

Summary
In aerobic environments, mutants of Escherichia coli that lack peroxidase and catalase activities (Hpx−) accumulate sub-micromolar concentrations of intracellular H2O2. We observed that in defined medium these strains constitutively expressed members of the Fur regulon. Iron-import proteins, which Fur normally represses, were fully induced. H2O2 may antagonize Fur function by oxidizing the Fur:Fe2+ complex and inactivating its repressor function. This is a potential problem, as in iron-rich environments excessive iron uptake would endanger H2O2-stressed cells by accelerating hydroxyl-radical production through the Fenton reaction. However, the OxyR H2O2-response system restored Fur repression in iron-replete LB medium by up-regulating the synthesis of Fur protein. Indeed, when the OxyR binding site upstream of fur was disrupted, Hpx− mutants failed to repress transporter synthesis, and they exhibited high levels of intracellular free iron. Mutagenesis and bacteriostasis resulted. These defects were eliminated by mutations or chelators that slowed iron import, confirming that dysregulation of iron uptake was the root problem. Thus aerobic organisms must grapple with a conundrum: how to monitor iron levels in oxidizing environments that might perturb the valence of the analyte. The induction of Fur synthesis by the OxyR response comprises one evolutionary solution to that problem.
Keywords: Iron, hydrogen peroxide, Fenton, Fur, OxyR
Introduction
Hydrogen peroxide (H2O2) is generated inside aerobic cells when reduced redox enzymes inadvertently transfer electrons to molecular oxygen (Imlay, 2003). While the rate is substantial, the impact is uncertain, because virtually all organisms contain high titers of peroxidases and catalases that rapidly degrade the H2O2. The estimated steady-state concentration of H2O2 in the model organism Escherichia coli is only 20 nM under laboratory growth conditions (Seaver and Imlay, 2001).
Organisms are also exposed to H2O2 from external sources. The chemical oxidation of compounds in cell environments can generate low-micromolar H2O2, and far higher doses arise from sources such as the oxidative burst of phagocytes, the cycling of redox-active antibiotics, and the excretion of H2O2 by lactic acid bacteria. Since H2O2 readily crosses membranes, cells that are exposed to these H2O2 sources must defend themselves against the stress that ensues.
Intracellular concentrations of H2O2 in excess of 100 nM are sufficient to induce the OxyR stress response system in E. coli (Aslund et al., 1999; Seaver and Imlay, 2001), implicitly defining the H2O2 level that evolution considers hazardous. It has been a challenge, however, to identify the biomolecules that such a small dose of H2O2 can damage. Part of the difficulty is the experimental design. When low concentrations of H2O2 are added to cell cultures, scavenging enzymes soon clear the H2O2, and so the stress is abbreviated and the impact upon growth processes cannot easily be studied. Millimolar doses can overwhelm these systems and are lethal to most organisms, but they are not physiological.
An alternative approach is to genetically eliminate the scavenging systems, thereby allowing the imposition of low, persistent H2O2 stress. We have used hydroperoxidase (Hpx−) mutants of E. coli, which lack its NADH peroxidase and two catalases, for this purpose. When these strains are grown in aerobic medium, H2O2 is formed by endogenous processes and equilibrates across the membrane, gradually rising to approximately 1 micromolar both inside and outside the cell. Substantial DNA damage accumulates due to the formation of hydroxyl radicals, as H2O2 reacts with unincorporated iron (Park et al., 2005):
| [1] |
In these mutants the H2O2 stress would be lethal except for the fact that OxyR triggers the induction of Dps (Almiron et al., 1992; Zhao et al., 2002), an iron-storage protein that partially suppresses DNA damage by sequestering iron in an unreactive form. Thus in aerobic media Hpx− oxyR and Hpx− dps mutants rapidly die (Park et al., 2005). OxyR also induces another dozen or so other proteins, many of whose roles are not yet clear (Zheng et al., 2001a; Zheng et al., 2001b).
Hpx− mutants also exhibit specific biosynthetic defects, suggesting that H2O2 poisons enzymes that belong to particular pathways. In the course of investigating those, we observed that the synthesis of superoxide dismutase is altered. We traced this phenotype to the ability of H2O2 to disrupt the function of the iron-homeostatic control protein Fur (Escolar et al., 1999; Braun, 2003). This report describes that effect and a mechanism whereby the OxyR system minimizes its impact.
Results
The fur regulon is derepressed in Hpx− mutants
Hydroperoxidase-deficient (Hpx−) mutants of E. coli lack both catalase and NADH peroxidase activities, and H2O2 consequently accumulates up to approximately 1 micromolar in culture media (Seaver and Imlay, 2001). Because H2O2 rapidly equilibrates across cell membranes, this concentration closely represents the concentration of H2O2 inside the cells. Several growth defects result, including deficiencies in the biosyntheses of aromatic and branched-chain amino acids (Jang and Imlay, 2007). These defects resemble those that are observed in mutants that lack superoxide dismutase (Carlioz and Touati, 1986), and so it was prudent for us to verify that the Hpx− mutants continue to synthesize normal levels of SOD. Unexpectedly, using several independent E. coli K-12 lineages, we found that Hpx− mutants actually contained substantially more SOD than did wild-type cells: 11.3 +/− 0.9 vs. 3.0 +/− 0.4 U/mg.
Activity gels revealed that the increase in total SOD activity was due to elevated levels of the manganese-containing SOD (MnSOD, encoded by sodA) (Fig. 1A). Strikingly, iron-containing SOD (FeSOD, sodB) was almost entirely absent.
Figure 1. MnSOD and FeSOD activities in Hpx− mutants.

(A) SOD activity gels. Congenic strains were grown aerobically in defined medium to an OD600 of 0.5. Lane 1: Wild type (MG1655); lane 2: ryhB mutant (EM1456); lane 3: Hpx− mutant (LC106); lane 4: Hpx− ryhB mutant (SMV36). (B) SOD activities. Extracts were prepared as in panel A, and SOD activity was determined by the xanthine-xanthine oxidase method. MG1655 (wild type), LC106 (Hpx−), SMV32 (sodB), SMV29 (Hpx− sodB), KCI420 (sodA), and SMV31 (Hpx− sodA), and SMV40 (Hpx− sodA ryhB).
