

Abstract
The function of connexins, which form gap junctions, can be rapidly modulated by degradation, because they have half-lives of only a few hours. Autophagy is a degradation pathway that has been implicated in several diseases and can be induced by cellular stresses such as starvation. We investigated the involvement of autophagy in proteolysis of the wild-type connexins CX50 and CX43, and a cataract-associated connexin mutant, CX50P88S, which forms cytoplasmic accumulations. We observed that cytoplasmic connexins were partially (cup-shaped) or completely (ring-shaped) enclosed by structures containing the autophagy-related protein LC3. Intracellular connexins also colocalized with p62, a protein that might serve as a cargo receptor for autophagic degradation. Starvation induced a decrease in connexin levels that was blocked by treatment with chloroquine, a lysosomal protease inhibitor, or by knockdown of the autophagy-related protein Atg5. These results demonstrate that autophagy can regulate cellular levels of wild-type connexins and imply that the persistence of accumulations of CX50P88S results from insufficient degradation capacity of constitutive autophagy.
Key words: Gap junction protein, Connexin-43, Connexin-50, Autophagy
Introduction
Cellular proteins are continually renewed by synthesis and degradation. One of the fundamental cellular mechanisms involved in protein degradation is macroautophagy, a process by which cells autodigest parts of their cytoplasm (Reggiori and Klionsky, 2005). This process involves sequestration of discrete portions of the cytosol into a double membrane vesicle, termed the autophagosome. Once completely formed, the cytoplasmic material trapped inside the autophagosome is delivered to a lysosome for degradation (Reggiori and Klionsky, 2005).
In mammalian cells, several proteins are involved in autophagy and its regulation (Thumm et al., 1994). Among these, the microtubule-associated protein 1 light chain 3 (LC3) is considered the most specific autophagosomal marker (Kabeya et al., 2000; Mizushima et al., 2004). LC3 is a mammalian homolog of the Aut7/Apg8 component of the yeast autophagosomal membrane. LC3 associates with the isolation membrane, and this association persists for most of the lifespan of the autophagosome including the early stages after fusion with the lysosome. Sequestosome-1 (p62) binds LC3 and ubiquitylated proteins and might serve as a cargo receptor for autophagic degradation (Bjørkøy et al., 2005; Pankiv et al., 2007).
We have considered the involvement of autophagy in degradation of gap junction proteins. Gap junctions are plasma membrane specializations that contain clusters of intercellular channels that allow passage of ions and small molecules. Gap junction channels are oligomeric assemblies of proteins called connexins. Unlike many other integral membrane proteins, most connexins have relatively short half-lives (1.5–5 hours). Both the ubiquitin–proteasomal and lysosomal pathways have been implicated in connexin turnover (for a review, see Berthoud et al., 2004). The relative contribution of these pathways to connexin proteolysis varies in different cell types (Berthoud et al., 2004) and can be affected by mutations within the connexin amino acid sequence (VanSlyke et al., 2000; Berthoud et al., 2003). Altered degradation of several disease-associated connexin mutants has been reported (VanSlyke et al., 2000; Berthoud et al., 2003). The half-lives of some connexin mutants is at least as short as that of the wild-type connexins; however, other mutants such as CX50P88S exhibit impaired degradation and form accumulations (Berthoud et al., 2003; Lichtenstein et al., 2009).
In the present study, we examined the involvement of autophagy in the degradation of two wild-type connexins, CX43 (which is expressed in many tissues and cell types) and CX50 (which is expressed only in the lens) and a mutant connexin, CX50P88S, which is associated with inherited congenital cataracts (Shiels et al., 1998).
Results
Starvation decreases levels of wild-type connexins
Autophagy can be induced by starvation. To elucidate whether autophagy has a role in the degradation of wild-type connexins, we starved cells expressing CX50 or CX43 and determined levels of connexin by immunoblotting. Starvation of HeLa cells stably expressing CX50 (HeLa-CX50) led to a time-dependent decrease in CX50 levels compared with levels in cells incubated under control conditions (Fig. 1A,B); levels of CX50 decreased after 2 and 6 hours of starvation (on average, 20% and 59%, respectively; n=3) whereas levels of β-tubulin remained relatively unchanged (Fig. 1A,B). Starvation similarly induced a time-dependent decrease in CX43 levels in NRK cells, which endogenously express CX43 (Fig. 1F,G). In these cells, levels of CX43 decreased after starvation for 2 and 6 hours (on average, 84% and 94%, respectively; n=3) (Fig. 1F,G).
Fig. 1.

Starvation decreases levels of wild-type CX50 and CX43. (A) Immunoblot detection of CX50 and β-tubulin in aliquots of total homogenates from HeLa-CX50 cells that were kept under control conditions (lane 1) or starved for 2 or 6 hours (lanes 2 and 3). (B) Levels of CX50 detected after starvation expressed as a percentage of the levels detected in non-starved cells (from three independent experiments). (C–E) Distribution of anti-CX50 immunoreactivity in HeLa-CX50ind cells maintained under normal growth conditions (C) or starved for 4 or 6 hours (D and E, respectively). (F) Immunoblot detection of CX43 in aliquots of total homogenates from NRK cells incubated in normal growth medium (lane 1) or starved by incubation in HBSS for 2 or 6 hours (lanes 2 and 3). The migration positions of the different phosphorylated forms of CX43 (NP, P1 and P2) are indicated. (G) Levels of CX43 detected after starvation expressed as a percentage of control (from three independent experiments). (H–J) Distribution of anti-CX43 immunoreactivity in NRK cells incubated in normal growth medium (H) or starved by incubation in HBSS for 2 or 6 hours (I and J, respectively). Scale bar: 15 μm (C–E), 20 μm (H–J).
In many cultured cells, connexins localize to appositional regions of the plasma membrane (corresponding to gap junction plaques) and within a perinuclear region (which probably corresponds to the Golgi compartment). To study the effects of starvation at the cellular level, the distribution of CX50 was assessed by immunofluorescence in induced HeLa-CX50ind cells grown under normal conditions or starved for 4 or 6 hours. Cells cultured under control conditions showed strong anti-CX50 immunoreactivity at appositional membranes and in perinuclear regions, as expected (Fig. 1C). By contrast, starved cells showed a dramatic decrease in anti-CX50 immunoreactivity at both subcellular locations (Fig. 1D,E). Similarly, although anti-CX43 immunoreactivity was prominent at the plasma membrane and in the perinuclear region of control cultures of NRK cells (Fig. 1H), starvation for 2 and 6 hours also reduced its intensity (at both sites) in a time-dependent manner (Fig. 1I,J).
CX50 and CX43 colocalize with autophagosomal markers
During autophagy, proteins destined for degradation become entrapped within the autophagosome. Therefore, we reasoned that, if autophagy participates in degradation of wild-type connexins, autophagosomal markers (e.g. LC3) should colocalize with intracellular connexin-containing structures. To test this hypothesis, HeLa-CX50 cells were starved and subjected to immunofluorescence using anti-CX50 and anti-LC3 antibodies. After starvation-induced autophagy, many cytoplasmic vesicular structures labeled with anti-LC3 antibodies were observed (Fig. 2B). Several such structures also contained anti-CX50 immunoreactivity (Fig. 2C,D), which was often surrounded by the anti-LC3 immunoreactivity (Fig. 2, arrows). The LC3-immunoreactive structures appeared cup-shaped or circular, probably reflecting different stages of autophagosome formation (i.e. isolation membranes before and after closure to form autophagosomal vesicles). Indeed, even though in many cases the structures delineated by the anti-LC3 antibodies were bigger than those labeled with anti-CX50 antibodies, on average 35% of the cytoplasmic CX50 colocalized with LC3 in cells starved for 4 hours (n=21).
