

Abstract
Cysteinyl leukotrienes and oxidative stress have both been implicated in bronchial asthma; however, there is no previous study that focused on the ability of oxidative stress to alter cysteinyl leukotriene generation. In this study, treatment of bone marrow-derived mast cells with prostaglandin D2 reduced their ability to generate leukotriene (LT) C4 upon calcium ionophore stimulation but had little effect on LTB4 generation. This effect could be reproduced by a selective agonist of the DP2 receptor, 15R-methyl prostaglandin D2 (15R-D2). 15R-D2 dose-dependently inhibited LTC4 generation with an IC50 of 2 μm, and the effect was not altered by a DP2/thromboxane antagonist or by a peroxisome proliferator-activated receptor-γ antagonist. 15R-D2 exerted its suppressive effect via a reduction in intracellular GSH, a mechanism that involved the conjugation of its non-enzymatic breakdown product to GSH. At 10 μm, 15R-D2 reduced LTC4 generation to 10%, intracellular GSH to 50%, and LTC4 synthase (LTC4S) activity to 33.5% of untreated cells without altering immunoreactive LTC4S protein expression or 5-lipoxygenase activity. The effects of 15R-D2 on LTC4S activity could be partially reversed by reducing reagent. The sulfhydryl-reactive oxidative agent diamide suppressed LTC4S activity and induced a reversible formation of covalent dimer LTC4S. LTC4S bearing a C56S mutation was resistant to the effect of diamide. Covalent dimer LTC4S was observed in nasal polyp biopsies, indicating that dimerization and inactivation of LTC4S can occur at the site of inflammation. These results suggest a cellular redox regulation of LTC4S function through a post-translational mechanism.
Keywords: Eicosanoid, Eicosanoid Biosynthesis, Enzyme Inactivation, Leukotriene, Oxidative Stress
Introduction
The cysteinyl leukotrienes (LT)2 LTC4, LTD4, and LTE4 are potent inflammatory lipid mediators. They are implicated in asthma (1–3) and allergic inflammation (4, 5). The biological actions of the cysteinyl leukotrienes as mediators of human asthma include contraction of bronchi (2), stimulation of mucous secretion from human airways in vitro (6), and induction of eosinophil infiltration into the airway of asthmatic individuals in vivo (7, 8). Biosynthesis of LTC4 occurs in limited numbers of effector cells, such as mast cell, basophils, eosinophils, and macrophages that express the 5-lipoxygenase (5-LO) pathway of enzymes as well as LTC4 synthase (LTC4S). In mast cells, LTC4 biosynthesis is initiated upon antigen cross-linking of the high affinity IgE receptors, which leads to influx of Ca2+ ion. An increase in intracellular Ca2+ activates calcium-dependent cytosolic phospholipase A2, which subsequently translocates to the perinuclear membrane and hydrolyzes the release of arachidonic acid from the membrane phospholipids (9). The released arachidonic acid interacts with the 5-LO-activating protein FLAP, an integral nuclear membrane protein (10), during presentation to 5-LO, which also translocates to the perinuclear membrane from both the cytosol and the nucleoplasm (11, 12). 5-LO converts arachidonic acid to 5-hydroperoxyeicosatetraenoic acid and subsequently to LTA4 (13). LTA4 either is hydrolyzed by cytosolic LTA4 hydrolase to form LTB4 (14) or is conjugated to GSH to form LTC4, the parent compound of cysteinyl leukotrienes (15, 16). This conjugation is the function of LTC4S (17).
LTC4S is an 18-kDa integral membrane protein located at the nuclear envelope of LTC4-generating cells. Cloning of the human and mouse LTC4S cDNAs revealed the highest amino acid identity to FLAP (17) and to bifunctional microsomal GST2 and GST3 (18). These proteins are all members of a superfamily of membrane-associated proteins involved in eicosanoid and glutathione metabolism (MAPEG) (18), which also includes microsomal PGE2 synthase-1 and microsomal GST1. LTC4 conjugates GSH to LTA4 to form LTC4, and in contrast to microsomal GST2 and GST3, LTC4S does not conjugate GSH to xenobiotics. Recently, it was demonstrated that LTC4S can also conjugate GSH to 5-oxohydroxyeicosatetraenoic acid to form FOG-7 (19). X-ray crystallography of human LTC4S with GSH shows that the LTC4S monomer has four transmembrane α-helices and forms a 3-fold symmetric trimer as a unit with functional domains across each interface with GSH resides in a U-shaped conformation within an interface between adjacent monomers (20, 21). This binding is stabilized by a loop structure at the top of the interface. X-ray crystallographic data suggest that LTA4 would fit into the interface so that Arg-104 of one monomer activates GSH to provide the thiolate anion that attacks C6 of LTA4 to form a thioether bond, and Arg-31 of the neighboring monomer would donate a proton to form a hydroxyl group at C5, resulting in the formation of 5S-hydroxy-6R,S-glutathionyl-7,9-trans-11,14-cis-eicosatetraenoic acid (LTC4). In addition, X-crystallography shows that Arg-51, Asn-55, Glu-58, Tyr-93, Tyr-97, and Arg-104 of LTC4S are involved in GSH binding.
