

Abstract
The identification of toxic Aβ species and/or the process of their formation is crucial for understanding the mechanism(s) of Aβ neurotoxicity in Alzheimer disease and also for the development of effective diagnostic and therapeutic interventions. To elucidate the structural basis of Aβ toxicity, we developed different procedures to isolate Aβ species of defined size and morphology distribution, and we investigated their toxicity in different cell lines and primary neurons. We observed that crude Aβ42 preparations, containing a monomeric and heterogeneous mixture of Aβ42 oligomers, were more toxic than purified monomeric, protofibrillar fractions, or fibrils. The toxicity of protofibrils was directly linked to their interactions with monomeric Aβ42 and strongly dependent on their ability to convert into amyloid fibrils. Subfractionation of protofibrils diminished their fibrillization and toxicity, whereas reintroduction of monomeric Aβ42 into purified protofibril fractions restored amyloid formation and enhanced their toxicity. Selective removal of monomeric Aβ42 from these preparations, using insulin-degrading enzyme, reversed the toxicity of Aβ42 protofibrils. Together, our findings demonstrate that Aβ42 toxicity is not linked to specific prefibrillar aggregate(s) but rather to the ability of these species to grow and undergo fibril formation, which depends on the presence of monomeric Aβ42. These findings contribute significantly to the understanding of amyloid formation and toxicity in Alzheimer disease, provide novel insight into mechanisms of Aβ protofibril toxicity, and important implications for designing anti-amyloid therapies.
Keywords: Alzheimer Disease, Amyloid, Cell Death, Neurodegeneration, Protein Self-assembly, Nucleated Polymerization, Oligomers
Introduction
Aggregation of amyloid-β (Aβ)2 peptides and deposition into neuritic plaques are hallmark features of Alzheimer disease (AD) neuropathology (1, 2). Therefore, research efforts during the past 3 decades have focused on elucidating the mechanisms of Aβ fibrillization, identifying toxic species, and developing strategies to inhibit and/or reverse Aβ amyloid formation and toxicity in vivo (3, 4).
Aβ peptides are produced as soluble monomers (5, 6) and undergo oligomerization and amyloid fibril formation via a nucleation-dependent polymerization process (7, 8). During the course of in vitro Aβ fibril formation, various nonfibrillar aggregation intermediates, collectively called soluble oligomers or protofibrils, have been shown to precede the emergence of fibrils. Increasing evidence from various sources points to Aβ oligomers/protofibrils as putative toxic species in AD pathogenesis and suggests that these species are potential therapeutic targets for treating AD (reviewed in Refs. 9, 10). Although the toxic oligomer hypothesis has emerged as one of the major current working hypotheses in AD research, the development of effective diagnostic tools and therapies on the basis of this hypothesis has yet to be realized (11–13). This is partially due to the fact that identification of a single toxic Aβ species that correlates with AD progression and severity remains elusive. Furthermore, the exact mechanisms by which these species contribute to Aβ toxicity in vivo and the nature of the toxic species are not yet fully understood. Recent evidence suggests that accelerating the process of Aβ fibrillization greatly enhances Aβ toxicity in vitro (14) and the spread of amyloid pathology in vivo (15–17).
Despite significant efforts by different groups to isolate specific intermediates along the amyloid formation pathway (12, 18–22), the inherent heterogeneity of the process and metastable nature of Aβ oligomers (11–13) have precluded the isolation of a single toxic species. Unless covalently cross-linked (23), Aβ oligomers do not exist as stable entities, i.e. they evolve into higher order aggregates and, if they are on-pathway intermediates, convert into fibrils (19). Therefore, it is plausible to assume that the structural dynamics of oligomers and factors that govern their interconversion and/or growth might influence some of the disease-related cytotoxic effects of Aβ. In other words, an ongoing polymerization process involving the elongation and growth of oligomers, rather than the formation of a stable oligomeric species, may be responsible for Aβ toxicity and neurodegeneration in AD.
To test this hypothesis, we developed different procedures to isolate Aβ species of defined size and morphology distribution (24), and we investigated their toxicity in different cell lines and primary neurons. We observed that crude Aβ42 preparations, containing a monomeric and heterogeneous mixture of Aβ42 oligomers and protofibrils, were more toxic than the purified monomeric protofibrillar fractions or fibrils. The toxicity of protofibrils was directly linked to their interactions with monomeric Aβ42 and strongly dependent on their ability to convert into amyloid fibrils.
Selective removal of the monomers, by SEC or by degradation with insulin-degrading enzyme (IDE), retarded the elongation of protofibrils, their fibrillization, and diminished protofibril toxicity toward cultured rat primary neurons, pheochromocytoma (PC12) cells, and neuroblastoma (SHSY5Y) cells. Similarly, we show that an ongoing Aβ42 polymerization process, rather than distinct Aβ42 aggregate states, also underlies previously reported alterations in astrocyte metabolic phenotypes (25). These findings contribute significantly to the understanding of amyloid formation and propagation in AD, provide novel insight into the mechanisms of Aβ protofibril toxicity, and carry important implications for designing anti-amyloid therapies.
EXPERIMENTAL PROCEDURES
Unless indicated otherwise, chemicals and reagents of analytical grade were purchased from Sigma. Aβ peptides were synthesized and purified by Dr. James I. Elliott, Yale University, New Haven, CT, as described previously (26). Chromatography columns (Table 1) were purchased from GE Healthcare except TSK-GEL G4000 PWXL (TSK4000), which was purchased from Tosoh Bioscience (Belgium). Highly purity distilled water was used to prepare buffers, and solutions were filtered and degassed by passing through vacuum-driven 0.22-μm stericup filtration units (Millipore, Switzerland) before use. Deoxy-d-glucose (2-[1,2-3H]glucose (2-[3H]DG), specific activity, 30–60 Ci/mmol) was obtained from ANAWA (Switzerland). DNase and papain were purchased from Sigma (catalog no. D4527 and P4762, respectively).
TABLE 1.
SEC fractionation of Aβ protofibrils and monomers
| Serial no. | SEC column | Separation range | PF fractions | M fractions |
|---|---|---|---|---|
| kDa | ||||
| 1. | Superdex 75 HR 10/30 | 3–70 | 1a | 1a |
| 2. | Superdex 200 GL 10/300 | 10–600 | 4, 5 | 1a |
| 3. | Superose 6 HR 10/30 | 5–5000 | 4–6 | 1a |
| 4. | TSK-GEL G4000PWXL | 10–1500 | 3, 4 | 1a |
| 5. | Superdex 75 CISb Superose 6 | 6–8 | 1a | |
| 6. | TSK-GEL G4000PWXL CISb Superose 6 | 6–8 | 1a |
a The fractions were combined.
b CIS means connected in series.
Preparation of Aβ42 Protofibrils
Crude Aβ42 protofibril (Aβ42 CR) solution was prepared as described previously (24, 27). Briefly, 1 mg of lyophilized Aβ42 was solubilized in 50 μl of 100% anhydrous DMSO in a 1.5-ml sterile microtube. Then 800 μl of high purity water was immediately added, and the pH was brought to ∼7.6 by adding 10 μl of 2 m Tris base, pH 7.6. The solution was always freshly prepared and used immediately.
Size Exclusion Chromatography
Separation of Aβ Protofibrils and Monomers
SEC fractionation was carried out using an ÄKTA Explorer FPLC (GE Healthcare) placed inside a cold (4 °C) chamber. SEC columns were thoroughly equilibrated with SEC running buffer (10 mm Tris-HCl, pH 7.4) prior to Aβ injections. Aβ (40 and 42) monomers and Aβ42 protofibrils were obtained by SEC as described previously (24, 27). To prepare Aβ monomers, 1 mg of lyophilized Aβ was dissolved in 1 ml of 6 m guanidine HCl solution and subsequently centrifuged (16,000 × g, 4 °C, 10 min). The supernatant was injected into a Superdex 75 HR 10/30 column, and Aβ was eluted at flow rate of 0.5 ml/min (1 ml/fraction). Aβ elution was monitored at absorbance wavelengths of A210, A254, and A280. To obtain both protofibrils (PF) and monomers (M), Aβ42 crude (CR) solution was prepared as described above. After centrifugation (16,000 × g, 4 °C, 10 min), the supernatant was fractionated on a Superdex 75 column as described above. The fractions eluting in the void volume were combined and labeled as protofibrils, whereas the fractions eluting under the 11–13-ml peak were combined and labeled as monomers. The PF and M fractions were further characterized by thioflavin-T (ThT) binding and transmission electron microscopy (TEM) as described below and used immediately for analytical SEC and toxicity studies.