The level of each isozyme can be quantified in extracts prepared from mutants that lack the other isozyme. The data indicated that in Hpx− mutants the MnSOD was induced approximately seven-fold above the levels that were present in Hpx+ strains (Fig. 1B). The FeSOD was almost undetectable.
MnSOD synthesis is controlled by at least four regulatory proteins. Transcription of the sodA gene is stimulated by the SoxRS system when cells are treated with redox-cycling drugs that trigger intracellular superoxide formation (Hassan and Fridovich, 1977; Ding and Demple, 1997; Gaudu et al., 1997), and other labs have shown that millimolar levels of H2O2 can also activate this system (Manchado et al., 2000; Zheng et al., 2001b). However, another SoxRS-inducible enzyme, glucose-6-phosphate dehydrogenase, was not induced in Hpx− mutants, and the MnSOD level in Hpx− mutants did not diminish when we introduced a ΔsoxRS null mutation (data not shown). Since two other regulators of sodA transcription--Fnr and Arc (Hassan and Sun, 1992; Compan and Touati, 1993)--should be deactivated in aerobic conditions, we suspected that the one remaining sodA regulator--the Fur repressor--might be inactivated in Hpx− mutants.
When E. coli is replete with iron, the Fur repressor binds Fe2+ and acquires activity as a repressor both of sodA and of multiple genes encoding iron-import systems (reviewed in Escolar et al., 1999, and Braun, 2003). Indeed, the introduction of a fur mutation into a wild-type strain increased MnSOD levels several-fold, while it did not further elevate MnSOD levels in the Hpx− strain (data not shown). This result supported the idea that endogenous H2O2 derepresses the Fur regulon when Hpx− mutants are grown in defined medium.
The Fur protein indirectly controls FeSOD synthesis as well (Masse and Gottesman, 2002; Geissmann and Touati, 2004). When the cell is replete with ferrous iron, Fe2+-metallated Fur protein represses the synthesis of the RyhB small RNA, which would otherwise trigger the degradation of sodB mRNA. In combination with the direct repression of MnSOD by Fur, this control mechanism directs the cell to synthesize FeSOD when it perceives that it has sufficient iron to activate it, and to synthesize MnSOD only when iron levels are inadequate. The addition of a ryhB null mutation restored the synthesis of FeSOD to Hpx− mutants (Fig. 1A, lane 4; Fig 1B, lane 7). It had no effect upon wild-type cells. Thus the altered regulation of SOD synthesis indicated that the Fur repressor was inactive in Hpx− mutants.
Intracellular hydrogen peroxide stimulates transcription of iron-import systems
If the Fur repressor is inactivated by H2O2, then the iron-uptake systems that Fur controls should also be derepressed. This prediction was tested by monitoring the expression of the iuc operon, which provides some E. coli strains with an additional siderophore biosynthetic pathway. Like other iron-transport systems, expression of iuc is directly repressed by the Fur:Fe2+ complex. An iucC::lacZ fusion was introduced into the Hpx+ and Hpx− strains, and expression was monitored in the same defined medium that was used in the studies of SOD synthesis. In the wild-type strain the fusion was only expressed when dipyridyl, an iron chelator, was added to the culture. However, the fusion was fully induced in the Hpx− strains even without the chelator (Fig. 2A). This result confirms that the Fur regulon is derepressed.
Figure 2. Expression of the Fur regulon in Hpx− mutants.

(A) Aerobic defined medium. (B) Aerobic LB medium. WT: wild-type for Hpx. DP: Dipyridyl added. The strains were MS100 (Hpx+ iucC::lacZ), MS102 (Hpx− iucC::lacZ), and SMV59 (Hpx− iucC::lacZ furNI). The results represent the averages of three independent experiments. C) Levels of fur RNA transcript (top gel) and 5S RNA (bottom gel). Lane 1: MG1655 (WT, Hpx+); lane 2: SMV55 (Hpx+ furNI); lane 3: KCI402 (Hpx+ fur::kan); lane 4: LC106 (Hpx−).
Under the conditions of these experiments, the intra- and extracellular H2O2 concentration rose to only 0.5–1.0 micromolar. An earlier study showed that these levels of H2O2 are sufficient to rapidly oxidize chelated ferrous iron through the Fenton reaction (Park et al., 2005). Because Fur repressor activity depends upon maintaining bound iron in the ferrous state, the oxidation of the Fur:Fe2+ complex provides a likely mechanism whereby H2O2 might derepress the Fur regulon (see Discussion).
The induction of fur by the OxyR system minimizes growth defects
The induction of iron-uptake systems seems like a misguided response for H2O2-stressed cells, since the rapid import of iron could make cells vulnerable to Fenton chemistry. Indeed, in Hpx− strains the OxyR response to H2O2 triggers the overexpression of the Fur protein, perhaps in an effort to shut the Fur regulon back off (Zheng et al., 1999). The iucC::lacZ data indicate that in defined medium this effort fails, presumably because iron availability is limited. The addition of 100 μM ferrous sulfate restored full repression (data not shown).
In complex medium, however, iron is more abundant and the gene fusion is fully repressed (Fig. 2B). Hpx− oxyR mutants are inviable in aerobic medium (Park et al., 2005. Therefore, to test whether the repression of iucC depends upon the OxyR-mediated induction of fur, we created a mutant strain in which the OxyR binding site upstream of the fur gene was selectively deleted (Materials and Methods). The resultant non-inducible fur allele (furNI) was fully functional in the absence of H2O2 stress, as Fur repressed iucC::lacZ in Hpx+ cells unless chelators were added to starve the cell for iron (data not shown). In addition, Northern-blot analysis confirmed that the basal transcription of fur was unaffected (Fig. 2). However, when the furNI allele was transduced into the Hpx− background, the iucC::lacZ fusion was significantly derepressed in LB medium, being expressed at a level between the repressed level of the Hpx− Fur+ strain and the fully-induced level of strains that are starved for iron (Fig. 2B). Therefore, during H2O2 stress the OxyR-dependent induction of Fur synthesis is needed to keep the Fur regulon repressed.