Recently, studies on prostaglandin biosynthesis have demonstrated that nitric oxide S-nitrosylates cytosolic phospholipase A2 (22) and COX2 (23), resulting in the activation of these two enzymes and an increase in prostaglandin synthesis. These results suggest that oxidative stress is able to induce post-translational modification of enzymes associated with eicosanoid biosynthesis. Although a decrease in intracellular GSH upon 1-chloro-2,4-dinitrobenzene (CDNB) treatment of human polymorphonuclear leukocytes has been shown to increase 5-LO activity (24) and to modify 5-LO activity by ERK2 and PKA phosphorylation (25, 26) and 5-LO translocation by MAPKAP2 phosphorylation (27), there is no information on the effect of oxidative stress on the post-translational modification of 5-LO or its pathway of enzymes. In this study, we examined the regulation of LTC4 biosynthesis by oxidative stress. On the basis of preliminary experiments indicating that prostaglandin (PG) D2, the major cyclooxygenase product of mast cells, could suppress LTC4 generation when applied exogenously to mouse bone marrow-derived mast cells (BMMCs), we unexpectedly recognized that PGD2 and the DP2 receptor agonist 15R-methyl-PGD2 (15R-D2) suppressed LTC4 generation by BMMCs through a reduction in intracellular GSH and suppression of LTC4S activity. In addition, diamide, a sulfhydryl-reactive oxidant, induced oxidative stress, suppressed LTC4S activity, and induced the formation of covalent dimer LTC4S, normally a noncovalent trimer. Importantly, the effect of oxidative stress was abolished in C56S mutant LTC4S, and the covalent dimer was present in nasal polyps of asthmatics, implying that LTC4S activity levels may be modified in vivo by oxidation-related dimerization.
EXPERIMENTAL PROCEDURES
Materials
15-Deoxy-Δ12,14-PGD2 (15d-D2), 15-deoxy-Δ12,14-PGJ2 (15d-J2), Δ12-PGJ2, 15R-D2, PGB2, PGD2, PGE2, PGF2α, GW 9662, Ramatroban, LTA4 methyl ester, and BW 245C were from Cayman Chemical (Ann Arbor, MI). Ionophore A23187 was from Calbiochem. FuGENE 6 transfection reagent was from Roche Applied Science. Horseradish peroxidase-conjugated anti-rabbit immunoglobulin was from Bio-Rad. Chemiluminescence substrate was from Pierce. Bovine serum albumin, diamide, CDNB, GSH, ionomycin, human GST, and buthionine sulfoximine (BSO) were from Sigma.
Culture of BMMCs
Bone marrow cells were collected from femurs and tibiae of BALB/c mice and cultured for 6–10 weeks in RPMI 1640 medium containing 10% fetal bovine serum, 2 mm l-glutamine, 0.1 mm nonessential amino acids, 100 units/ml penicillin, 100 μg/ml streptomycin, and 1% culture supernatant from CHO cells expressing mouse interleukin-3. The culture medium for the BMMCs was changed every week, and the cell density was adjusted to 5 × 105/ml at every passage. After 4 weeks, >97% of the cells were BMMCs as assessed by staining with toluidine blue.
Ionophore-stimulated 5-LO Product Generation
BMMCs were first treated with PGD2, 15R-D2, or vehicle (ethanol) for up to 48 h. Cells were then harvested, washed, and suspended in Hanks' buffer containing calcium, magnesium, and 2 mg/ml bovine serum albumin (buffer A). Stimulation of BMMCs were initiated by the addition of ionophore (2 μm), followed by incubation at 37 °C for 15 min. Reactions were terminated by the addition of 3 volumes of methanol containing 400 ng/ml PGB2. Samples were analyzed for 5-LO product formation by reverse-phase (RP) HPLC with a Beckman System Gold Model 126 pump and Model 168 diode array UV detector using a programmed gradient and solvent system as described previously (28) and are expressed as picomoles/106 cells.
Enzyme Assay
To assay for LTC4S activity, BMMCs were first pelleted by centrifugation and then resuspended in 200 μl of assay buffer (50 mm HEPES and 20 mm MgCl2, pH 7.6) and lysed by microtip sonication. The cell lysates were incubated with 20 μm LTA4 methyl ester and 10 mm GSH in assay buffer at room temperature for 10 min. Samples were analyzed by RP-HPLC (17). To assay for 5-LO activity, BMMCs were lysed in 5-LO assay buffer (0.1 m Tris, 30 mm KH2PO4, and 1.5 mm EDTA), and the reactions were initiated with the addition of 50 μm arachidonic acid, 2 mm ATP, and 1 mm CaCl2 and incubated at 37 °C for 15 min. Reaction products were analyzed by RP-HPLC.
Western Blot Analysis
Cells were resuspended in lysis buffer (10 mm Tris-HCl, pH 7.6, 100 mm NaCl, 50 mm NaF, 100 μm Na3VO4, 50 μg/ml PMSF, 10 μg/ml aprotinin, and 10 μg/ml leupeptin), boiled in SDS-PAGE sample buffer under reducing and nonreducing conditions, and separated on Tris/glycine-14% SDS-polyacrylamide gel (Novex). After transfer to a PVDF membrane, the blot was blocked with 4% nonfat dry milk and incubated with affinity-purified anti-LTC4S IgG (1 μg/ml) for 2 h. After washing, the blot was incubated with horseradish peroxidase-linked secondary antibody for 1 h, washed, and developed with chemiluminescence substrate. For nasal polyps, samples were minced in lysis buffer, homogenized, and boiled in SDS-PAGE sample buffer under reducing and nonreducing conditions.
Real-time PCR
To determine the effect of 15R-D2 on LTC4S mRNA, we examine the time-dependent changes in LTC4S mRNA by real-time PCR. Cells were treated with 10 μm 15R-D2 for 0, 3, 6, and 24 h and harvested, and total RNA were then isolated with TRIzol reagent (Invitrogen). The relative LTC4S mRNA level was determined by real-time PCR (GeneDirect, Princeton, NJ) and by comparing the values of the ratio of the LTC4S gene product to the 18 S housekeeping gene product.