Fig. 2.

LC3 colocalizes with intracellular wild-type CX50. (A–C) Differential interference contrast image (A), and corresponding immunofluorescence images obtained using anti-LC3 (B) and anti-CX50 (C) antibodies in HeLa-CX50 cells after starvation for 4 hours. (D) Overlap between anti-LC3 and anti-CX50 immunoreactivities. Arrows indicate cytoplasmic CX50-immunoreactive structures that are surrounded by ring- or cup-shaped LC3-immunopositive structures; CX50 immunoreactivity at the plasma membrane is indicated by arrowheads. Scale bar: 20 μm.
Because lysosomes are involved in macroautophagy-mediated protein degradation, we tested whether the starvation-induced decrease in connexin levels was dependent on the activity of lysosomal proteases. HeLa-CX50 cells treated with the lysosomal protease inhibitor, chloroquine, had on average 127% of the CX50 levels detected in cells cultured under normal growth conditions (n=3) (Fig. 3A,B). CX50 levels fell to 26% of control levels in starved HeLa-CX50 cells (n=3), but CX50 levels only decreased to 74% in starved cells that had been treated with chloroquine (n=3) (Fig. 3A,B). By comparison, levels of β-tubulin in cells pretreated with chloroquine remained relatively unchanged (Fig. 3A).
Fig. 3.

The starvation-induced decrease in levels of wild-type connexins is partially prevented by pretreatment with the lysosomal inhibitor chloroquine. (A) Immunoblot showing the levels of CX50 and β-tubulin in aliquots of total homogenates from HeLa-CX50 cells that were cultured in normal growth medium (lanes 1 and 2) or in HBSS for 6 hours (lanes 3 and 4) and left untreated (lanes 1 and 3) or treated with 100 μM chloroquine (CLQ) for 6.5 hours (lanes 2 and 4). (B) Levels of CX50 (expressed as a percentage of the values determined in cells grown in normal growth medium without CLQ) obtained from three independent experiments performed under the conditions described in A. (C) Immunoblot detection of CX43 in aliquots of total homogenates from NRK cells cultured in normal growth medium (lanes 1 and 2) or starved for 6 hours (lanes 3 and 4) that were left untreated (lanes 1 and 3) or treated with 100 μM chloroquine for 6.5 hours (lanes 2 and 4). (D) Levels of CX43 (expressed as a percentage of control, lane 1) detected in cells treated as described in C in three independent experiments.
In NRK cells, lysosomal inhibition with chloroquine led to more than a twofold increase in levels of CX43 over the values detected in cells cultured under control conditions (Fig. 3C,D). Starvation of NRK cells for 6 hours led to a marked decrease in levels of CX43; the remaining levels of CX43 represented 10% of the control values (n=3) (Fig. 3C,D). Pretreatment of cells with chloroquine before starvation reduced the decrease in CX43 levels; cells cultured under these conditions had 59% of the CX43 levels detected under control conditions (n=3) (Fig. 3C,D).
Inhibition of lysosomal degradation during starvation-induced autophagy would be expected to result in localization of LC3 within larger vesicles and to increase colocalization of wild-type connexins with LC3. To test these predictions, we determined the localization of CX50 and LC3 in HeLa-CX50 cells that were transiently transfected with GFP–LC3 and that were untreated or treated with chloroquine. Under control conditions, several GFP–LC3-labeled vesicular structures were observed in the cytoplasm of transfected cells (Fig. 4B), and anti-CX50 immunoreactivity localized mainly at appositional membranes (Fig. 4C, arrows); we occasionally observed cytoplasmic structures containing closely associated CX50 and GFP–LC3 fluorescence (Fig. 4D), suggesting some degradation of CX50 by constitutive autophagy. Three-dimensional reconstruction of a z-series of confocal images of structures with overlapping fluorescent signals revealed that CX50-containing structures were surrounded by GFP–LC3-labeled material (Fig. 4B–D, insets). When HeLa-CX50 cells were starved, several of the intracellular CX50-containing vesicular structures were also labeled with GFP–LC3 (Fig. 4F–H). Chloroquine treatment of HeLa-CX50 cells cultured in normal growth medium resulted in an increase in the number and size of GFP–LC3-labeled vesicles compared with untreated cells (Fig. 4, compare panels B and J). Under these conditions, wild-type CX50 localized in intracellular vesicles (Fig. 4K) as expected (Berthoud et al., 2004); some of these structures were also labeled with GFP–LC3 (Fig. 4L). When chloroquine-treated cells were starved, overlap between the two signals was more extensive than that observed in cells that were only starved (Fig. 4N–P). After starvation for 2 hours, 47% of the remaining CX50 overlapped with GFP–LC3 (n=20), but pretreatment with chloroquine before starvation resulted in 75% colocalization of CX50 with GFP–LC3 (n=20).
Fig. 4.

Starvation and chloroquine treatments increase the extent of colocalization of LC3 with intracellular CX50. (A–P) Confocal images of HeLa-CX50 cells transiently transfected with a plasmid encoding GFP–LC3 and subjected to immunofluorescence using rabbit anti-CX50 and Cy3-conjugated goat anti-rabbit IgG antibodies. Cells were incubated in the absence (A–H) or presence (I–P) of chloroquine (CLQ) for 24 hours in normal growth medium (A–D and I–L) or starved by incubation in HBSS for the final 2 hours (E–H and M–P). The insets in B–D show deconvolved images of cytosolic accumulations co-labeled with GFP–LC3 and anti-CX50 antibodies at higher magnification obtained by spectral two-photon confocal microscopy. Scale bar: 25 μm (A–D), 20 μm (E–P).
Similar experiments were performed on NRK cells to examine the effects of starvation and lysosomal inhibition on possible colocalization of LC3 and CX43. NRK cells were transiently transfected with GFP–LC3, and the distribution of CX43 was studied. Under normal growth conditions, CX43 localized mostly at appositional membranes (Fig. 5B); we observed some overlap between cytosolic CX43 and the GFP–LC3-labeled structures (Fig. 5A–C), suggesting that some CX43 underwent constitutive autophagy. In CX43 immunopositive structures that colocalized with GFP–LC3 fluorescence (such as the one indicated by the arrow in Fig. 5A–C), the GFP–LC3 fluorescence surrounded the anti-CX43 immunoreactivity as revealed by the z-series of confocal images (Fig. 5D). When NRK cells were starved for 2 hours, no increase in colocalization between the two signals was observed (Fig. 5E–G), most likely because a significant proportion of CX43 had already undergone degradation (as demonstrated by immunoblotting, Fig. 1F,G). The frequency of overlap between GFP–LC3 and CX43, and the sizes of structures containing both proteins increased in chloroquine-treated cells (Fig. 5H–J); the percentage of cytoplasmic CX43 that colocalized with GFP–LC3 in starved cells increased from 25% (n=20) to 48% (n=17) when cells were pretreated with chloroquine.
Fig. 5.

