

Abstract
Orthologous positions of 55 genes associated with height in four human populations were located on the bovine genome. Single nucleotide polymorphisms close to eight of these genes were significantly associated with stature in cattle (Bos taurus and Bos indicus). This suggests that these genes may contribute to controlling stature across mammalian species.
UNDERSTANDING the genetic basis for variation in complex traits has been advanced by many genome-wide association studies (GWAS) conducted in humans (Donnelly 2008) and, to a lesser extent, in other species (e.g., Karlsson et al. 2007). However, there is still considerable debate on the interpretation of these studies. Most of the single nucleotide polymorphisms (SNPs) associated with complex traits account for a very small proportion of the genetic variance in the trait (Visscher 2008). This could be because they are in incomplete linkage disequilibrium (LD) with the causative polymorphism, because the causative polymorphism has a small effect on the trait, or because the minor allele at this polymorphism is rare. The first and third of these explanations tend to apply to the same cases because a rare causative allele cannot be in complete LD with a common allele at a SNP on a standard chip. Other explanations include genotype by environment interactions, epistasis, and allelic heterogeneity. It has even been suggested that most of these associations are artifacts caused by cryptic stratification of the sample of individuals used (McClellan and King 2010). One of the reasons for skepticism is that there is often no known mechanism linking the genes found by a GWAS to the complex trait with which they are associated.
There are many examples of traits affected by single genes where mutations in the same gene cause a similar phenotype in different species. Classic examples include genes with a role in pigmentation, such as the Mc1r gene responsible for fair hair color in humans (Valverde et al. 1995) and coat or feather color in species as diverse as horse, pig, and chicken (Andersson 2003). For polygenic traits, where there are many genes with small-to-moderate effect, there is little information on the extent to which the orthologous genes cause variation in different mammalian species. However, there is evidence to suggest a high degree of conservation of certain gene classes among mammalian species, e.g., milk protein genes and mammary genes (Lemay et al. 2009). Evidence that the same genes are involved in controlling a complex trait would be important for several reasons. First, it would act as the ultimate validation study because stratification is unlikely to cause the same artifact in different species. Second, it would be strong evidence for a physiological connection between a gene in LD with an associated SNP and the trait. Third, it would provide some insight into the forces controlling genetic variation in complex traits.
Stature is an easy-to-measure phenotype used as a model complex trait in humans (Visscher 2008) and also measured in some domesticated cattle breeds (Barwick et al. 2009). In four separate GWAS of human stature, a total of 58 loci were significantly associated with stature in Caucasians (Gudbjartsson et al. 2008; Lettre et al. 2008; Weedon et al. 2008) and a further 15 loci have been reported in a Korean population (Kim et al. 2010). To test if these loci were controlling stature in cattle, we first identified the positions of orthologous genes that could be matched to Bovine Genome Build 4.0 in the NCBI database (http://www.ncbi.nlm.nih.gov/projects/genome/guide/cow/). We were able to successfully map the positions of 55 genes (some genes were close neighbors and were considered to be at the same location). There were 879 SNPs that were 500 kbp either side of these genes on the BovineSNP50 BeadChip (Illumina, San Diego, CA; Matukumalli et al. 2009), which comprises around 50,000 approximately equally spaced SNPs. The names of the orthologous genes and their bovine map positions are presented in supporting information, Table S1. We tested these SNPs for their effect on bovine stature in two cattle populations (dairy and beef cattle). The dairy data included 1832 Holstein dairy bulls with estimated breeding values for stature (hip height) based on at least 80 milking daughters per sire. The beef data set comprised hip height on 1224 Brahman and tropical composite beef heifers (Bos taurus × Bos indicus) measured at the end of the first post-weaning wet season when the heifers were aged ∼18 months (Barwick et al. 2009). We observed a skew in the quantile-quantile plot (see Figure 1), which indicates that more SNPs are associated with stature than would be expected by chance. Of 879 SNPs tested, 10 and 12 were associated with stature (P < 0.001) in dairy and beef data sets, respectively (Table 1).
