

Abstract
Class switch DNA recombination (CSR) from IgM to IgG and IgA is crucial for antiviral immunity. Follicular B cells undergo CSR upon engagement of CD40 by CD40 ligand on CD4+ T cells. This T cell-dependent pathway requires 5–7 days, which is too much of a delay to block quickly replicating pathogens. To compensate for this limitation, extrafollicular B cells rapidly undergo CSR through a T cell-independent pathway that involves innate Ag receptors of the TLR family. We found that a subset of upper respiratory mucosa B cells expressed TLR3 and responded to viral dsRNA, a cognate TLR3 ligand. In the presence of dsRNA, mucosal B cells activated NF-κB, a transcription factor critical for CSR. Activation of NF-κB required TRIF (Toll/IL-1R domain-containing protein inducing IFN-β), a canonical TLR3 adapter protein, and caused germline transcription of downstream CH genes as well as expression of AID (activation-induced cytidine deaminase), a DNA-editing enzyme essential for CSR. Subsequent IgG and IgA production was enhanced by BAFF (B cell-activating factor of the TNF family), an innate mediator released by TLR3-expressing mucosal dendritic cells. Indeed, these innate immune cells triggered IgG and IgA responses upon exposure to dsRNA. By showing active TLR3 signaling and ongoing CSR in upper respiratory mucosa B cells from patients with CD40 signaling defects, our findings indicate that viral dsRNA may initiate frontline IgG and IgA responses through an innate TLR3-dependent pathway involving BAFF.
Class switching from IgM to IgG and IgA is essential to neutralize viruses systemically and at mucosal sites of entry (1). Most Ags initiate class switching by up-regulating CD40 ligand (CD40L)3 on CD4+ T cells (2). Engagement of CD40 on B cells by CD40L triggers a germinal center (GC) reaction that fosters Ig gene diversification through class switch DNA recombination (CSR) and somatic hypermutation (3). CSR substitutes the CH region of IgM with that of IgG, IgA or IgE, thereby modulating the effector functions of Abs without changing their specificity for Ag (4). Somatic hypermutation increases the affinity of Abs for Ag by introducing point mutations in the V(D)J genes encoding the VH and VL regions of Abs (5).
Although generating memory B cells and plasma cells expressing high-affinity Abs (6), T cell-dependent B cell responses require 5–7 days, which is too much of a delay to quickly neutralize replicating pathogens. To compensate for this limitation, specialized B cell subsets such as B-1 and splenic marginal zone B cells rapidly produce IgM as well as class-switched IgG and IgA through a T cell-independent (TI) pathway involving B cell-activating factor of the TNF family (BAFF; also know as BLyS) and a proliferation-inducing ligand (APRIL) (7–9). This TI pathway yields not only low-affinity and polyreactive Abs encoded by unmutated V(D)J genes (10–12) but also high-affinity carbohydrate-specific Abs encoded by mutated V(D)J genes, at least in humans (13). By recognizing highly conserved microbial molecular patterns, including neutralizing epitopes on viral envelopes, TI Ab responses would provide a first line of defense against infections (11, 14–16).
Growing evidence indicates that dendritic cells (DCs) play an important role in the initiation of TI Ab responses. DCs release innate B cell-stimulating and Ab-inducing factors, including BAFF and APRIL, after sensing microbes through TLRs (17–21). In addition to stimulating various DC subsets (22–24), TLRs activate B cells (19, 25). A typical TLR with powerful B cell-activating functions is TLR9, an innate Ag receptor that binds hypomethylated deoxycytidylate-phosphate-deoxyguanylate (CpG)-rich DNA from viruses and bacteria (26–28). Although sufficient to stimulate CSR (29–31), CpG DNA requires BAFF or APRIL to induce optimal IgG and IgA production (29, 32). Of note, BAFF and APRIL are released by myeloid DCs (mDCs) in response to IFN-α, which is in turn produced by plasmacytoid dendritic cells (pDCs) exposed to TLR9 ligands (23, 33–35). Thus, TI IgG and IgA responses to viruses may involve a cross-talk between B cells and various DC subsets via TLRs.
TLR9 initiates CSR through NF-κB (29, 30). This transcription factor is retained in an inactive cytoplasmic form by IκB (inhibitor of NF-κB) proteins (36). By recruiting MyD88, TNFR-associated factor 6 (TRAF6) and activating IL-1R-activated kinases (IRAKs), TLR9 activates IκBα kinase (IKK), which phosphorylates IκB (22). The ensuing IκB degradation permits nuclear translocation of NF-κB. Binding of NF-κB to κB sites on intronic (I) promoters upstream of targeted CH genes initiates transcription of these genes (29, 30). This early CSR event is followed by the recruitment of activation-induced cytidine deaminase (AID) to switch (S) regions intercalated between an I exon and the CH gene (2, 4). AID promotes IgG CSR by favoring the formation of double-strand DNA breaks within targeted S regions (2–4). Of note, TLR9 signaling via NF-κB not only initiates germline IH-CH transcription but also up-regulates the expression of AID (29).
Although most TLRs signal via MyD88, the viral dsRNA receptor TLR3 requires the adapter protein Toll/IL-1R domain-containing protein inducing IFN-β (TRIF) (22, 37, 38). In mice, DCs and B cells express TLR3 and undergo TRIF-dependent activation of NF-κB upon exposure to viral dsRNA (37). Human TLR3 is positive in mDCs and in some B cells (20, 33, 39, 40), raising the possibility that viral dsRNA triggers TI Ab responses in humans as it does in mice (37, 41).
We found that human upper respiratory mucosa B cells expressed functional TLR3. In the presence of dsRNA, these B cells initiated germline CH gene transcription through an NF-κB-dependent signaling pathway requiring TRIF but not MyD88. Up-regulation of AID expression, CSR as well as IgG and IgA production, involved IL-10 and BAFF, two B cell-stimulating factors released by TLR3-expressing mDCs upon exposure to dsRNA. These in vitro findings were complemented by in vivo data showing active TLR3 signaling and ongoing CSR as well as IgG and IgA production in the mDC-rich subepithelial region of the upper respiratory tract of patients with CD40 signaling defects.
Materials and Methods
B cells
Human B cells were negatively selected from peripheral blood and tonsillar mononuclear cells with a commercially available kit (Miltenyi Biotec). Buffy coats from healthy donors were purchased at the New York Blood Center (New York, NY) and used to isolate PBMCs. Tonsillar mononuclear cells were obtained from tissue specimens of patients undergoing tonsillectomy for tonsillar hypertrophy. The Institutional Review Board of Weill Medical College of Cornell University (New York, NY) approved the use of tonsil specimens for this study, and patients provided informed consent. IgD+ B cells were magnetically sorted by the incubation of total B cells with a biotinylated IADB6 mAb to IgD (Southern Biotechnologies) and streptavidin microbeads (Miltenyi Biotec) specifically designed to minimize BCR cross-linking. All sorting procedures were done on ice. Tonsillar IgD+CD38−, IgD+CD38+, IgD−CD38+, and IgD−CD38− B cell subsets were sorted as reported (42). 2E2 is a TLR3-expressing subclone of CL-01, a human IgD+ B cell line that initiates germline Ig gene transcription after exposure to appropriate stimuli (29).
Dendritic cells
mDCs were obtained from peripheral blood monocytes. Briefly, monocytes were sorted with a biotinylated mAb to CD14 (Serotec) and streptavidin microbeads and then cultured for 6 days in RPMI 1640 medium (Invitrogen) supplemented with 5% human AB serum (Sigma-Aldrich), 1,000 U/ml GM-CSF (Berlex Laboratories), 1,000 U/ml IL-4, and 25 ng/ml TNF-α (R&D Systems). Every 2 days, 400 μl of medium was removed from each well and replaced by 500 μl of fresh medium with appropriate cytokines. After 6 days, >95% of the cells in culture expressed DC-specific Ags, including CD11c, DEC-205, and DC-SIGN (DC-specific ICAM-3-grabbing nonintegrin).
Cultures and reagents
Cultures were conducted in complete RPMI 1640 medium supplemented with 10% bovine serum. Recombinant human BAFF (Alexis Biochemicals), IL-10 (Schering-Plough), IFN-α (Sigma-Aldrich), and soluble trimeric CD40L (Immunex) were used at 250 ng/ml, 50 ng/ml, 200 ng/ml, and 250 ng/ml, respectively. A polyclonal Ab (pAb) to the μ-chain of surface Ig (Caltag Laboratories) and polyinosinic/cytidylic acid (poly(I:C)) were used at 2 μg/ml and 10 μg/ml, respectively, unless otherwise indicated.
Flow cytometry
Monocytes, mDCs, and B cells were stained with the fluorescein-, PE-, or allophycocyanin-conjugated mAbs 1D6 to BAFF, eB-h209 to DC-SIGN (eBioscience), HL3 to CD11c, MoP9 to CD14, 3/23 to CD40, 16-10A1 to CD80, HB-15e to CD83, GL-1 to CD86, M5-115 to HLA-II, MG38 to DEC-205, 19.2 to mannose receptor, HIT2 to CD38, 2H7 to CD20, IA6-2 to IgD (BD Pharmingen), and TLR.7 to TLR3 (Imgenex). Intracellular TLR3 and AID stainings were performed with B cells treated with a 0.2% saponin permeabilization buffer. AID was detected with pAb H80 (Santa Cruz Biotechnologies). Preliminary experiments showed that pAb H80 stained AID-positive Ramos B cells but not AID-negative 293 epithelial cells. At least 1 × 104 viable cells were acquired with a FACScalibur analyzer (BD Pharmingen). Histogram shifts were analyzed with the Kolmogorov-Smirnov test included in the CellQuest software (BD Pharmingen) and were considered statistically significant at a p < 0.001.
ELISAs
Total IgG, IgA, and IgM Abs as well as BAFF and IL-10 were detected by standard ELISAs as reported (18, 20). Readings were done at 450 nm.
B cell proliferation and survival assays
B cells (1 × 106/ml) were incubated for 10 min. at 37°C in prewarmed PBS containing 0.1% BSA and 1 μM CFSE (Renovar). Staining was quenched by adding 5 volumes of ice-cold culture medium and through incubation on ice for 5 min. Then, CFSE-stained B cells were washed twice with fresh medium, resuspended in complete medium, and finally seeded for in vitro culture. Proliferation was measured through flow cytometry by quantifying the percentage of B cells that had diluted CFSE after 6 days. Viability was measured through flow cytometry by quantifying the percentage of B cells impermeable to 7-amino-actinomycin D (7-AAD) (eBioscience).