Chloroquine treatment increases colocalization of CX43 and LC3 in NRK cells. (A–J) Confocal images of NRK cells transiently transfected with DNA encoding GFP–LC3 and subjected to immunofluorescence using anti-CX43 antibodies. Cells were incubated in the absence (A–G) or presence (H–J) of chloroquine for 4 hours in normal growth conditions (A–C) or starved for the final 2 hours (E–J). (D) Deconvolved z-series of images obtained by spectral two-photon confocal microscopy of the structure indicated by the arrow in A–C. Scale bar: 17 μm (A–C), 1 μm (D), 25 μm (E–G), 20 μm (H–J).
To examine further the involvement of autophagy in connexin degradation in different cell types, we performed experiments in mouse embryonic fibroblasts (MEFs), which endogenously express CX43. Similarly to the results obtained in HeLa-CX50 and NRK cells, starvation of MEFs resulted in a time-dependent decrease in CX43 levels (data not shown). Under control conditions, CX43 localized to gap junction plaques and to the cytoplasm (Fig. 6A); some of the intracellular structures containing CX43 appeared doughnut-shaped after deconvolution, suggesting that they were internalized gap junctions. Under control conditions, we consistently observed cells containing LC3-immunopositive vesicular structures (Fig. 6B), indicating that some constitutively active autophagy was occurring; some of these structures also contained CX43 (Fig. 6C,D). Starvation of MEFs for 1 hour led to a decrease in CX43 immunofluorescence intensity and an increase in the number of cells with LC3-immunopositive vesicles (Fig. 6E–H). The colocalization of the two signals was 33% in cells exhibiting constitutively active autophagy (22–53%; n=21) and 41% in cells undergoing starvation-induced autophagy (21–67%; n=18). Similarly to the results obtained after transfection of NRK cells with GFP–LC3, deconvolution of the images obtained in MEFs showed intracellular CX43 surrounded by LC3 (Fig. 6D,H).
Fig. 6.

LC3 colocalizes with CX43 in mouse embryonic fibroblasts. (A–H) Confocal images show the immunolocalization of CX43 (A,E) and LC3 (B,F) in mouse embryonic fibroblasts incubated under control conditions (A–D) or starved for 1 hour (E–H). Colocalization of the two signals is shown in the merged panels (C and G). The boxed regions are shown at higher magnification after deconvolution in D and H. Scale bar: 16 μm (A–C,E–G) and 2 μm (D,H).
To test whether connexins colocalized with p62, which has been implicated in linking ubiquitylated proteins to the autophagic machinery (Bjørkøy et al., 2005; Pankiv et al., 2007), we performed double-label immunofluorescence in HeLa-CX50 cells and MEFs under control conditions. p62 localized to small vesicles whose abundance varied among different cells of the same culture (supplementary material Fig. S1). In HeLa-CX50 cells, many of the p62-containing vesicles were also positive for CX50; most of the colocalization between the two signals occurred in the perinuclear region, whereas no colocalization of p62 and CX50 at gap junctional plaques was observed (supplementary material Fig. S1A–C). Similarly, in MEFs, intracellular CX43 colocalized with p62-containing vesicles, and no colocalization was observed at gap junctional plaques (supplementary material Fig. S1E–G). The close association of p62 with cytoplasmic connexins was particularly evident in deconvolved images (supplementary material Fig. S1D,H).
We further analyzed connexin and gap junction degradation by examining the ultrastructure of MEFs grown under control conditions and after 1 hour of starvation by transmission electron microscopy. Under control conditions, gap junction structures were found between adjacent cells (Fig. 7A, arrows). Profiles of internalized gap junctions were observed in the cytoplasm. Some profiles of partially disrupted internalized gap junctions (recognized by their pentalaminar pattern) were observed inside membranous structures, which probably represent secondary lysosomes (Fig. 7B). When MEFs were starved for 1 hour, many autophagic vacuoles were observed in the cytoplasm. Some internalized gap junctions enclosed by double membrane structures were also observed (Fig. 7C,D); these double membrane structures were located in close proximity to rough ER membranes, a hallmark of autophagosomes (Eskelinen, 2009).
Fig. 7.

Internalized gap junctions are found in double membrane structures in mouse embryonic fibroblasts. (A) Electron micrograph showing gap junctions (arrows) at the appositional membranes of mouse embryonic fibroblasts. (B) Electron micrograph showing an internalized gap junction structure within a membrane-surrounded compartment, which probably corresponds to a secondary lysosome or autolysosome. Note that the pentalaminar pattern characteristic of gap junctions is being lost (arrow) in the internalized structure, probably because it is undergoing degradation. (C) Electron micrograph obtained from mouse embryonic fibroblasts starved by incubation in HBSS for 1 hour showing a double membrane structure, probably an autophagosome (arrow), that encloses an internalized gap junction in close proximity to rough endoplasmic reticulum (short arrow). (D) Electron micrograph showing the internalized gap junction enclosed within the autophagosome at higher magnification; the rough endoplasmic reticulum membrane is indicated by the short arrow. Nu, nucleus. Scale bar: 200 nm (A,C), 100 nm (B), 50 nm (D).
Inhibition of autophagy decreases the effects of starvation on wild-type connexins
Because autophagy depends on the function of several Atg (autophagy-related) proteins, autophagy-mediated degradation of connexins should also depend on these proteins. Therefore, we examined the levels of CX50 in HeLa-CX50 cells that were treated with Atg5 siRNA to reduce levels of Atg5. Under normal growth conditions, knockdown of Atg5 (confirmed by an 89% decrease in levels of the Atg5–Atg12 complex; 84–94%; n=3; Fig. 8A) increased levels of CX50 to 150–170% of that detected under control conditions (Fig. 8A), further suggesting the involvement of constitutive autophagy in the degradation of CX50. Starvation of HeLa-CX50 cells that had been transfected with Atg5 siRNA showed a less-pronounced decrease in CX50 levels than starved cells transfected with a non-targeting control siRNA (61% vs 82% compared with their respective non-starved control cells; n=3) (Fig. 8A,B). Levels of β-tubulin remained relatively constant (Fig. 8A). These results implicate the participation of Atg5-dependent autophagy in the starvation-induced degradation of CX50.
Fig. 8.

Reduced levels or absence of Atg5 prevent the starvation-induced decrease in connexin levels. (A) Immunoblot detection of CX50, Atg5 and β-tubulin in HeLa-CX50 cells 120 hours after transfection with non-targeting control siRNA (lanes 1 and 2) or Atg5 siRNA (lanes 3 and 4) cultured under control conditions (lanes 1 and 3) or starved for the last 6 hours (lanes 2 and 4). (B) CX50 levels (expressed as a percentage of control) determined in cells treated with non-targeting control siRNA (left) or in cells transfected with Atg5 siRNA (right) and starved for 6 hours (from three independent experiments). (C) Immunoblot detection of CX43 and Atg12 in wild-type and Atg5 KO MEFs cultured under control conditions (lanes 1 and 3) or starved for 4 hours (lanes 2 and 4). (D) Graphs depict the results of two independent experiments performed as described in C; levels of CX43 are expressed as a percentage of control.
We also studied the importance of Atg5 for the starvation-induced degradation of CX43 by performing experiments on wild type and Atg5-knockout (Atg5 KO) MEFs. After starvation for 4 hours, levels of CX43 were reduced by 43–56% compared with values in wild-type MEFs cultured under control conditions (Fig. 8C,D). By contrast, little or no decrease in the level of CX43 was observed when Atg5 KO MEFs (which do not express Atg5 protein) were starved for 4 hours (81–113% of the values detected under control conditions) (Fig. 8C,D). As expected, no band corresponding to the Atg5–Atg12 complex was detected in Atg5 KO MEFs (Fig. 8C).