Strikingly, Hpx− strains exhibited growth defects that increased in parallel with their inability to synthesize Fur repressor (Fig. 3). Upon dilution into aerobic medium, all strains grew well for several generations. Strains with a wild-type fur allele grew continuously. In contrast, after two hours of growth, strains that lacked the OxyR binding site upstream of fur exhibited a prolonged lag and some loss of viability. Finally, strains bearing a fur::kan null allele lagged even longer, and their viability declined tenfold during the lag. The eventual outgrowth of these mutants may be due to the OxyR-mediated induction and action of Dps, an iron-sequestration protein, as Hpx− dps mutants do not recover from a growth lag (Park et al., 2005). In all cases the growth defects were fully eliminated by the addition of a plasmid containing the wild-type fur allele but not by a control vector (data not shown).
Figure 3. Growth behavior of Hpx− strains in LB medium.

(A) Growth after strains were subcultured into LB medium at time zero. Hpx− mutant (LC106, circles), Hpx− furNI (SMV57, squares), Hpx− fur::kan (KCI323, triangles). Dashed lines: 2 mM desferrioxamine was included in the medium of the Hpx− furNI (squares) or Hpx− fur (triangles) mutants. (B) Number of viable cells during the experiment of panel A. Viability was determined at time points by anaerobic plating and colony enumeration.
Cell-permeable iron chelators can prevent the intracellular formation of hydroxyl radicals by binding iron in a redox-inactive form (Imlay et al., 1988). In fact, the addition of desferrioxamine to the growth medium eliminated the growth lag of the Hpx− fur null mutant (Fig. 3A, dashed line) and the Hpx− furNI mutant (not shown). Cell death was also prevented (not shown).
Hydroxyl radicals will damage whatever biomolecules they encounter, but the target of greatest consequence is typically DNA. One morphological consequence of DNA damage in E. coli is the appearance of cell filaments, which arise when this bacterium cannot replicate its DNA and therefore postpones septation. Hpx− mutants exhibit some filamentation (Fig. 4). Filamentation was more extensive during the growth lab of Hpx− furNI mutants, suggesting that derepression of the Fur regulon accelerated DNA damage. Filaments were not formed when desferrioxamine was added.
Figure 4. Filamentation of Hpx− furNI cells.
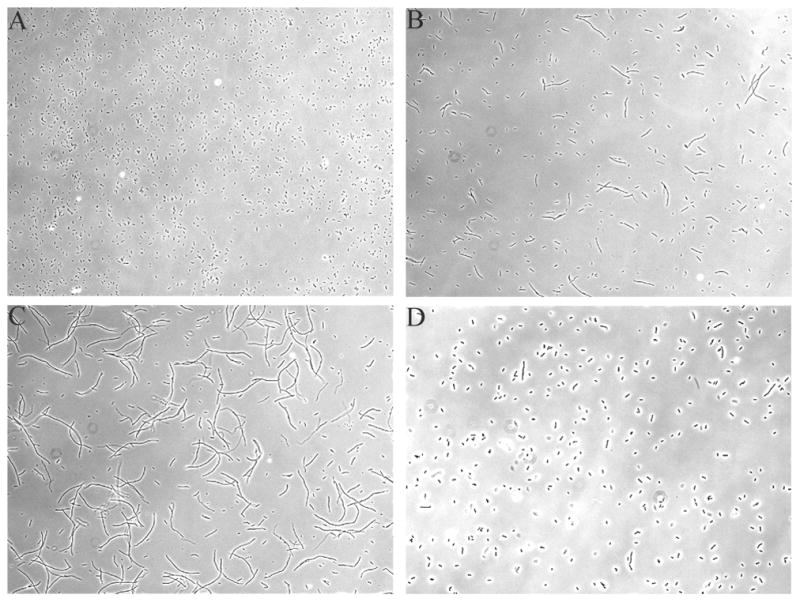
Photographs were obtained 6 hrs into the aerobic growth period. (A) MG1655 (wild type), (B) LC106 (Hpx−), (C) SMV57 (Hpx− furNI), and (D) SMV57 (Hpx− furNI) with 20 mM desferrioxamine in the medium. KCI323 (Hpx− fur::kan) exhibited morphologies similar to that of panels C and D.
To independently test whether DNA damage was occurring, mutation rates were measured in wild-type (Hpx+ Fur+), Hpx− Fur+, Hpx− furNI, and Hpx− fur::kan strains. Cultures were grown into exponential phase anaerobically, shifted into aerobic medium for four hours, and then plated on anaerobic trimethoprim plates for quantitation of the thyA mutations that had occurred during the period of aeration. Wild-type cells cultures contained 1 × 10−6 thyA mutants per viable cell. The Hpx− mutant had 9-fold more mutants, whereas the rates were 110 and 650-fold higher in the Hpx− furNI and the Hpx− fur::kan strains, respectively. These results suggest that derepression of the fur regulon substantially exacerbates the high rate of DNA damage that already occurs in Hpx− mutants.
OxyR must induce fur to avoid excessively high levels of intracellular iron
The unincorporated iron that catalyzes DNA damage can be quantified inside intact cells by EPR spectroscopy (Woodmansee and Imlay, 2002). Desferrioxamine is added to aerobic cultures, and intracellular desferrioxamine-iron chelates exhibit a signal at 4.3 G. Desferrioxamine captures iron that is loosely bound to biomolecules, including DNA; however, desferrioxamine does not appear to remove iron that has been incorporated into metalloproteins.
The unincorporated (free) iron levels were substantially higher inside Hpx− mutants than wild-type cells (Fig. 5). Consistent with our expectation, levels were particularly high in Hpx− strains that carry the furNI allele.
Figure 5. Intracellular concentrations of unincorporated iron.