Subfractionation of Aβ42 Protofibrils
Aβ CR solution was prepared and centrifuged as described above. The supernatant was injected into either a single SEC column with a higher molecular weight separation range than Superdex 75 or a combination of SEC columns connected in series (Table 1) and fractionated as described above. For experimental purposes, the fractions corresponding to monomeric elution were combined. However, the fractions corresponding to protofibrillar fractions were kept separate and labeled according to their elution position (Table 1). The fractions were further characterized by ThT binding and TEM and used immediately for analytical SEC and toxicity studies.
Aβ Concentration Determinations
Aβ concentration in fractions was determined by UV absorbance at 280 nm (A280) using the theoretical molar extinction coefficient 1490 m−1 cm−1 (28).
Preparation of Fibrils and Sonicated Fibrils
Aβ42 F were prepared by incubating the CR solution (200 μm) at 37 °C with gentle shaking for 24–48 h. Fibril formation was confirmed by ThT binding and TEM. To generate fibril seeds (SF), 100–300-nm-long fibrillar structures, the fibrils were sonicated on ice using a Vibra-CellTM instrument (Sonics and Materials, Inc.) equipped with a fine tip (five times, 20-s pulses, amplitude 40%, output watts 6, 20-s delay between successive pulses). Subsequently, the quality of SF preparation and fibril fragmentation was confirmed by TEM.
Analytical SEC (Ana-SEC)
Ana-SEC was carried out to assess the efficiency of fractionation and heterogeneity in each of the purified protofibrillar fractions. For this purpose, an analytical SEC column Superose 6 pc 3.2/30 was connected to a Waters Separation Module 2795 equipped with a photodiode array detector (Waters). Aliquots (50 μl) of the Aβ fractions were injected into the column and eluted at flow rate of 0.05 ml/min. Aβ elution was monitored at UV A210, A254, and A280.
Cell Culture Toxicity Studies
Primary Neuronal Cell Cultures
Primary cortical neurons were prepared from Sprague-Dawley rats (Charles River Laboratories, L'Arbresle, France) at postnatal day 1 essentially as described previously (29). After removing the meninges, the cortical tissue was cut into small pieces and enzymatically disrupted in dissociation buffer (papain, CaCl2, EDTA, and HEPES; 30 min; 37 °C). The DNase was added for 10 min. Cortex pieces were then washed three times in prewarmed complete growth medium (neurobasal + 10% FCS + penicillin/streptomycin + l-glutamine; Invitrogen) and then triturated seven times in 5 ml of medium. Cells were filtered through a 0.45-μm cell strainer, counted, and plated onto 96-well, poly-l-lysine-precoated plates (30,000 cells/200 μl/well). After 90 min of incubation at 37 °C, the medium was replaced by astrocyte-conditioned growth medium. On the 4th day in vitro (DIV 4), 2.5 μm cytarabine (Sigma) was added.
Rat Pheochromocytoma (PC12) and Human Neuroblastoma (SHSY5Y) Cell Cultures
PC12 cells were cultured in DMEM (Invitrogen 41966-029) supplemented with 1% penicillin/streptomycin (Invitrogen) and 2 μm human recombinant insulin (Invitrogen). The cells were plated in 96-well transparent BD Falcon plates (30,000 cells/200 μl/well) and allowed to grow in a cell culture incubator (37 °C; ambient humidity; 5% CO2) for 24–48 h. SHSY5Y cells (40,000 cells/100 μl/well) were cultured in a similar fashion as PC12.
Aβ Toxicity Studies in Primary Neuronal Cultures
Primary neurons were treated with the following Aβ42 preparations: 1) Aβ42 crude; 2) Aβ42 fibrils; 3) SEC purified protofibrils and monomers; and 4) 1:1 molar mixtures of protofibrils and monomers. For this purpose, on DIV 7 half of the culture medium was replaced by complete growth medium containing Aβ preparations (0.2 (v/v) medium dilution; Aβ preparations and the buffer vehicle contained 140 mm NaCl and 10 μm final Aβ concentration). After 24 h of treatment, neuronal cell viability was assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction assay as described below.
Aβ Toxicity Studies in PC12 and SHSY5Y Cell Cultures
Toxicity of Aβ42 Crude, Fibrils, Protofibrils, and Monomers
After 24–48 h in culture, half of the culture medium was removed and replaced with the indicated Aβ preparations. For this purpose, Aβ preparations were added to the supplemented 10× DMEM (Table 2) at a 9:1 (v/v) ratio. PC12 and SHSY5Y cells were treated with final Aβ concentration of ∼10 μm. After 24 h of treatment, cell viability was assessed by MTT reduction assay. Medium aliquots were obtained for ThT binding and TEM.
TABLE 2.
Supplemented 10× DMEM for delivery of Aβ fractions for toxicity assays
| Serial no. | Component (stock) | Volume | Concentration |
|---|---|---|---|
| μl | |||
| 1. | DMEM (10×, Sigma D2429) | 690 | |
| 2. | l-Glutamine (200 mm) | 200 | 40 mm |
| 3. | Insulin (690 μm) | 30 | 20.7 μm |
| 4. | Glucose (5 m) | 40 | 250 mma |
| 5. | Folic acid (2.25 mm) in NaHCO3 (11 m) | 40 | 90 and 440 mm, respectively |
| Total volume | 1000 μl | ||
a Sigma 10× DMEM already contains 10 g/liter glucose. Therefore, more glucose is added in order to bring the concentration to 45 g/liter (250 mm).
Toxicity of Aβ42 Protofibril/Monomer Mixtures and Protofibril/Fibril Mixtures
Mixtures of SEC-purified Aβ42 monomers, protofibrils fractions (Table 1), and sonicated fibrils were prepared in supplemented 10× DMEM (Table 2). Briefly, the following mixtures were prepared (molar ratios): 1) protofibrils and monomers (1:1); 2) protofibrils and sonicated fibrils (4:1); 3) monomers and sonicated fibrils (4:1); 4) protofibrils, monomers, and sonicated fibrils (2:2:1); and 5) protofibrils and monomers, and the monomers were incubated with insulin-degrading enzyme (R & D Systems catalog no. 2496-ZN) for 20 min at room temperature (Aβ: recombinant human IDE, 20:1) and then added to protofibrils. PC12 and SHSY5Y cells were treated with mixtures 1–5 (final Aβ concentration of ≈10 μm), and after 24 h, viability was assessed by MTT reduction assay. Medium aliquots were obtained for ThT binding and TEM.
Time Course of Aβ42 Toxicity
PC12 cells were cultured in phenol red-free DMEM (Invitrogen catalog no. 21063-029) supplemented with 1% penicillin/streptomycin (Invitrogen) and 2 μm human recombinant insulin (Invitrogen). PC12 cells were treated with 10 μm Aβ42 crude solution. Medium aliquots were obtained at selected time points and analyzed by ThT binding and TEM. In parallel, cell viability was assessed by MTT reduction and lactate dehydrogenase release in the media as described below.
Cell Viability Assays
MTT Reduction Assay
The viability of Aβ-treated cells was assessed by MTT reduction assay using a commercial kit (Promega, catalog no. G4000) according to the manufacturer's instructions. The absorbance was measured at 570 nm (λ690 nm was used as a reference wavelength) in a Safire2 microplate reader (TECAN). The signal was expressed as percentage of the A570–690 count from the vehicle-treated cells (100%).
Lactate Dehydrogenase Release Assay
The lactate dehydrogenase release assay was carried out on medium aliquots using a commercial kit (Sigma TOX-7) according to the manufacturer's instructions. The lactate dehydrogenase content in the medium of the vehicle treated cells was expressed as 100%.
Primary Astrocyte Cell Cultures
Primary cortical astrocyte cultures were prepared as established previously using 1–2-day-old Swiss Albino mice (OF1, Charles River Laboratories) (25). After removing the meninges, cortices were isolated and dissociated by passage through needles of decreasing gauges (1.2 × 40 mm, 0.8 × 40 mm, and 0.5 × 16 mm) using a 10-ml syringe. Cells were seeded on polyornithine-coated 35 × 10-mm culture dishes in growth medium (Dulbecco's modified Eagle's medium (DMEM), Sigma catalog no. D7777, supplemented with 10% fetal calf serum, 44 mm NaHCO3, and 1% antibiotic/antimycotic solution (Invitrogen)) and incubated (37 °C; 5% CO2, 95% air). On DIV 5, the medium was renewed with fresh medium and then renewed twice a week until use. On DIV 21, cells were treated for 24 h with Aβ preparations (in 10 mm Tris-HCl, pH 7.4) diluted in the growth medium (0.2 (v/v) dilution), for 24 h. 2-[3H]DG utilization (uptake and phosphorylation) by Aβ-treated cells was assessed as outlined below.