Accumulations of the mutant connexin CX50P88S are associated with autophagy markers and are degraded after starvation
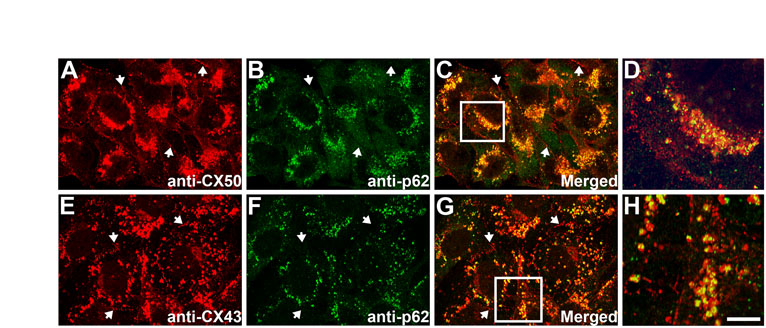
Autophagy has been implicated in several diseases associated with accumulation of mutant proteins such as Huntington's disease and Alzheimer's disease (Huang and Klionsky, 2007). To test the possible association of CX50P88S, a mutant CX50 that forms accumulations, with autophagosomes, HeLa-CX50P88S(Cys)4 cells were first labeled with ReAsH (resorufin-based arsenical hairpin) and subsequently incubated with MDC (monodansylcadaverine), a marker for autophagic vacuoles (Biederbick et al., 1995). ReAsH and MDC showed similar staining patterns with a high degree of overlap (Fig. 9A–C). Moreover, when HeLa-CX50P88S(Cys)4 cells were transiently transfected with GFP–LC3 and then labeled with ReAsH, several of the ReAsH-labeled CX50P88S(Cys)4 accumulations also labeled with GFP–LC3 (Fig. 9D–I). Deconvolution of images from z-series of individual double-labeled structures revealed that the volume enclosed by LC3 was larger than that labeled with ReAsH (Fig. 9D–I, insets). When the GFP–LC3 fluorescence formed a U- or cup-shaped structure, three-dimensional volume projection and optical sectioning in the x–z plane revealed a layer of GFP–LC3 closely associated with a layer of ReAsH with little, if any, intermingling (Fig. 9D–F, insets); when the GFP–LC3 fluorescence demarcated a vesicular structure, the ReAsH-labeled material was enclosed within the GFP–LC3-labeled vesicle (Fig. 9I, insets). Colocalization of the CX50 mutant accumulations with endogenous LC3 was also observed when HeLa-CX50P88S(Cys)4 cells were labeled with ReAsH and then subjected to immunofluorescence using anti-LC3 antibodies (Fig. 9J–L); indeed, 92% of the ReAsH-labeled structures colocalized with LC3 (n=71 cells). Consistent with the results obtained after transient transfection with GFP–LC3, some double-labeled accumulations (Fig. 9J–L, arrow) had cup-shaped structures, which were suggestive of pre-autophagosomes (Fig. 9J–L, inset).
Fig. 9.

CX50P88S cytoplasmic accumulations colocalize with autophagosome markers and p62. (A–C) Photomicrographs of HeLa-CX50P88S(Cys)4 cells labeled with ReAsH and then with MDC (shown in green). (D–I) Confocal images of HeLa-CX50P88S(Cys)4 cells transiently transfected with a DNA construct coding for GFP–LC3 and labeled with ReAsH 48 hours after transfection. The insets in D–I show deconvolved images of accumulations labeled with GFP–LC3 and ReAsH. The smaller inset in F corresponds to an x–z profile of the z-series of confocal images through the accumulation volume at the level indicated by the white line in the larger inset above. The inset on the upper right corner of I shows the deconvolved image of the accumulation indicated by the arrow in G–I at higher magnification. The inset on the left shows other examples. (J–L) Confocal images of HeLa-CX50P88S(Cys)4 cells labeled with ReAsH and subjected to immunofluorescence using anti-LC3 antibodies. The accumulations indicated by the arrows are shown at higher magnification in the insets. (M–O) Confocal images of HeLa-CX50P88S show the localization of CX50P88S (M) and p62 (N) after double-label immunofluorescence using anti-CX50 and anti-p62 antibodies. The superposition of the two signals is shown in O. Arrows indicate colocalization of p62 with CX50P88S accumulations. Scale bar: 27 μm (A–C), 25 μm for (D–F), 1 μm (insets in D–F), 22 μm (G–I), 38 μm (J–L), 43 μm (M–O).
Because p62 has been implicated in the formation and clearance of protein aggregates (Bjørkøy et al., 2005; Pankiv et al., 2007), we studied whether it colocalized with the cytoplasmic accumulations of CX50P88S. Double-immunofluorescence studies of HeLa-CX50P88S cells demonstrated colocalization of p62 with the cytoplasmic accumulations of CX50P88S (Fig. 9M–O).
To prove that starvation also led to degradation of CX50P88S, we determined its levels by immunoblotting in HeLa-CX50P88S cells incubated in normal growth medium or starved. A substantial decrease (64% on average, n=3) in the level of CX50P88S was observed after 24 hours of starvation compared with that in cells grown under control conditions (Fig. 10A,B). By contrast, levels of the cytoplasmic protein β-tubulin decreased by 24% (9–33%; n=3) in cells starved for 24 hours. The changes in CX50P88S levels were associated with a marked decrease in anti-CX50 immunoreactivity as assessed by immunofluorescence of similarly treated HeLa-CX50P88S(Cys)4 cells (Fig. 10C–F).
Fig. 10.

Starvation decreases the levels of CX50P88S. (A) Immunoblot detection of CX50P88S and β-tubulin in HeLa-CX50P88S cells incubated in normal growth medium (Control) or starved for 24 hours (Starved). (B) CX50P88S levels (expressed as a percentage of control) determined in cells starved for 24 hours in three independent experiments. (C–F) Distribution of CX50P88S in HeLa-CX50P88S(Cys)4 cells cultured under normal growth conditions or starved for 24 hours and subjected to immunofluorescence using anti-CX50 antibodies. Scale bar: 20 μm.
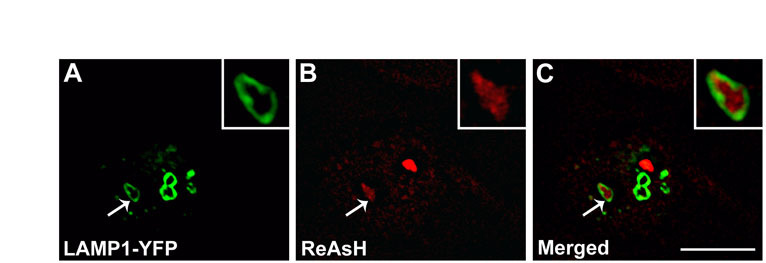
To test the association of CX50P88S accumulations with lysosomes, HeLa-CX50P88S(Cys)4 cells were transiently transfected with lysosome-associated membrane protein 1 (LAMP1)–YFP and labeled with ReAsH. Analysis of these samples by confocal microscopy revealed that in some cases, LAMP1-YFP-containing structures surrounded the ReAsH-labeled CX50P88S accumulations (supplementary material Fig. S2A–C).
