

Abstract
Background
Adoptive immunotherapy with antigen-specific effector T-cell (TE) clones is often limited by poor survival of the transferred cells. We describe here a Macaca nemestrina model for studying transfer of T-cell immunity.
Methods
We derived, expanded, and genetically marked CMV-specific CD8+ TE clones with surface markers expressed on B cells. TE cells were adoptively transferred, and toxicity, persistence, retention of introduced cell-surface markers, and phenotype of the persisting T cells was evaluated.
Results
CD8+ TE clones were efficiently isolated from distinct memory precursors and gene-marking with CD19 or CD20 permitted in vivo tracking by quantitative PCR. CD19 was a more stable surface-marker for tracking cells in vivo and was used to re-isolate cells for functional analysis. Clonally derived CD8+ TE cells differentiated in vivo to phenotypically and functionally heterogeneous memory T-cell subsets.
Conclusions
These studies demonstrate the utility of Macaca nemestrina for establishing principles for T-cell therapeutics applicable to humans.
Keywords: Immunotherapy, lymphocyte, gene transfer
Introduction
Adoptive T-cell therapy, in which disease fighting T cells are isolated, propagated to large numbers in vitro, and then transferred back to patients is a rapidly advancing treatment modality for cancer and infectious diseases [8, 14, 20, 40]. This approach has restored immunity to viruses in immunodeficient patients [17, 32, 37, 39, 49] and produced antitumor responses in a subset of cancer patients when combined with lymphodepleting chemotherapy and high-dose interleukin (IL)-2 [10, 11]. In human trials, the efficacy of transferred effector T cells (TE) correlated with their persistence in vivo [38]. Although T-cell therapy has shown promise, many components of this complex strategy require optimization including the type of T cell selected for therapy, manipulation of the host environment, and the cytokine regimens that most effectively facilitate persistence and function of transferred T cells.
The availability of an animal model that is highly predictive for human translation could significantly improve the clinical efficacy of T-cell therapy. Inbred mouse strains have proven valuable for uncovering basic immunological mechanisms, but mouse studies of adoptive T-cell transfer have not always translated to humans. This could reflect the different culture conditions used to propagate murine T cells and/or intrinsic differences in memory T cells (TM) as a consequence of the evolutionary distance (∼65 million years) and disparity in the life-span between humans and mice [9, 27]. Old world monkeys, including macaques, have the closest evolutionary relationship to humans among approachable animal models, and the difference in life-span is less profound [29]. Additionally, human and macaque T cells share multiple markers of T-cell phenotype, differentiation, and regulation [26, 29, 30, 33]. A disadvantage of the macaque model for studies treating malignant disease is the lack of a tumor model to analyze the antitumor efficacy of transferred T cells. However, macaques are susceptible to viruses such as cytomegalovirus (CMV) that are targets of immunotherapy in humans [22, 33, 34, 52] and can provide a useful model to define strategies for isolating antigen-specific T cells, determine safety, and characterize the durability and quality of immunity achieved by adoptive transfer.
We have studied the adoptive transfer of antigen-specific CD8+ TE clones in Macaca nemestrina (M. nemestrina) using CMV as a model antigen [7]. The model was developed to employ culture conditions and cell doses identical to those used in human trials of T-cell therapy [35, 37, 49, 51]. In prior work, we showed that CMV-specific TE clones derived from the small subset of CD62L+ central memory T cells (TCM), but not from CD62L− effector memory T cells (TEM) survived long-term after transfer and reverted to both TCM and TEM phenotypes in vivo [7]. Here we describe the methodology for isolating, genetically modifying, and re-infusing macaque antigen-specific TE cells, with subsequent monitoring for safety, persistence, and function.