Figure 1.—

Quantile-quantile plot of P-values of 879 SNPs that were 500 kbp either side of 55 orthologous genes found to be associated with height in human populations (Lettre et al. 2008; Gudbjartsson et al. 2008; Weedon et al. 2008; Kim et al. 2010). Using the dairy and beef data sets, the phenotype (stature) was regressed on each SNP by using a mixed model that included pedigree (ASReml, Gilmour et al. 2006) and the same approach as Pryce et al. (2010). The model used was  , where y is the vector of phenotypes, 1n s a vector of 1s of length n animals, W is a vector allocating SNP effects where each SNP was fitted individually, g is the fixed effect of the SNP, u is the vector of polygenic residual breeding values not accounted for by the effect of the SNP sampled from the distribution N(0,
, where y is the vector of phenotypes, 1n s a vector of 1s of length n animals, W is a vector allocating SNP effects where each SNP was fitted individually, g is the fixed effect of the SNP, u is the vector of polygenic residual breeding values not accounted for by the effect of the SNP sampled from the distribution N(0,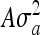 ) where A is the additive relationship matrix constructed from the pedigree,
) where A is the additive relationship matrix constructed from the pedigree, 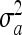 is the additive genetic variance, and e is the vector of random deviates. The purpose of including pedigree is to control for relationships and population structure. Observed and expected P-values would fall on the gray solid line if there were no association. The dashed horizontal line is the threshold selected for significance (P < 0.001). Note that a 1-Mbp window was used from which to select SNPs because, in contrast to humans, where LD is expected to persist over only 10s of kilobase pairs (Tenesa et al. 2007), non-zero levels of LD have been observed up to 1 Mbp in cattle (Bovine Hapmap Consortium 2009).
is the additive genetic variance, and e is the vector of random deviates. The purpose of including pedigree is to control for relationships and population structure. Observed and expected P-values would fall on the gray solid line if there were no association. The dashed horizontal line is the threshold selected for significance (P < 0.001). Note that a 1-Mbp window was used from which to select SNPs because, in contrast to humans, where LD is expected to persist over only 10s of kilobase pairs (Tenesa et al. 2007), non-zero levels of LD have been observed up to 1 Mbp in cattle (Bovine Hapmap Consortium 2009).
TABLE 1.
Dairy and beef cattle SNPs associated with stature within 500 kbp of genes associated with human height
| Gene associated with height in humans | Bovine chromosome | Orthologous start and stop position of gene (bp) in cattle | Position of significant SNPs (bp) | Dairy |
Beef |
||
|---|---|---|---|---|---|---|---|
| P-value | Effect size (%)a | P-value | Effect size (%)a | ||||
| HMGA2b,c,d | 5 | 51,740,850–51,890,260 | 51,770,120 | 0.69 | 0.01 | 2.7 × 10−10 | 4.03 |
| 52,214,922 | 0.72 | 0.00 | 2.0 × 10−5 | 1.38 | |||
| LCORL/NCAPGc,d,e | 6 | 38,153,047–38,199,154 | 38,256,889 | 0.05 | 0.34 | 1.1 × 10−4 | 0.97 |
| 38,326,147 | 0.45 | 0.07 | 5× 10−5 | 1.07 | |||
| 38,479,643 | 5.68 × 10−4 | 0.40 | 0.76 | 0.01 | |||
| 38,500,209 | 5.68 × 10−4 | 1.53 | 0.76 | 0.01 | |||
| 38,558,526 | 2.57 × 10−4 | 0.50 | 0.27 | 0.09 | |||
| FBP2d | 8 | 85,344,905–85,387,652 | 84,943,316 | 8.61 × 10−4 | 0.06 | 0.01 | 0.41 |
| PTCH1c | 8 | 86,551,192–86,621,018 | 86,576,819 | 0.60 | 0.03 | 9.9 × 10−4 | 0.70 |
| PAPPAb | 8 | 110,722,545–110,982,014 | 111,423,182 | 8.53 × 10−4 | 1.86 | 0.66 | 0.01 |
| GPR126b | 9 | 82,555,167–82,704,883 | 82,609,868 | 0.59 | 0.02 | 7.7 × 10−4 | 0.87 |
| PLAG1, CHCHD7, RDHE2b,d,e | 14 | 23,219,718–23,221,723PLAG1 | 22,720,374 | 0.19 | 0.33 | 1.2 × 10−5 | 1.47 |
| 22,768,981 | 0.02 | 1.14 | 9.7 × 10−6 | 1.50 | |||
| 23,265,198–23,271,074CHCHD7 | 22,803,367 | 3.27 × 10−4 | 2.53 | 9.9 × 10−6 | 1.50 | ||
| 22,838,802 | 0.11 | 0.55 | 4.1 × 10−6 | 1.62 | |||
| 23,365,427–23,398,159RDHE2 | 22,967,675 | 0.19 | 0.29 | 9.0 × 10−5 | 1.26 | ||
| 23,519,449 | 8.63 × 10−5 | 2.83 | 5.5 × 10−6 | 1.66 | |||
| CABLES1d,e | 24 | 34,619,497–34,683,911 | 34,436,971 | 4.35 × 10−4 | 0.55 | 0.08 | 0.24 |
| 34,457,809 | 5.86 × 10−4 | 1.47 | 1.00 | 0.00 | |||
| 34,637,479 | 3.59 × 10−4 | 0.57 | 0.22 | 0.12 | |||
P-values that are < 0.001 are shown in bold-face; P-values that are > 0.001 are shown in non-bold-face
Effect size (percentage of variation explained) (2pqβ2)/VA, where p and q are allele frequencies, β is the SNP solution estimate, and VA is the genetic variance of stature.