2-[3H]DG Utilization
2-[3H]DG utilization experiments were conducted as described previously (25). The media used for 2-[3H]DG uptake were pre-equilibrated at 37 °C, 5% CO2 and 95% air. To initiate the 2-[3H]DG assay, the medium was replaced by 2 ml of serum-free medium (DMEM (D5030, Sigma) supplemented with 5 mm glucose, 44 mm NaHCO3, and 1% of antibiotic/antimycotic solution) containing 1 μCi/ml 2-[3H]DG. The cells were incubated for 20 min at 37 °C, 5% CO2 and 95% air. The assay was terminated by aspiration of the culture medium and washing the cells three times with 4 ml of ice-cold PBS. Cells were then lysed by adding 2 ml of 10 mm NaOH containing 0.1% Triton X-100. Then 500-μl aliquots, in duplicates, were assayed for radioactivity using liquid scintillation counting. The results were calculated by subtracting from the total counts the portion that was not inhibited by the glucose transporter inhibitor cytochalasin B (25 μm), which was added 20 min prior to and along the 2-[3H]DG incubation. 2-[3H]DG utilization was calculated as femtomoles per dish and then expressed as percentage of the vehicle control.
ThT Binding Assay
ThT binding assay was performed by mixing 80 μl of Aβ preparations (in SEC buffer or cell culture media) with 20 μl of ThT (100 μm) and 10 μl of glycine-NaOH, pH 8.5 (500 mm), in a Nunc 384-well fluorescence plate (100 μl/well). The ThT fluorescence of each sample was measured in an Analyst AD fluorometer (Molecular Devices, Switzerland) at excitation and emission wavelengths of λ450 nm and λ485 nm, respectively.
TEM Sample Grid Preparation and Image Acquisition
A 5–10-μl droplet of sample containing Aβ (Aβ preparations or cell culture media) was deposited on a 200 mesh Formvar-coated TEM grid (EM Sciences) and was allowed to settle for ∼60 s. The excess solution was wicked away by gently applying a piece of blotting paper to the edge of the grid. Then a 10-μl droplet of 2% uranyl acetate was deposited on the grid and allowed to settle for ∼60 s. The excess solution was removed as above. Finally, the grid was vacuum-dried by gently applying the vacuum probe close to the grid edges. Image acquisition was carried out using a Phillips CM10 microscope operated at an acceleration voltage of 80 kV.
RESULTS
SEC Subfractionation Retards Elongation of Protofibrils and Decreases Toxicity
We and others have shown that toxic Aβ protofibril preparations consist of a heterogeneous mixture of high molecular weight aggregates of various sizes and morphologies (12, 24, 30). To determine whether the cytotoxicity of protofibrils is associated with specific type of aggregates or depends on the dynamics of inter-conversion among different aggregate species, we subjected a crude Aβ42 protofibril preparation (Aβ42 CR) to fractionation by SEC (12, 24). Then we investigated the cellular toxicity of individual Aβ42 fractions, enriched with distinct protofibrillar aggregates, using cultured PC12 and SHSY5Y cells. Subfractionation of Aβ42 protofibrils on a Superose 6 column resulted in 4–6 fractions corresponding to the elution of protofibrils (Fig. 1A). Only the first four protofibril fractions (F1–F4) had sufficient Aβ content (≥25 μm) to be useful for toxicity experiments. The monomers eluted later in two fractions (F5 and F6) that were separate from protofibrils (Fig. 1A). Reinjection of the purified Aβ species onto an Ana-SEC column showed that only F1 and F4 exhibited distinct elution profile for the protofibril peak, suggesting marked differences in the size distribution of the aggregates in these fractions (Fig. 1B). Soon after SEC fractionation, F1 consisted of large aggregates and clusters of protofibrils (Fig. 1F), whereas F3 was predominantly enriched with short curvilinear and spherical protofibrils (Fig. 1G). The monomer fraction (F5) was devoid of any visible aggregates (Fig. 1H). Ana-SEC of the monomer fractions (F5 and F6) showed a single narrow elution peak (data not shown).
FIGURE 1.

Fibril formation and toxicity of subfractionated Aβ42 protofibrils. A, subfractionation of Aβ42 protofibrils (F1–F4) and isolation of monomers (F5 and F6) on a Superose 6 SEC column. B, analytical SEC of protofibrillar fractions (F1–F4) on a Superose 6 pc 3.2/30 column. C, cell viability (MTT reduction assay) of cultured PC12 and SHSY5Y cells after 24 h of treatment with 10 μm Aβ42 CR protofibrils, protofibrils fractions (F1–F4), and monomers (F5) (one-way ANOVA, n = 9, *, p < 0.05; **, p < 0.01, mean ± S.D.). D, ThT binding over time by 10 μm Aβ42 CR protofibrils, protofibril fractions (F1–F4), and monomers (F5) in supplemented 10× DMEM (96 h of incubation; 37 °C, mean ± S.D.). E–L, representative TEM images of Aβ42 (CR, 0 and 96 h, respectively) (E and I), F1 (0 and 96 h, respectively) (F and J), F3 (0 and 96 h, respectively) (G and K), and F5 (0 and 96 h, respectively) (H and L) in aliquots of the culture medium (scale bar = 200 nm) are shown. (a.u., arbitrary units; Veh, buffer vehicle.)
Cell viability assessments of Aβ-treated cultured PC12 and SHSY5Y cells revealed that crude Aβ42 protofibrils (CR) decreased viability by ∼40–45% (p < 0.01), whereas the toxicity of Aβ42 SEC fractions (protofibrils and monomers) did not exceed ∼10–20% (Fig. 1C). Aβ40 monomers were also tested as control and consistently showed only a slight, but not significant, effect on the cell viability (∼5–10%, p > 0.05; Fig. 1C). To determine whether the observed toxicity correlated with the changes in Aβ42 aggregation state in individual fractions, we sampled the cell culture medium at different time points and probed the aggregation state of Aβ using ThT and TEM. Only the crude protofibrils exhibited a significant and gradual rise in ThT binding and reached a maximum within 24–48 h. In contrast, none of the purified protofibril fractions (F1–F4) showed a comparable rise in ThT signal even after 96 h of incubation. ThT binding by the monomer fraction (F5), after 96 h of incubation, did not exceed that of the protofibrils (Fig. 1D).
On the basis of these observations, we hypothesized that the loss of fibrillization led to attenuation of Aβ toxicity and that the toxicity in crude Aβ42 protofibrils (CR) was mediated by an ongoing process of Aβ polymerization rather than the formation of specific/stable aggregation state(s). In support of this hypothesis, analysis of the culture medium by TEM revealed that only the crude Aβ42 protofibrils (CR) formed extensive mature fibrils (>1 μm long and high ThT binding) during the course of the experiments (Fig. 1I). Even after 96 h of incubation, mature fibrils were not observed in culture medium containing the purified protofibril fractions F1 and F3 (Fig. 1, J and K, respectively). Instead, clusters of elongated curvilinear protofibrils were observed. The monomer fraction (F5) did show some fibril-like structures (Fig. 1L), but they were not as extensive as those seen in the case of the crude Aβ42 protofibrils (Fig. 1I). Subsequent subfractionation experiments, using combinations of SEC column connected in series (Table 1), confirmed these findings and revealed that the fibrillization and toxicity of crude Aβ42 protofibrils were closely linked processes (supplemental Figs. 1 and 2).
Separation of Monomers from Protofibrils Retards Aβ42 Fibril Formation and Reduces the Toxicity of Protofibrils
Some of the possible explanations for the reduced fibrillization and toxicity of Aβ42 protofibrils upon SEC subfractionation include the following: 1) the presence of a small number of fibrils in the crude preparations that are lost upon SEC fractionation; 2) disruption of the structure of toxic Aβ species upon interaction with the SEC column matrices; 3) disruption of the dynamics of inter-conversion among different protofibril species; or 4) retardation of protofibril growth due to the removal of excess free Aβ monomers. To investigate these possibilities, we compared the aggregation and toxic properties of crude Aβ protofibrils (CR) with those of monomers (M), fibrils (F), and the protofibrils (PF) that were not subfractionated and simply separated from monomers. When Aβ42 CR was fractionated on a Superdex 75 SEC column, all protofibrils eluted in the void volume without further separation (cutoff ≥70 kDa) and monomers eluted later in the included volume (Fig. 2A), as reported previously (27, 30). We have verified that these conditions yield protofibrils and monomer fractions that are free of fibrils (24). The PF fraction contains a heterogeneous mixture of different quaternary structures. Cell viability assessments of Aβ-treated primary neuron cultures, PC12 cells, and SHSY5Y cells revealed that 24 h of treatment with Aβ42 CR decreased the viability of PC12 and SHSY5Y cells by ≥40% (p < 0.01) and of primary neurons by ∼50% (p < 0.01) (Fig. 2C). The purified, but not subfractionated, PF decreased cell viability by ∼20–30% (p < 0.05), and Aβ42 M exhibited only a slight effect (∼5–20%, p > 0.05) (Fig. 2C). Treatment of cells with preformed F, obtained by incubation of monomers at the same concentration, also impaired cell viability by ∼10–20% (p > 0.05) but was remarkably less toxic than crude protofibrils or purified PF.