Tissue samples
Tonsil tissue samples were obtained from donors with tonsillar hypertrophy and from 1 patient with hyper-IgM (HIGM) syndrome. This patient was an 11-mo old male with recurrent bacterial infections, intermittent anemia, mild neutropenia, thrombocytopenia, chronic splenomegaly, hepatomegaly, polyclonal B cell hyperplasia, elevated serum IgM (2880 mg/dL; normal range: 26–105), reduced total serum IgG (270 mg/dL; normal range: 277–746), reduced total serum IgA (11 mg/dL; normal range: 12–73), and reduced tetanus-specific serum IgG. Lymphocyte subpopulations were normal and phenotypic and molecular studies failed to identify common HIGM-associated gene defects. Intestinal tissue sections were obtained from a HIGM3 patient carrying a mutated TNFRSF5 gene encoding CD40. This CD40-deficient patient corresponds to “patient 1” described elsewhere (43). The Institutional Review Board of Weill Medical College of Cornell University approved the use of tissue specimens for this study and patients provided informed consent.
Immunohistochemistry
Paraformaldehyde-fixed frozen tissue sections 5-μm thick were stained with various combinations of the following primary Abs to human Ags: fluorescein-conjugated mouse mAb IA6-2 to IgD (BD Pharmingen); biotin-conjugated goat F(ab′)2 pAb 2032-01 to IgD, 2042-01 to IgG, and 2050-09 to IgA (Southern Biotechnologies); unconjugated mouse pAb MIB-1 to Ki-67 (Dako); unconjugated rat mAb 5B11 to CD123 (Serotec); unconjugated rat pAb EK2-5G9 to AID (Ascenion); biotin-conjugated mouse mAb 1D6 to BAFF (eBioscience); unconjugated rabbit pAb to BAFF (Upstate Biotechnologies); biotin-conjugated mouse mAb TLR3.7 to TLR3 (HyCult Biotechnology); and unconjugated rabbit pAb to a phosphorylated tyrosine residue (Tyr759) of TLR3 involved in TLR3 signaling (Imgenex). Control primary Abs with irrelevant binding activity included fluorescein-conjugated, biotin-conjugated, or unconjugated mouse IgG1 mAbs, unconjugated rat IgG2a and IgG2b mAbs, biotin-conjugated goat F(ab′)2 pAb, and unconjugated rabbit pAb. Slides were incubated with the following secondary reagents: indodicarbocyanine-conjugated anti-mouse pAb, rhodamine-conjugated anti-mouse pAb (Jackson ImmunoResearch Laboratories), Alexa Fluor 546/488-conjugated anti-goat pAb, Alexa Fluor 647-conjugated anti-rabbit pAb, and cyanine 3/5-conjugated or rhodamine-conjugated streptavidin (Molecular Probes). Nuclei were visualized with 4′,6′-diamidino-2-phenylindole (DAPI) (Boehringer Mannheim). Slides were applied with Slow Fade reagent (Molecular Probes). Images were acquired with an Axiovert 200M microscope (Carl Zeiss) supplied with an RTE/CCD-1300-Y/HS camera (Princeton Instruments) and × 5/0.15 numerical aperture (NA) dry, 10×/0.50 NA dry Ph1, ×40/1.3 NA oil DIC III, and 63×/1.4 NA oil objective lenses (Carl Zeiss). Acquired images were processed with Adobe Photoshop software CS2 for Macintosh, version 9 (Adobe Systems).
RT-PCR and quantitative real-time RT-PCR (QRT-PCR)
Total RNAs were prepared using TRIzol (Invitrogen). cDNA was synthesized from total RNA as described (18). TLR3, TLR4, TLR9, TNFSF13B, and ACTB transcripts were amplified by RT-PCR for 25 cycles as reported (20, 29, 44). QRT-PCR analyses were done in triplicate on an ABI PRISM 7900HT sequence detection system (Applied Biosystems) with the SYBR Green PCR kit from Applied Biosystems as instructed by the manufacturer. The amount of mRNA was normalized relative to the amount of ACTB mRNA. The generation of amplification products of only the correct size was confirmed by dissociation curve and agarose gel electrophoresis. The relative level of expression (RE) for a specific gene were calculated according to the equation: REn = 2−(ΔCtn − ΔCt1), ΔCt = Ct/test gene − Ct/ACTB, where Ct is cycle threshold, n is specific sample, and 1 is the sample with the lowest expression level. The following primer pairs were used: ACTB, 5′-GGATGCAGAAGGAGATCACT-3′ (forward) and 5′-CGATCCACACGGAGTACTTG-3′ (reverse); AICDA, 5′-AGAGGCGTGACAGTGCTACA-3′ (forward) and 5′-TGTAGCGGAGGAAGAGCAAT-3′ (reverse); Iμ-Cμ, 5′-GTGATTAAGGAGAAACACTTTGAT-3′ (forward) and 5′-AGACGAGGGGGAAAAGGGTT-3′ (reverse); Iγ1-Cγ1, 5′-GACCTGAGCTCAGGAGGCAGCAGAGACC-3′ (forward) and 5′-GAAGACCGATGGGCCCTTGGTGGA-3′ (reverse); TLR3, 5′-TGACTGAACTCCATCTCATGTCC-3′ (forward) and 5′-CCATTATGAGACAGATCTAATGTG-3′ (reverse); and TNFSF13b, 5′-ACCGCGGGACTGAAAATCT-3′(forward) and 5′-CACGCTTATTTCTGCTGTTCTGA-3′ (reverse).
Genomic DNA PCRs
Genomic DNA was extracted by using the QIAmp DNA mini kit (Qiagen). Deletional Sγ-Sμ switch circles and β-actin were amplified from 500 ng of genomic DNA as described. The conditions were denaturation for 1 min. at 94°C, annealing for 1 min. at 68°C, and extension for 4 min at 72°C for two rounds of 30 cycles. Before each PCR, DNA was denaturated for 5 min at 94° C. The identity of PCR products with switch circles was confirmed by DNA sequencing.
Southern blots
PCR products were fractionated onto agarose gels, transferred overnight to nylon membranes, and hybridized with radiolabeled probes as described (18, 45, 46). Sγ-Sμ switch circles were hybridized with a probe recognizing the recombined Sμ region (18). Iγ-Cμ and Iμ-Cμ transcripts were hybridized with a probe encompassing nt 1–250 of the first Cμ exon (18).
Luciferase (Luc) reporter assays
IgD+ 2E2 B cells (20 × 106 cells/ml) were transfected by electroporation with 40 μl of plasmid DNA-Tris-EDTA solution (pH 8.0) containing 10 μg of Iγ1-Luc or NF-κB2-Luc reporter plasmid expressing firefly Luc and 200 ng of control pRL-TK reporter plasmid expressing Renilla Luc under control of the thymidine kinase promoter (Promega). Firefly and Renilla activities were measured after 48 h with the Dual luciferase assay system (Promega). The Luc activity of Iγ3-Luc and NF-κB2-Luc reporter plasmids was normalized to that of the cotransfected pRL-TK control plasmid and expressed as fold induction, i.e., normalized Luc activity of extracts from stimulated B cells/normalized Luc activity of extracts from unstimulated B cells.
Immunoblots
Equal amounts of cytoplasmic or nuclear proteins were fractionated onto a 10% SDS polyacrylamide gel and transferred onto nylon membranes (Bio-Rad Laboratories). After blocking, membranes were probed with primary pAbs to actin (I-19) (Santa Cruz Biotechnologies) or phosphoserine 32/phosphoserine 36-IκBα (5A5) (Cell Signaling Technology). Then, membranes were washed and incubated with an appropriate peroxidase-conjugated secondary polyclonal Ab (Santa Cruz Biotechnologies). Proteins were detected with an ECL detection system (Amersham Biosciences).
Statistical analysis
Values were calculated as mean SD for at least three separate experiments done in triplicate. The significance of differences between experimental variables was determined with the paired Student's t test.
Results
Upper respiratory mucosa B cells express TLR3
The respiratory mucosa constitutes a major site of entry for viruses. In mice, B cells can initiate quick IgG and IgA responses to viruses in a TI fashion (14, 47–49). These responses are thought to involve TLRs, including TLR3, which binds viral dsRNA (20, 22, 37, 41, 50, 51). In initial experiments, we took advantage of immunohistology to determine the expression of TLR3 in B cells from human tonsils, an upper respiratory district heavily exposed to viruses. TLR3 was abundant in mucosa-associated GCs, which are typically populated by actively class switching/postswitched IgD− B cells (Fig. 1A). Although more variable, TLR3 expression was positive in the mantle zone of mucosa-associated follicles, which is comprised of preswitched IgD+ B cells, and in the mucosal subepithelium, which contains both postswitched IgD− and preswitched IgD+ B cells (Fig. 1B). Of note, scattered follicular mantle and subepithelial B cells expressed as much TLR3 as GC B cells. Appropriate controls confirmed the specificity of TLR3 tissue staining (Fig. 1, C–E).
FIGURE 1.
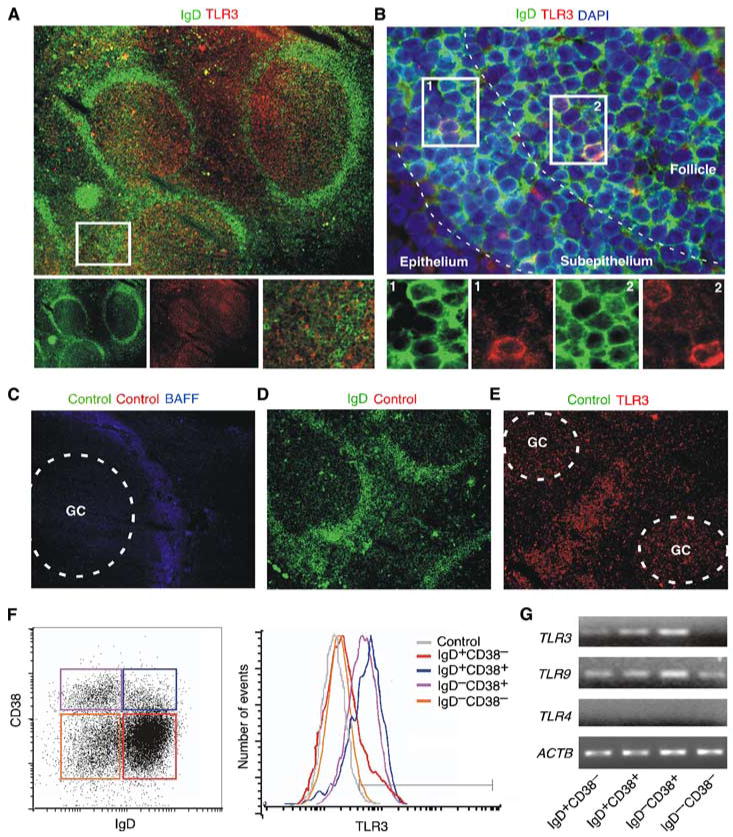
Upper respiratory mucosa B cells express TLR3. A, Immunofluorescence analysis of tonsillar mucosa stained for IgD (green) and TLR3 (red). The left and middle bottom panels show individual IgD and TLR3 stainings. The right bottom panel corresponds to the boxed area in the upper panel. Original magnification, ×5. B, Follicular and subepithelial B cells from the tonsillar mucosa stained for IgD (green), TLR3 (red), and DAPI (blue). Bottom panels show individual or merged TLR3, IgD, and DAPI stainings from the boxed areas in the upper panel. Original magnification, ×40. C–E, Tonsillar tissue specimens stained with various control Abs as detailed in Material and Methods, including Abs with irrelevant binding activity. The Ab to BAFF stains the interfollicular area. Both control and anti-BAFF Abs were labeled with appropriate secondary reagents. Dashed lines indicate GCs. Original magnification, ×10. F, Left dot plot shows flow cytometric analysis of surface IgD and CD38 on CD19-gated B cells from tonsils. Red, blue, pink, and orange gates correspond to naive IgD+CD38− B cells, founder GC/activated IgD+CD38+ B cells, GC IgD−CD38+ cells, and memory IgD−CD38− B cells. Right histograms show cytosolic TLR3 expression in individual B cell subsets. The gray histogram is a control with an isotype-matched Ab to an irrelevant Ag. G, TLR3, TLR9, TLR4, and ACTB transcripts RT-PCR amplified from individual tonsillar B cell subsets. A–G show one of five experiments yielding similar results.