Strains were grown in aerobic LB medium for four hours, and intracellular iron concentrations were measured using EPR spectroscopy. (A) Representative EPR spectra of the strains, normalized to cell density. (B) Quantification of the intracellular iron concentrations. The values are the averages of three independent experiments. The strains used were MG1655 (wild type), LC106 (Hpx−), SMV57 (Hpx− furNI) and KCI323 (Hpx− fur::kan).
To establish a direct connection between excessive iron import and the growth defects of the fur mutants, we tested whether the defects could be avoided if import were slowed. EDTA limits the availability of iron for the transport systems, and it almost completely suppressed the problems, even for Hpx− mutants bearing the fur null allele (Fig. 6). The same result was achieved by the introduction of a tonB mutation. The TonB protein powers iron uptake through several high-affinity systems that Fur would normally repress in this medium (Wiener, 2005). We deduce that H2O2 stress endangers cells by inactivating Fur-dependent repression of TonB-dependent iron import systems and that OxyR induces Fur in order to partially correct this situation.
Figure 6. Role of iron import systems in the growth defects of fur mutants.
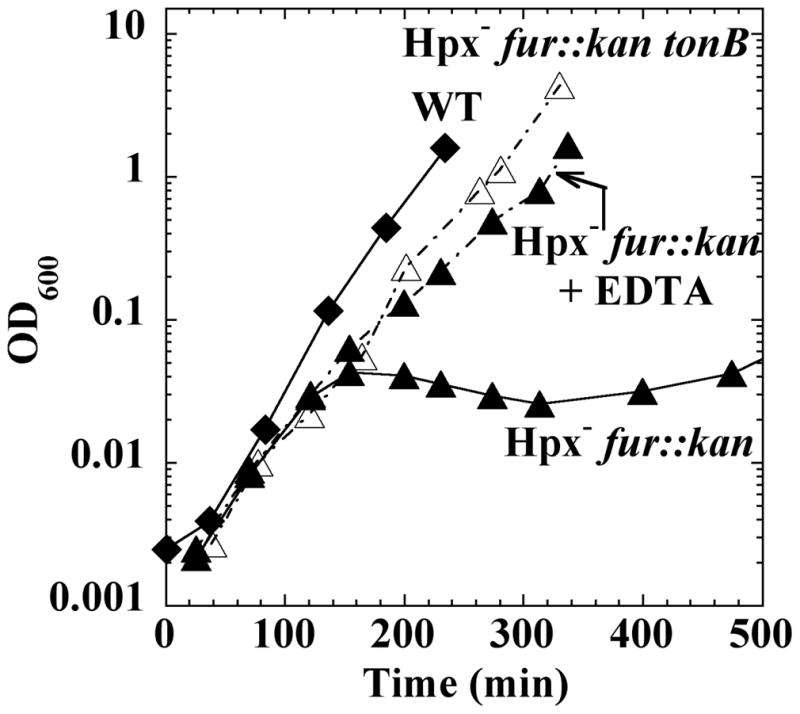
Cultures were grown in aerobic LB medium. Hpx− fur::kan (KCI323, filled triangles), Hpx− fur::kan tonB (AW510, open triangles), and wild-type (MG1655, diamonds) strains are shown. Where indicated, EDTA was included in the medium.
Discussion
Organisms monitor intracellular Fe2+ levels to ensure iron sufficiency. Because hydrogen peroxide efficiently converts ferrous iron to its ferric form, bacteria are vulnerable to the error of mistaking the presence of H2O2 for the absence of iron. This misperception may have disastrous consequences, if their response is to increase iron uptake. To avoid this error, E. coli has evolved the OxyR system, which detects H2O2 and attempts to correct the cell in two ways: by increasing the cellular titer of Fur protein, in hopes that the amount of Fur:Fe2+ complex will reach a level that turns import systems back off; and by synthesizing large amounts of Dps, an iron-storage protein that sequesters the ferrous iron that uptake systems may inappropriately import. During periods of chronic, sub-micromolar oxidative stress, both strategies are critical, as Hpx− oxyR (Park et al., 2005) and Hpx− furNI mutants both exhibit high rates of mutagenesis and toxicity.
The reactivity of the Fur:Fe2+ complex with H2O2 is a simple example of Fenton chemistry. Initial investigations of the Fenton reaction were conducted by chemists, who worked at acidic pH in order to ensure the solubility of the ferrous iron. Under those conditions they determined the rate constant of the reaction to be 76 M−1 s−1 (Walling, 1975). This value is relatively low, and when it entered the biological literature some workers inferred that, at physiological levels of H2O2, Fenton chemistry was unlikely to occur quickly enough to comprise a significant threat (discussed by Halliwell and Gutteridge, 1990). However, when ferrous iron is complexed by anionic ligands--either hydroxide anion in neutral-pH solutions, or by protein or nucleic acid ligands--the reaction rate is elevated by orders of magnitude (Rush et al., 1990; Park et al., 2005). By extrapolation from measurements made at lower temperatures, we estimate that at neutral pH the rate constant for the Fenton reaction with hexaqueous or ATP-bound iron is 20,000–30,000 M−1 s−1 at 37° C. Therefore, when 0.5 μM H2O2 is present, as in the experiments reported here, the half-time of Fe2+ oxidation will be about 1 min. Thus cell damage through Fenton chemistry is a real hazard, including the DNA damage. These doses of H2O2 also rapidly inactivate some enzymes that contain [4Fe-4S] clusters, through an analogous reaction between H2O2 and a solvent-exposed iron atom (Jang and Imlay, 2007). Strikingly, at this point each of the phenotypes of Hpx− mutants has been tracked to reactions between H2O2 and iron.
These studies affirm a long-standing hypothesis: aerobic organisms evolved H2O2-scavenging enzymes to avoid being poisoned by endogenous H2O2. However, their evolution did not solve the problem of oxidative stress completely. Because the cell membrane is a weak barrier to H2O2, it does not permit a large outside-to-inside concentration gradient (Seaver and Imlay, 2001); therefore, wild-type cells need encounter only ~ 5 μM H2O2 in their environment for the intracellular levels to rise to the same 1 μM dose that damaged DNA, inactivated enzymes, and disrupted iron homeostasis in the Hpx− mutants. It is for this reason that H2O2 is a weapon of choice among competing organisms.