Measurement of Intracellular GSH
Intracellular GSH levels were measured by enzymatic conjugation of CDNB to GSH present in the mast cell lysate to form glutathionyl-S-dinitrophenyl (GS-DNP). Mast cells were lysed with 200 μl of 100 mm Tris-HCl, pH 8.0, and then incubated with 200 μm CDNB at room temperature for 30 min in the presence of 1 unit of GST from human placenta. Reactions were terminated by the addition of 10 μl of acetic acid (17.4 m) and 3 volumes of methanol containing 400 ng/ml PGB2 and then analyzed by RP-HPLC for GS-DNP levels, with the ultraviolet absorbance monitored at 280 and 340 nm (for GS-DNP). Intracellular GSH was calculated by comparing the peak area of GS-DNP with that of a standard curve generated by incubating known amounts of GSH (125–4000 pmol) with 200 μm CDNB and 1 unit of GST under identical conditions and is expressed as picomoles/106 cells.
Effect of BSO and Diamide
To examine the effect of the oxidizing condition on the ability of BMMCs to generate 5-LO products, BMMCs were pretreated with either 200 μm BSO, a GSH synthesis inhibitor, for 24 h or with 300 μm diamide, a sulfhydryl-reactive oxidative agent, for 30 min to deplete their intracellular GSH. Cells were then harvested and separated into three aliquots. One aliquot of cells was suspended in buffer A and stimulated with 2 μm ionophore to determine the ability to generate 5-LO products, and the other two aliquots were lysed and assayed for LTC4S activity and GSH levels, respectively.
Metabolism of PGD2 and 15R-D2
BMMCs were incubated with 10 μm PGD2 or 10 μm 15R-D2 for 24 h and then analyzed by RP-HPLC for product profiles. For non-enzymatic conversion of these prostanoids, we incubated PGD2 and 15R-D2 with culture medium alone for 24 h and then analyzed the product profiles.
Effect of C56S and C82V Mutations on Diamide-induced Suppression of Enzyme Activity and Covalent Dimer Formation
To determine whether Cys-56 of LTC4S is involved in covalent dimer formation, we transfected wild-type, C56S, and C82V plasmid cDNAs into CHO cells by lipofection using FuGENE 6 reagent. Two days after transfection, cells were harvested, washed, and then incubated with 300 μm diamide. After 30 min, cells were lysed and assayed for LTC4S activity and molecular size.
RESULTS
PGD2 and PGE2 Inhibit LTC4 Generation in BMMCs
Like cysteinyl leukotrienes, PGD2 is abundant in allergic inflammation and a product of activated mast cells. No previous study had addressed whether PGD2 could modify the generation of LTC4. In addition, as LTC4S catalyzes the conjugation of LTA4 to GSH, we were also interested in determining whether changing intracellular GSH through oxidative stress will affect LTC4 generation. Because PGD2 has previously been shown to increase LTC4 generation by mouse eosinophils (29), and its cyclopentenone metabolite has been proposed as a potential endogenous regulator of redox-sensitive transcription factors (30) and has been shown to deplete intracellular GSH (31), we thus examined the effect of PGD2 on LTC4 generation by BMMCs. BMMCs were incubated with 10 μm PGD2 for 48 h, and for specificity purposes, we also included 10 μm PGE2 and 10 μm PGF2α for comparison. After harvesting, cells were stimulated with 2 μm calcium ionophore A23187 for 10 min. As shown in Fig. 1, whereas both PGD2 and PGE2 inhibited LTC4 generation by >70 and 65%, respectively, PGF2α had no effect. We concentrated on the effect of PGD2, as it is also a mast cell-derived product. None of these prostaglandins affected the biosynthesis of 12-hydroxyheptadecatrienoic acid (data not shown).
FIGURE 1.

Effect of PGD2, PGE2, and PGF2α on ionophore-stimulated LTC4 generation by BMMCs. BMMCs were treated with and without (control (CNT)) prostaglandins (10 μm) for 48 h, harvested, and stimulated with the calcium ionophore A23187 (2 μm) for 15 min, and LTC4 levels were analyzed by HPLC and are expressed as picomoles/106 cells (mean ± S.E., n = 4). *, p < 0.01.
Inhibitory Effect of PGD2 Does Not Act through the DP1 or DP2 Receptor or Peroxisome Proliferator-activated Receptor-γ (PPARγ)
To investigate the possible receptor involved in the suppression of LTC4 generation by PGD2, BMMCs were incubated with the DP1 agonist BW 245C or the DP2 agonists 15R-D2 and 15-deoxy-Δ12,14-PGD2 (15d-D2; also a PPARγ agonist), and we then examined the ionophore-stimulated generation of 5-LO products. As shown in Fig. 2A, the effect of PGD2 could be mimicked by the DP2 agonists 15R-D2 and 15d-D2 but not by the DP1 agonist BW 245C. To further investigate whether the suppressive effect is mediated through the DP2 receptor or PPARγ, we examined the effect of the DP2 antagonist Ramatroban and the PPARγ antagonist GW 9662 using 15R-D2 as an agonist. As shown in Fig. 2B, the effect of 15R-D2 on LTC4 generation was not inhibited by the DP2 antagonist Ramatroban or by the PPARγ antagonist GW 9662, suggesting that the inhibitory effect of PGD2 or 15R-D2 is not mediated through DP1, DP2, or PPARγ. Because the effect of PGD2 could be reproduced by 15R-D2, we performed subsequent experiments using 15R-D2 as an agonist.
FIGURE 2.

Effect of 15R-D2, BW 245C, and 15d-D2 (10 μm each) on LTC4 generation by BMMCs (A) and effect of Ramatroban (10 μm) and GW 9662 (10 μm) on the suppressive effect of 15R-D2 (10 μm) on A23187-stimulated LTC4 generation. Cells were treated with 10 μm DP agonists for 48 h and then stimulated with A23187 for 15 min (A) or treated with 15R-D2 (15R) in the absence or presence of Ramatroban (RAMA) and GW 9662 (GW) for 48 h and then stimulated with 2 μm A23187 for 15 min (B). Data are expressed as picomoles/106 cells. *, p < 0.01 versus the control (CNT; mean ± S.E., n = 6); #, p < 0.02 versus 15R-D2 (mean ± S.E., n = 5).