The expression of TLR3 by tonsillar B cells was further investigated by flow cytometry and RT-PCR. Flow cytometry required permeabilization of B cells due to the predominant cytosolic topography of TLR3. In general, preswitched IgD+CD38− B cells, a naive subset colonizing the mantle zone of mucosa-associated follicles (52, 53), expressed low but significant levels of TLR3 protein and TLR3 transcripts (Fig. 1, F and G). TLR3 protein and TLR3 transcripts were more abundant in preswitched IgD+CD38+ B cells, which comprise a founder GC subset from the outer rim of the GC as well as activated B cells from the extrafollicular area, including the subepithelium (52–54). TLR3 protein and TLR3 transcripts were also abundant in postswitched IgD−CD38+ B cells, which populate the GC (52, 53). In contrast, postswitched memory IgD−CD38− B cells, which colonize the perifollicular and epithelial areas of mucosa-associated follicles (52, 53), expressed little or no TLR3 protein or TLR3 transcripts. In agreement with prior studies (39, 40), all mucosal B cell subsets expressed TLR9 but lacked TLR4 transcripts. These data indicate that B cell subsets from the upper respiratory mucosa express distinct TLR3 expression patterns.
Mucosal B cells proliferate in response to viral dsRNA
Having shown TLR3 expression by upper respiratory mucosal B cells, we evaluated the ability of these B cells to mount a functional response to poly(I:C), a synthetic analog of viral dsRNA. We chose to analyze B cell proliferation, because this function is effectively elicited by TLR ligands (25). To optimize dsRNA-induced B cell proliferation, B cells were also incubated with BAFF, a TLR-inducible B cell-stimulating factor present in the microenvironment of the upper respiratory mucosa (20). Flow cytometry-based assays measuring CFSE dilution showed that a fraction of naive IgD+CD38− B cells proliferated in the absence of poly(I:C) or BAFF (Fig. 2A), perhaps as a result of their prior exposure to microenvironmental cues in vivo. A larger fraction of founder-GC/activated IgD+CD38+ B cells and GC IgD−CD38+ B cells proliferated in the absence of poly(I:C) or BAFF. These findings were confirmed by immunohistology, which demonstrated the presence of the nuclear proliferation Ag Ki-67 not only in the majority of IgD− B cells within the GC, but also in scattered IgD+ B cells dwelling in the follicular mantle and in extrafollicular areas (Fig. 2B).
FIGURE 2.
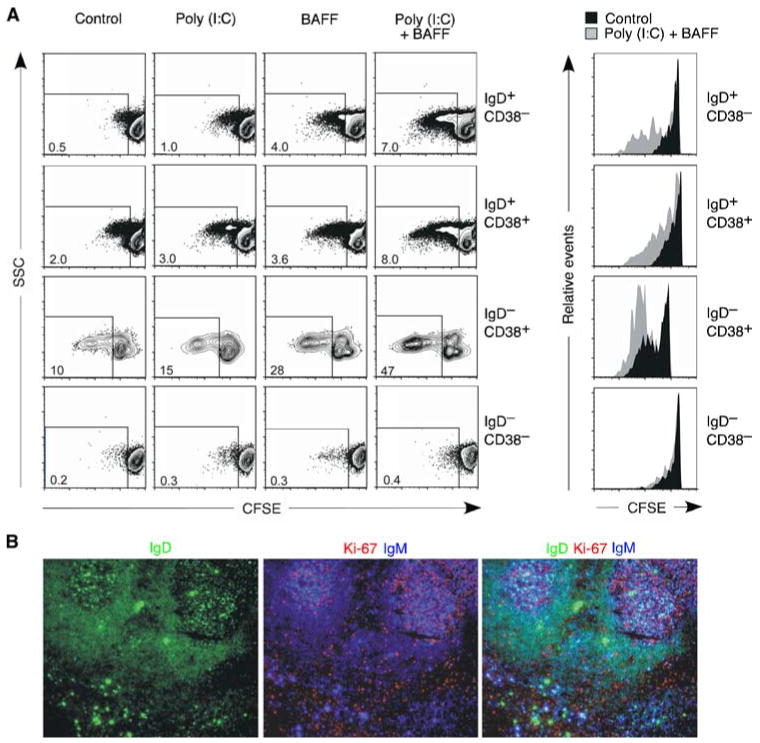
Upper respiratory mucosa B cells proliferate in response to dsRNA. A, Flow cytometric analysis of CFSE-labeled tonsillar naive IgD+CD38−, founder GC/activated IgD+CD38+, GC IgD−CD38+, and memory IgD−CD38− B cell subsets cultured with or without poly(I:C) and/or BAFF. Dot plots indicate percentage of B cells showing reduced CFSE expression over a 6-day culture. Histograms show CFSE dilution by control unstimulated (solid filled histograms) or poly(I:C)- and BAFF-stimulated (solid gray histograms) B cells gated as shown in the dot plots on the left. B, Immunofluorescence analysis of tonsillar mucosa stained for IgD (green), Ki-67 (red), and IgM (blue). Original magnification, ×5. Figure shows one of three experiments yielding similar results.
Poly(I:C) combined with BAFF increased the proliferation of a variable fraction of naive, founder GC/activated, and GC B cells, whereas poly(I:C) alone or BAFF alone induced no or little proliferation. As shown by subsequent experiments (see below), the proliferative effect of poly(I:C) and BAFF was genuine as it was dissociated from the ability of these stimuli to increase B cell survival. Finally, consistent with their inability to express TLR3, memory IgD−CD38− B cells did not proliferate in response to poly(I:C) alone or combined with BAFF. These data indicate that upper respiratory mucosal B cell subsets express a functional TLR3.
Mucosal B cells up-regulate AID and undergo CSR in response to viral dsRNA
Having shown that upper respiratory mucosal B cells express a functional TLR3, we wondered whether dsRNA was capable of inducing CSR. First, we evaluated the expression of AID protein, a hallmark of ongoing CSR, in permeabilized tonsillar B cells by flow cytometry. We found that AID expression was mostly confined to the IgD− fraction of tonsillar B cells (Fig. 3A), thereby confirming the specificity of our flow cytometry-based assay. As expected, these postswitched/switching IgD−AID+ B cells coexpressed CD38 (not shown), indicating their origin from the GC (52, 55). Then, we verified the ability of poly(I:C) to induce AID expression in the naive and founder GC fractions of preswitched IgD+ B cells. These experiments were conducted in the presence of IL-10, as this cytokine is abundantly expressed by mucosal mDCs and was shown in prior studies to induce CSR in cooperation with various TLR ligands (18, 20, 29, 44, 46). In the presence of poly(I:C) and IL-10, a subset of naive IgD+CD38− B cells up-regulated the expression of AID (Fig. 3B). This up-regulation was less evident in IgD+CD38+ B cells, perhaps because these cells had already received CSR-inducing signals in vivo. Indeed, some IgD+CD38+ B cells constitutively expressed AID. Alternatively, IgD+CD38− B cells may contain subpopulations highly responsive to viral dsRNA. Consistent with this possibility, the CD27+ fraction of tonsillar IgD+CD38− B cells, which dwells in the subepithelial area and constitutes the tonsillar counterpart of splenic marginal zone B cells (13, 54), expressed more TLR3 than founder GC/activated IgD+CD38+ B cells (not shown). As shown by subsequent experiments (see below), poly(I:C) and IL-10 did not significantly enhance the baseline proliferation or the survival of IgD+CD38− B cells, further indicating the ability of dsRNA to deliver genuine CSR-inducing signals.
FIGURE 3.
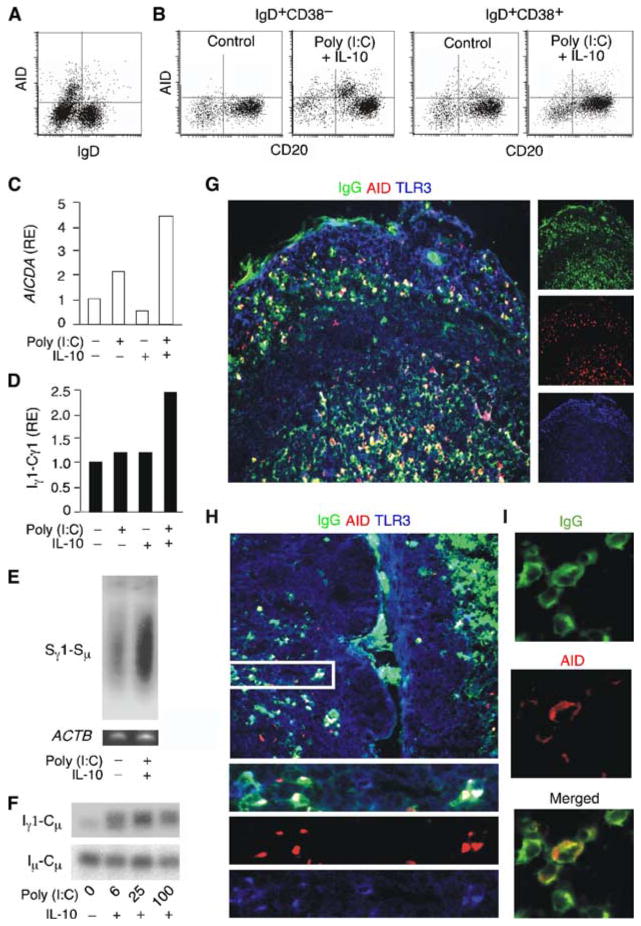
Upper respiratory mucosa B cells undergo CSR in response to dsRNA in vitro. A, Flow cytometric analysis of surface IgD and intracellular AID in purified tonsillar B cells. B, Expression of surface CD20 and intracellular AID in naive IgD+CD38− B cells and founder GC/activated IgD+CD38+ B cells from tonsils. B cells were incubated in the presence or absence of poly(I:C) and IL-10. Control indicates unstimulated B cells. C and D, QRT-PCR of AICDA transcripts and germline Iγ1-Cγ1 transcripts from tonsillar IgD+ B cells cultured with or without poly(I:C) and/or IL-10 for 48 h. AICDA was normalized to ACTB mRNA, whereas Iγ1-Cγ1 was normalized to germline Iμ-Cμ mRNA, which is constitutively expressed by B cells. RE indicates relative expression. E, Southern blot of extrachromosomal Sγ1-Sμ switch circles PCR amplified from the genomic DNA of tonsillar IgD+ B cells cultured as in C and D for 4 days. Genomic β-actin was PCR-amplified as loading control. F, Southern blot of Iγ1-Cμ switch circle transcripts RT-PCR amplified from tonsillar IgD+ B cells cultured as in A and B for 4 days. Constitutively produced germline Iμ-Cμ transcripts were RT-PCR amplified as loading control. G–I, Immunofluorescence analysis of different compartments of the tonsillar mucosa stained for IgG (green), AID (red), and TLR3 (blue). Subepithelial B cells coexpressing IgG, AID, and TLR3 are shown at a higher magnification in H (bottom panels correspond to boxed area) and I. Original magnifications, ×5 (large panels in G and H), ×10 (lower panels in H), and ×63 (panels in I). Panels A–I show one of three experiments yielding similar results.