We used a randomized permutation test to evaluate how likely it was to retrieve this number of SNPs with P < 0.001 by chance. We repeated the tests between bovine SNPs and stature by selecting 55 genes at random (SNPs in the 1-Mbp region surrounding each gene) and counted the number of associations that were P < 0.001. Figure 2 shows the distribution of 10,000 random tests (for each data set). Only 0.19% and 0.38% of the randomized permutation tests in beef and dairy cow data, respectively, had fractions equal to or exceeding the results observed for stature. Therefore, it is unlikely that the results reported here for stature in dairy and beef cattle arose by chance. Note that the genes identified as stature orthologs were included in the data set used for the randomized permutation test. However, the same pattern of results was observed when the stature orthologs were excluded.
Figure 2.—

A frequency distribution of the proportion of SNPs with a P < 0.001 effect on stature in dairy and beef cattle data sets in 10,000 control tests. To make the tests comparable to the results reported in Figure 1, 55 genes were randomly selected from 16,850 bovine genes that were orthologs of human genes downloaded from BioMart (http://www.ensembl.org/biomart/index.html). The SNPs for the random control tests were in a 1-Mbp region centered on each of the 55 genes. The vertical lines represent the number of bovine SNPs with P < 0.001 association with stature in each data set. The percentage of control tests with an equivalent or greater proportion of significant SNPs associated with stature in the beef and dairy cow data was 0.19 and 0.38%, respectively. For each test, the number of SNPs significantly (P < 0.001) associated with stature was counted. The number of significant SNPs divided by the number of SNPs tested is represented along the x-axis.
Of 55 orthologous genes tested, the SNPs that were associated with stature in cattle were close to 10 genes and 8 genomic regions (Table 1). Six of these had significant SNPs in either beef or dairy cattle, while significant SNPs in both dairy and beef cattle were observed in two regions containing the gene NCAPG and a cluster on chromosome 14 (PLAG1, CHCHD7, and RDHE2). NCAPG has previously been associated with fetal growth (Eberlein et al. 2009) and carcass size (Setoguchi et al. 2009) in cattle and is thought to have a role in cell division (Murphy and Sarge 2008). The strongest signal in our study was observed for a SNP close to HMGA2 in the beef data set (2.7 × 10−10). This gene has consistently been found to be associated with stature in human studies (Visscher 2008), and it is interesting that it seems likely that it has a conserved role in cattle, too. There were only two SNPs that had associations with stature in both beef and dairy cattle data sets. This is not surprising as the SNP density of the Illumina BovineSNP50 BeadChip is insufficient to expect the phase of LD to persist across breeds as diverse as beef and dairy cattle (de Roos et al. 2008).
These results provide a unique confirmation of the significant associations found in human GWAS for height. The power to detect associations in the cattle experiments described is not high (there were <2000 animals in each study) so associations with all the SNPs tested is not expected. Furthermore, the physiological control of height is probably not identical in the two species. However, by using a series of 10,000 randomized permutation tests, we were able to show that achieving the number of associations listed in Table 1 by chance would be very unlikely (Figure 2). Together, these results add considerable weight to the conclusion that several genes identified in human GWAS for stature also have a conserved role in the physiology of growth in cattle, including a role in regulating cell division and cell cycle.
If many complex traits have a similar architecture in different species, then humans could in fact be used as a model for identifying genes for complex traits in non-model species such as primates, marsupials, and cetaceans. Comparing GWAS across species, as done here, will aid understanding of gene pathways that contribute to complex traits.
Acknowledgments
We thank colleagues from our funding organizations for their assistance in a variety of ways. We are grateful for funding from the Cooperative Research Centre for Beef Genetic Technologies, Armidale, New South Wales, Australia, and the Cooperative Research Centre for Innovative Dairy Products, Melbourne, Australia.
Supporting information is available online at http://www.genetics.org/cgi/content/full/genetics.110.123943/DC1.
Available freely online through the author-supported open access option.