FIGURE 2.

Toxicity of isolated Aβ42 monomers, protofibrils, and fibrils toward cultured cells. A, fractionation of Aβ42 CR protofibrils on a Superdex 75 SEC column to separate PF and M. B, analytical SEC of protofibrillar fractions (PF) from A on a Superose 6 pc 3.2/30 column. It is noteworthy that some of the monomers observed in the PF fractions are due to PF dissociation during the SEC separation. C, cell viability (MTT reduction assay) of cultured rat primary neurons, PC12 cells, and SHSY5Y cells after 24 h of treatment with 10 μm Aβ42 CR, PF, M, and F (one-way ANOVA, n = 6 (neurons) and n = 9 (PC12 and SHSY5Y); *, p < 0.05; **, p < 0.01, mean ± S.D.). D, ThT binding over time by 10 μm Aβ42 CR protofibrils, fractionated PF, M, F, and Aβ40 M in supplemented 10× DMEM (24 h of incubation; 37 °C, mean ± S.D.). E–L, representative TEM images of Aβ42 CR (0 and 24 h, respectively) (E and I), Aβ42 PF (0 and 24 h, respectively) (F and J), Aβ42 M (0 and 24 h, respectively) (G and K), and Aβ42 F (0 and 24 h, respectively) (H and L) in aliquot of the culture medium (scale bar = 200 nm) are shown. (a.u., arbitrary units; Veh, buffer vehicle.)
As observed earlier (Fig. 1D), the Aβ42 CR protofibrils exhibited substantial rise in ThT binding within 24 h (Fig. 2D). As a control, mature preformed F were also added to the culture medium and showed a ThT signal similar to crude Aβ42 protofibrils at 24 h. These observations confirmed the substantial fibrillization of Aβ42 CR during the time interval of toxicity experiments (24 h). TEM analysis of culture medium revealed that Aβ42 CR preparations formed extensive mature fibrils within 24 h (Fig. 2I, compare with the image of preformed fibrils in Fig. 2H). Neither M nor purified PF exhibited a comparable rise in ThT binding over time (Fig. 2D). This is consistent with TEM observations that revealed the absence of mature fibrils in the culture medium and only clusters of protofibrils after 24 h of incubation (Fig. 2, J and K, respectively).
Addition of Monomers to Protofibrils Enhances Aβ42 Fibrillization and Toxicity
Our results suggest that SEC fractionation of protofibrils reduces, but does not completely eliminate, their toxicity and demonstrate that toxicity of Aβ42 protofibrils is strongly linked to their ability to undergo fibrillization and form mature fibrils. Therefore, we wanted to investigate if the slow fibrillization of protofibrils, and thus their reduced toxicity, were due to the removal of excess monomers and/or fibrils by SEC. To this end, we reintroduced Aβ42 monomers, or preformed fibrils, to purified PF (obtained using a Superdex 75 column) or subfractionated protofibril fractions (F1–F4, obtained using a Superose 6 column), and we assessed the fibrillization and toxicity of each fraction and mixtures thereof. We hypothesized that the addition of free monomers and/or preformed fibril seeds would enhance both protofibril fibrillization and toxicity. Accordingly, we observed that reintroduction of Aβ42 monomers to purified PF resulted in an increase in fibril formation and enhanced protofibril toxicity (supplemental Fig. 2). To confirm these findings, we added purified monomers to the subfractionated Aβ42 protofibrils (F1–F4). Mixtures of monomers and protofibril fractions (F1–F4) exhibited higher ThT binding over time (Fig. 3A) as compared with fractions containing protofibrils only (Fig. 1D). However, it became apparent that the extent of ThT binding in the monomer/protofibril mixtures was not as robust as seen in the case of Aβ42 CR (Fig. 1D), suggesting a weak seeding capacity of subfractionated protofibrils. Treatment of cells with mixtures of Aβ42 monomers and subfractionated protofibrils (F1–F4) impaired cell viability by ∼25–35% (Fig. 3B, p < 0.05), which was comparable with that seen with Aβ42 crude (≥40%, Fig. 2C). TEM analysis of the culture medium revealed that addition of Aβ42 monomers to protofibrillar fractions (F1–F4) induced substantial fibril formation (supplemental Fig. 3, A and B, e.g. F1+M and F3+M, respectively, data for F2+M and F4+M is not shown). The monomer fraction (F5), after 96 h of incubation, showed the presence of weakly ThT binding immature fibrils (Fig. 3A and supplemental Fig. 3C).
FIGURE 3.

Effect of monomer addition on fibril formation and toxicity of subfractionated protofibrils. A, ThT binding over time by 1:1 molar mixtures of Aβ42 protofibrils fractions (F1–F5) obtained from Superose 6 with Aβ42 monomers in supplemented 10× DMEM (10 μm Aβ42, 96 h of incubation; 37 °C, mean ± S.D.). B and C, cell viability (MTT reduction assay) of cultured PC12 and SHSY5Y cells after 24 h of treatment with 1:1 molar mixture of Aβ42 protofibrils fractions (F1–F5) and Aβ42 monomers (10 μm Aβ; one-way ANOVA, n = 9, *, p < 0.05; **, p < 0.01, mean ± S.D.). C, monomers were pretreated with IDE before addition to the fractions F1–F5 (Aβ:IDE, 20:1, w/w). D, cell viability (MTT reduction assay) of cultured PC12 and SHSY5Y cells 24 h after treatment with mixtures of sonicated Aβ42 fibrils (2 μm) with SEC protofibrils fractions F1–F5 (8 μm) (one-way ANOVA, n = 9; *, p < 0.05; **, p < 0.01, mean ± S.D.) (CR, crude Aβ42 protofibrils; a.u., arbitrary units; Veh, buffer vehicle).
Taken together, if Aβ42 fibrillization and toxicity are closely linked processes, and if these processes require the availability of Aβ monomers, then selective degradation of monomers should significantly retard protofibril elongation and reduce their toxicity. To examine this hypothesis, we sought to induce selective removal of Aβ42 monomers using IDE, an enzyme that selectively degrades Aβ monomers but not oligomers or fibrils (31–33). IDE was added to a solution of Aβ42 monomers, and this mixture was then added to subfractionated protofibril fractions (F1–F4) in supplemented 10× DMEM (see under “Experimental Procedures”). As anticipated, the addition of IDE reversed the effect of Aβ42 monomers in enhancing protofibril toxicity (compare Fig. 3, C with B). The toxicity of protofibrils (F1–F4) mixed with IDE-treated Aβ42 monomers was similar to the toxicity seen with protofibrillar fractions (F1–F4) alone (compare Fig. 3C with 1C). Similarly, we observed that addition of IDE to toxic crude Aβ42 preparations resulted in selective degradation of the monomers and diminished the cytotoxicity and fibrillization of these preparations, albeit to a lesser extent than monomer/protofibril mixtures (supplemental Fig. 4).
Next we wanted to determine whether the presence of a small amount of fibrillar aggregates in the crude preparation, which might have been lost upon SEC fractionation, also contributed to the enhanced toxicity of this preparation relative to the SEC-purified protofibrils and monomers. To investigate this, we introduced a small amount (2 μm) of sonicated Aβ42 fibrils (SF) to subfractionated Aβ42 protofibrils (F1–F4). We found that the addition of sonicated fibrils did not lead to any significant enhancement of protofibril toxicity (Fig. 3D), regardless of differences in the size distribution between the various protofibril fractions. TEM analysis revealed that, even after 96 h of incubation, these fractions predominantly contained mixtures of fibrillar seeds and protofibrils (supplemental Fig. 3, D (F1+SF) and E (F3+SF)). In contrast, addition of SF to the monomer fraction (F5) resulted in significantly increased amyloid formation as evidenced by the presence of mature fibrils (supplemental Fig. 3F), but the toxicity was relatively unaffected (Fig. 3D). These findings may indicate that monomer-protofibril interactions, rather than monomer-fibril interactions, are the key determinant of Aβ42 toxicity. Interestingly, the addition of the mixtures of M and fibrillar seeds (SF) to the protofibril fractions, and not fibrillar seeds (SF) only, induced robust toxicity in subfractionated protofibrils (supplemental Fig. 5B).