Material and methods
Animals and sample acquisition
Adult M. nemestrina were housed at the Washington National Primate Research Center, under American Association for Accreditation of Laboratory Animal Care certified conditions. The Institutional Review Board and Institutional Animal Care and Use Committee approved the protocols that were followed. Animal care personnel monitored the clinical status of the animals throughout the experimental protocol. Complete blood count (CBC) and serum chemistry were measured in accredited clinical laboratories.
Cytokine flow cytometry (CFC) assay for detection of CMV-specific T cells
CMV+ macaques were identified using a CFC assay that detects CMV-specific T cells by stimulating peripheral blood mononuclear cells (PBMC) with pools of 15-mer peptides with an 11 amino acid (aa) overlap that spanned the 558 aa sequence of the rhesus CMV (rhCMV) immediate early (IE)-1 protein, or with a pp65B and IE-2 peptide previously identified as antigenic in macaques and kindly provided by Dr. L. Picker (Oregon Health Sciences University) [33]. The 137 peptides that comprised the panel were arranged in an analytic grid consisting of 24 pools, with 11-12 peptides per pool (Fig. 1A). CFC was performed as described [7, 24]. In some experiments, CMV-specific CD8+ TE clones were stimulated for 6 hours with peptide-pulsed (1 μg/mL) antigen-presenting cells and examined by CFC.
Fig. 1. Identification of CMV-reactive CD8+ T cells in peripheral blood lymphocytes from M. nemestrina.
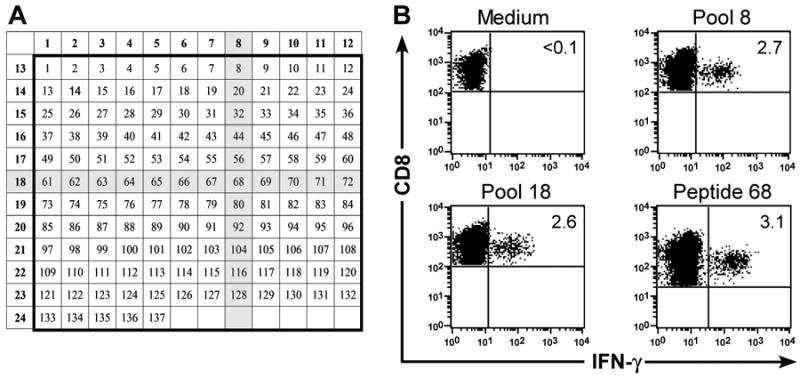
(A) Arrangement of 137 15-mer peptides spanning the sequence of the rhCMV IE-1 protein. The shaded areas correspond to the peptides present in the two positive pools (8; 18) in a representative epitope mapping experiment. PBMC obtained from a 23 macaques were examined for the presence of CMV-specific T-cell responses by CFC. (B) CFC detects IFN-γ production by CMV-specific CD8+ T cells after stimulation of PBMC with CMV IE-1 peptides in pool 8 (upper right panel) and 18 (lower left panel). Stimulation of PBMC with the corresponding peptide 68 (lower right panel) confirmed that sequences within the single peptide shared by both pools 8 and 18 stimulated IFN-γ-production by CD8+ T cells. PBMC stimulated with medium alone (upper left panel) served as a negative control. Data are gated on CD3+CD8+ cells.
Retroviral vectors
A truncated CD19 gene (ΔCD19) encoding the extracellular and transmembrane domains and four aa of the cytoplasmic tail and the full-length CD20 gene were amplified by RT-PCR from cDNA generated from M. nemestrina PBMC, and cloned into plasmid pMP71pre as described [7, 12]. Retrovirus supernatant was produced in the retrovirus-packaging cell-line Phoenix Galv grown in Dulbecco's modified Eagle medium with 10% heat-inactivated fetal bovine serum (Gemini, West Sacramento, CA) [18]. Stably transduced packaging cells were generated by transfecting retroviral constructs into Phoenix Galv cells using Fugene G (Roche Diagnostics, Indianapolis, IN) according to the manufacturer's instruction, and then purifying transduced cells by fluorescence-activated cell sorting on a FACS Vantage Becton Dickinson Instrument (BD Biosciences, BDB, San Diego, CA) after staining with allophyocyanin (APC)-conjugated CD19 monoclonal antibody (mAb) (J4.119, Immunotech Coulter, Marseille Cedex, France) or CD20-APC (clone 2H7, BDB). Supernatants from the sort-purified packaging cells were tested for transduction of primary macaque T cells, and cell lines that provided >10% transduction were cryopreserved in aliquots as a master cell bank for subsequent short-term expansion for production of retroviral supernatant for transduction of T cells for in vitro studies and adoptive transfer.