If the OxyR system evolved to defend cells against external H2O2, then even scavenger-proficient (Hpx+) cells that bear the furNI allele should exhibit sensitivity to H2O2. No impact was expected or observed under usual experimental conditions, as moderately dense cultures scavenge micromolar H2O2 from the media very rapidly (data not shown). Therefore we measured the outgrowth of furNI and Δfur (Hpx+) strains when they were diluted to 103 cfu/ml into LB medium spiked with micromolar H2O2. In all trials, the recovery of Δfur mutants lagged substantially, as they achieved a measurable absorbance approximately three generations after wild-type cells. In each of five trials the furNI mutants trailed the wild-type strain by an average of 0.5 generations--but the variation among triplicate samples was too large to provide statistical significance. We have not identified the source of the data dispersion, so at present this result must be regarded as uncertain. It is probable that bacteria confront protracted H2O2 stress in natural environment, but the difficulty of replicating this exposure is the reason that we employed Hpx− strains in this study.
Interestingly, Lee and Helmann have shown that Bacillus subtilis has exploited the reactivity of protein-bound ferrous iron to create an alternative mechanism of H2O2 sensing (Lee and Helmann, 2006). PerR is an H2O2 sensor that plays the same role in B. subtilis as does OxyR in E. coli, as it regulates many of the same defensive proteins, including homologues of catalase, Ahp, and Dps. However, PerR structurally resembles Fur, and it binds ferrous iron in a similar coordination environment. As H2O2 levels rise, the oxidation of the bound iron atom inactivates the repressor activity of the protein, allowing induction of the genes that it otherwise represses. In PerR, the Fenton reaction triggers oxidation of histidyl ligands of the iron cofactor. It is not yet clear whether upon oxidation the E. coli Fur protein is similarly modified. In any case, the ferric ion that is produced in Fur is expected to dissociate from the protein, as the failure of ferric iron to adhere to the binding site is the reason that many workers have used stable Mn2+ as a cofactor during aerobic in vitro experiments.
The mammalian IRE-binding protein may represent an evolutionary adaptation that avoids the Fur problem and allows cells to distinguish oxidative stress from iron deficiency (reviewed by Cairo et al., 2002). This aconitase-like protein has an exposed [4Fe-4S] cluster that cannot be formed when iron is scarce; in that situation the apoprotein binds to mRNAs, stabilizing those that encode iron-uptake proteins and destabilizing those that encode iron-storage proteins. As mentioned, H2O2 can directly oxidize and destabilize such clusters. However, when [4Fe-4S] clusters are oxidized, a [3Fe-4S] form, rather than an apoprotein, is generated (Brown et al., 2002; Jang and Imlay, 2007). Experiments to date suggest that the residual cluster is fairly stable and that it continues to occupy the cleft that must be empty for IRE-binding protein to bind RNA. When iron is available, the cluster is directly remetallated to the native [4Fe-4S] form (Djaman et al., 2004). As a result, the oxidized protein should not behave like the apoprotein, and the presence of H2O2 would not be mistaken for the absence of iron.
Experimental procedures
Chemicals and strains
Desferrioxamine (deferoxamine mesylate), diethylenetriaminepentaacetic acid (DETAPAC), dipyridyl, o-nitrophenyl B-D-galactopyranoside (ONPG), casamino acids, xanthine, xanthine oxidase, ferric chloride, thymidine, and trimethoprim were from Sigma. Nitroblue tetrazolium was from Fisher, and manganese chloride was from Aldrich.
Strains
Strains used in this study are listed in Table 1. All constructions in Hpx− (i.e., katE katG ahp) backgrounds were performed in an anaerobic chamber to ensure that suppressor mutations were not selected during outgrowth. Transductions and matings were by standard procedures (Miller, 1972). Mutant alleles were selected by the associated antibiotic resistance. Mutations in soxS and ryhB were verified by PCR analysis; in sodA and sodB, by activity gel (Beauchamp and Fridovich, 1971); in fur, by the co-transduced cam allele. Transconjugates that received the pColV plasmid bearing the iucC::lacZ allele were identified by the induction of B-galactosidase when grown in LB medium containing 1 mM dipyridyl.
Table 1.
