dubreuil Y., Brugg B., Miquel M. C. (2005) Eur. J. Neurosci. 21, 2311–2319 [DOI] [PubMed] [Google Scholar]