In vitro culture of CMV-specific CD8+ T-cell clones and gene-transfer
Isolation of CMV-specific CD8+ TE clones from defined TM subsets
CD8+ TCM and TEM subsets were purified by cell-sorting or magnetic bead selection [7]. Briefly, aliquots of PBMC were stained with anti-CD8 (RPA-T8) and anti-CD62L (SK11) monoclonal antibodies (mAbs) and sorted on a Vantage instrument (BDB) into a CD62L+CD8+ fraction containing TCM and a CD62L−CD8+ TEM fraction. In some experiments, CD62L+CD8+ T cells were isolated using a CD8+ T-cell isolation kit, followed by positive selection with a CD62L mAb and immunomagnetic beads (Miltenyi Biotec, Auburn, CA). Sorted subsets were resuspended in RPMI-1640 supplemented with 25 mM HEPES, L-glutamine, 25 μM 2-mercaptoethanol (Invitrogen, Carlsbad, CA), and 10% human serum (Gemini: T-cell media) and co-cultured for 7 days with autologous monocytes and the cognate CMV peptide (1 μg/mL). IL-2 (10 U/mL; Chiron, Emeryville, CA) was added on day three. CD8+ TE clones were isolated by plating at 0.3 cells/well in 96-well round-bottom plates with 7.5×104 γ-irradiated autologous PBMC and 1×104 γ-irradiated B-lymphoblastoid lymphocytes (LCL), peptide antigen (1 μg/mL) and 50 U/mL IL-2 [7]. After 12-14 days, a 30-μL aliquot from each well with visible growth was tested for recognition of 51Cr-labeled peptide-pulsed or unpulsed target cells [37].
In vitro stimulation and retroviral gene-transfer
To restimulate CMV-reactive TE clones, aliquots were stimulated with anti-CD3 (SP34; 20 ng/mL, BDB) and anti-CD28 (9.3; 1 μg/mL, FHCRC) mAbs, 25×106 γ-irradiated human PBMC (3500 rad), 5×106 γ-irradiated human LCL (8000 rad), and IL-2 (50 U/mL) [7]. For retroviral gene-transfer, T cells were exposed to ΔCD19 or CD20 retrovirus supernatant at a ratio of 1:4 (T cell media: retrovirus supernatant) with IL-2 (50 U/mL) and polybrene (5 μg/mL; Chemicon, Billerica, MA) on days two and three after stimulation, centrifuged at 1000g for one hour at 32°C, and incubated overnight after each exposure [3]. The cells were then washed, cultured in T-cell media containing IL-2, and selected for ΔCD19 or CD20-expression by immunomagnetic bead-selection as described (Miltenyi) [7]. After 14 days of culture, T cells were used for functional assays or cryopreserved in aliquots as a cell-bank.