Time-dependent Effect of 15R-D2 on the Ability of BMMCs to Generate LTC4 and LTC4S Protein or mRNA Expression
When 15R-D2 was incubated with BMMCs, it caused a time-dependent decrease in LTC4 generation when stimulated with the calcium ionophore. When BMMCs were treated with 15R-D2 for 24 h, there was a >75% reduction in ionophore-stimulated LTC4 generation (Fig. 3A), with little additional reduction when treated for 48 h (data not shown). Thus, we examined the dose-dependent effect of 15R-D2 over 24 h of treatment.
FIGURE 3.

Time-dependent effect of 15R-D2. BMMCs were incubated with 10 μm 15R-D2 for the indicated times, washed, and separated into four aliquots. One aliquot was stimulated with A23187 (2 μm) to determine LTC4 generation, and a second aliquot was used for LTC4S activity determination (A). One aliquot was used for immunoreactive LTC4S protein expression under reducing conditions (B). The last aliquot was used for LTC4S mRNA expression (C) (mean ± S.E., n = 4). Me, methyl ester.
To determine whether 15R-D2 reduced cellular LTC4S activity through reduced LTC4S transcription and protein expression, we incubated BMMCs with 10 μm 15R-D2 for 0, 3, 6, and 24 h. LTC4S mRNA expression was then analyzed by real-time PCR, and protein expression was examined by immunoblot analysis. As shown in Fig. 3 (B and C), 15R-D2 did not alter the expression of either the mRNA or immunoreactive LTC4S protein. This result indicates that 15R-D2 does not reduce the cellular LTC4 activity of BMMCs through a reduction in LTC4S gene transcription and protein expression, thus suggesting a post-translational modification of LTC4S.
Dose-dependent Effect of 15R-D2
To examine the dose-dependent ability of 15R-D2 to suppress LTC4 generation and its effect on 5-LO activity, we incubated BMMCs with various concentrations of 15R-D2 for 24 h and then performed various assays. As shown in Fig. 4A, 15R-D2 dose-dependently reduced ionophore-stimulated LTC4 generation by BMMCs with >90% inhibition at 10 μm 15R-D2. With 10 μm 15R-D2, the mean LTC4 generation by BMMCs was 10.0 ± 2.2% (mean ± S.E., n = 8) of untreated BMMCs. An IC50 of ∼2 μm was observed for the inhibition of LTC4 generation by 15R-D2. There were no significant changes in all-trans-diastereoisomers of LTB4 or LTB4 or 5-hydroxyeicosatetraenoic acid generation (Fig. 2A), suggesting that 15R-D2 selectively reduces the ability of BMMCs to generate LTC4 without affecting the 5-LO activity. To confirm that 15R-D2 did not affect 5-LO enzyme activity, both the 5-LO and LTC4S activities were measured in BMMC lysates obtained from BMMCs that were treated with various concentrations of 15R-D2 and from untreated cells. As shown in Fig. 4 (B and C), 15R-D2 did not affect cellular 5-LO activity but dose-dependently reduced LTC4S activity. The mean LTC4S activity of 15R-D2-treated BMMCs was 33.5 ± 3.5% (mean ± S.E., n = 8) (Fig. 3B) of the control cells, thus confirming that the reduction in LTC4S activity upon 15R-D2 treatment contributed to the decrease in ionophore-stimulated LTC4 generation. Furthermore, the effect of 15R-D2 on LTC4 generation was not restricted to the calcium ionophore A23187, as 15R-D2 also suppressed LTC4 generation induced by ionomycin and by cross-linking of IgE. 15R-D2-reduced ionomycin (2 μm) stimulated LTC4 generation to 8.6 ± 2.9% (mean ± S.E., n = 10) of the untreated cells. In two experiments, 10 μm 15R-D2 also reduced LTC4 generation induced by cross-linking of IgE to 10.6 and 11.8% of the untreated cells, respectively.
FIGURE 4.

Dose-dependent effect of 15R-D2. Cells were treated with the indicated concentrations of 15R-D2 (in 2 μl of ethanol) or with an equal volume of vehicle (0 μm 15R-D2) for 24 h, washed, and split into four aliquots. One aliquot was stimulated with A23187 (2 μm) to examine LTC4 generation (n = 8) (A), one for LTC4S activity (n = 8) (B), one for 5-lipoxygenase activity (n = 8) (C), and one for intracellular GSH measurement (n = 5) (D) as described under “Experimental Procedures.” ME, methyl ester.
15R-D2 Reduces Intracellular GSH Levels in BMMCs
15R-D2 inhibited LTC4 generation without a significant effect on LTC4S protein expression or 5-LO activity, suggesting that 15R-D2 may affect LTC4 generation by altering the intracellular levels of GSH, the second substrate of the LTA4 conjugation reaction. Thus, we investigated the level of intracellular GSH by assaying the ability of the cell lysate to enzymatically conjugate CDNB to endogenous GSH to form GS-DNP at pH 8.0 by adding excess CDNB and exogenous GST. As shown in Fig. 4D, 15R-D2 dose-dependently decreased the intracellular GSH levels as indicated by a reduction in the amount of GS-DNP generated when cell lysates were incubated with CDNB. At 10 μm 15R-D2, the mean GSH level of treated BMMCs was 43.9 ± 5.8% (mean ± S.E., n = 5) of the control cells.