| Strain | Genotype | Reference |
|---|---|---|
| BW381 | As GC4468 plus soxS::Tn10 | (Tsaneva and Weiss, 1990) |
| SMV37 | As LC106 plus soxS::Tn10 | P1(BW381) × LC106 |
| LC106 | ΔahpCF’ kan::’ahpF Δ (katG17::Tn10)1 Δ(katE12::Tn10)1 | (Seaver and Imlay, 2004) |
| GI69 | zdg-299::Tn10 | (Grogan and Cronan, 1984) |
| JI132 | (sodA::Mud PR13)25 (sodB::kan)1-Δ2 | Lab stock |
| KK183 | As JI132 plus zdg-299::Tn10 | P1(G169) × JI132 |
| SMV29 | As LC106 plus (sodB::kan)1-Δ2 | P1(KK183) × LC106 |
| SMV32 | As MG1655 plus (sodB::kan) 1-Δ2 | P1(KK183) × MG1655 |
| SMV31 | As LC106 plus (sodA::Mud PR13)25 | P1(KK183) × LC106 |
| KCI420 | As MG1655 plus (sodA::Mud PR13)25 | P1(PN134) × MG1655 |
| RZ8457 | As MG1655, As MG1655, Δfnr (Bsm-Mlu)::Ω(SpR), zcj::Tn10 | P. Kiley |
| SMV33 | As MG1655 plus Δfnr (Bsm-Mlu)::Ω(SpR), zcj::Tn10 | P1(RZ8457) × MG1655 |
| SMV34 | As LC106 plus Δfnr (Bsm-Mlu) )::Ω(SpR), zcj::Tn10 | P1(RZ8457) × LC106 |
| EM1456 | As MG1655 lacx74 Δ ara714 ryhB::cam with pNM12 | E. Masse |
| SMV36 | As LC106 plus rhyB::cam | P1(EM1456) × LC106 |
| KK1445 | zhh-5::Tn10 | E. coli Genetic Stock Center |
| SMV380 | As EM1456 plus zhh-5::Tn10 | P1(KK1445) × EM1456 |
| SMV40 | As SMV31 plus ryhB::cam~zhh-5::Tn10 | P1(SMV380) × SMV31 |
| CGSC6391 | Hfr valS uxuBA zbf-507::Tn10 (near 15.7′) trp-49 (Am), lacZ125 (Am), relA1, spot1, l- | E. coli Genetic stock center |
| BW25113 | Plus pKD46 | (Datsenko and Wanner, 2000) |
| SP81 | As BW25113 plus zbf-507::Tn10 | P1(CGSC6391) × BW25113 |
| SP82 | As SP81 plus fur::camNI | This work |
| SP84 | As BW25113 plus furNI~zbf-507::Tn10 (asnB-507::Tn10) | This work |
| SMV49 | As MG1655 plus fur::camNI~Tn10 | P1(SP82) × MG1655 |
| SMV50 | As LC106 plus fur::camNI~Tn10 | P1(SP82) × LC106 |
| SMV52 | As MG6155 plus fur::camNI | P1(SP82) × MG1655 |
| SMV53 | As LC106 plus fur::camNI | P1(SP82) × LC106 |
| SMV55 | As MG1655 plus furNI~zbf-507::Tn10 | P1(SP84) × MG1655 |
| SMV57 | As LC106 plus furNI~zbf-507::Tn10 | P1(SP84) × LC106 |
| BN407 | As AB1157 pColV::lacZ (iucC::lacZ) | (Bagg and Neilands, 1985) |
| SMV59 | As SMV57 plus pcolV::lacZ(iucC::lacZ) | BN407 × SMV57 |
| MS100 | As MG1655 plus pcolV::lacZ(iucC::lacZ) | BN407 × MG1655 |
| MS102 | As LC106 plus pcolV::lacZ(iucC::lacZ) | BN407 × LC106 |
| KK204 | As AB1157 plus fur::kan | Lab stock |
| KK210 | fur::kan~ zbf-507::Tn10. | Lab stock |
| KCI323 | As LC106 plus fur::kan~zbf-507::Tn10 | P1(KK210) × LC106 |
| AW25113 | As BW25113 plus tonB::cat | This work |
| AW510 | As KCI323 plus tonB::cat | P1(AW25113) × KCI323 |
To create plasmids overexpressing the wild-type fur allele, primers that flank the fur gene and its promoter region (709,200–710,400) were designed with EcoRI and XbaI sites. The PCR product was inserted into the pWKS30 plasmid. The chromosomal furNI allele, in which the OxyR binding site is removed and replaced with an flp scar sequence, and the tonB mutant were generated by standard methods (Datsenko and Wanner, 2000). The resultant mutations were confirmed by PCR preparation and sequencing. In the furNI mutant allele bases 710,094–710,129 have been removed, corresponding to positions −43 to −78 upstream of the transcriptional start site. The deletion removes the OxyR binding site (Zheng et al., 1999) but does not alter the −10/−35 promoter region or the 5′ UTR of the transcript.
Growth curves and survival of strains
Defined medium consisted of minimal A salts (Miller, 1972) supplemented with 0.2% glucose and 0.2% casamino acids, and complex medium consisted of Luria broth (LB) (Miller, 1972). Overnight cultures were grown in anaerobic medium inside an anaerobic chamber (Coy Laboratory Products, Inc.). They were then diluted to approximately 0.010 OD600 in fresh anaerobic medium and grown in the chamber for 3–4 generations. For studies of aerobic stress, these exponentially growing cells were then subcultured either to an initial OD600 of 0.001 for experiments in LB medium or to an OD600 of 0.015 for studies in defined medium. Under these conditions Amplex red/HRP methods (Seaver and Imlay, 2001) show that the concentration of H2O2 in defined medium gradually rises in several hours to about 0.8 micromolar ((Park et al., 2005) and data not shown). The concentration of H2O2 in LB medium could not be directly determined, because components of the medium interfere with the H2O2 assay; however, data suggest that photochemical reactions between room light and chromophores in the medium gradually create 1–5 micromolar H2O2 (unpublished data).
Cells were photographed using a phase contrast microscope after six hours of growth in aerobic LB medium. The viability of aerobic cultures was monitored by transferring aliquots into the anaerobic chamber. The cultures were then diluted in LB medium and plated in anaerobic top agar on anaerobic LB plates. Colonies were counted after 24 hrs.
Enzyme assays
Aerobic cultures were grown in defined medium to an OD600 of 0.5. For measurements of SOD activity, extracts were then prepared and assayed by the xanthine/xanthine oxidase method (McCord and Fridovich, 1969) or visualized on activity gels (Beauchamp and Fridovich, 1971) by standard procedures. In some cases, extracts were first subjected to partial denaturation and renaturation in the presence of metals to ensure the full activation of metal-specific SOD isozymes (Privalle and Fridovich, 1988). B-galactosidase activity was measured in extracts prepared in the same buffer (Miller, 1972). Total protein was determined using a dye-binding assay (Pierce).
RNA analysis
Anaerobic cultures in LB medium were diluted into aerobic LB medium and grown for 1 hour. RNA was extracted, isolated, and analyzed by Northern blot as described (Masse and Gottesman, 2002). A biotinylated DNA probe containing the fur gene sequence 5′-AGCGTTACTTTCACCCCAGCTTTCTTTAGGGCGGTATTGT- 3′ was used to probe for the fur RNA transcript.