Then, we used RT-PCR-based assays to verify whether poly(I:C) and IL-10 were capable of inducing additional molecular hallmarks of ongoing CSR. These experiments were conducted with preswitched IgD+ B cells inclusive of both naive and founder GC/activated fractions, because we had already established that poly(I:C) and IL-10 predominantly target naive B cells. Furthermore, we reasoned that regardless of their phenotype and activation state, IgD+ B cells contain genomic switch regions in germline configuration and therefore are suitable for in vitro assays measuring the induction of CSR. In the presence of poly(I:C) alone or combined with IL-10, tonsillar IgD+ B cells up-regulated the expression of AID (AICDA) transcripts (Fig. 3C) as well as that of germline Iγ-Cγ transcripts (Fig. 3D), an early event in IgG CSR (4). These effects were not induced by IL-10 alone. Tonsillar IgD+ B cells exposed to poly(I:C) and IL-10 induced extrachromosomal Sγ-Sμ switch circular genomic DNA (Fig. 3E) as well as switch Iγ-Cμ circle transcripts (Fig. 3F), two hallmarks of ongoing IgG CSR (18, 56). Similar culture conditions induced germline Iα-Cα transcripts, extrachromosomal Sα-Sμ switch circular genomic DNA, and switch Iα-Cμ circle transcripts (not shown), which are hallmarks of ongoing IgA CSR. These data strongly suggest that mucosal B cells undergo CSR in response to TLR3 engagement by viral dsRNA.
Mucosal B cells exhibit active TLR3 signaling and ongoing CD40-independent CSR
We next wished to reinforce our in vitro data by providing in vivo evidence of TLR3-mediated CSR. Prior findings indicate that TLR ligands elicit TI IgG and IgA responses in the subepithelial district of the respiratory mucosa (20). Thus, we used immunohistology to verify the expression of AID by class-switched B cells populating the subepithelium of the upper respiratory mucosa. We reasoned that the presence of AID in these B cells would strongly indicate in situ CSR (56). We found that the subepithelial and epithelial areas of the tonsillar mucosa contained IgG+TLR3+AID+ B cells (Fig. 3, G–I) as well as IgA+TLR3+AID+ B cells (not shown). Having detected TLR3 expression and ongoing CSR in subepithelial and epithelial B cells, we asked whether these B cells receive CD40-independent signals from TLR3. To establish this point, we took advantage of mucosal specimens from HIGM patients.
HIGM syndromes form a heterogeneous group of congenital disorders characterized by impaired IgG and IgA responses and a compensatory increase of IgM (57). HIGM1 (type 1 HIGM) and HIGM3 (type 3) are associated with CD40L and CD40 deficiencies, respectively, whereas HIGM2 (type 2) is caused by AID deficiency (57). Finally, HIGM4 (type 4) is caused by various intrinsic B cell defects, including CD40 signaling defects, whereas HIGM5 (type 5) involves a deficiency of uracil DNA glycosylase, an enzyme downstream of AID (57). We performed immunohistological studies on mucosal tissue specimens from patients with HIGM3 and HIGM4, two entities associated with impaired GC formation as well as defective IgG and IgA class switching (57). In these studies we looked for subepithelial B cells expressing class-switched isotypes as well as AID and phosphorylated TLR3 (pTLR3), a hallmark of active TLR3 signaling (58).
In healthy control subjects, IgG+AID+pTLR3+ B cells (Fig. 4A) and IgA+AID+pTLR3+ B cells (not shown) were detected in both the GC and subepithelial areas of the tonsillar mucosa. In the HIGM4 patient, IgG+AID+pTLR3+ B cells (Fig. 4B) and IgA+AID+pTLR3+ B cells (not shown) were readily identified in the subepithelial area but not in the GC. Indeed, despite being hyperplastic, the mucosa-associated follicles from the HIGM4 patient lacked GCs. The lack of functional GCs could be inferred not only from the absence of class-switched isotypes and AID in the follicular area, but also from the absence of Bcl-6 (not shown), a hallmark of the GC reaction. Finally, IgG+AID+pTLR3+ B cells (Fig. 4C) and IgA+AID+pTLR3+ B cells (not shown) were also detected in the intestinal mucosa of the HIGM3 patient. Finally, IgG+AID+pTLR3+ B cells and IgA+AID+pTLR3+ B cells were detected in the intestinal mucosa from two patients with AIDS (not shown), a condition causing early and massive loss of mucosal CD4+ T cells (21, 59). Thus, in agreement with the in vitro data, these in vivo findings suggest that TLR3 engagement by viral dsRNA triggers frontline CSR through an innate pathway that remains operative in the absence of GCs, CD40L, or CD4+ T cells.
FIGURE 4.
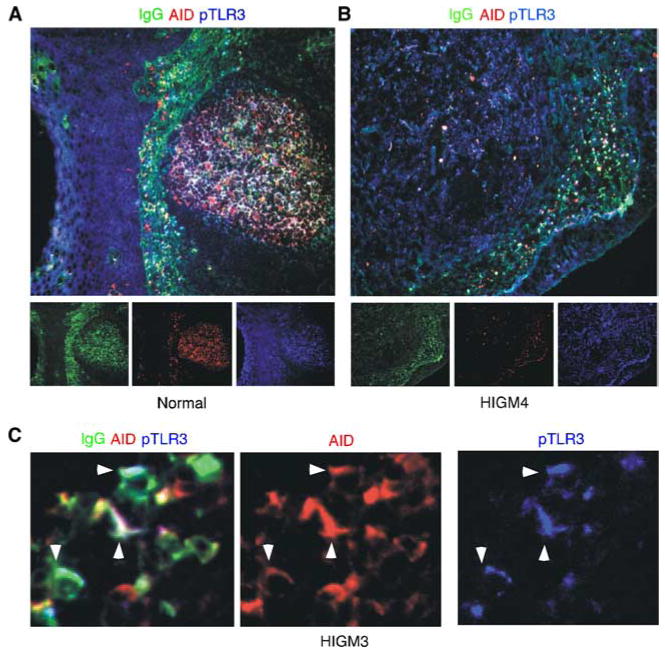
Mucosal B cells from patients with CD40 signaling defects exhibit active TLR3 signaling and CSR. A and B, Immunofluorescence analysis of the tonsillar mucosa from a healthy subject and a HIGM4 patient. Tissue specimens were stained for IgG, AID, and pTLR3. Original magnification, ×10. C, Mucosal B cells from the colon lamina propria of an HIGM3 patient stained as in A and B. The arrowheads point to B cells coexpressing IgG, AID, and pTLR3. Original magnification, ×10. A–C show one of at least three experiments yielding similar results.
Mucosal B cells initiate CSR through a TRIF-dependent pathway in response to viral dsRNA
Having shown that mucosal B cells actively undergo CSR upon in vitro exposure to dsRNA, we wondered whether dsRNA exerts this effect through a canonical TLR3 pathway. First, we performed immunoblotting and reporter assays to evaluate whether B cells exposed to viral dsRNA activate NF-κB, a transcription factor essential for CSR (2). Activation of NF-κB requires phosphorylation of its inhibitor IκBα (36). We found that tonsillar preswitched IgD+ B cells up-regulated IκBα phosphorylation upon exposure to increasing amounts of poly(I:C) (Fig. 5A). Similarly, poly(I:C) induced transcriptional activation of an NF-κB-dependent minimal gene promoter in an IgD+ 2E2 B cell (Fig. 5B), a TLR3-expressing subclone of the CL-01 B cell line (29). In these B cells, poly(I:C) induced transcriptional activation of the Iγ1 promoter, a cis-regulatory Cγ1 gene sequence highly dependent on NF-κB (29, 60). This effect required TLR3, because enforced expression of dominant negative (DN)-TRIF inhibited poly(I:C)-induced NF-κB-dependent gene transcription, whereas DN-MyD88 did not (Fig. 5C). Thus, when exposed to viral dsRNA, mucosal B cells activate NF-κB and initiate class switching by activating a TLR3-mediated signaling pathway involving TRIF.
FIGURE 5.
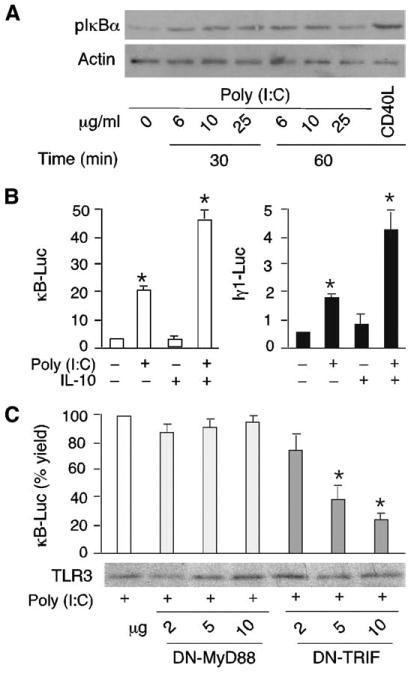
Upper respiratory mucosa B cells activate NF-κB via TLR3 upon exposure to dsRNA. A, Immunoblots showing phosphorylated IκBα (pIκBα) and actin (loading control) proteins in tonsillar IgD+ B cells incubated with or without poly(I:C) or CD40L (positive control). B, Luciferase activity from IgD+ 2E2 B cells transfected with κB2-Luc (open histograms) or Iγ1-Luc (solid histograms) reporter vectors and cultured with or without poly(I:C) and/or IL-10 for 48 h. Data are presented as fold induction as detailed in Materials and Methods. C, κB2-Luc activity from IgD+ 2E2 B cells incubated with poly(I:C) in the presence of DN-MyD88 or DN-TRIF expression vectors for 48 h. Data are presented as a percentage of yield (fold activation with expression vector/fold activation without expression vector × 100). Gel shows TLR3 immunoblotting. A shows one of three experiments yielding similar results, whereas B and C summarize three experiments (bars indicate SD and * indicates p < 0.05 vs B cells incubated with medium alone or without DN plasmid).