BSO Reduces LTC4 Generation
To confirm that 15R-D2 reduces ionophore-stimulated generation of LTC4 by BMMCs through a reduction in intracellular GSH, we compared the effect of BSO, a glutathione synthase inhibitor, with that of 15R-D2 on ionophore-stimulated LTC4 generation. When BMMCs treated with 15R-D2 were stimulated with ionophore, LTC4S activity was reduced to 15.2 ± 5.9% (mean ± S.E., n = 3) of the control BMMCs. Treatment with BSO reduced LTC4 generation to undetectable levels. Similarly, BSO reduced intracellular GSH levels to 0.57 ± 0% (mean ± S.E., n = 3) of the control cells.
Mechanism of Reduction in Intracellular GSH by 15R-D2
Because cyclopentenone metabolites of PGD2 have previously been shown to be able to reduce intracellular GSH through conjugation to GSH and because mast cells have been shown to be poor producers of reactive oxygen species (32), we examined the metabolism of 15R-D2 and PGD2 in BMMCs. When PGD2 was incubated with BMMCs or with culture medium alone for 24 h and then analyzed by HPLC for its metabolites present in the culture medium, we found three major non-enzymatic metabolites that corresponded to synthetic standards of Δ12-PGJ2, 15d-J2, and 15d-D2, respectively, in both the BMMC culture medium (Fig. 5A) and the medium with no BMMCs (Fig. 5B). However, there was an additional polar metabolite (compound X) in the HPLC chromatograph of BMMC culture medium that was absent in the medium-alone sample. Furthermore, the relative peak heights of Δ12-PGJ2 (also monitored at its UV maximum of 244 nm; chromatograph not shown) and 15d-J2 culture media were nearly identically with or without BMMCs, whereas the peak height of 15d-D2 in the medium with BMMCs was 24.9 ± 1.82% (mean ± S.E., n = 3) lower than that with no BMMCs. Similarly, incubation of 15R-D2 with BMMCs and culture medium alone yielded metabolite profiles identical to those of PGD2 with three 15R-methyl derivatives of the corresponding PGD2 metabolites (data not shown). We thus examined whether compound X is derived from 15d-D2 or from 15d-J2. We first incubated 15d-D2 and 15d-J2) with and without GSH for 4 h at 37 °C and then analyzed the polar metabolite formation, as they are readily available commercially. Both 15d-D2 and 15d-J2 were conjugated to GSH non-enzymatically to form polar metabolites with a similar retention time in HPLC in the presence of GSH (data not shown). However, only 15d-D2 was converted to a polar metabolite with a HPLC retention time identical to that of compound X when incubated with BMMCs (Fig. 6, A and B). There was no metabolism of 15d-J2 observed when it was incubated with BMMCs (data not shown). These results suggest that compound X is a PGD2 metabolite derived from the conjugation of 15d-D2 to GSH and that 15R-D2 spontaneously degrades to form 15R-derivatives of 15d-D2, which are then conjugated to GSH, and lowers intracellular GSH. The chemical structure of compound X is not known and is being elucidated. This result is very similar to that reported for the conjugation of 9-deoxy-Δ9,12(E)-PGD2 to GSH (31).
FIGURE 5.

HPLC chromatographs of the culture media obtained from incubations of 10 μm PGD2 with (A) and without (B) BMMCs monitored at 310 nm. The identities of peaks 1–3 are Δ12-PGJ2, 15d-D2, and 15d-J2, respectively. The unknown polar PGD2 metabolite is labeled X. mAU, milliabsorbance units.
FIGURE 6.

HPLC analysis of the culture media obtained from incubations of 10 μm 15d-D2 with (A) and without (B) BMMCs. The unknown polar metabolite (X) corresponds to that obtained from PGD2 incubation with BMMCs. mAU, milliabsorbance units.
Effect of 15R-D2 on LTC4S Is Partially Reversible
Because β-mercaptoethanol (β-ME) counters the effect of 15R-D2 on LTC4 generation, we examined whether β-ME can rescue LTC4S activity 24 h after 15R-D2 treatment. BMMCs were treated with 10 μm 15R-D2 for 24 h, harvested, washed, and incubated with and without 10 mm β-ME. After 20 min, cells were washed once and then assayed for LTC4S activity. Treatment of BMMCs with 15R-D2 for 24 h reduced LTC4S activity to 29.9 ± 8.5% (mean ± S.E., n = 4) of the control cells. LTC4S activity increased to 65.4 ± 9.8% with β-ME treatment. These results suggest that the effect of 15R-D2 on LTC4S is at least a partially reversible process.
Effect of Diamide on LTC4S
Our demonstration that a reduction in intracellular GSH suppresses LTC4S function suggested that oxidative stress may also exert an effect similar to that of 15R-D2. Thus, we utilized the sulfhydryl-reactive oxidant diamide to acutely induce oxidative stress and to examine the enzyme activity of LTC4S. Incubation of diamide with BMMCs for 30 min reduced LTC4S activity by >75% (data not shown). To further study the effect of oxidative stress on LTC4S, we examine the effect of diamide on CHO cells transfected with wild-type and mutant LTC4S cDNAs. When diamide was incubated with CHO cells transfected with wild-type LTC4S cDNA, it reduced LTC4S function to 22.8 ± 3.4% (mean ± S.E., n = 4) of the control cells. In addition, diamide induced the formation of covalent dimer LTC4S in a nonreducing gel (Fig. 7A). In contrast, C56S mutant LTC4S retained >91% of the enzyme activity (n = 2) with diamide treatment and no covalent dimer formation (Fig. 7A). Mutation of the only other cysteine residue (Cys-82) did not protect enzyme activity from diamide treatment and formation of a covalent dimer (Fig. 7A). The effect of diamide was also reversible, as diamide-treated CHO cells that were subsequently incubated with 10 mm β-ME restored LTC4S activity to 89.2 ± 9.1% (n = 4) of the control enzyme, and there was no covalent dimer formation.
FIGURE 7.