To determine whether the observed findings are specific to Aβ42, we carried out fibrillization and toxicity experiments using different preparations of Aβ40 (crude, monomers, protofibrils, and mixtures thereof). The data show that at the concentrations similar to those used in the Aβ42 (5–10 μm) studies, we did not observe significant difference in terms of toxicity between monomeric, protofibrillar, or crude preparations of Aβ40 (supplemental Fig. 6). These findings are consistent with the lack of changes in the ThT signal and fibrillization of these samples, thus supporting our hypothesis that Aβ toxicity is also linked to its ability to undergo fibril formation. Consistent with this hypothesis, significant toxicity (30–40%) was only observed when Aβ40 undergoes fibrillization (≥50 μm). These findings point to the processes of protofibril-monomer interactions during maturation and elongation of Aβ protofibrils/oligomers, rather than relatively stable and discrete oligomers species, as the primary processes that mediate Aβ toxicity.
Viability of the Cultured Cells Is Critically Impaired before the Emergence of Mature Aβ42 Fibrils
To better understand the relationship between the process of amyloid formation and Aβ42 toxicity, we treated cultured PC12 cells with Aβ42 CR and sought to correlate the Aβ42 aggregation state in the culture medium with cell viability at different time points. Fig. 4A shows that Aβ42 CR exhibited a gradual rise in ThT binding within the first 9 h and then remained relatively unchanged. Interestingly, a gradual and early decline in MTT reduction by Aβ42 CR-treated cells was also observed during the initial 6 h (Fig. 4B, solid line, ≥30% decrease compared with time 0, p < 0.01), whereas a significant rise of lactate dehydrogenase in the culture medium (≥80%, p < 0.005 compared with time 0) was only observed after ∼6 h of incubation (Fig. 4B, dashed line). TEM analysis of the culture medium at ∼3 h showed clusters of protofibrils and spherical aggregates (Fig. 4C). At ∼6 h, the protofibrils were more elongated and had an immature fibril-like appearance (Fig. 4D). Mature fibrils started to appear at ∼9 h (Fig. 4E) and were more abundant at ∼24 h (Fig. 4F). These observations suggested that impairment of cell viability, indicated by a decline in the reduction of MTT, a marker for mitochondrial metabolism (34), was affected by the process of Aβ42 protofibril elongation, in the presence of free monomers, rather than the formation of mature fibrils. In other words, toxicity is mediated by nonfibrillar Aβ42 aggregates formed as a result of an ongoing process of protofibril elongation, whereas the emergence of mature, high ThT binding fibrils is a later event.
FIGURE 4.
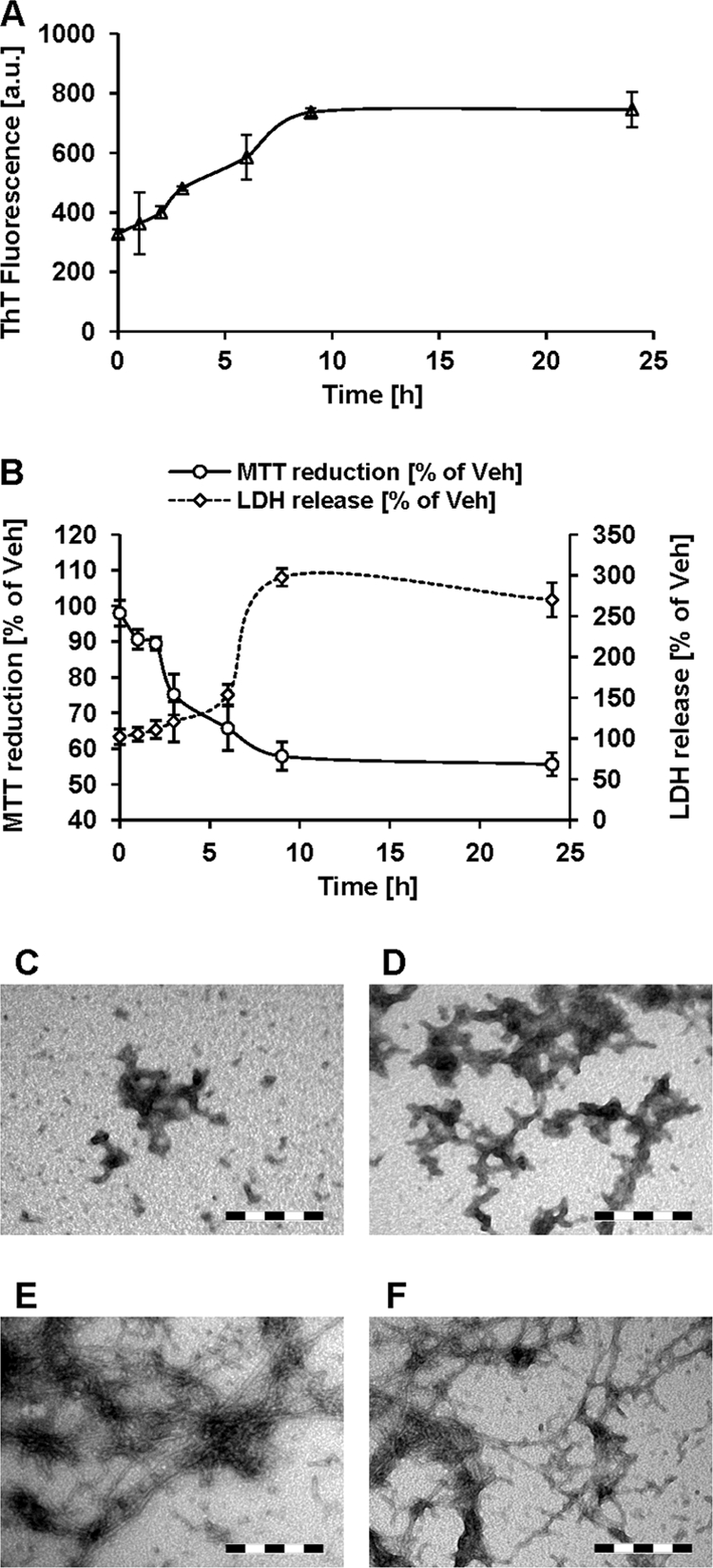
Time course of Aβ42 fibril formation and toxicity. A, ThT binding over time by 10 μm Aβ42 CR protofibrils in DMEM (37 °C; mean ± S.D.). B, PC12 cell viability (MTT reduction assay and quantification of lactate dehydrogenase release in the medium) after treatment with 10 μm Aβ42 CR. For the MTT assay, at the indicated time points, the cultured medium was completely removed and replaced with MTT assay solution. For lactate dehydrogenase release assay, aliquots of the phenol red-free culture medium were removed from each well at the indicated time points. The data are expressed as the percentage of the buffer vehicle (Veh) (mean ± S.D.). C–F, representative TEM images of Aβ42 CR in culture medium aliquots obtained at 3 h (C), 6 h (D), 9 h (E), and 24 h (F) (scale bar in C–F = 200 nm).
Crude Aβ42 Preparations, but Not Monomers, Protofibrils, or Fibrils Alone, Increased Glucose Utilization by Cultured Astrocytes
We have previously reported that treatment of cultured astrocytes with Aβ42, at comparable concentrations as used for cell viability measurements in the experiments described above, does not induce significant cell death but alters the astrocyte metabolic activity (25). We observed that Aβ42 significantly enhances glucose utilization (uptake and phosphorylation), as assessed by 2-[3H]DG uptake, by cultured astrocytes as compared with Aβ40 (25). We sought to investigate whether this phenomenon was attributable to particular Aβ42 assembly state(s) i.e. monomers, protofibrils, or fibrils, and/or was influenced by the dynamics of the Aβ42 aggregation process. Treatment of cultured astrocytes with defined Aβ aggregates (fibrils and SEC-purified monomers and protofibrils) had a negligible effect on glucose utilization by astrocytes (Fig. 5). However, treatment with an Aβ42 CR preparation, which contained substantial amounts of protofibril-abundant monomers, caused a significant (p < 0.005) increase in glucose utilization (Fig. 5). These observations suggest that the dynamics of the Aβ42 fibrillization process bear important implications for not only neuronal viability but also neuron-glia metabolic coupling in AD pathogenesis.
FIGURE 5.
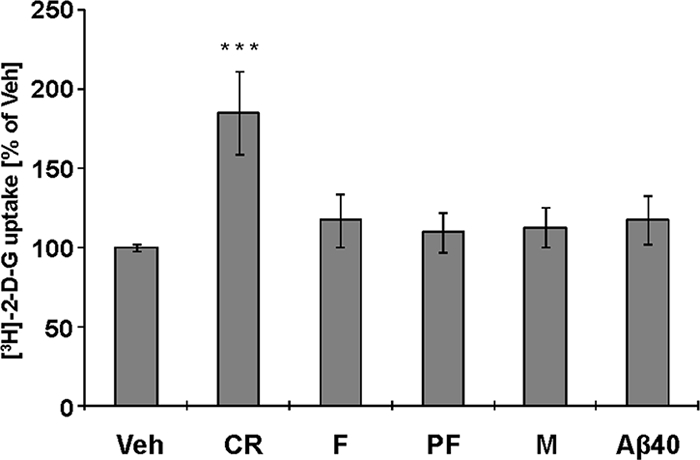
Effect of Aβ42 fibril formation on glucose utilization by cultured astrocytes. 2-[3H]DG utilization by cultured (DIV 21) mouse astrocytes after 24 h of treatment with Aβ42 CR protofibrils, F, fractionated PF, and M (10 μm final Aβ concentration). Monomeric Aβ40 (10 μm) was included for comparison (n = 16 from eight independent experiments, ANOVA followed by a Dunnett's post hoc test; ***, p < 0.005, mean ± S.E.; Veh, buffer vehicle).