In vitro expansion and adoptive transfer of CMV-specific CD8+ T-cell clones
Aliquots of CMV-specific CD8+ TE clones were thawed from the cell-banks and stimulated in 75-cm2 flasks with anti-CD3/CD28 mAbs, γ-irradiated PBMC and LCL feeder cells as described [7]. IL-2 (50 U/mL) was added one, five, eight, and 11 days after stimulation. Growth was measured by counting viable cells by trypan-blue staining. Cultures were split once the density exceeded 1.5×106 T cells/mL. After 14 days, the T cells were harvested into 250-mL centrifuge tubes, washed three times, and resuspended in 0.9% NaCl-solution supplemented with 2% autologous serum. T cells (3-5×108/kg) were administered intravenously over 30 minutes after ketamine sedation. An aliquot of each cell-product was tested for sterility, phenotype, and CMV-specific function. Blood was collected prior to infusion, every other day for one week after transfer and weekly thereafter. An inguinal lymph node (LN) biopsy and bone marrow (BM) aspirate was obtained 14 days after transfer.
Flow cytometry and cell-sorting
Cells were surface-labeled with the following fluorochrome-conjugated mAbs (obtained from BDB unless noted): CD3 (SP34-2), CD8, CD28 (CD28.2), CD62L, CCR7, CD95 (DX2), CD20 (2H7; BDB or eBioscience, San Diego, CA), CD19 and CD127 (Beckman Coulter, Miami, FL). Analyses were performed using a FACSCalibur and CellQuest Software (BDB). Multiparameter flow-cytometry was performed on a 3-laser BDB-LSR-II instrument using Pacific Blue, AmCyan, fluorescein isothiocyanate, phycoerythrin, phycoerythrin-Cy7, allophycocyanin, peridinin-chlorophyll protein-Cy5.5, or allophycocyanin-Cy7 as fluorescent parameters [2, 48]. Data were analyzed using FlowJo software (Treestar, Ashland, OR). The analysis included lineage-defining markers (CD8, CD3, CD19) and phenotyping markers (CD95, CCR7, CD62L, CD28, or CD127). Samples for intracellular staining of Ki-67 were fixed in Cytofix/Cytoperm solution (20 minutes, 4°C), before washing, permeabilization, and mAbs-labeling in Perm/Wash-Buffer (BDB).
Re-isolation experiments
ΔCD19+CD8+CD62L+ and ΔCD19+CD8+CD62L− T cells were re-isolated from post-infusion PBMC samples by cell-sorting on a Vantage BDB instrument after staining with anti-CD8, anti-CD19, and anti-CD62L mAbs. Sorted cells were stimulated with anti-CD3/CD28 mAbs, γ-irradiated PBMC and LCL, and IL-2 (50 U/mL) as described above.
Stability assay
Flow cytometry was used for assessing the stability of the ΔCD19 or CD20 marker. Aliquots of transduced T cells were cultured for 14 days and then plated at 2×106 cells/well in T-cell media supplemented with IL-15 (0.1 ng/mL; R&D Systems, Minneapolis, MN). Cytokine and half-medium exchanges were performed twice weekly. After four weeks of rest, aliquots were stimulated for 6 days with anti-CD3 (20 ng/mL) and anti-CD28 (1 μg/mL) mAbs, and IL-2 (50 U/mL) and evaluated by flow cytometry for transgene-expression after staining with mAbs to CD3, CD8, CD19 or CD20 [4]. To assess the marker gene stability on transferred T cells, aliquots of post-infusion PBMC were examined by flow cytometry for transgene-expression before and 4 days after stimulation with CD3/28 mAbs.
Cytotoxicity assays
Cytotoxicity was measured as described [4, 6, 7]. Autologous 51Cr-labeled target T cells were pulsed overnight with peptide antigen (1-5 μg/mL) or medium alone, washed three times, and used as targets to assess lysis by CMV-specific CD8+ TE. Specific lysis was calculated using the standard formula [36].