BAFF enhances viral dsRNA-induced IgG and IgA production in mucosal B cells exposed to dsRNA
Prior studies indicate that TLR ligands activate B cells in cooperation with innate immune mediators released by DCs, including BAFF and IFN-α (18, 20, 29, 32, 61–63). Thus, we set up ELISAs to establish whether viral dsRNA cooperates with BAFF or IFN-α to induce IgG and IgA production in B cells from the upper respiratory mucosa. Compared with medium alone, IL-10 alone, or poly(I:C) alone, a combination of poly(I:C) and IL-10 induced more IgG and IgA production but comparable IgM production in tonsillar IgD+ B cells (Fig. 6, A–C). Flow cytometric assays measuring dilution of CFSE and permeability to 7-AAD established that this induction was not associated with an increased proliferation and survival of IgD+ B cells (Fig. 6, D and E) whether they had a naive or founder GC/activated phenotype (not shown). These findings indicate that IL-10 augments the class switch-inducing activity of viral dsRNA.
FIGURE 6.
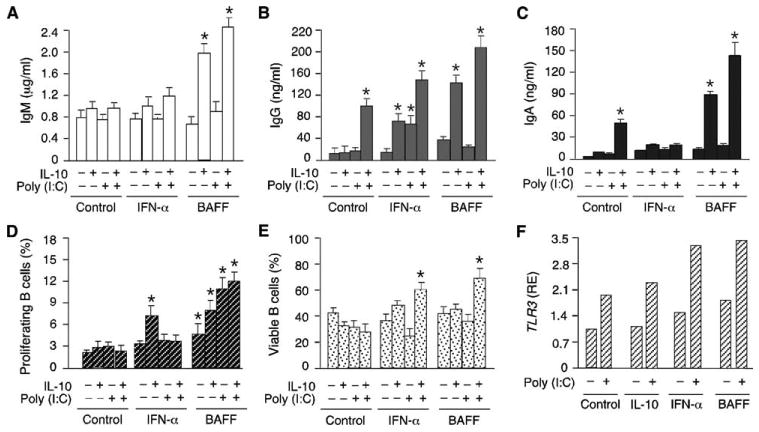
Upper respiratory mucosal B cells use BAFF to enhance IgG and IgA responses induced by dsRNA. A–C, ELISAs of IgM, IgA, and IgG secreted by tonsillar IgD+ B cells cultured with or without poly(I:C) and/or IL-10 and in the presence or absence of IFN-α or BAFF for 8 days. Control indicates no IFN-α or BAFF. D, Flow cytometric analysis of proliferation of tonsillar IgD+ B cells cultured for 6 days as in A–C. Proliferation was measured by calculating the proportion of B cells with down-regulated CFSE expression. E, Flow cytometric analysis of viable tonsillar IgD+ B cells cultured for 6 days as in A–C. Viability was determined by calculating the proportion of B cells impermeable to 7-AAD. F, QRT-PCR of TLR3 transcripts from tonsillar IgD+ B cells cultured with or without poly(I:C) and in the presence or absence of IL-10, IFN-α, or BAFF for 24 h. TLR3 mRNA was normalized to ACTB mRNA and RE indicates relative expression. Control indicates no IL-10, IFN-α, or BAFF. A–E summarize three experiments (bars indicate SD and * indicates p < 0.05 vs B cells incubated with medium alone), whereas F shows one of three experiments yielding similar results.
IFN-α increased the production of IgG but not that of IgA by tonsillar IgD+ B cells exposed to poly(I:C) alone or combined with IL-10. Induction of IgG was not associated with increased B cell proliferation and survival, indicating that IFN-α has a genuine IgG class switch-inducing effect when combined with viral dsRNA. As for BAFF, this innate immune mediator up-regulated the production of both IgG and IgA by tonsillar IgD+ B cells exposed to poly(I:C) and IL-10. Such up-regulation was coupled with increased B cell proliferation and survival, indicating that BAFF promotes the expansion of B cells that have undergone class switching in response to poly(I:C) and IL-10. Finally, QRT-PCR-based assays showed that tonsillar IgD+ B cells exposed to IL-10, IFN-α, or BAFF up-regulated the expression of TLR3 transcripts (Fig. 6F), suggesting that these soluble factors enhance viral dsRNA-induced B cell responses by making B cells more responsive to dsRNA.
Mucosal TLR3-expressing mDCs up-regulate the CSR-inducing factor BAFF in response to viral dsRNA
Various DC subsets release BAFF, IL-10, and/or IFN-α upon exposure to microbial TLR ligands (18, 20, 29, 33). Having shown that BAFF, IL-10, and IFN-α up-regulate viral dsRNA-induced IgG and IgA responses in vitro, we wondered whether upper respiratory mucosal B cells interact with DCs in vivo. By using immunohistology, we were able to detect abundant BAFF expression in the epithelial and subepithelial areas of the tonsillar mucosa (Fig. 7A). These districts contained TLR3+ BAFF+ mDCs that were extensively interacting with B cells, including IgD+ B cells. Mucosal IgD+ B cells were also in contact with CD123+BAFF+ (TLR3−) pDCs, suggesting that both mDC and pDC subsets participate in the initiation of frontline Ab responses to viral dsRNA. Then, we set up QRT-PCR- and ELISA-based experiments to verify whether mDCs up-regulate BAFF production in response to viral dsRNA. In the presence of poly(I:C), mDCs up-regulated the expression of BAFF-encoding TNFSF13b transcripts as well as the release of soluble BAFF protein (Fig. 7B). In addition, these mDCs up-regulated the expression of IL-10 transcripts as well as the release of soluble IL-10 protein (Fig. 7C). Consistent with these findings, poly(I:C)-activated mDCs induced IgG and IgA production in IgD+ B cells (Fig. 7D).
FIGURE 7.
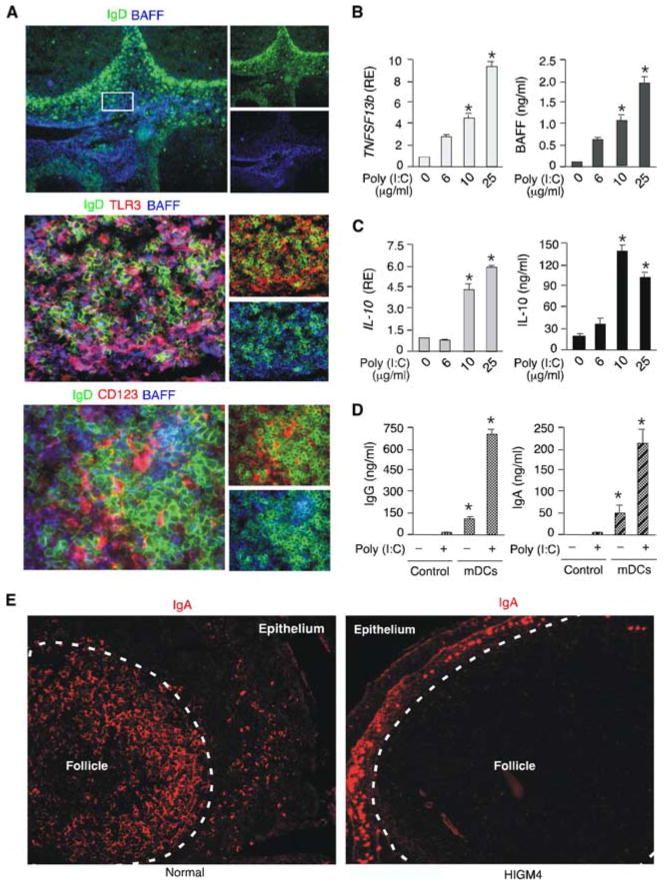
Upper respiratory mucosal mDCs and pDCs release BAFF upon exposure to dsRNA. A, Immunofluorescence analysis of tonsillar mucosa stained for IgD (green), TLR3 (red) or CD123 (red), and BAFF (blue). The boxed area in the upper panel corresponds to the subepithelial district containing the TLR3+ mDCs and the CD123+ pDCs shown in the middle and bottom left panels, respectively. Original magnification, ×5 (upper panel) and ×40 (middle and bottom panels). B and C, QRT-PCR of TNFSF13b and IL-10 transcripts and ELISAs of BAFF and IL-10 proteins from mDCs cultured with or without grading amounts of poly(I:C) for 24 h. TNFSF13b and IL-10 mRNAs were normalized to ACTB mRNA, and RE indicates relative expression. D, ELISAs of IgG and IgA from tonsillar IgD+ B cells incubated for 8 days with or without poly(I:C) in the presence or absence of mDCs. Control indicates no mDCs. E, Immunofluorescence analysis of the tonsillar mucosa from a healthy subject and a HIGM4 patient. Tissue specimens were stained for IgA. Original magnification, ×10. A and E show one of three experiments yielding similar results, whereas B–D summarize three experiments (bars indicate SD and * indicates p < 0.05 vs B cells incubated with medium alone and in the absence of mDCs.
The contribution of TLR3-activated mDCs to frontline TI IgA responses was further supported by immunohistological studies showing IgA-producing B cells in the subepithelial district of the upper respiratory mucosa of a patient with HIGM4 (Fig. 7E). Unlike healthy controls, the HIGM4 patient completely lacked IgA-producing B cells in mucosa-associated follicles. These results paralleled those showing conserved subepithelial IgG production in the same HIGM4 patient. Overall, our data indicate that viral dsRNA can initiate TI IgG and IgA responses not only directly by activating TLR3-expressing B cells, but also indirectly by inducing BAFF, IL-10, and IFN-α production in TLR3+ mDCs. Additional viral molecules or mDC-derived mediators would trigger BAFF and IFN-α release from TLR3− pDCs.
Discussion
We have reported in this article that B cells from the upper respiratory mucosa expressed TLR3 and underwent TRIF-dependent activation of the CSR-inducing transcription factor NF-κB in response to dsRNA. In the presence of IL-10, viral dsRNA elicited germline CH gene transcription, AICDA expression, and CSR as well as IgG and IgA production in a CD40-independent fashion. The secretion of IgG and IgA was further enhanced by BAFF, an innate factor produced by mDCs in response to dsRNA. Consistent with these in vitro data, in vivo findings showed active TLR3 signaling as well as ongoing IgG and IgA CSR and production in the mDC-rich subepithelial regions of the upper respiratory and intestinal mucosal tracts of HIGM patients with CD40 signaling defects. Thus, we propose that viral dsRNA triggers frontline TI IgG and IgA responses by activating mucosal B cells through an innate pathway involving TLR3.