DISCUSSION
The identification of toxic Aβ species and/or the process of their formation is crucial for understanding the mechanism(s) of Aβ neurotoxicity in AD and development of effective diagnostic and therapeutic interventions (35). Several lines of evidence implicate prefibrillar Aβ oligomers, including protofibrils, as the primary neurotoxins in AD (9, 36). Aiming to identify specific toxic Aβ species, we and others have developed reproducible protocols to isolate prefibrillar aggregates of defined size and morphology distributions (22, 24, 30). Herein, we extended our separation protocols to further subfractionate the prefibrillar oligomers and evaluated their fibrillization and toxicity compared with the monomeric and fibrillar preparations of Aβ42. We demonstrate that the crude Aβ42 preparations, containing monomeric and heterogeneous mixture of Aβ42 oligomers, are more toxic than purified monomeric, protofibrillar fractions or fibrils. Subfractionation of protofibril preparations, to separate different protofibril species, resulted in further attenuation of their toxicity (Figs. 1 and 2). The diminished toxicity of purified and subfractionated Aβ42 protofibrils was strongly linked to their reduced fibrillization, thus indicating that Aβ42 toxicity is not necessarily linked to specific prefibrillar aggregate(s) but rather to the ability of these species to grow in the presence of monomeric Aβ42. Consistent with this hypothesis, reintroduction of monomeric Aβ42 into these fractions restored amyloid formation and enhanced their toxicity to comparable levels as observed with the crude preparations. Furthermore, selective removal of monomeric Aβ42 from these preparations using IDE reversed the toxicity of Aβ42 protofibrils (Fig. 3). In parallel, we demonstrate that the crude Aβ42 preparations were more potent in inducing metabolic alterations in cultured astrocytes as compared with the purified monomers, protofibrils, and fibrils (Fig. 5). Finally, we also demonstrate that promoting the fibrillization of Aβ40, the major Aβ variant in vivo, also results in increased cytotoxicity to the cultured cells (supplemental Fig. 6). Together, these findings provide strong evidence that Aβ toxicity is linked to an ongoing Aβ polymerization process and is greatly reduced when the polymerization process is slowed down by the selective removal/degradation of Aβ monomers (Figs. 1–3).
Monomer-Protofibril Interactions Are Key Determinants of Aβ42 Fibrillization and Toxicity
Consistent with a nucleated polymerization process, the fibrillization of Aβ42 protofibrils, or lack thereof, was strongly influenced by the availability of monomers. Cell toxicity experiments revealed that Aβ42 toxicity was enhanced or reduced depending on the fibrillization propensity of the protofibril or monomer preparations (Figs. 1–3). This is in agreement with observations that Aβ variants that exhibit a high aggregation propensity, e.g. Aβ42 or Aβ-arctic, also induce greater toxicity in cultured neurons as compared with slower aggregating Aβ variants, e.g. Aβ40 (37–39). Similarly, accelerating the fibrillogenesis of soluble Aβ40 preparations by adding exogenous Aβ40 fibrils also enhances their toxicity (14). Our data suggest that the fibrillization and toxicity of Aβ42 protofibrils is dependent on the presence of monomers and not on fibrillar forms of Aβ. The presence of fibrillar seeds contributes, but is not essential, to the toxicity seen with mixtures of Aβ42 protofibrils and monomers (supplemental Fig. 5).
Conversion of Aβ protofibrils into fibrils occurs by incorporation of monomers into the growing ends. Therefore, removal of monomers from solution is expected to enhance protofibril stability and reduce the rate of their fibrillization. Our findings suggest that kinetic stabilization of protofibrils would reduce their toxicity. This hypothesis is also supported by the observations that enhancing the kinetic stability of Aβ42 protofibrils by the addition of Aβ40 monomers significantly reduces their toxicity toward cultured neurons (27). Similarly, small molecules that stabilize Aβ protofibrils in vitro have also been shown to improve behavioral performance in APP transgenic mice (40). The critical requirement for Aβ monomers to maintain Aβ fibrillization, and associated toxicity in vitro and in vivo, is also underscored by the overexpression of IDE in APP transgenic animals. These studies revealed that overexpressing IDE in these animals ameliorates plaque pathology, improves behavioral performance, and prevents premature death (32, 41). Therefore, the concentration of monomeric Aβ plays important roles in modulating the amyloid formation and toxicity of Aβ peptides. Intriguingly, we found that IDE was less efficient at improving the survival of Aβ42 crude treated cells as compared with the cells treated with monomer/protofibril mixtures (compare Fig. 3C with supplemental Fig. 4B). We speculate that this can possibly result from the preferential incorporation of Aβ42 monomers into protofibrils and/or binding of the IDE to Aβ oligomers but less efficient degradation due to the secondary structure elements (42).
The exact mechanisms by which the process of amyloid formation contributes to Aβ toxicity and neurodegeneration in AD remain unknown. Our results suggest that the formation of the toxic Aβ species could occur on site and is mediated by other cellular factors that interact with, or mediate the interactions between, protofibrils and monomers or respond to their fibrillization. Given that Aβ species were administered extracellularly, it is plausible to postulate that fibrillization events take place on the cell membrane where the process of amyloid formation triggers downstream intracellular cascade of events culminating in cell death. This hypothesis is supported by the findings that association of Aβ to membrane gangliosides also promotes Aβ fibrillization on the cell surface and induces toxicity (43). Alternatively, it is also possible that certain cell surface receptors are activated due to the binding of protofibrils (44) or their fibrillization, which triggers cellular pathways compromising neuronal survival capabilities.
In addition to Aβ40, Wogulis et al. (14) also demonstrated that mixtures of soluble (nonfibrillar) and insoluble (fibrillar) fractions of IAPP were more toxic to the cultured neurons than fibrillar and nonfibrillar fractions alone. A recent compelling model for IAPP toxicity directly links the processes of amyloid aggregation on membrane surfaces with cell-membrane disruption and the onset of toxicity (45, 46). In other words, as the fibril develops on the membrane surface, the structural integrity of the membrane is simultaneously compromised. In an elegant study, Engel et al. (45) demonstrated the synchronization of the kinetic profiles for human IAPP-fibril growth, which was monitored by ThT fluorescence, and the induction of dye leakage from coincubated mixed 1,2-dioleoyl-sn-glycero-3-phosphocholine/1,2-dioleoyl-sn-glycero-3-phospho-l-serine vesicles. The two profiles were characterized by a lag phase of ∼3 h followed by a sigmoidal transition to human IAPP fibrils and near-complete dye leakage from the vesicles. This result indicates that it is the process of fibril formation on membrane surfaces that is responsible for abolishing the membrane barrier function. Cryogenic electron microscopy images of 1,2-dioleoyl-sn-glycero-3-phosphocholine/1,2-dioleoyl-sn-glycero-3-phospho-l-serine large unilamellar vesicles coincubated with human IAPP showed distortion and pinching of regions of the membrane in contact with fibrils, whereas vesicles incubated in the presence of nonamyloidogenic mouse IAPP remained unperturbed (45). As a mechanistic model, the authors proposed that IAPP-fibril growth on the membrane occurred concomitantly with a forced change in membrane curvature and weakened lipid packing, which enabled the leakage of intravesicular contents. Interestingly, this notion is consistent with our findings, which indicate that it is not one specific oligomeric state that induces cell death and toxicity but rather the dynamic process of fibril formation.
Alternatively, the uptake and internalization of soluble Aβ (monomers and protofibrils) may impair cellular energy metabolism (intracellular toxicity). This is supported by some recent reports demonstrating that Aβ in culture medium is internalized and aggregated intracellularly into fibrils, eventually causing membrane disruption (47, 48). Further studies are required to dissect the structural and molecular mechanisms underlying the critical role of protofibril-monomer interactions in Aβ toxicity. In addition to enhancing neuronal vulnerability, the process of Aβ amyloid formation also seems to interfere with the regulation of glial energy metabolism as indicated by enhanced glucose utilization by astrocytes (Fig. 5). Importantly, such metabolic changes in astrocytes are associated with deleterious consequences for neuronal viability (25) and bear important implications for neuron-glia metabolic coupling (49). Thus, our results demonstrate that accelerated Aβ amyloidogenesis triggers a host of mechanisms, involving both neurons and glia that converge on triggering neuronal dysfunction and eventual demise.