Fluorescent-probe PCR
PCR was performed using a quantitative real-time PCR (qPCR) assay [6, 7]. DNA was isolated from PBMC or T cells using a QIAamp DNA Kit (Qiagen, Valencia, CA). For the titration experiments to determine the sensitivity of the ΔCD19 or CD20 qPCR and to prepare the standard curve, we employed serial 1:10 dilutions of DNA isolated from the ΔCD19+ or CD20+ T cells (corresponding to 1×105 T cells) into aliquots of DNA obtained from pre-infusion PBMC or untreated control animals (corresponding to 1×105 PBMC/reaction). For the in vivo tracking studies, aliquots of DNA (0.3-1 μg; corresponding to approximately 50,000–150,000 cell equivalents) were obtained from PBMC. We used generally 1 μg DNA per reaction except at early time points when the frequency was high, to ensure the amount of input DNA was above the threshold of detection. DNA was amplified in a 50 μL-reaction with TaqMan Gene-Expression Master Mix, and PCR primers and a fluorescent-tagged probe designed to detect a unique sequence encompassing the junction of the retroviral vector and the CD20 or ΔCD19 gene (Applied Biosystems, Foster City, CA). Amplifications were performed in duplicates (42 cycles) on an ABI Prism 7900-HT Real-time PCR System and Sequence Detection System 2.2.2 software (Applied Biosystems).
Results
Identification of CMV-specific CD8+ T-cell responses
CMV infection is widely prevalent in M. nemestrina colonies and CMV-immune macaques maintain a TM response to viral antigens including the viral IE-1 protein [33]. To facilitate the isolation of CMV-specific T cells, we used an intracellular CFC assay to detect CD8+ T cells in PBMC that produced interferon (IFN)-γ in response to stimulation with an overlapping peptide panel corresponding to the rhCMV IE-1 protein or to individual pp65B or IE-2 peptides previously shown to be immunogenic (Fig. 1A) [7, 33]. This approach allows the identification of antigenic epitopes without knowledge of the complex MHC type in each animal [50]. We examined PBMC from 23 macaques by CFC (Fig. 1B) and detected IFN-γ+ IE-1-specific CD8+ T cells in 12 animals at frequencies ranging from 0.12%-4.4% of CD8+ T cells, and pp65B or IE-2 -specific T cells in 6 additional animals (Table 1). Two animals had responses both to peptides in the IE-1 panel and either pp65B or IE-2. The minimal essential peptide for the IE-1 responses was deduced by testing a series of nonamer peptides derived from the 15-mer sequence used for mapping (Table 1). The IE-1 peptide KKGDIKDRV was previously described and recognized by T cells in six animals [7]. Collectively, the results identified a panel of CMV peptides that facilitated the detection of CMV-specific CD8+ T cell responses in the overwhelming majority (78%) of the M. nemestrina in this colony.
Table 1. Detection of CMV-specific CD8+ T cell responses in M. nemestrina.
| No. of animals | Target specificity of rhCMV-specific CD8+ T cells | Frequency of CMV-specific CD8+ T cells (%) | |
|---|---|---|---|
| Peptide nomenclature | Amino acid residue | ||
| 6 | IE-1 #52 | KKGDIKDRV | 0.17;0.21;0.22;0.38;1.7;4.4 |
| 4 | pp65B #23 | NPTDRPIPT | 0.15;0.15;1.0;1.4 |
| 1 | IE-2 #29-30 | KPTDSMSQR | 0.9 |
| 3 | IE-2 #81-82 | ATTRSLEYK | 0.25;2.2;10.7 |
| 2 | IE-1 #60 | EEHVKLFFK | 0.2;0.49 |
| 1 | IE-1 #68 | KLDDEQKEV | 3.1 |
| 1 | IE-1 #77 | KNDEAMLGMHTPITM | 1.7 |
| 1 | IE-1 #80 | DQVRVLILY | 2.0 |
| 1 | IE-1 #137 | SKSLHPMQTRSKSDK | 0.12 |
Isolation and expansion of CMV-specific CD8+ T-cell clones from distinct TM subsets
Antigen-specific T cells are isolated and expanded in vitro to generate sufficient numbers of TE cells for adoptive T-cell therapy. Recent studies in macaques and mice suggest the TM subset from which the TE cells are derived influences their capacity to persist in vivo after adoptive transfer [7, 23]. Two broad phenotypic subsets of memory cells termed TCM and TEM are identified, differing in homing, function, and transcriptional and epigenetic programming [41]. In macaques and humans, TCM and TEM can be distinguished from each other based on differential expression of CD62L or CCR7, and from TN based on differential expression of CD95 (Fig. 2A) [33]. We used these markers to analyze the frequency of TN, TEM, and TCM subsets in CD8+ T cells from cryopreserved PBMC samples obtained from 18 healthy macaques. We employed CCR7 for this analysis because CD62L may be shed upon cryopreservation and thawing. CD8+ TCM comprised a mean of 2.2% ± 1.3% (mean ± SD; range: 0.78%-5.3%) of CD8+ T cells (Fig. 2B). By contrast, CD8+ TEM and TN were more prevalent and comprised 60.5% ± 32.2% (8.2%-95.2%) and 36.1% ± 31.7% (3.4%-90.2%) of CD8+ T cells, respectively (Fig. 2B).