A large body of evidence indicates that mouse B cells can generate quick Ab responses without requiring help from CD4+ T cells through CD40L (14, 47–49). Abs resulting from this TI pathway may play an important role in the initial control of infections by quickly replicating pathogens (7, 14). In addition to IgM, early TI B cell responses yield class-switched IgG and IgA Abs, particularly at mucosal sites of entry (15). Although IgA is more abundant than IgG in the digestive tract, IgG is more abundant than IgA in the respiratory tract, a mucosal district heavily exposed to viruses (64). TI production of polyreactive IgA (and IgM) Abs may mediate steady-state containment of commensal microorganisms (65, 66), whereas TI production of polyreactive IgG (and IgA) Abs would blunt infections by pathogens, including viruses, at a very early stage (47–49, 67–69). Strikingly, TI Ab responses can even generate immune protection against viruses (14, 47, 48).
Mucosal TI Ab responses are less well characterized in humans. Human B cells can undergo CD40-independent IgG and IgA production upon exposure to BAFF and APRIL, two CD40L-related molecules produced by various innate immune cells and stromal cells, including DCs, macrophages, and epithelial cells (18, 20, 21). Tonic release of BAFF by innate and stromal cells is thought to provide survival signals essential for the homeostasis of the peripheral B cell compartment (70–72). In the presence of microbial TLR ligands, innate and stromal cells further up-regulate the release of BAFF as well as that of APRIL and thereafter acquire CSR-licensing functions (18, 20, 21, 51). Ultimately, BAFF and APRIL trigger CSR by engaging the transmembrane activator, calcium modulator, and cyclophilin ligand interactor (TACI), a receptor expressed on the surface of B cells (73–76). In humans, the key in vivo role of BAFF and APRIL is clearly documented by recent evidence showing that patients expressing TACI mutants develop selective IgA deficiency or common variable immunodeficiency, a disorder associated with impaired IgM, IgG, and IgA production (77, 78). Despite these studies, the role of BAFF and APRIL in mucosal Ab responses remains unclear.
We found that mDCs from the upper respiratory mucosa produced BAFF and closely interacted with B cells. Interestingly, these mucosal mDCs were also in close contact with pDCs, a DC subset with professional IFN-α-producing activity (23). Similarly as mDCs, mucosal pDCs expressed BAFF, suggesting that TI IgG and IgA responses require a close cooperation between distinct DC subsets. Consistent with this possibility, mDCs up-regulate the release of BAFF upon exposure to paracrine IFN-α (18). As shown by others (33), mDCs contained abundant TLR3 and up-regulated the production of BAFF in response to viral dsRNA. In addition, viral dsRNA up-regulated mDC release of IL-10, a cytokine critically involved in class switching and Ab production (18, 20, 21, 84–87). By inducing BAFF and IL-10, viral dsRNA conferred CD40-independent IgG- and IgA-inductive properties to mDCs, further indicating that TLR3 may be involved in the initiation of TI class switching in the upper respiratory district.
In addition to mDCs, viral dsRNA activated upper respiratory mucosa B cells. Indeed, tonsillar B cells expressed TLR3 and underwent proliferation and CSR in response to dsRNA. This TLR3 ligand activated the transcription factor NF-κB and thereafter initiated germline transcription of downstream CH genes, including Cγ1, in a CD40-independent fashion. Of note, activation of NF-κB required TRIF but not MyD88, indicating that viral dsRNA stimulates class switching through a canonical TLR3-dependent pathway. In the presence of IL-10, dsRNA also triggered AID expression and CSR as well as IgG and IgA production. BAFF further increased IgG and IgA production by augmenting the survival of class-switched B cells generated by viral dsRNA and IL-10. In this regard it must be noted that, in addition to inducing class switching via TACI (18, 74), BAFF delivers powerful survival signals via the BAFF receptor, also known as BR-3 (71).
Interestingly, BAFF would also make upper respiratory mucosa B cells more responsive to viral dsRNA. Indeed, tonsillar B cells up-regulated TLR3 expression upon exposure to dsRNA. A similar effect was induced by other DC-derived stimuli, including IL-10 and IFN-α. Consistent with this observation, both IL-10 and IFN-α were capable of enhancing the induction of IgA and/or IgG production by viral dsRNA.
Our data suggest that human B cells from the upper respiratory mucosa can undergo TI IgG and IgA production through an innate TLR3 pathway involving BAFF. Indeed, we detected abundant BAFF-producing mDCs and pDCs in the subepithelial area of the tonsillar mucosa. These BAFF-producing DCs were in close proximity with IgG+ and (not shown) IgA+ B cells expressing AID, pTLR3, hallmarks of ongoing CSR and active TLR3 signaling, respectively (56, 58). These recently class-switched B cells were conserved in the subepithelium of HIGM patients with CD40 signaling defects. The mucosa-associated lymphoid follicles from the same patients lacked IgG, IgA, and AID expression as well as recognizable GCs, further confirming the TI nature of subepithelial IgG and IgA responses.
These findings are consistent with prior observations showing that IgG and IgA Abs are still detectable in the circulation of HIGM patients with defects in CD40 signaling (79). We argue that some of these Abs may originate from mucosal TI responses involving TLRs, including TLR3. More generally, the present and previous data (20, 21) indicate that, similar to CD40-deficient mice (80), CD40-deficient humans partially retain TI IgG and IgA responses in the mucosal lymphoid tissue. As indicated by recent studies conducted in mice (81–83), such TI Ab responses may generate limited affinity maturation and immune memory and may therefore confer some degree of immune protection not only against against commensals, but also against some pathogens.
In conclusion, we propose that RNA viruses trigger frontline TI IgM production as well as class switching by stimulating mucosal B cell subsets through a TLR3-dependent pathway involving multiple innate signals including BAFF. Unmutated and/or mutated IgM, IgG, and IgA Abs produced through this TI pathway could limit viral penetration across mucosal surfaces by neutralizing conserved conformational or linear epitopes embedded within envelope glycoproteins (16). It is tempting to speculate that such TI Ab responses contribute to the innate resistance of certain subjects to a highly infectious RNA virus such as HIV (88).
Acknowledgments
We thank Yifang Liu and Meimei Shan (Weill Medical College of Cornell University, New York, NY) for processing tissue samples and generating mDCs.
Footnotes
This work was supported by National Institutes of Health Grants R01 AI057653, R01 AI057653-S1, R01 AI074378 (to A.C.), and T32 AI07621 (to W.X.), by an Irma T. Hirschl Scholar Award (to A.C.), by a Cancer Research Institute Fellowship (to P.S.), and by funds from Fondazione C. Golgi and Centro Immunodeficienze Mario Di Martino (to A.P.).
Abbreviations used in this paper: CD40L, CD40 ligand; 7-AAD, 7-amino-actinomycin; AID, activation-induced cytidine deaminase; APRIL, a proliferation-inducing ligand; BAFF, B cell activating factor of the TNF family; CpG DNA, DNA with deoxycytidylate-phosphate-deoxyguanylate motifs; CSR, class switch DNA recombination; DAPI, 4′,6′-diamidine-2-phenylindole; DC, dendritic cell; DN, dominant negative; GC, germinal center; HIGM, hyper-IgM; I, intronic; Luc, luciferase; mDC, myeloid DC; NA, numerical aperture; pAb, polyclonal Ab; pDC, plasmacytoid DC; poly(I:C), polyinosinic/cytidylic acid; pTLR3, phosphorylated TLR3; QRT-PCR, quantitative real-time RT-PCR; S, switch; TACI, transmembrane activator, calcium modulator, and cyclophilin ligand interactor; TI, T cell independent; TRIF, Toll/IL-1R domain-containing protein inducing IFN-β.
Disclosures: The authors have no financial conflict of interest.
References
- 1.Burton DR. Antibodies, viruses, and vaccines. Nat Rev Immunol. 2002;2:706–713. doi: 10.1038/nri891. [DOI] [PubMed] [Google Scholar]
- 2.Stavnezer J. Antibody class switching. Adv Immunol. 1996;61:79–146. doi: 10.1016/s0065-2776(08)60866-4. [DOI] [PubMed] [Google Scholar]
- 3.Honjo T, Kinoshita K, Muramatsu M. Molecular mechanism of class switch recombination: linkage with somatic hypermutation. Annu Rev Immunol. 2002;20:165–196. doi: 10.1146/annurev.immunol.20.090501.112049. [DOI] [PubMed] [Google Scholar]
- 4.Chaudhuri J, Alt FW. Class-switch recombination: interplay of transcription, DNA deamination, and DNA repair. Nat Rev Immunol. 2004;4:541–552. doi: 10.1038/nri1395. [DOI] [PubMed] [Google Scholar]
- 5.Odegard VH, Schatz DG. Targeting of somatic hypermutation. Nat Rev Immunol. 2006;6:573–583. doi: 10.1038/nri1896. [DOI] [PubMed] [Google Scholar]
- 6.McHeyzer-Williams MG, Ahmed R. B cell memory and the long-lived plasma cell. Curr Opin Immunol. 1999;11:172–179. doi: 10.1016/s0952-7915(99)80029-6. [DOI] [PubMed] [Google Scholar]
- 7.Fagarasan S, Honjo T. T-independent immune response: new aspects of B cell biology. Science. 2000;290:89–92. doi: 10.1126/science.290.5489.89. [DOI] [PubMed] [Google Scholar]
- 8.MacLennan I, Vinuesa C. Dendritic cells, BAFF, and APRIL: innate players in adaptive antibody responses. Immunity. 2002;17:235–238. doi: 10.1016/s1074-7613(02)00398-9. [DOI] [PubMed] [Google Scholar]
- 9.Macpherson AJ, Lamarre A. BLySsful interactions between DCs and B cells. Nat Immunol. 2002;3:798–800. doi: 10.1038/ni0902-798. [DOI] [PubMed] [Google Scholar]
- 10.Casali P, Schettino EW. Structure and function of natural antibodies. Curr Top Immunol. 1996;120:167–179. doi: 10.1007/978-3-642-85226-8_17. [DOI] [PubMed] [Google Scholar]