Implications for Therapies in AD
The possibility that an ongoing Aβ polymerization process, rather than specific aggregate of defined size or structure, strongly determines Aβ neurotoxicity has important implications for understanding the role of Aβ aggregation in AD pathogenesis and the design of anti-Aβ therapeutics. Our results provide direct evidence linking Aβ toxicity to the growth and fibrillization of Aβ oligomers in the presence of Aβ monomers. These studies suggest that anti-Aβ therapeutic strategies, many of which are currently being tested in animal models or in clinical trials in AD patients, hold potential as promising disease-modifying interventions. These approaches are based on the following: 1) reducing the production of Aβ monomers (50–52); 2) sequestering Aβ monomers and oligomers to promote their clearance (53, 54); 3) degrading of Aβ monomers and oligomers (41, 55); and 4) preventing Aβ oligomers from stably binding to the neuronal membranes (40, 56).
Reducing the monomer concentration in vivo is likely to attenuate Aβ toxicity not only by preventing the formation of additional toxic oligomers but also by reducing the growth of circulating oligomers, including protofibrils, and promoting their clearance from brain. Proof-of-principle experiments clearly show that even minimal (∼15%) reduction in monomer Aβ production have profound effects on amyloid pathology and related deficits in APP transgenic animals (50–52). In the absence of free monomers, the unstable nuclei would cease to grow and possibly disintegrate. Experimental evidence for the latter stipulation can be found in reports showing that sequestration of Aβ monomers using a small engineered protein (ZAβ3) (57) led to gradual dissolution of preexisting oligomers in vitro and promotes amyloid clearance in vivo (57). The administration of therapeutic anti-Aβ antibodies that sequester Aβ monomers and oligomers or Aβ vaccination to generate such antibodies in the host prevents amyloid formation (53, 58), reduces plaque burden in older plaque-bearing mice (54, 58), and improves behavioral performance (59, 60).
Development of small molecules that inhibit amyloid formation by stabilizing the monomeric state of Aβ has not been successful due to the conformational heterogeneity and flexibility of the protein. Our findings suggest that small molecules targeting the growth of oligomers/protofibrils and fibrils, by preventing monomer incorporation into the growing ends of these species, would constitute a more effective strategy to block and/or reverse amyloid formation and toxicity in vivo. Blocking oligomer growth by capping the ends or binding to their surfaces can be achieved at substoichiometric levels (27), whereas molecules that stabilize the monomers to prevent self-assembly would have to be used at stoichiometric amounts.
Some studies have shown that Aβ immunotherapy improves behavioral performance in APP transgenic animals without decreasing total (soluble and insoluble) Aβ burden (59, 61). Although the latter observations seem to challenge the relevance of the Aβ amyloid cascade hypothesis to AD pathogenesis, our findings serve to reconcile such discrepancies and suggest that the amyloid cascade hypothesis has yet to be disproved. Our data support the notion that a modest reduction in soluble Aβ (monomers and oligomers) and the disruption of further nucleation and/or polymerization events would neutralize protofibril toxicity, facilitate amyloid clearance from brain, and abrogate the spreading of amyloid pathology. In conclusion, targeting the nucleated polymerization of amyloid-forming proteins offers an exciting framework for the development of anti-amyloid therapies for a host of neurodegenerative diseases characterized by the pathological accumulation of protein aggregates, including AD, Parkinson disease, and prion diseases. We hope that these findings will stimulate further research into understanding the role of the process of amyloid formation in neurodegenerative diseases and the development of in vitro and in vivo mechanistic models to design and evaluate intervention strategies.
Supplementary Material
Acknowledgments
We thank Jia Cui, Xiaqin Sun, John Perrin, and Yvan Varisco for helping with SEC fractionation, preparation of primary neuronal cultures, and/or cell culture toxicity assays.
This work was supported by the Swiss Federal Institute of Technology Lausanne and Grant 310000-110027 from the Swiss National Foundation.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–6.
- Aβ
- amyloid-β
- AD
- Alzheimer disease
- TEM
- transmission electron microscopy
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- SEC
- size exclusion chromatography
- 2-DG
- 2-deoxy-d-glucose
- ANOVA
- analysis of variance
- ThT
- thioflavin-T
- CR
- crude Aβ42
- DIV
- day in vitro
- M
- monomer
- F
- fibril
- PF
- protofibrils
- IDE
- insulin-degrading enzyme
- SF
- sonicated fibril
- Ana-Sec
- analytical size exclusion chromatography
- APP
- amyloid precursor protein
- IAPP
- islet amyloid polypeptide.
REFERENCES
- 1. Glenner G. G., Wong C. W., Quaranta V., Eanes E. D. (1984) Appl. Pathol. 2, 357–369 [PubMed] [Google Scholar]
- 2. Merz P. A., Wisniewski H. M., Somerville R. A., Bobin S. A., Masters C. L., Iqbal K. (1983) Acta Neuropathol. 60, 113–124 [DOI] [PubMed] [Google Scholar]
- 3. Selkoe D. J., Schenk D. (2003) Annu. Rev. Pharmacol. Toxicol. 43, 545–584 [DOI] [PubMed] [Google Scholar]
- 4. Hardy J., Selkoe D. J. (2002) Science 297, 353–356 [DOI] [PubMed] [Google Scholar]
- 5. Selkoe D. J. (2001) Physiol. Rev. 81, 741–766 [DOI] [PubMed] [Google Scholar]
- 6. Sisodia S. S. (1992) Ann. N.Y. Acad. Sci. 674, 53–57 [DOI] [PubMed] [Google Scholar]
- 7. Jarrett J. T., Lansbury P. T., Jr. (1993) Cell 73, 1055–1058 [DOI] [PubMed] [Google Scholar]
- 8. Harper J. D., Lieber C. M., Lansbury P. T., Jr. (1997) Chem. Biol. 4, 951–959 [DOI] [PubMed] [Google Scholar]
- 9. Haass C., Selkoe D. J. (2007) Nat. Rev. Mol. Cell Biol. 8, 101–112 [DOI] [PubMed] [Google Scholar]
- 10. Walsh D. M., Selkoe D. J. (2004) Protein Pept. Lett. 11, 213–228 [DOI] [PubMed] [Google Scholar]
- 11. Kodali R., Wetzel R. (2007) Curr. Opin. Struct. Biol. 17, 48–57 [DOI] [PubMed] [Google Scholar]
- 12. Lashuel H. A., Hartley D. M., Petre B. M., Wall J. S., Simon M. N., Walz T., Lansbury P. T., Jr. (2003) J. Mol. Biol. 332, 795–808 [DOI] [PubMed] [Google Scholar]
- 13. Hepler R. W., Grimm K. M., Nahas D. D., Breese R., Dodson E. C., Acton P., Keller P. M., Yeager M., Wang H., Shughrue P., Kinney G., Joyce J. G. (2006) Biochemistry 45, 15157–15167 [DOI] [PubMed] [Google Scholar]
- 14. Wogulis M., Wright S., Cunningham D., Chilcote T., Powell K., Rydel R. E. (2005) J. Neurosci. 25, 1071–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kane M. D., Lipinski W. J., Callahan M. J., Bian F., Durham R. A., Schwarz R. D., Roher A. E., Walker L. C. (2000) J. Neurosci. 20, 3606–3611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Eisele Y. S., Bolmont T., Heikenwalder M., Langer F., Jacobson L. H., Yan Z. X., Roth K., Aguzzi A., Staufenbiel M., Walker L. C., Jucker M. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 12926–12931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Meyer-Luehmann M., Coomaraswamy J., Bolmont T., Kaeser S., Schaefer C., Kilger E., Neuenschwander A., Abramowski D., Frey P., Jaton A. L., Vigouret J. M., Paganetti P., Walsh D. M., Mathews P. M., Ghiso J., Staufenbiel M., Walker L. C., Jucker M. (2006) Science 313, 1781–1784 [DOI] [PubMed] [Google Scholar]
- 18. Kayed R., Pensalfini A., Margol L., Sokolov Y., Sarsoza F., Head E., Hall J., Glabe C. (2009) J. Biol. Chem. 284, 4230–4237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Harper J. D., Wong S. S., Lieber C. M., Lansbury P. T. (1997) Chem. Biol. 4, 119–125 [DOI] [PubMed] [Google Scholar]