Discussion
Autophagy is a proteolytic pathway that shows a constitutively low level of activity under normal conditions and is altered in pathological conditions (Huang and Klionsky, 2007; Meijer and Codogno, 2009). Increases in autophagic proteolytic activity can help cells adjust to environmental changes such as starvation (Mitchener et al., 1976). In this study, we have demonstrated by biochemical and colocalization studies that autophagy participates in the degradation of both wild-type and mutant connexins. We have also shown internalized gap junctions inside double membrane vesicles. Based on our results and previous ultrastructural studies, we propose a hypothetical model for the degradation of connexins by autophagy (Fig. 11). We will separately consider the aspects of the autophagic degradation of wild-type and mutant connexins below.
Fig. 11.

Diagram of the hypothesized autophagic process for connexins. Proposed sequence of events involved in the degradation of wild-type (A) or mutant (B) connexins through autophagy based on results presented in this study and published ultrastructural studies. (A) Wild-type connexins in the cytoplasm present in vesicles from the secretory pathway or in internalized gap junctions (IGJs) are entrapped by the LC3-containing isolation membrane into an autophagosome. The autophagosome then fuses with a lysosome (L) forming an autolysosome. Lysosomal enzymes degrade lipid and protein components of the entrapped material. Differences in cell types and the kinetics of the different steps might determine the frequency with which connexin-containing structures can be observed in autophagosomes. (B) Cytoplasmic accumulations of mutant connexin (red) are entrapped by the isolation membrane into an autophagosome that then fuses with a lysosome (L) forming an autolysosome. After starvation, the mutant connexin in accumulations is degraded by lysosomal proteases. Under normal growth conditions, the levels of constitutive autophagy are insufficient to degrade the mutant protein and might contribute to the formation of large accumulations by bringing small accumulations together. LC3 is represented by gray dots on membrane surfaces. Nu, nucleus; rER, rough endoplasmic reticulum.
Autophagy and wild-type connexins
Our studies showed that cytoplasmic immunoreactive connexin (CX43 or CX50) was partially (cup-shaped) or completely (ring-shaped) enclosed by LC3-containing structures. The differences in shape probably reflect different stages in autophagosome formation (i.e. isolation membranes before and after closure to form autophagosomal vesicles). These results are consistent with the interpretation that connexins targeted for subsequent degradation are sequestered by an LC3-containing membrane under basal conditions or after stimulation of autophagy by starvation.
The increase in levels of CX50 detected after siRNA-mediated knockdown of Atg5 implies that steady-state levels of CX50 are regulated (at least in part) through Atg5-dependent autophagy under control conditions. This might also be true for CX43 and other wild-type connexins, because we observed colocalization of cytoplasmic wild-type connexins with p62, a protein that might facilitate targeting of ubiquitylated proteins to the authophagic machinery by binding both ubiquitylated proteins and LC3 (Bjørkøy et al., 2005; Pankiv et al., 2007). Ubiquitylation of CX43 and of the chicken ortholog of CX50 have been demonstrated (Laing and Beyer, 1995; Yin et al., 2008). Thus, it is possible that p62 targets wild-type connexins for autophagic degradation through direct binding.
Our results showed that chloroquine partly blocked the decrease in levels of wild-type connexins induced by starvation, implying that the lysosome is involved in the starvation-induced degradation of wild-type connexins. Our immunofluorescence studies showed an increase in the size of LC3-containing vesicles and in the frequency of their association with wild-type connexins in cells treated with chloroquine. The increased colocalization of connexins with autophagosomal markers following treatment with lysosomal inhibitors probably results from accumulation of the connexin in autolysosomes. These results are in agreement with those obtained in neonatal rat cardiac myocytes by Hesketh and colleagues (Hesketh et al., 2010), who observed sporadic colocalization between intracellular CX43 and GFP–LC3 under baseline culture conditions and a significant increase in colocalization in cells treated with chloroquine.
The Atg5 dependence of the starvation-induced decrease in CX43 and CX50 levels further supports the involvement of autophagy in degradation of wild-type connexins. The reduction in CX50 levels that occurred in starved HeLa-CX50 cells despite transfection with Atg5 siRNA (Fig. 8) might reflect the persistence of a low level of Atg5-dependent autophagy because of incomplete knock-down of Atg5. However, it might also indicate that part of the observed connexin degradation occurs through Atg5-independent autophagy. A fraction of the starvation-induced degradation of wild-type connexins observed in our study could also be independent of lysosomal protease activity, because chloroquine did not fully prevent the starvation-induced decrease in connexin levels.
Several studies have provided evidence consistent with autophagic degradation of gap junctions or connexins in the tissues of intact animals. Ultrastructural studies have shown internalized gap junctions enclosed within double membrane structures in sections from rat liver, incisor and fetal epidermis (Pfeifer, 1980; Hayward and Kent, 1983; Sasaki and Garant, 1986) and equine hoof wall (Leach and Oliphant, 1984). In the failing heart, double membranes were observed in close association with internalized gap junctions and were suggested to be putative isolation membranes (Hesketh et al., 2010). In the hearts of starved mice, CX43 immunoreactivity decreased at appositional membranes, whereas it increased intracellularly (McLachlan et al., 2009). LC3-positive vesicles were observed in the hearts of starved mice expressing GFP–LC3 (Mizushima et al., 2004).
Taken together, our results demonstrate the involvement of constitutive and induced autophagy in the degradation of wild-type connexins. Moreover, our studies are the first to provide immunological and molecular evidence for the involvement of endogenously expressed autophagic components in this process. Our immunofluorescence microscopy studies show that starvation (an inducer of autophagy) leads to substantial loss of connexin from the plasma membrane (Fig. 1), implicating autophagy in the degradation of gap junction plaques. In support of this conclusion, we observed internalized gap junctions surrounded by double membrane structures that were similar to those observed in ultrastructural studies (Pfeifer, 1980; Hayward and Kent, 1983; Leach and Oliphant, 1984; Sasaki and Garant, 1986). The participation of autophagy in the degradation of plasma membrane proteins is not without precedent, because autophagic degradation of internalized GABAA receptors has been reported (Rowland et al., 2006). Our results also show a decrease in connexin immunostaining in the perinuclear region; this suggests the participation of autophagy in the degradation of connexins contained in intracellular compartments of the secretory pathway (although a contribution from decreased protein synthesis cannot be discarded). Indeed, a transport pathway has been described that directs CX32R142W in PC12 cells from post-ER compartments and CX43 in MDA-MB-231vCx43 cells from early secretory compartments to lysosomes for degradation (VanSlyke et al., 2000; Qin et al., 2003).
We have considered our results in the context of the previous studies and developed a hypothetical model for degradation of wild-type connexins (Fig. 11A). In this model, connexin-containing vesicles derived from the secretory pathway or from internalized (annular) gap junctions are sequestered by LC3-containing isolation membranes, followed by fusion with lysosomes and degradation of connexins by lysosomal enzymes (Fig. 11A). Previous studies have shown that the relative involvement of the lysosome in connexin degradation varies in different cell types, as demonstrated by differences in the levels and turnover of connexins, or their subcellular distribution after treatment with lysosomal inhibitors (Laing et al., 1997; Laing et al., 1998; Beardslee et al., 1998; Qin et al., 2003; Thomas et al., 2003). Our current data suggest that the contribution of Atg5-dependent autophagy to the constitutive and starvation-induced degradation of wild-type connexins differs between cell types. Thus, the importance of autophagy for connexin degradation and the sources of the connexin targeted for autophagic degradation are likely to differ depending on cell type and nutrient or growth conditions.