EPR measurements
Unincorporated iron was measured by EPR spectroscopy (Woodmansee and Imlay, 2002). Cells were grown in 1 liter of freshly autoclaved LB aerobically as described above. MG1655 was harvested at an OD600nm of 0.1, while Hpx− strains were harvested 4 hours into the growth curve. Cells were pelleted by centrifugation at 8,000 rpm for 5 mins at 4°C. The cell paste was resuspended in 9 mls of LB, and 1 mL of 0.1 M DETAPAC (pH 7.0) and 1 mL of 0.2 M desferrioxamine (pH 8.0) were added sequentially. DETAPAC is a strong cell-impermeant chelator that blocks further iron import, while desferrioxamine is a cell-penetrating iron chelator that captures unincorporated intracellular ferrous iron and triggers its oxidation to the EPR-active ferric form. At the millimolar doses of desferrioxamine that were used in these experiments, its import does not depend upon the Fhu system and therefore is not affected by the status of the fur gene. The culture was then incubated aerobically at 37°C for 15 mins. The cells were then centrifuged at 4° C, and the pellet was washed twice with ice-cold 20 mM Tris-Cl (pH 7.4) and then resuspended in 200 μl of the same ice cold buffer containing 10% glycerol. Most of the dense cell suspension (200 μl) was added to the EPR tube and promptly frozen on dry ice. Another aliquot was diluted, and its absorbance was measured. The use of a factor that transforms absorbance into cell volume (1 ml of 1 OD cells comprises 0.47 μl cytoplasmic volume (Imlay and Fridovich, 1991)) allowed subsequent calculations of intracellular iron concentrations. Iron standards were prepared and EPR measurements were conducted according to previous methods.
Measurement of mutagenesis
Cells were grown and inoculated into aerobic LB medium as described above; parallel cultures were grown in anaerobic LB medium. Four hours into the growth curve, aliquots of cultures were diluted into minimal A salts in the anaerobic chamber. They were then aliquotted into F-top agar containing catalase and either thymidine (to determine total viable cells) or thymidine plus trimethoprim (TMP; to determine thyA mutants) and spread onto defined-medium plates. Colonies were quantified after 24 hrs. Subsequent experiments verified that trimethoprim-resistant colonies could replate on defined medium only if thymidine was supplied, consistent with a thyA genotype (Miller, 1972).
Acknowledgments
We thank Mark Nilges of the University of Illinois EPR Center and Cari Vanderpool of our department for their expert experimental guidance, Eric Masse for providing strains, and Snehalkumar Patel and Michel Bellini for the use of their phase contrast microscope. This work was supported by GM49640 from the National Institutes of Health.
References
- Almiron M, Link AJ, Furlong D, Kolter R. A novel DNA-binding protein with regulatory and protective roles in starved Escherichia coli. Genes & Development. 1992;6:2646–2654. doi: 10.1101/gad.6.12b.2646. [DOI] [PubMed] [Google Scholar]
- Aslund F, Zheng M, Beckwith J, Storz G. Regulation of the OxyR transcriptional factor by hydrogen peroxide and the cellular thiol-disulfide status. Proc Natl Acad Sci USA. 1999;96:6161–6165. doi: 10.1073/pnas.96.11.6161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagg A, Neilands JB. Mapping of a mutation affecting regulation of iron uptake systems in Escherichia coli K-12. J Bacteriol. 1985;161:450–453. doi: 10.1128/jb.161.1.450-453.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauchamp C, Fridovich I. Superoxide dismutase: improved assays and an assay applicable to acrylamide gels. Anal Biochem. 1971;44:276–287. doi: 10.1016/0003-2697(71)90370-8. [DOI] [PubMed] [Google Scholar]
- Braun V. Iron uptake by Escherichia coli. Front Biosci. 2003;1:s1409–1421. doi: 10.2741/1232. [DOI] [PubMed] [Google Scholar]
- Brown NM, Kennedy MC, Antholine WE, Eisenstein RS, Walden WE. Detection of a [3Fe-4S] cluster intermediate of cytosolic aconitase in yeast expressing iron regulatory protein 1. Insights into the mechanism of Fe-S cluster cycling. J Biol Chem. 2002;277:7246–7254. doi: 10.1074/jbc.M110282200. [DOI] [PubMed] [Google Scholar]
- Cairo G, Recalcati S, Pietrangelo A, Minotti G. The iron regulatory proteins: targets and modulators of free radical reactions and oxidative damage. Free Rad Biol Med. 2002;32:1237–1243. doi: 10.1016/s0891-5849(02)00825-0. [DOI] [PubMed] [Google Scholar]
- Carlioz A, Touati D. Isolation of superoxide dismutase mutants in Escherichia coli: is superoxide dismutase necessary for aerobic life? EMBO J. 1986;5:623–630. doi: 10.1002/j.1460-2075.1986.tb04256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compan I, Touati D. Interaction of six global transcription regulators in expression of manganese superoxide dismutase in Escherichia coli K-12. J Bacteriol. 1993;175:1687–1696. doi: 10.1128/jb.175.6.1687-1696.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H, Demple B. In vivo kinetics of a redox-regulated transcriptional switch. Proc Natl Acad Sci USA. 1997;94:8445–8449. doi: 10.1073/pnas.94.16.8445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djaman O, Outten FW, Imlay JA. Repair of oxidized iron-sulfur clusters in Escherichia coli. J Biol Chem. 2004;279:44590–44599. doi: 10.1074/jbc.M406487200. [DOI] [PubMed] [Google Scholar]
- Escolar L, Perez-Martin J, de Lorenzo V. Opening the iron box; transcriptional metalloregulation by the Fur protein. J Bacteriol. 1999;181:6223–6229. doi: 10.1128/jb.181.20.6223-6229.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudu P, Moon N, Weiss B. Regulation of the soxRS oxidative stress regulon. Reversible oxidation of the Fe-S center of SoxR in vivo. J Biol Chem. 1997;272:5082–5086. doi: 10.1074/jbc.272.8.5082. [DOI] [PubMed] [Google Scholar]