SDS-PAGE immunoblot analysis of cell lysates obtained from CHO cells transfected with wild-type (lanes 1 and 2), C56S mutant (lanes 3 and 4), and C82V mutant (lanes 5 and 6) LTC4S cDNAs treated with diamide (lanes 2, 4, and 6) and untreated controls (lanes 1, 3, and 5) (A) and of purified LTC4S treated with and without diamide (B). SDS-PAGE was performed under nonreducing conditions. CNT, control.
To determine whether oxidative stress similarly affects purified LTC4S, we incubated purified LTC4S with diamide and then assayed its conjugating activity and analyzed the formation of the covalent dimer by Western blotting. Similar to the transfected CHO cells, diamide treatment suppressed the enzyme activities of purified LTC4S by >90% (data not shown) as well as the formation of a covalent dimer (Fig. 7B). These results indicate that oxidative stress can reversibly suppress LTC4S function at least in part through induction of disulfide bridging of Cys-56 from neighboring monomers and the formation of an enzymatically inactive covalent dimer.
To determine whether the covalent dimer exists in vivo, we examined nasal polyps from asthmatic subjects, which have been reported to contain large numbers of infiltrated activated eosinophils (33). Western blot analyses demonstrated the presence of a 34-kDa covalent dimer LTC4S (Fig. 8) under nonreducing conditions, similar to wild-type LTC4S treated with diamide. In addition, the presence of covalent dimer LTC4S was also observed in nasal polyps obtained from a normal individual. These results suggest that dimerization of LTC4S monomers can occur in vivo under oxidative stress.
FIGURE 8.
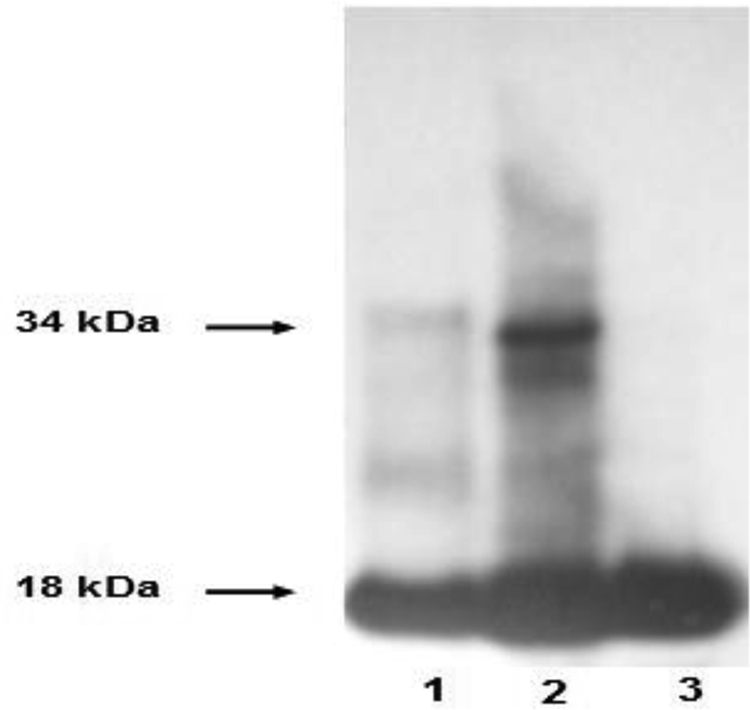
Immunoblot analysis of the molecular size of LTC4S. Shown are a nasal polyp biopsy obtained from an aspirin-sensitive asthmatic patient under nonreducing (lane 2) and reducing (lane 3) conditions and a LTC4S plasmid-transfected COS cell (lane 1).
DISCUSSION
This study has demonstrated that PGD2, the DP2 agonist 15R-D2, and the PPARγ agonist 15d-J2 suppress ionophore-stimulated LTC4 generation. The suppressive effect is a receptor-mediated event, as it was not block by either the DP2 antagonist Ramatroban or the PPARγ antagonist GW 9662 (Fig. 2B), and on the contrary, these antagonists significantly enhanced the suppressive effect of 15R-D2 on LTC4 generation. This potentiation effect has not been investigated further. The effect of 15R-D2 is not mediated through an alteration of transcription or protein expression, as neither was affected by 15R-D2 (Fig. 3). Because reduction in intracellular GSH has previously been shown to affect various cell functions, including gene transcription and cell proliferation (34, 35), we thus examined if the suppressive effect could be mediated through a reduction in intracellular GSH, a second substrate for LTC4S. We demonstrated that the DP2 agonist 15R-D2 reduces intracellular GSH in BMMCs (Fig. 4D) and reduces the ability of these cells to synthesize cysteinyl leukotrienes in response to ionophore stimulation as well as suppression of LTC4S activity through post-translational modification (Fig. 3, A and B, and Fig. 4). The mechanism by which 15R-D2 reduces intracellular GSH is at least partially through conjugation of GSH to its non-enzymatic breakdown product and does not involve any known PGD2 receptor (Figs. 5 and 6). When 15R-D2 was incubated with BMMCs, it non-enzymatically converted to 15R-derivatives of Δ12-PGJ2, 15d-J2, and 15d-D2. These determinations were based on the identical product profiles when PGD2 was utilized (Fig. 5) and identical HPLC retention times compared with available authentic standards of these PGD2 metabolites. Of these three cyclopentenone metabolites, both 15d-J2 and 15d-D2 conjugated to GSH non-enzymatically, with 15d-J2 being a better substrate for conjugation (data not shown). When incubated with BMMCs, however, a GSH-conjugated metabolite was observed in only incubations of 15d-D2 with BMMCs (Fig. 6). No metabolism was observed with 15d-J2 (data not shown), suggesting that the unknown polar metabolite X is a conjugated product of 15d-D2. The reason for the difference in metabolism of 15d-D2 and 15d-J2 by BMMCs is not known. It could be that uptake of these eicosanoids requires a membrane carrier and that there is an uptake carrier in BMMCs (36) for 15d-D2 but not for 15d-J2. The ability to non-enzymatically break down to cyclopentenone metabolites may account for the observed ability of PGD2 and PGE2 to suppress LTC4 generation, whereas PGF2α did not (Fig. 1). Furthermore, the ability of PGD2 to reduce intracellular GSH did not alter the biosynthesis of 12-hydroxyheptadecatrienoic acid, indirectly suggesting that there is no change in thromboxane synthesis, as 12-hydroxyheptadecatrienoic acid is an equal molar byproduct of thromboxane synthesis (37). We also examined if the reduction in intracellular GSH could be a result of an increase in reactive oxygen species generation, but our experiments showed that BMMCs produced negligible amounts of reactive oxygen species upon A23187 and phorbol 12-myristate 13-acetate stimulation (data not shown), an observation that is in agreement with that of Swindle et al. (32). In addition, the NADPH oxidase inhibitor diphenyleneiodonium sulfate did not overcome the effect of 15R-D2 (data not shown), thus confirming that the effect of 15R-D2 is not mediated through an increase in reactive oxygen species generation.