Autophagy and CX50P88S
We have previously demonstrated that the cataract-associated mutant, CX50P88S, forms ER-derived cytosolic accumulations that are long-lived and show impaired degradation (Berthoud et al., 2003; Lichtenstein et al., 2009). The presence of these accumulations and their persistence in the lens may cause light scattering and cataracts. In this study, we showed colocalization of MDC, LC3, p62 and LAMP1 with CX50P88S. These observations suggest that autophagy contributes to the degradation of the mutant connexin. Colocalization of protein aggregates with the cargo receptor for autophagic degradation, p62, has been observed in neurodegenerative diseases and liver disorders (Kuusisto et al., 2001; Zatloukal et al., 2002). It has been proposed that p62 is involved in the formation and the clearance of protein aggregates (Bjørkøy et al., 2005; Pankiv et al., 2007). Our data suggest that p62 is involved in targeting CX50P88S for autophagic degradation and might contribute to the formation of its multilamellar accumulations under normal growth conditions and to their clearance during starvation.
The hypothetical sequence of events leading to degradation of CX50P88S by autophagy after prolonged starvation is presented as a model in Fig. 11B. Cytoplasmic accumulations of CX50P88S are entrapped by the isolation membrane, leading to colocalization of CX50P88S and LC3 (pre-autophagosome/autophagosome stage). Then, these autophagosomes fuse with lysosomes (autolysosome stage) resulting in overlap of CX50P88S with LAMP1, while the LC3 is degraded or recycled to participate in formation of new autophagosomes. CX50P88S and LAMP1 colocalization is lost as LAMP1 is recycled (or degraded) and CX50P88S is degraded. The presence of some CX50P88S-containing structures that do not label with either LAMP1 or LC3 implies that not all accumulations in a cell undergo autophagy at the same time.
The impaired CX50P88S degradation observed under normal nutrient conditions suggests that CX50P88S is only subject to a very low level of constitutive autophagy. Mutant proteins such as CX50P88S that can reach the plasma membrane when cells are incubated at reduced temperatures are believed to be misfolded. It has been reported that accumulation of disease-associated misfolded proteins can induce autophagy, perhaps triggered by ER stress (Yorimitsu et al., 2006; Marazziti et al., 2009). However, even if this were the case in cells expressing CX50P88S, the levels of autophagy attained under normal nutrient conditions must be insufficient to degrade and clear the cytosol of accumulations of the mutant CX50.
Relevance of autophagy to connexin function
Autophagy is a process that can be regulated and that has key roles in development, tissue and organ homeostasis, and many diseases (Levine and Klionsky, 2004; Huang and Klionsky, 2007; Mizushima et al., 2008; Meijer and Codogno, 2009). Based on the results presented here, we conclude that autophagy is a degradation pathway for wild-type and mutant connexins. Because levels of autophagy can be modified in response to different physiological and pathological stimuli, regulation of autophagy might lead to gap junction remodeling and alterations in connexin functions, including intercellular communication. Thus, autophagy might contribute to the gap junction and connexin abnormalities observed in various diseases, including cancer, diabetes, heart failure and cataracts.
Materials and Methods
Chemicals
All chemicals were obtained from Sigma (St Louis, MO), unless otherwise specified.
DNA constructs
The GFP–LC3 plasmid was a gift from Noboru Mizushima (National Institute for Basic Biology, Okazaki, Japan). The LAMP1–YFP plasmid was a gift from Norma Andrews (Yale University School of Medicine, New Haven, CT).
Cell culture
HeLa cells stably transfected and constitutively expressing CX50 (HeLa-CX50), CX50P88S (HeLa-CX50P88S) or CX50P88S(Cys)4 [HeLa-CX50P88S(Cys)4] or HeLa cells stably transfected with CX50 under the control of a ponasterone-A-inducible promoter (HeLa-CX50ind) have been described previously (Berthoud et al., 2003; Lichtenstein et al., 2009; Minogue et al., 2009). Transient transfection with plasmids encoding GFP–LC3 or LAMP1–YFP was performed using Lipofectin Transfection Reagent (Invitrogen, Carlsbad, CA). HeLa cells were grown in MEM supplemented with non-essential amino acids, 10% fetal bovine serum, 2 mM glutamine, 100 U/ml penicillin G and 10 μg/ml streptomycin sulfate. For HeLa-CX50, HeLa-CX50P88S and HeLa-CX50P88S(Cys)4 cells, the growth medium was supplemented with 1 mg/ml Geneticin (Invitrogen). Normal rat kidney cells (NRK-52E, ATCC CRL 1571) were grown in DMEM supplemented with non-essential amino acids, 10% fetal bovine serum, 2 mM glutamine, 100 U/ml penicillin G and 10 μg/ml streptomycin sulfate. Wild-type and Atg5-knockout MEFs (a gift from Aimee Edinger from UC Irvine, Irvine, CA with approval from Noboru Mizushima) were grown in DMEM supplemented with 10% fetal bovine serum, 100 U/ml penicillin G and 10 μg/ml streptomycin sulfate.
Antibodies
Rabbit polyclonal anti-CX50 antibodies have been previously characterized (Berthoud et al., 2003). Mouse monoclonal anti-CX50 antibody was obtained from Invitrogen. Rabbit polyclonal anti-CX43 antibodies and mouse monoclonal anti-β-tubulin antibody were obtained from Sigma. Mouse monoclonal anti-CX43 antibody was obtained from Chemicon (Temecula, CA). Rabbit polyclonal anti-LC3, anti-Atg5 and anti-Atg12 antibodies were obtained from Cell Signaling Technology (Danvers, MA). Guinea pig polyclonal anti-p62 antibodies were obtained from PROGEN Biotechnik (Heidelberg, Germany). Horseradish-, Cy2- and Cy3-conjugated goat anti-rabbit or anti-mouse IgG antibodies, and Cy2-conjugated donkey anti-guinea pig antibodies were obtained from Jackson ImmunoResearch (West Grove, PA).
Cell treatment
Starvation
Cells were grown to about 40–50% confluence. Then, the growth medium was replaced with fresh growth medium for control cells or with Hanks balanced salt solution (HBSS; Invitrogen) for starved cells (two rinses in HBSS before being placed in HBSS). The cells were then incubated in HBSS for various times.
Chloroquine
Cells were grown to about 40–50% confluence. Then, the culture growth medium was replaced with normal growth medium containing 100 μM chloroquine. In the case of starved cells, after at least 30 minutes in chloroquine-containing medium, cells were rinsed twice with HBSS before adding HBSS containing 100 μM chloroquine. The cells were returned to the incubator for various times.
siRNA transfection
HeLa-CX50 cells were subcultured onto 60 mm dishes. The next day, they were transfected with siRNAs. siRNAs for transfection were prepared as follows: Atg5 siGENOME SMART pool or siGENOME Non-Targeting siRNA pool #1 (Thermo Fisher Scientific Dharmacon Products, Lafayette, CO) was added to 200 μl serum-free OPTI-MEM (Invitrogen). 30 μl Lipofectin Transfection Reagent were added to this solution and it was mixed by gentle flicking and incubated for 15 minutes. After this time, the volume was brought to 4 ml with OPTI-MEM so that the final concentration of siRNA was 50 nM. Cells were rinsed once with OPTI-MEM before incubating them in the 4 ml siRNA/transfection reagent mix. 3 hours later, bovine serum albumin was added to a final concentration of 10%, and the cells were incubated overnight. The next day, the medium was replaced with normal growth medium without antibiotics. The cells were grown for a total of 5 days. On the fifth day, the growth medium was replaced with fresh growth medium for control cells or with HBSS for starved cells. The cells were starved for 6 hours.