- Geissmann TA, Touati D. Hfq, a new chaperoning role: binding to messenger RNA determines access for small RNA regulator. EMBO J. 2004;23:396–405. doi: 10.1038/sj.emboj.7600058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grogan DW, Cronan JE. Genetic characterization of the Escherichia coli cyclopropane fatty acid (cfa) locus and neighboring loci. Mol Gen Genet. 1984;196:367–372. doi: 10.1007/BF00328074. [DOI] [PubMed] [Google Scholar]
- Halliwell B, Gutteridge JMC. Role of free radicals and catalytic metal ions in human disease: an overview. Meth Enzymol. 1990;186:1–85. doi: 10.1016/0076-6879(90)86093-b. [DOI] [PubMed] [Google Scholar]
- Hassan HM, Fridovich I. Regulation of the synthesis of superoxide dismutase in Escherichia coli. Induction by methyl viologen. J Biol Chem. 1977;252:7667–7672. [PubMed] [Google Scholar]
- Hassan HM, Sun HCH. Regulatory roles of Fnr, Fur, and Arc in expression of manganese-containing superoxide dismutase in Escherichia coli. Proc Natl Acad Sci USA. 1992;89:3217–3221. doi: 10.1073/pnas.89.8.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imlay JA, Chin SM, Linn S. Toxic DNA damage by hydrogen peroxide through the Fenton reaction in vivo and in vitro. Science. 1988;240:640–642. doi: 10.1126/science.2834821. [DOI] [PubMed] [Google Scholar]
- Imlay JA, Fridovich I. Assay of metabolic superoxide production in Escherichia coli. J Biol Chem. 1991;266:6957–6965. [PubMed] [Google Scholar]
- Imlay JA. Pathways of oxidative damage. Ann Rev Microbiol. 2003;57:395–418. doi: 10.1146/annurev.micro.57.030502.090938. [DOI] [PubMed] [Google Scholar]
- Jang S, Imlay JA. Micromolar intracellular hydrogen peroxide disrupts metabolism by damaging iron-sulfur enzymes. J Biol Chem. 2007;282:929–937. doi: 10.1074/jbc.M607646200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JW, Helmann JD. The PerR transcription factor senses H2O2 by metal-catalyzed histidine oxidation. Nature. 2006;440:363–367. doi: 10.1038/nature04537. [DOI] [PubMed] [Google Scholar]
- Manchado M, Michan C, Pueyo C. Hydrogen peroxide activates the SoxRS regulon in vivo. J Bacteriol. 2000;182:6842–6844. doi: 10.1128/jb.182.23.6842-6844.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masse E, Gottesman S. A small RNA regulates the expression of genes involved in iron metabolism in Escherichia coli. Proc Natl Acad Sci USA. 2002;99:4620–4625. doi: 10.1073/pnas.032066599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCord JM, Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein) J Biol Chem. 1969;244:6049–6055. [PubMed] [Google Scholar]
- Miller JH. Experiments in Molecular Genetics. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1972. [Google Scholar]
- Park S, You X, Imlay JA. Substantial DNA damage from submicromolar intracellular hydrogen peroxide detected in Hpx− mutants of Escherichia coli. Proc Natl Acad Sci USA. 2005;102:9317–9322. doi: 10.1073/pnas.0502051102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Privalle CT, Fridovich I. Inductions of superoxide dismutases in Escherichia coli under anaerobic conditions. Accumulation of an inactive form of the manganese enzyme. J Biol Chem. 1988;263:4274–4279. [PubMed] [Google Scholar]
- Rush JD, Maskos Z, Koppenol WH. Distinction between hydroxyl radical and ferryl species. Meth Enzymol. 1990;186:148. doi: 10.1016/0076-6879(90)86104-4. [DOI] [PubMed] [Google Scholar]
- Seaver LC, Imlay JA. Hydrogen peroxide fluxes and compartmentalization inside growing Escherichia coli. J Bacteriol. 2001;183:7182–7189. doi: 10.1128/JB.183.24.7182-7189.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaver LC, Imlay JA. Are respiratory enzymes the primary sources of intracellular hydrogen peroxide? J Biol Chem. 2004;279:48742–48750. doi: 10.1074/jbc.M408754200. [DOI] [PubMed] [Google Scholar]
- Tsaneva IR, Weiss B. soxR, a locus governing a superoxide response regulon in Escherichia coli K-12. J Bact. 1990;172:4197–4205. doi: 10.1128/jb.172.8.4197-4205.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walling C. Fenton’s reagent revisited. Accounts Chem Res. 1975;8:125–131. [Google Scholar]
- Wiener MC. TonB-dependent outer membrane transport: going for Baroque? Curr Opin Struct Biol. 2005;15:294–400. doi: 10.1016/j.sbi.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Woodmansee AL, Imlay JA. Quantitation of intracellular free iron by electron paramagnetic resonance spectroscopy. Meth Enzymol. 2002;349:3–9. doi: 10.1016/s0076-6879(02)49316-0. [DOI] [PubMed] [Google Scholar]
- Zhao G, Ceci P, Ilari A, Giangiacomo L, Laue TM, Chiancone E, Chasteen ND. Iron and hydrogen peroxide detoxification properties of DNA-binding protein from starved cells. A ferritin-like DNA-binding protein of Escherichia coli. J Biol Chem. 2002;277:27689–27696. doi: 10.1074/jbc.M202094200. [DOI] [PubMed] [Google Scholar]
- Zheng M, Doan B, Schneider TD, Storz G. OxyR and SoxRS regulation of fur. J Bacteriol. 1999;181:4639–4643. doi: 10.1128/jb.181.15.4639-4643.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng M, Wang X, Doan B, Lewis KA, Schneider TD, Storz G. Computation-directed identification of OxyR DNA binding sites in Escherichia coli. J Bacteriol. 2001a;183:4571–4579. doi: 10.1128/JB.183.15.4571-4579.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng M, Wang X, Templeton LJ, Smulski DR, LaRossa RA, Storz G. DNA microarray-mediated transcriptional profiling of the Escherichia coli response to hydrogen peroxide. J Bacteriol. 2001b;183:4562–4570. doi: 10.1128/JB.183.15.4562-4570.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]