- 20. Walsh D. M., Lomakin A., Benedek G. B., Condron M. M., Teplow D. B. (1997) J. Biol. Chem. 272, 22364–22372 [DOI] [PubMed] [Google Scholar]
- 21. Bitan G., Kirkitadze M. D., Lomakin A., Vollers S. S., Benedek G. B., Teplow D. B. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 330–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lambert M. P., Barlow A. K., Chromy B. A., Edwards C., Freed R., Liosatos M., Morgan T. E., Rozovsky I., Trommer B., Viola K. L., Wals P., Zhang C., Finch C. E., Krafft G. A., Klein W. L. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 6448–6453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bitan G. (2006) Methods Enzymol. 413, 217–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jan A., Hartley D. M., Lashuel H. A. (2010) Nat. Protoc. 5, 1186–1209 [DOI] [PubMed] [Google Scholar]
- 25. Allaman I., Gavillet M., Bélanger M., Laroche T., Viertl D., Lashuel H. A., Magistretti P. J. (2010) J. Neurosci. 30, 3326–3338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sato T., Kienlen-Campard P., Ahmed M., Liu W., Li H., Elliott J. I., Aimoto S., Constantinescu S. N., Octave J. N., Smith S. O. (2006) Biochemistry 45, 5503–5516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jan A., Gokce O., Luthi-Carter R., Lashuel H. A. (2008) J. Biol. Chem. 283, 28176–28189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pace C. N., Vajdos F., Fee L., Grimsley G., Gray T. (1995) Protein Sci. 4, 2411–2423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Meberg P. J., Miller M. W. (2003) Methods Cell Biol. 71, 111–127 [DOI] [PubMed] [Google Scholar]
- 30. Walsh D. M., Hartley D. M., Kusumoto Y., Fezoui Y., Condron M. M., Lomakin A., Benedek G. B., Selkoe D. J., Teplow D. B. (1999) J. Biol. Chem. 274, 25945–25952 [DOI] [PubMed] [Google Scholar]
- 31. Walsh D. M., Klyubin I., Fadeeva J. V., Rowan M. J., Selkoe D. J. (2002) Biochem. Soc. Trans. 30, 552–557 [DOI] [PubMed] [Google Scholar]
- 32. Leissring M. A., Farris W., Chang A. Y., Walsh D. M., Wu X., Sun X., Frosch M. P., Selkoe D. J. (2003) Neuron 40, 1087–1093 [DOI] [PubMed] [Google Scholar]
- 33. Leissring M. A. (2008) J. Biol. Chem. 283, 29645–29649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Morgan D. M. (1998) Methods Mol. Biol. 79, 179–183 [DOI] [PubMed] [Google Scholar]
- 35. Lansbury P. T., Lashuel H. A. (2006) Nature 443, 774–779 [DOI] [PubMed] [Google Scholar]
- 36. Selkoe D. J. (2008) Behav. Brain Res. 192, 106–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Whalen B. M., Selkoe D. J., Hartley D. M. (2005) Neurobiol. Dis. 20, 254–266 [DOI] [PubMed] [Google Scholar]
- 38. Nilsberth C., Westlind-Danielsson A., Eckman C. B., Condron M. M., Axelman K., Forsell C., Stenh C., Luthman J., Teplow D. B., Younkin S. G., Näslund J., Lannfelt L. (2001) Nat. Neurosci. 4, 887–893 [DOI] [PubMed] [Google Scholar]
- 39. Pike C. J., Burdick D., Walencewicz A. J., Glabe C. G., Cotman C. W. (1993) J. Neurosci. 13, 1676–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hawkes C. A., Deng L. H., Shaw J. E., Nitz M., McLaurin J. (2010) Eur. J. Neurosci. 31, 203–213 [DOI] [PubMed] [Google Scholar]
- 41. Farris W., Mansourian S., Chang Y., Lindsley L., Eckman E. A., Frosch M. P., Eckman C. B., Tanzi R. E., Selkoe D. J., Guenette S. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 4162–4167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Morelli L., Llovera R., Gonzalez S. A., Affranchino J. L., Prelli F., Frangione B., Ghiso J., Castano E. M. (2003) J. Biol. Chem. 278, 23221–23226 [DOI] [PubMed] [Google Scholar]
- 43. Okada T., Ikeda K., Wakabayashi M., Ogawa M., Matsuzaki K. (2008) J. Mol. Biol. 382, 1066–1074 [DOI] [PubMed] [Google Scholar]
- 44. Yan S. D., Chen X., Fu J., Chen M., Zhu H., Roher A., Slattery T., Zhao L., Nagashima M., Morser J., Migheli A., Nawroth P., Stern D., Schmidt A. M. (1996) Nature 382, 685–691 [DOI] [PubMed] [Google Scholar]
- 45. Engel M. F., Khemtémourian L., Kleijer C. C., Meeldijk H. J., Jacobs J., Verkleij A. J., de Kruijff B., Killian J. A., Höppener J. W. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 6033–6038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Friedman R., Pellarin R., Caflisch A. (2009) J. Mol. Biol. 387, 407–415 [DOI] [PubMed] [Google Scholar]
- 47. Friedrich R. P., Tepper K., Rönicke R., Soom M., Westermann M., Reymann K., Kaether C., Fändrich M. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 1942–1947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hu X., Crick S. L., Bu G., Frieden C., Pappu R. V., Lee J. M. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 20324–20329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Magistretti P. J. (2006) J. Exp. Biol. 209, 2304–2311 [DOI] [PubMed] [Google Scholar]
- 50. Lanz T. A., Himes C. S., Pallante G., Adams L., Yamazaki S., Amore B., Merchant K. M. (2003) J. Pharmacol. Exp. Ther. 305, 864–871 [DOI] [PubMed] [Google Scholar]
- 51. McConlogue L., Buttini M., Anderson J. P., Brigham E. F., Chen K. S., Freedman S. B., Games D., Johnson-Wood K., Lee M., Zeller M., Liu W., Motter R., Sinha S. (2007) J. Biol. Chem. 282, 26326–26334 [DOI] [PubMed] [Google Scholar]
- 52. Barten D. M., Guss V. L., Corsa J. A., Loo A., Hansel S. B., Zheng M., Munoz B., Srinivasan K., Wang B., Robertson B. J., Polson C. T., Wang J., Roberts S. B., Hendrick J. P., Anderson J. J., Loy J. K., Denton R., Verdoorn T. A., Smith D. W., Felsenstein K. M. (2005) J. Pharmacol. Exp. Ther. 312, 635–643 [DOI] [PubMed] [Google Scholar]
- 53. McLaurin J., Cecal R., Kierstead M. E., Tian X., Phinney A. L., Manea M., French J. E., Lambermon M. H., Darabie A. A., Brown M. E., Janus C., Chishti M. A., Horne P., Westaway D., Fraser P. E., Mount H. T., Przybylski M., St George-Hyslop P. (2002) Nat. Med. 8, 1263–1269 [DOI] [PubMed] [Google Scholar]
- 54. DeMattos R. B., Bales K. R., Cummins D. J., Dodart J. C., Paul S. M., Holtzman D. M. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 8850–8855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kanemitsu H., Tomiyama T., Mori H. (2003) Neurosci. Lett. 350, 113–116 [DOI] [PubMed] [Google Scholar]
- 56. Aisen P. S., Gauthier S., Vellas B., Briand R., Saumier D., Laurin J., Garceau D. (2007) Curr. Alzheimer Res. 4, 473–478 [DOI] [PubMed] [Google Scholar]
- 57. Luheshi L. M., Hoyer W., de Barros T. P., van Dijk Härd I., Brorsson A. C., Macao B., Persson C., Crowther D. C., Lomas D. A., Ståhl S., Dobson C. M., Härd T. (2010) PLoS Biol. 8, e1000334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schenk D., Barbour R., Dunn W., Gordon G., Grajeda H., Guido T., Hu K., Huang J., Johnson-Wood K., Khan K., Kholodenko D., Lee M., Liao Z., Lieberburg I., Motter R., Mutter L., Soriano F., Shopp G., Vasquez N., Vandevert C., Walker S., Wogulis M., Yednock T., Games D., Seubert P. (1999) Nature 400, 173–177 [DOI] [PubMed] [Google Scholar]
- 59. Janus C., Pearson J., McLaurin J., Mathews P. M., Jiang Y., Schmidt S. D., Chishti M. A., Horne P., Heslin D., French J., Mount H. T., Nixon R. A., Mercken M., Bergeron C., Fraser P. E., St George-Hyslop P., Westaway D. (2000) Nature 408, 979–982 [DOI] [PubMed] [Google Scholar]
- 60. Morgan D., Diamond D. M., Gottschall P. E., Ugen K. E., Dickey C., Hardy J., Duff K., Jantzen P., DiCarlo G., Wilcock D., Connor K., Hatcher J., Hope C., Gordon M., Arendash G. W. (2000) Nature 408, 982–985 [DOI] [PubMed] [Google Scholar]
- 61. Dodart J. C., Bales K. R., Gannon K. S., Greene S. J., DeMattos R. B., Mathis C., DeLong C. A., Wu S., Wu X., Holtzman D. M., Paul S. M. (2002) Nat. Neurosci. 5, 452–457 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.