Immunofluorescence
Cells grown on coverslips or on four-well LAB-TEK Nalge Nunc International chamber slides (Thermo Fisher Scientific, Rochester, NY) were fixed in 4% paraformaldehyde in PBS for 15 minutes. For HeLa-CX50P88S(Cys)4, cells were labeled with ReAsH as previously described (Lichtenstein et al., 2009) before fixation. For single staining with anti-CX43 or anti-CX50 antibodies, fixed cells were then permeabilized in 1% Triton X-100 in PBS for 15 minutes and blocked in 4% normal goat serum, 1% Triton X-100 in PBS for 10 minutes at room temperature. Immunofluorescence was performed using primary antibodies and Cy2- or Cy3-conjugated secondary antibodies as previously described (Berthoud et al., 2003). For double staining using anti-LC3 antibodies, immunofluorescence was performed following the manufacturer's instructions. For double staining using anti-p62 antibodies, fixed cells were permeabilized using 0.1% Triton X-100 in PBS and immunofluorescence was performed as described (Bjørkøy et al., 2009).
Specimens were studied with a Zeiss Plan Apochromat 40× objective (NA 1.0) in an Axioplan 2 microscope (Carl Zeiss, München, Germany) equipped with a mercury lamp. Images were captured with a Zeiss Axiocam digital camera using Zeiss AxioVision software. Confocal images were obtained using a Leica SP2 AOBS laser scanning confocal microscope or a Leica SP5 spectral two-photon confocal microscope (Leica Microsystems, Bannockburn, IL) and a 63× (NA 1.4) or 100× (NA 1.4) oil objective using the factory settings for excitation and emission wavelengths of Cy2, Cy3, YFP and GFP fluorescence. Images were collected by sequential scanning using single laser-line excitation to eliminate bleeding from one channel into the other. Images illustrating the overlap of the fluorescent signals were generated using ImageJ software (http://rsbweb.nih.gov/ij/) or Adobe Photoshop (Adobe Systems, San Jose, CA).
Colocalization of two fluorescent signals in individual cells was quantified using ImageJ. A selection area was drawn around a cell (excluding membrane staining in the case of wild-type connexins), and the fluorescence corresponding to cytosolic connexin- and LC3-positive structures was identified using particle analysis; then, the connexin particles that colocalized with LC3-positive structures were identified using the ‘colocalization finder’ plug-in. The percentage of the total connexin particles that colocalized with LC3 particles was calculated. The results are presented as the average of the percentage colocalization obtained in a number of cells (n).
Immunoblotting
Cells were harvested in PBS, 4 mM EDTA containing complete EDTA-free protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN) and centrifuged at 13,000 g for 5 minutes. Pellets were resuspended in harvesting buffer and sonicated. To detect Atg5–Atg12 complex, cells from sister cultures were harvested in 1× lysis solution (Cell Signaling Technology); homogenates were further fractionated by centrifugation at 14,000 g for 10 minutes. Aliquots from cell homogenates (CX43 and CX50) or supernatants (Atg5 or Atg12) were resolved on 9% SDS-containing polyacrylamide gels, transferred to Immobilon P membranes (Millipore, Billerica, MA) and subjected to immunoblotting using anti-CX43, anti-CX50, anti-Atg5 or anti-Atg12 antibodies as previously described (Berthoud et al., 2003). Owing to some cell loss at later time points, aliquots from HeLa-CX50, MEF and HeLa-CX50P88S cell homogenates containing equal amounts of total protein were loaded in each lane. Because NRK cells had no cell loss, lanes were loaded with equal volume aliquots from homogenates of these cells (representing a fixed fraction of the total). The intensities of the bands were quantified by densitometry using Adobe Photoshop. Values are reported as a percentage of the control values.
Thin-section electron microscopy
MEFs cultured on two-well LAB-TEK Nalge Nunc International chamber slides (Thermo Fisher Scientific) were left untreated or starved by incubation in HBSS for 1 hour. Then, cells were rinsed in 1× PBS and fixed in 2% glutaraldehyde, 4% paraformaldehyde in 0.1 M sodium cacodylate, pH 7.3 for 2 hours at room temperature. Cells were subsequently washed with sodium cacodylate buffer three times for 5 minutes each, postfixed with 1% osmium tetroxide in sodium cacodylate buffer for 1 hour at room temperature, washed twice with sodium cacodylate buffer for 5 minutes, rinsed once in maleate buffer pH 5.1, stained with 1% uranyl acetate in maleate buffer for 1 hour, and washed in maleate buffer three times for 5 minutes each. Then, the specimens were dehydrated through increasing concentrations of ethanol followed by propylene oxide. After that, the specimens were infiltrated with propylene oxide:Spurr resin mixtures of increasing proportions of Spurr resin followed by six 1 hour changes of 100% Spurr resin. After polymerization of the Spurr resin at 60°C for 1–2 days, ultrathin sections (90 nm) were cut on a Reichert-Jung Ultracut-E ultramicrotome (Vienna, Austria) and contrasted with uranyl acetate and lead citrate. Specimens were examined with a FEI Tecnai F30 scanning transmission electron microscope (Hillsboro, OR) equipped with a Gatan CCD camera (Pleasanton, CA) at 300 kV.
Supplementary Material
Acknowledgments
This work was supported by a grant from the Children's Research Foundation (to A.L.) and grant NIH EY08368 (to E.C.B.). Deposited in PMC for release after 12 months.