Both 15R-D2 and oxidative stress induced by diamide caused a possible post-translational modification of LTC4S, as they suppressed LTC4S activity without affecting protein expression (Figs. 3 and 7). Diamide also induced the formation of inactive covalent dimer LTC4S (Fig. 7). The suppressive effect of 15R-D2 on LTC4S activity can only be partially reversed with reducing agent, whereas the effect of diamide is totally reversible. Although the rationale for the difference in reversibility between 15R-D2 and diamide is not known, one possible explanation may be that the cyclopentenone metabolite of 15R-D2 can directly modify Cys-56 of LTC4S, an effect that has been reported for the p50 subunit of NFκB using 15d-J2 (38). Unlike the disulfide bridge of the covalent dimer, the metabolite-modified Cys-56 cannot be reduced by reducing agent, and therefore, that portion of LTC4S is irreversibly inhibited.
Importantly, our experiments suggest that cellular LTC4S may exist in two different forms, an enzymatically active form and an inactive form, and that the relative ratio of these two forms will depend on the oxidative state of the cells. Thus, our findings that 15R-D2 alters cell function through a reduction in cellular GSH further suggest that the cellular redox state may control the homeostasis of inflammatory cell function and subsequently the inflammatory processes. In a preliminary experiment with one nasal polyp, we also observed a 3-fold increase in LTC4S activity after it was treated with the reducing agent β-ME (data not shown). This observation, together with the demonstration of the presence of inactive covalent dimer LTC4S in a nasal polyp biopsy (Fig. 8), suggests that the cellular oxidation/reduction state of the tissue can modify its ability to generate 5-LO products and that tissue immunohistochemistry alone is not sufficient in determining the true capacity of the tissue in leukotriene biosynthesis in situ.
Acknowledgment
We are thankful to Dr. Joshua Boyce for helpful comments on the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants HL36110, HL082695, and HL090630.
- LT
- leukotriene
- 5-LO
- 5-lipoxygenase
- LTC4S
- LTC4 synthase
- CDNB
- 1-chloro-2,4-dinitrobenzene
- PG
- prostaglandin
- BMMC
- bone marrow-derived mast cell
- 15R-D2
- 15R-methyl-PGD2
- 15d-D2
- 15-deoxy-Δ12,14-PGD2
- 15d-J2
- 15-deoxy-Δ12,14-PGJ2
- BSO
- buthionine sulfoximine
- RP
- reverse-phase
- GS-DNP
- glutathionyl-S-dinitrophenyl
- PPARγ
- peroxisome proliferator-activated receptor-γ
- β-ME
- β-mercaptoethanol.
REFERENCES
- 1. Samuelsson B. (1983) Science 220, 568–575 [DOI] [PubMed] [Google Scholar]
- 2. Dahlén S. E., Hedqvist P., Hammarström S., Samuelsson B. (1980) Nature 288, 484–486 [DOI] [PubMed] [Google Scholar]
- 3. Weiss J. W., Drazen J. M., Coles N., McFadden E. R., Jr., Weller P. F., Corey E. J., Lewis R. A., Austen K. F. (1982) Science 216, 196–198 [DOI] [PubMed] [Google Scholar]
- 4. Henderson W. R., Jr., Lewis D. B., Albert R. K., Zhang Y., Lamm W. J., Chiang G. K., Jones F., Eriksen P., Tien Y. T., Jonas M., Chi E. Y. (1996) J. Exp. Med. 184, 1483–1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Henderson W. R., Jr., Tang L. O., Chu S. J., Tsao S. M., Chiang G. K., Jones F., Jonas M., Pae C., Wang H., Chi E. Y. (2002) Am. J. Respir. Crit. Care Med. 165, 108–116 [DOI] [PubMed] [Google Scholar]
- 6. Marom Z., Shelhamer J. H., Bach M. K., Morton D. R., Kaliner M. (1982) Am. Rev. Respir. Dis. 126, 449–451 [DOI] [PubMed] [Google Scholar]
- 7. Laitinen L. A., Laitinen A., Haahtela T., Vilkka V., Spur B. W., Lee T. H. (1993) Lancet 341, 989–990 [DOI] [PubMed] [Google Scholar]
- 8. Diamant Z., Hiltermann J. T., van Rensen E. L., Callenbach P. M., Veselic-Charvat M., van der Veen H., Sont J. K., Sterk P. J. (1997) Am. J. Respir. Crit. Care Med. 155, 1247–1253 [DOI] [PubMed] [Google Scholar]
- 9. Dennis E. A. (1994) J. Biol. Chem. 269, 13057–13060 [PubMed] [Google Scholar]
- 10. Dixon R. A., Diehl R. E., Opas E., Rands E., Vickers P. J., Evans J. F., Gillard J. W., Miller D. K. (1990) Nature 343, 282–284 [DOI] [PubMed] [Google Scholar]
- 11. Brock T. G., Paine R., 3rd, Peters-Golden M. (1994) J. Biol. Chem. 269, 22059–22066 [PubMed] [Google Scholar]