- 11.Bendelac A, Bonneville M, Kearney JF. Autoreactivity by design: innate B and T lymphocytes. Nat Rev Immunol. 2001;1:177–186. doi: 10.1038/35105052. [DOI] [PubMed] [Google Scholar]
- 12.Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant autoantibody production by early human B cell precursors. Science. 2003;301:1374–1377. doi: 10.1126/science.1086907. [DOI] [PubMed] [Google Scholar]
- 13.Weller S, Braun MC, Tan BK, Rosenwald A, Cordier C, Conley ME, Plebani A, Kumararatne DS, Bonnet D, Tournilhac O, et al. Human blood IgM “memory” B cells are circulating splenic marginal zone B cells harboring a prediversified immunoglobulin repertoire. Blood. 2004;104:3647–3654. doi: 10.1182/blood-2004-01-0346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Szomolanyi-Tsuda E, Welsh RM. T-cell-independent antiviral antibody responses. Curr Opin Immunol. 1998;10:431–435. doi: 10.1016/s0952-7915(98)80117-9. [DOI] [PubMed] [Google Scholar]
- 15.Fagarasan S, Honjo T. Intestinal IgA synthesis: regulation of frontline body defences. Nat Rev Immunol. 2003;3:63–72. doi: 10.1038/nri982. [DOI] [PubMed] [Google Scholar]
- 16.Haynes BF, Fleming J, St Clair EW, Katinger H, Stiegler G, Kunert R, Robinson J, Scearce RM, Plonk K, Staats HF, et al. Cardiolipin polyspecific autoreactivity in two broadly neutralizing HIV-1 antibodies. Science. 2005;308:1906–1908. doi: 10.1126/science.1111781. [DOI] [PubMed] [Google Scholar]
- 17.Nardelli B, Belvedere O, Roschke V, Moore PA, Olsen HS, Migone TS, Sosnovtseva S, Carrell JA, Feng P, Giri JG, Hilbert DM. Synthesis and release of B-lymphocyte stimulator from myeloid cells. Blood. 2001;97:198–204. doi: 10.1182/blood.v97.1.198. [DOI] [PubMed] [Google Scholar]
- 18.Litinskiy MB, Nardelli B, Hilbert DM, He B, Schaffer A, Casali P, Cerutti A. DCs induce CD40-independent immunoglobulin class switching through BLyS and APRIL. Nat Immunol. 2002;3:822–829. doi: 10.1038/ni829. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 19.Cerutti A, Qiao X, He B. Plasmacytoid dendritic cells and the regulation of immunoglobulin heavy chain class switching. Immunol Cell Biol. 2005;83:554–562. doi: 10.1111/j.1440-1711.2005.01389.x. [DOI] [PubMed] [Google Scholar]
- 20.Xu W, He B, Chiu A, Chadburn A, Shan M, Buldys M, Ding A, Knowles DM, Santini PA, Cerutti A. Epithelial cells trigger frontline immunoglobulin class switching through a pathway regulated by the inhibitor SLPI. Nat Immunol. 2007;8:294–303. doi: 10.1038/ni1434. [DOI] [PubMed] [Google Scholar]
- 21.He B, Xu W, Santini PA, Polydorides AD, Chiu A, Estrella J, Shan M, Chadburn A, Villanacci V, Plebani A, et al. Intestinal bacteria trigger T cell-independent immunoglobulin A2 class switching by inducing epithelial-cell secretion of the cytokine APRIL. Immunity. 2007;26:812–826. doi: 10.1016/j.immuni.2007.04.014. [DOI] [PubMed] [Google Scholar]
- 22.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 23.Colonna M, Trinchieri G, Liu YJ. Plasmacytoid dendritic cells in immunity. Nat Immunol. 2004;5:1219–1226. doi: 10.1038/ni1141. [DOI] [PubMed] [Google Scholar]
- 24.Steinman RM, Hemmi H. Dendritic cells: translating innate to adaptive immunity. Curr Top Microbiol Immunol. 2006;311:17–58. doi: 10.1007/3-540-32636-7_2. [DOI] [PubMed] [Google Scholar]
- 25.Peng SL. Signaling in B cells via Toll-like receptors. Curr Opin Immunol. 2005;17:230–236. doi: 10.1016/j.coi.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 26.Krieg AM, Yi AK, Matson S, Waldschmidt TJ, Bishop GA, Teasdale R, Koretzky GA, Klinman DM. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature. 1995;374:546–549. doi: 10.1038/374546a0. [DOI] [PubMed] [Google Scholar]
- 27.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 28.Bauer S, Kirschning CJ, Hacker H, Redecke V, Hausmann S, Akira S, Wagner H, Lipford GB. Human TLR9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proc Natl Acad Sci USA. 2001;98:9237–9242. doi: 10.1073/pnas.161293498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.He B, Qiao X, Cerutti A. CpG DNA induces IgG class switch DNA recombination by activating human B cells through an innate pathway that requires TLR9 and cooperates with IL-10. J Immunol. 2004;173:4479–4491. doi: 10.4049/jimmunol.173.7.4479. [DOI] [PubMed] [Google Scholar]
- 30.Lin L, Gerth AJ, Peng SL. CpG DNA redirects class-switching towards “Th1-like” Ig isotype production via TLR9 and MyD88. Eur J Immunol. 2004;34:1483–1487. doi: 10.1002/eji.200324736. [DOI] [PubMed] [Google Scholar]
- 31.Jegerlehner A, Maurer P, Bessa J, Hinton HJ, Kopf M, Bachmann MF. TLR9 signaling in B cells determines class switch recombination to IgG2a. J Immunol. 2007;178:2415–2420. doi: 10.4049/jimmunol.178.4.2415. [DOI] [PubMed] [Google Scholar]
- 32.Katsenelson N, Kanswal S, Puig M, Mostowski H, Verthelyi D, Akkoyunlu M. Synthetic CpG oligodeoxynucleotides augment BAFF- and APRIL-mediated immunoglobulin secretion. Eur J Immunol. 2007;37:1785–1795. doi: 10.1002/eji.200636800. [DOI] [PubMed] [Google Scholar]
- 33.Kadowaki N, Ho S, Antonenko S, Malefyt RW, Kastelein RA, Bazan F, Liu YJ. Subsets of human dendritic cell precursors express different Toll-like receptors and respond to different microbial antigens. J Exp Med. 2001;194:863–869. doi: 10.1084/jem.194.6.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bauer M, Redecke V, Ellwart JW, Scherer B, Kremer JP, Wagner H, Lipford GB. Bacterial CpG-DNA triggers activation and maturation of human CD11c−, CD123+ dendritic cells. J Immunol. 2001;166:5000–5007. doi: 10.4049/jimmunol.166.8.5000. [DOI] [PubMed] [Google Scholar]
- 35.Liu YJ. IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu Rev Immunol. 2005;23:275–306. doi: 10.1146/annurev.immunol.23.021704.115633. [DOI] [PubMed] [Google Scholar]
- 36.Siebenlist U, Brown K, Claudio E. Control of lymphocyte development by nuclear factor-κB. Nat Rev Immunol. 2005;5:435–445. doi: 10.1038/nri1629. [DOI] [PubMed] [Google Scholar]
- 37.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 38.Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 39.Bernasconi NL, Onai N, Lanzavecchia A. A role for Toll-like receptors in acquired immunity: up-regulation of TLR9 by BCR triggering in naive B cells and constitutive expression in memory B cells. Blood. 2003;101:4500–4504. doi: 10.1182/blood-2002-11-3569. [DOI] [PubMed] [Google Scholar]
- 40.Bourke E, Bosisio D, Golay J, Polentarutti N, Mantovani A. The toll-like receptor repertoire of human B lymphocytes: inducible and selective expression of TLR9 and TLR10 in normal and transformed cells. Blood. 2003;102:956–963. doi: 10.1182/blood-2002-11-3355. [DOI] [PubMed] [Google Scholar]
- 41.Gavin AL, Hoebe K, Duong B, Ota T, Martin C, Beutler B, Nemazee D. Adjuvant-enhanced antibody responses in the absence of Toll-like receptor signaling. Science. 2006;314:1936–1938. doi: 10.1126/science.1135299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cerutti A, Zan H, Kim EC, Shah S, Schattner EJ, Schaffer A, Casali P. Ongoing in vivo immunoglobulin class switch DNA recombination in chronic lymphocytic leukemia B cells. J Immunol. 2002;169:6594–6603. doi: 10.4049/jimmunol.169.11.6594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ferrari S, Giliani S, Insalaco A, Al-Ghonaium A, Soresina AR, Loubser M, Avanzini MA, Marconi M, Badolato R, Ugazio AG, et al. Mutations of CD40 gene cause an autosomal recessive form of immunodeficiency with hyper IgM. Proc Natl Acad Sci USA. 2001;98:12614–12619. doi: 10.1073/pnas.221456898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.He B, Qiao X, Klasse PJ, Chiu A, Chadburn A, Knowles DM, Moore JP, Cerutti A. HIV-1 envelope triggers polyclonal Ig class switch recombination through a CD40-independent mechanism involving BAFF and C-type lectin receptors. J Immunol. 2006;176:3931–3941. doi: 10.4049/jimmunol.176.7.3931. [DOI] [PubMed] [Google Scholar]
- 45.Cerutti A, Kim EC, Sha S, Zan H, Schattner EJ, Schaffer A, Casali P. Dysregulation of CD30+ T cells by leukemia impairs isotype switching in normal B cells. Nat Immunol. 2001;2:150–156. doi: 10.1038/84254. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 46.He B, Raab-Traub N, Casali P, Cerutti A. EBV-encoded latent membrane protein 1 cooperates with BAFF/BLyS and APRIL to induce T cell-independent Ig heavy chain class switching. J Immunol. 2003;171:5215–5224. doi: 10.4049/jimmunol.171.10.5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Franco MA, Greenberg HB. Immunity to rotavirus in T cell deficient mice. Virology. 1997;238:169–179. doi: 10.1006/viro.1997.8843. [DOI] [PubMed] [Google Scholar]
- 48.Lee BO, Rangel-Moreno J, Moyron-Quiroz JE, Hartson L, Makris M, Sprague F, Lund FE, Randall TD. CD4 T cell-independent antibody response promotes resolution of primary influenza infection and helps to prevent reinfection. J Immunol. 2005;175:5827–5838. doi: 10.4049/jimmunol.175.9.5827. [DOI] [PubMed] [Google Scholar]
- 49.Heer AK, Shamshiev A, Donda A, Uematsu S, Akira S, Kopf M, Marsland BJ. TLR signaling fine-tunes anti-influenza B cell responses without regulating effector T cell responses. J Immunol. 2007;178:2182–2191. doi: 10.4049/jimmunol.178.4.2182. [DOI] [PubMed] [Google Scholar]
- 50.Beisner DR, Ch'en IL, Kolla RV, Hoffmann A, Hedrick SM. Cutting edge: innate immunity conferred by B cells is regulated by caspase-8. J Immunol. 2005;175:3469–3473. doi: 10.4049/jimmunol.175.6.3469. [DOI] [PubMed] [Google Scholar]