Footnotes
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/124/6/910/DC1
References
- Beardslee M. A., Laing J. G., Beyer E. C., Saffitz J. E. (1998). Rapid turnover of connexin43 in the adult rat heart. Circ. Res. 83, 629-635 [DOI] [PubMed] [Google Scholar]
- Berthoud V. M., Minogue P. J., Guo J., Williamson E. K., Xu X., Ebihara L., Beyer E. C. (2003). Loss of function and impaired degradation of a cataract-associated mutant connexin50. Eur. J. Cell Biol. 82, 209-221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthoud V. M., Minogue P. J., Laing J. G., Beyer E. C. (2004). Pathways for degradation of connexins and gap junctions. Cardiovasc. Res. 62, 256-267 [DOI] [PubMed] [Google Scholar]
- Biederbick A., Kern H. F., Elsässer H. P. (1995). Monodansylcadaverine (MDC) is a specific in vivo marker for autophagic vacuoles. Eur. J. Cell Biol. 66, 3-14 [PubMed] [Google Scholar]
- Bjørkøy G., Lamark T., Brech A., Outzen H., Perander M., Øvervatn A., Stenmark H., Johansen T. (2005). p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cells death. J. Cell Biol. 171, 603-614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjørkøy G., Lamark T., Pankiv S., Øvervatn A. B., Johansen T. (2009). Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 452, 181-197 [DOI] [PubMed] [Google Scholar]
- Eskelinen E.-L. (2008). Fine structure of the autophagosome. Methods Mol. Biol. 445, 11-28 [DOI] [PubMed] [Google Scholar]
- Hayward A. F., Kent A. P. (1983). Gap junctions in the epidermis of fetal rats studied by transmission electron microscopy. J. Ultrastruct. Res. 84, 182-193 [DOI] [PubMed] [Google Scholar]
- Hesketh G. G., Shah M. H., Halperin V. L., Cooke C. A., Akar F. G., Yen T. E., Kass D. A., Machamer C. E., Van Eyk J. E., Tomaselli G. F. (2010). Ultrastructure and regulation of lateralized connexin43 in the failing heart. Circ. Res. 106, 1153-1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J., Klionsky D. J. (2007). Autophagy and human disease. Cell Cycle 6, 1837-1849 [DOI] [PubMed] [Google Scholar]
- Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., Yoshimori T. (2000). LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720-5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuusisto E., Salminen A., Alafuzoff I. (2001). Ubiquitin-binding protein p62 is present in neuronal and glial inclusions in human tauopathies and synucleinopathies. NeuroReport 12, 2085-2090 [DOI] [PubMed] [Google Scholar]
- Laing J. G., Beyer E. C. (1995). The gap junction protein connexin43 is degraded via the ubiquitin proteasome pathway. J. Biol. Chem. 270, 26399-26403 [DOI] [PubMed] [Google Scholar]
- Laing J. G., Tadros P. N., Westphale E. M., Beyer E. C. (1997). Degradation of connexin43 gap junctions involves both the proteasome and the lysosome. Exp. Cell Res. 236, 482-492 [DOI] [PubMed] [Google Scholar]
- Laing J. G., Tadros P. N., Green K., Saffitz J. E., Beyer E. C. (1998). Proteolysis of connexin43-containing gap junctions in normal and heat-stressed cardiac myocytes. Cardiovasc. Res. 38, 711-718 [DOI] [PubMed] [Google Scholar]
- Leach D. H., Oliphant L. W. (1984). Degradation of annular gap junctions of the equine hoof wall. Acta Anat. 120, 214-219 [DOI] [PubMed] [Google Scholar]
- Levine B., Klionsky D. J. (2004). Development by selfdigestion: molecular mechanisms and biological functions of autophagy. Dev. Cell 6, 463-477 [DOI] [PubMed] [Google Scholar]
- Lichtenstein A., Gaietta G. M., Deerinck T. J., Crum J., Sosinsky G. E., Beyer E. C., Berthoud V. M. (2009). The cytoplasmic accumulations of the cataract-associated mutant, Connexin50P88S, are long-lived and form in the endoplasmic reticulum. Exp. Eye Res. 88, 600-609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marazziti D., Di Pietro C., Golini E., Mandillo S., Matteoni R., Tocchini-Valentini G. P. (2009). Induction of macroautophagy by overexpression of the Parkinson's disease-associated GPR37 receptor. FASEB J. 23, 1978-1987 [DOI] [PubMed] [Google Scholar]
- McLachlan C. S., Almsherqi Z. A., Mossop P., Suzuki J., Leong S. T., Deng Y. (2009). Down regulation of immuno-detectable cardiac connexin-43 in BALB/c mice following acute fasting. Int. J. Cardiol. 136, 99-102 [DOI] [PubMed] [Google Scholar]
- Meijer A. J., Codogno P. (2009). Autophagy: regulation and role in disease. Crit. Rev. Clin. Lab. Sci. 46, 210-240 [DOI] [PubMed] [Google Scholar]
- Minogue P. J., Tong J. J., Arora A., Russell-Eggitt I., Hunt D. M., Moore A. T., Ebihara L., Beyer E. C., Berthoud V. M. (2009). A mutant connexin50 with enhanced hemichannel function leads to cell death. Invest. Ophthalmol. Vis. Sci. 50, 5837-5845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchener J. S., Shelburne J. D., Bradford W. D., Hawkins H. K. (1976). Cellular autophagocytosis induced by deprivation of serum and amino acids in HeLa cells. Am. J. Pathol. 83, 485-492 [PMC free article] [PubMed] [Google Scholar]
- Mizushima N., Yamamoto A., Matsui M., Yoshimori T., Ohsumi Y. (2004). In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell 15, 1101-1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N., Levine B., Cuervo A. M., Klionsky D. J. (2008). Autophagy fights disease through cellular self-digestion. Nature 451, 1069-1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankiv S., Clausen T. H., Lamark T., Brech A., Bruun J.-A., Outzen H., Øvervatn A., Bjørkøy G., Johansen T. (2007). p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 282, 24131-24145 [DOI] [PubMed] [Google Scholar]
- Pfeifer U. (1980). Autophagic sequestration of internalized gap junctions in rat liver. Eur. J. Cell Biol. 21, 244-246 [PubMed] [Google Scholar]
- Qin H., Shao Q., Igdoura S. A., Alaoui-Jamali M. A., Laird D. W. (2003). Lysosomal and proteasomal degradation play distinct roles in the life cycle of Cx43 in gap junctional intercellular communication-deficient and -competent breast tumor cells. J. Biol. Chem. 278, 30005-30014 [DOI] [PubMed] [Google Scholar]
- Reggiori F., Klionsky D. J. (2005). Autophagosomes: biogenesis from scratch? Curr. Opin. Cell Biol. 17, 415-422 [DOI] [PubMed] [Google Scholar]
- Rowland A. M., Richmond J. E., Olsen J. G., Hall D. H., Bamber B. A. (2006). Presynaptic terminals independently regulate synaptic clustering and autophagy of GABAA receptors in Caenorhabditis elegans. J. Neurosci. 26, 1711-1720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T., Garant P. R. (1986). Fate of annular gap junctions in the papillary cells of the enamel organ in the rat incisor. Cell Tissue Res. 246, 523-530 [DOI] [PubMed] [Google Scholar]
- Shiels A., Mackay D., Ionides A., Berry V., Moore A., Bhattacharya S. (1998). A missense mutation in the human connexin50 gene (GJA8) underlies autosomal dominant “zonular pulverulent” cataract, on chromosome 1q. Am. J. Hum. Genet. 62, 526-532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas M. A., Zosso N., Scerri I., Demaurex N., Chanson M., Staub O. (2003). A tyrosine-based sorting signal is involved in connexin43 stability and gap junction turnover. J. Cell Sci. 116, 2213-2222 [DOI] [PubMed] [Google Scholar]
- Thumm M., Egner R., Koch B., Schlumpberger M., Straub M., Veenhuis M., Wolf D. H. (1994). Isolation of autophagocytosis mutants of Saccharomyces cerevisiae. FEBS Lett. 349, 275-280 [DOI] [PubMed] [Google Scholar]
- VanSlyke J. K., Deschenes S. M., Musil L. S. (2000). Intracellular transport, assembly, and degradation of wild-type and disease-linked mutant gap junction proteins. Mol. Biol. Cell 11, 1933-1946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X., Liu J., Jiang J. X. (2008). Lens fiber connexin turnover and caspase-3-mediated cleavage are regulated alternately by phosphorylation. Cell Commun. Adhes. 15, 1-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorimitzu T., Nair U., Yang Z., Klionsky D. J. (2006). Endoplasmic reticulum stress triggers autophagy. J. Biol. Chem. 281, 30299-30304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zatloukal K., Stumptner C., Fuchsbichler A., Heid H., Schnoelzer M., Kenner L., Kleinert R., Prinz M., Aguzzi A., Denk H. (2002). p62 is a common component of cytoplasmic inclusions in protein aggregation diseases. Am. J. Pathol. 160, 255-263 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}