References
- Andersson, L., 2003. Melanocortin receptor variants with phenotypic effects in horse, pig, and chicken. Ann. NY Acad. Sci. 994 313–318. [DOI] [PubMed] [Google Scholar]
- Barwick, S. A., M. L. Wolcott, D. J. Johnston, H. M. Burrow and M. T. Sullivan, 2009. Genetics of ‘wet’ and ‘dry’ seasons and their relationships with steer performance in two tropical beef genotypes. Anim. Prod. Sci. 49 367–382. [Google Scholar]
- Bovine Hapmap Consortium, 2009. Genome-wide survey of SNP variation uncovers the genetic structure of cattle breeds. Science 24 528–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Roos, A., B. Hayes, R. Spelman and M. Goddard, 2008. Linkage disequilibrium and persistence of phase in Holstein–Friesian, Jersey and Angus cattle. Genetics 179 1503–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly, P., 2008. Progress and challenges in genome-wide association studies in humans. Nature 456 728–731. [DOI] [PubMed] [Google Scholar]
- Eberlein, A., A. Takasuga, K. Setoguchi, R. Pfuhl, K. Flisikowski et al., 2009. Dissection of genetic factors modulating fetal growth in cattle indicates a substantial role of the non-SMC condensin I complex subunit G (NCAPG) gene. Genetics 183 728–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmour, A. R., B. J. Gogel, B. R. Cullis, S. J. Welham and R. Thompson, 2006. ASReml User Guide Release 2.0. VSN International, Hemel Hempstead, UK.
- Gudbjartsson, D. F., G. B. Walters, G. Thorleifsson, H. Stefansson, B. V. Halldorsson et al., 2008. Many sequence variants affecting diversity of adult human height. Nat. Genet. 40 609–615. [DOI] [PubMed] [Google Scholar]
- Karlsson, E. K., I. Baranowska, C. M. Wade, N. H. C. Salmon Hillbertz, M. C. Zody et al., 2007. Efficient mapping of Mendelian traits in dogs through genome-wide association. Nat. Genet. 39 1321–1328. [DOI] [PubMed] [Google Scholar]
- Kim, J.-J., H.-I. Lee, T. Park, K. Kim, J.-E. Lee et al., 2010. Identification of 15 loci influencing height in a Korean population. J. Hum. Genet. 55 27–31. [DOI] [PubMed] [Google Scholar]
- Lemay, D. G., D. J. Lynn, W. F. Martin, M. C. Neville, T. M. Casey et al., 2009. The bovine lactation genome: insights into the evolution of mammalian milk. Genome Biol. 10 R43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lettre, G., A. U. Jackson, C. Gieger, F. R. Schumacher, S. I. Berndt et al., 2008. Identification of ten loci associated with height highlights new biological pathways in human growth. Nat. Genet. 40 584–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matukumalli, L., C. Lawley, R. Schnabel, J. Taylor, M. Allan et al., 2009. Development and characterization of a high density SNP genotyping assay for cattle. PLoS ONE 4(4): e5350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClellan, J., and M-C. King, 2010. Genetic heterogeneity in human disease. Cell 141 210–217. [DOI] [PubMed] [Google Scholar]
- Murphy, L. A., and K. D. Sarge, 2008. Phosphorylation of CAP-G is required for its chromosomal DNA localization during mitosis. Biochem. Biophys. Res. Commun. 377 1007–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryce, J. E., S. Bolormaa, A. J. Chamberlain, P. J. Bowman, K. Savin et al., 2010. A validated genome wide association study in 2 dairy cattle breeds using variable length haplotypes. J. Dairy Sci. 93 3331–3345. [DOI] [PubMed] [Google Scholar]
- Setoguchi K., M. Furuta, T. Hirano, T. Nagao, T. Watanabe et al., 2009. Cross-breed comparisons identified a critical 591-kb region for bovine carcass weight QTL (CW-2) on chromosome 6 and the Ile-442-Met substitution in NCAPG as a positional candidate. BMC Genet. 4 10–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenesa, A., P. Navarro, B. J. Hayes, D. L. Duffy, G. M. Clarke et al., 2007. Recent human effective population size estimated from linkage disequilibrium. Genome Res. 17 520–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valverde, P., E. Healy, I. Jackson, J. L. Rees and A. J. Thody, 1995. Variants of the melanocyte-stimulating hormone-receptor gene are associated with fair hair and skin in humans. Nat. Genet. 11 328–330. [DOI] [PubMed] [Google Scholar]
- Visscher, P. M., 2008. Sizing up human height variation. Nat. Genet. 40 489–490. [DOI] [PubMed] [Google Scholar]
- Weedon, M. N., H. Lango, C. M. Lindgren, C. Wallace, D. M. Evans et al., 2008. Genome wide association study identifies 20 loci that influence human height. Nat. Genet. 39 1245–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]