- 51.Kato A, Truong-Tran AQ, Scott AL, Matsumoto K, Schleimer RP. Airway epithelial cells produce B cell-activating factor of TNF family by an IFN-β-dependent mechanism. J Immunol. 2006;177:7164–7172. doi: 10.4049/jimmunol.177.10.7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu YJ, Barthelemy C, de Bouteiller O, Arpin C, Durand I, Banchereau J. Memory B cells from human tonsils colonize mucosal epithelium and directly present antigen to T cells by rapid up-regulation of B7-1 and B7-2. Immunity. 1995;2:239–248. doi: 10.1016/1074-7613(95)90048-9. [DOI] [PubMed] [Google Scholar]
- 53.Liu YJ, Malisan F, de Bouteiller O, Guret C, Lebecque S, Banchereau J, Mills FC, Max EE, Martinez-Valdez H. Within germinal centers, isotype switching of immunoglobulin genes occurs after the onset of somatic mutation. Immunity. 1996;4:241–250. doi: 10.1016/s1074-7613(00)80432-x. [DOI] [PubMed] [Google Scholar]
- 54.Dono M, Zupo S, Leanza N, Melioli G, Fogli M, Melagrana A, Chiorazzi N, Ferrarini M. Heterogeneity of tonsillar subepithelial B lymphocytes, the splenic marginal zone equivalents. J Immunol. 2000;164:5596–5604. doi: 10.4049/jimmunol.164.11.5596. [DOI] [PubMed] [Google Scholar]
- 55.Liu YJ, de Bouteiller O, Arpin C, Briere F, Galibert L, Ho S, Martinez-Valdez H, Banchereau J, Lebecque S. Normal human IgD+ IgM− germinal center B cells can express up to 80 mutations in the variable region of their IgD transcripts. Immunity. 1996;4:603–613. doi: 10.1016/s1074-7613(00)80486-0. [DOI] [PubMed] [Google Scholar]
- 56.Kinoshita K, Harigai M, Fagarasan S, Muramatsu M, Honjo T. A hallmark of active class switch recombination: transcripts directed by I promoters on looped-out circular DNAs. Proc Natl Acad Sci USA. 2001;98:12620–12623. doi: 10.1073/pnas.221454398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Durandy A, Peron S, Fischer A. Hyper-IgM syndromes. Curr Opin Rheumatol. 2006;18:369–376. doi: 10.1097/01.bor.0000231905.12172.b5. [DOI] [PubMed] [Google Scholar]
- 58.Sarkar SN, Peters KL, Elco CP, Sakamoto S, Pal S, Sen GC. Novel roles of TLR3 tyrosine phosphorylation and PI3 kinase in double-stranded RNA signaling. Nat Struct Mol Biol. 2004;11:1060–1067. doi: 10.1038/nsmb847. [DOI] [PubMed] [Google Scholar]
- 59.Hel Z, McGhee JR, Mestecky J. HIV infection: first battle decides the war. Trends Immunol. 2006;27:274–281. doi: 10.1016/j.it.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 60.Schaffer A, Cerutti A, Zan H, Casali P. The evolutionary conserved sequence upstream of the human Sγ3 region is a functional promoter: synergistic activation by CD40 ligand and IL-4 via distinct cis regulatory elements. J Immunol. 1999;162:5327–5336. [PubMed] [Google Scholar]
- 61.Le Bon A, Schiavoni G, D'Agostino G, Gresser I, Belardelli F, Tough DF. Type I interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity. 2001;14:461–470. doi: 10.1016/s1074-7613(01)00126-1. [DOI] [PubMed] [Google Scholar]
- 62.Groom JR, Fletcher CA, Walters SN, Grey ST, Watt SV, Sweet MJ, Smyth MJ, Mackay CR, Mackay F. BAFF and MyD88 signals promote a lupuslike disease independent of T cells. J Exp Med. 2007;204:1959–1971. doi: 10.1084/jem.20062567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Treml LS, Carlesso G, Hoek KL, Stadanlick JE, Kambayashi T, Bram RJ, Cancro MP, Khan WN. TLR stimulation modifies BLyS receptor expression in follicular and marginal zone B cells. J Immunol. 2007;178:7531–7539. doi: 10.4049/jimmunol.178.12.7531. [DOI] [PubMed] [Google Scholar]
- 64.Woof JM, Mestecky J. Mucosal immunoglobulins. Immunol Rev. 2005;206:64–82. doi: 10.1111/j.0105-2896.2005.00290.x. [DOI] [PubMed] [Google Scholar]
- 65.Macpherson AJ, Lamarre A, McCoy K, Harriman GR, Odermatt B, Dougan G, Hengartner H, Zinkernagel RM. IgA production without μ or δ chain expression in developing B cells. Nat Immunol. 2001;2:625–631. doi: 10.1038/89775. [DOI] [PubMed] [Google Scholar]
- 66.Macpherson AJ, Uhr T. Induction of protective IgA by intestinal dendritic cells carrying commensal bacteria. Science. 2004;303:1662–1665. doi: 10.1126/science.1091334. [DOI] [PubMed] [Google Scholar]
- 67.Sha Z, Compans RW. Induction of CD4+ T-cell-independent immunoglobulin responses by inactivated influenza virus. J Virol. 2000;74:4999–5005. doi: 10.1128/jvi.74.11.4999-5005.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yao Q, Zhang R, Guo L, Li M, Chen C. Th cell-independent immune responses to chimeric hemagglutinin/simian human immunodeficiency virus-like particles vaccine. J Immunol. 2004;173:1951–1958. doi: 10.4049/jimmunol.173.3.1951. [DOI] [PubMed] [Google Scholar]
- 69.Mozdzanowska K, Furchner M, Zharikova D, Feng J, Gerhard W. Roles of CD4+ T-cell-independent and -dependent antibody responses in the control of influenza virus infection: evidence for noncognate CD4+ T-cell activities that enhance the therapeutic activity of antiviral antibodies. J Virol. 2005;79:5943–5951. doi: 10.1128/JVI.79.10.5943-5951.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mackay F, Schneider P, Rennert P, Browning J. BAFF and APRIL: a tutorial on B cell survival. Annu Rev Immunol. 2003;21:231–264. doi: 10.1146/annurev.immunol.21.120601.141152. [DOI] [PubMed] [Google Scholar]
- 71.Schneider P. The role of APRIL and BAFF in lymphocyte activation. Curr Opin Immunol. 2005;17:282–289. doi: 10.1016/j.coi.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 72.Miller JP, Stadanlick JE, Cancro MP. Space, selection, and surveillance: setting boundaries with BLyS. J Immunol. 2006;176:6405–6410. doi: 10.4049/jimmunol.176.11.6405. [DOI] [PubMed] [Google Scholar]
- 73.von Bulow GU, van Deursen JM, Bram RJ. Regulation of the T-independent humoral response by TACI. Immunity. 2001;14:573–582. doi: 10.1016/s1074-7613(01)00130-3. [DOI] [PubMed] [Google Scholar]
- 74.Castigli E, Wilson SA, Scott S, Dedeoglu F, Xu S, Lam KP, Bram RJ, Jabara H, Geha RS. TACI and BAFF-R mediate isotype switching in B cells. J Exp Med. 2005;201:35–39. doi: 10.1084/jem.20032000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chiu A, Xu W, He B, Dillon SR, Gross JA, Sievers E, Qiao X, Santini P, Hyjek E, Lee JW, et al. Hodgkin lymphoma cells express TACI and BCMA receptors and generate survival and proliferation signals in response to BAFF and APRIL. Blood. 2007;109:729–739. doi: 10.1182/blood-2006-04-015958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Darce JR, Arendt BK, Wu X, Jelinek DF. Regulated expression of BAFF-binding receptors during human B cell differentiation. J Immunol. 2007;179:7276–7286. doi: 10.4049/jimmunol.179.11.7276. [DOI] [PubMed] [Google Scholar]
- 77.Castigli E, Wilson SA, Garibyan L, Rachid R, Bonilla F, Schneider L, Geha RS. TACI is mutant in common variable immunodeficiency and IgA deficiency. Nat Genet. 2005;37:829–834. doi: 10.1038/ng1601. [DOI] [PubMed] [Google Scholar]
- 78.Salzer U, Chapel HM, Webster AD, Pan-Hammarstrom Q, Schmitt-Graeff A, Schlesier M, Peter HH, Rockstroh JK, Schneider P, Schaffer AA, et al. Mutations in TNFRSF13B encoding TACI are associated with common variable immunodeficiency in humans. Nat Genet. 2005;37:820–828. doi: 10.1038/ng1600. [DOI] [PubMed] [Google Scholar]
- 79.Jain A, Ma CA, Lopez-Granados E, Means G, Brady W, Orange JS, Liu S, Holland S, Derry JM. Specific NEMO mutations impair CD40-mediated c-Rel activation and B cell terminal differentiation. J Clin Invest. 2004;114:1593–1602. doi: 10.1172/JCI21345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bergqvist P, Gardby E, Stensson A, Bemark M, Lycke NY. Gut IgA class switch recombination in the absence of CD40 does not occur in the lamina propria and is independent of germinal centers. J Immunol. 2006;177:7772–7783. doi: 10.4049/jimmunol.177.11.7772. [DOI] [PubMed] [Google Scholar]
- 81.Weller S, Faili A, Garcia C, Braun MC, Le Deist FF, de Saint Basile GG, Hermine O, Fischer A, Reynaud CA, Weill JC. CD40-CD40L independent Ig gene hypermutation suggests a second B cell diversification pathway in humans. Proc Natl Acad Sci USA. 2001;98:1166–1170. doi: 10.1073/pnas.98.3.1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Alugupalli KR, Leong JM, Woodland RT, Muramatsu M, Honjo T, Gerstein RM. B1b lymphocytes confer T cell-independent long-lasting immunity. Immunity. 2004;21:379–390. doi: 10.1016/j.immuni.2004.06.019. [DOI] [PubMed] [Google Scholar]
- 83.Obukhanych TV, Nussenzweig MC. T-independent type II immune responses generate memory B cells. J Exp Med. 2006;203:305–310. doi: 10.1084/jem.20052036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Defrance T, Vanbervliet B, Briere F, Durand I, Rousset F, Banchereau J. Interleukin 10 and transforming growth factor β cooperate to induce anti-CD40-activated naive human B cells to secrete immunoglobulin A. J Exp Med. 1992;175:671–682. doi: 10.1084/jem.175.3.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dubois B, Vanbervliet B, Fayette J, Massacrier C, Van Kooten C, Briere F, Banchereau J, Caux C. Dendritic cells enhance growth and differentiation of CD40-activated B lymphocytes. J Exp Med. 1997;185:941–951. doi: 10.1084/jem.185.5.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cerutti A, Zan H, Schaffer A, Bergsagel LP, Harindranath N, Max EE, Casali P. CD40 ligand and appropriate cytokines induce switching to IgG, IgA, and IgE and coordinated germinal center-like phenotype differentiation in a human monoclonal IgM+IgD+ B cell line. J Immunol. 1998;160:2145–2157. [PMC free article] [PubMed] [Google Scholar]
- 87.Cerutti A, Schaffer A, Zan H, Liou HC, Goodwin RG, Casali P. CD30 is a CD40-inducible molecule that negatively regulates CD40-mediated immunoglobulin class switching in non-antigen-selected human B cells. Immunity. 1998;9:247–256. doi: 10.1016/s1074-7613(00)80607-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mazzoli S, Trabattoni D, Lo Caputo S, Piconi S, Ble C, Meacci F, Ruzzante S, Salvi A, Semplici F, Longhi R, et al. HIV-specific mucosal and cellular immunity in HIV-seronegative partners of HIV-seropositive individuals. Nat Med. 1997;3:1250–1257. doi: 10.1038/nm1197-1250. [DOI] [PubMed] [Google Scholar]