- 12. Ford-Hutchinson A. W., Gresser M., Young R. N. (1994) Annu. Rev. Biochem. 63, 383–417 [DOI] [PubMed] [Google Scholar]
- 13. Rouzer C. A., Matsumoto T., Samuelsson B. (1986) Proc. Natl. Acad. Sci. U.S.A. 83, 857–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Funk C. D., Rådmark O., Fu J. Y., Matsumoto T., Jörnvall H., Shimizu T., Samuelsson B. (1987) Proc. Natl. Acad. Sci. U.S.A. 84, 6677–6681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Penrose J. F., Gagnon L., Goppelt-Struebe M., Myers P., Lam B. K., Jack R. M., Austen K. F., Soberman R. J. (1992) Proc. Natl. Acad. Sci. U.S.A. 89, 11603–11606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nicholson D. W., Ali A., Vaillancourt J. P., Calaycay J. R., Mumford R. A., Zamboni R. J., Ford-Hutchinson A. W. (1993) Proc. Natl. Acad. Sci. U.S.A. 90, 2015–2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lam B. K., Penrose J. F., Freeman G. J., Austen K. F. (1994) Proc. Natl. Acad. Sci. U.S.A. 91, 7663–7667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jakobsson P. J., Morgenstern R., Mancini J., Ford-Hutchinson A., Persson B. (1999) Protein Sci. 8, 689–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hevko J. M., Murphy R. C. (2002) J. Biol. Chem. 277, 7037–7043 [DOI] [PubMed] [Google Scholar]
- 20. Ago H., Kanaoka Y., Irikura D., Lam B. K., Shimamura T., Austen K. F., Miyano M. (2007) Nature 448, 609–612 [DOI] [PubMed] [Google Scholar]
- 21. Martinez Molina D., Wetterholm A., Kohl A., McCarthy A. A., Niegowski D., Ohlson E., Hammarberg T., Eshaghi S., Haeggström J. Z., Nordlund P. (2007) Nature 448, 613–616 [DOI] [PubMed] [Google Scholar]
- 22. Xu L., Han C., Lim K., Wu T. (2008) J. Biol. Chem. 283, 3077–3087 [DOI] [PubMed] [Google Scholar]
- 23. Kim S. F., Huri D. A., Snyder S. H. (2005) Science 310, 1966–1970 [DOI] [PubMed] [Google Scholar]
- 24. Hatzelmann A., Ullrich V. (1987) Eur. J. Biochem. 169, 175–184 [DOI] [PubMed] [Google Scholar]
- 25. Werz O., Bürkert E., Fischer L., Szellas D., Dishart D., Samuelsson B., Rådmark O., Steinhilber D. (2002) FASEB J. 16, 1441–1443 [DOI] [PubMed] [Google Scholar]
- 26. Luo M., Jones S. M., Phare S. M., Coffey M. J., Peters-Golden M., Brock T. G. (2004) J. Biol. Chem. 279, 41512–41520 [DOI] [PubMed] [Google Scholar]
- 27. Werz O., Klemm J., Samuelsson B., Rådmark O. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 5261–5266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kanaoka Y., Maekawa A., Penrose J. F., Austen K. F., Lam B. K. (2001) J. Biol. Chem. 276, 22608–22613 [DOI] [PubMed] [Google Scholar]
- 29. Mesquita-Santos F. P., Vieira-de-Abreu A., Calheiros A. S., Figueiredo I. H., Castro-Faria-Neto H. C., Weller P. F., Bozza P. T., Diaz B. L., Bandeira-Melo C. (2006) J. Immunol. 176, 1326–1330 [DOI] [PubMed] [Google Scholar]
- 30. Kim E. H., Surh Y. J. (2006) Biochem. Pharmacol. 72, 1516–1528 [DOI] [PubMed] [Google Scholar]
- 31. Atsmon J., Freeman M. L., Meredith M. J., Sweetman B. J., Roberts L. J., 2nd (1990) Cancer Res. 50, 1879–1885 [PubMed] [Google Scholar]
- 32. Swindle E. J., Hunt J. A., Coleman J. W. (2002) J. Immunol. 169, 5866–5873 [DOI] [PubMed] [Google Scholar]
- 33. Adamjee J., Suh Y. J., Park H. S., Choi J. H., Penrose J. F., Lam B. K., Austen K. F., Cazaly A. M., Wilson S. J., Sampson A. P. (2006) J. Pathol. 209, 392–399 [DOI] [PubMed] [Google Scholar]
- 34. Fratelli M., Goodwin L. O., Ørom U. A., Lombardi S., Tonelli R., Mengozzi M., Ghezzi P. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 13998–14003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hadzic T., Li L., Cheng N., Walsh S. A., Spitz D. R., Knudson C. M. (2005) J. Immunol. 175, 7965–7972 [DOI] [PubMed] [Google Scholar]
- 36. Banu S. K., Lee J., Satterfield M. C., Spencer T. E., Bazer F. W., Arosh J. A. (2008) Endocrinology 149, 219–231 [DOI] [PubMed] [Google Scholar]
- 37. Okuno T., Iizuka Y., Okazaki H., Yokomizo T., Taguchi R., Shimizu T. (2008) J. Exp. Med. 205, 759–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cernuda-Morollón E., Pineda-Molina E., Cañada F. J., Pérez-Sala D. (2001) J. Biol. Chem. 276, 35530–35536 [DOI] [PubMed] [Google Scholar]