Fig. 2. Derivation of macaque CMV-specific CD8+ T cells from TEM or TCM subsets.
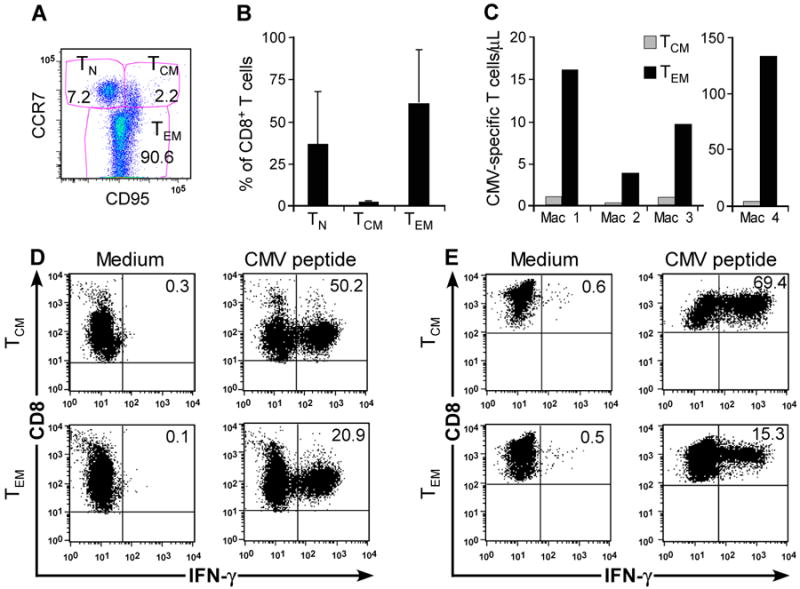
(A) Flow cytometry showing CD8+ T-cell subsets in macaque PBMC including TCM (CCR7+CD95+), TEM (CCR7−CD95+), and TN (CCR7+CD95−). (B) Frequency of CD8+ TCM, CD8+ TEM, and CD8+ TN (%) in peripheral blood CD8+ lymphocytes. Aliquots of PBMC obtained from 18 healthy macaques were stained with mAbs binding to CD8, CD3, CD95, and CCR7 and examined by flow cytometry after gating on CD3+CD8+ T cells. Mean and SD is shown. (C) CMV IE-specific T cells are present in distinct CD8+ TM subsets. CFC assay for IFN-γ+CD8+ T cells specific for CMV in sort-purified CD8+CD62L+ and CD8+CD62L− T-cell subsets obtained from 4 macaques. The absolute number of CMV-specific CD8+ TCM or TEM/μL peripheral blood was determined by calculating the absolute number of CD3+CD8+ T cells/μL of blood (% of CD8+ T cells/lymphocyte subset × lymphocyte count/μL blood/100). Subsequently, the absolute number of the TEM, TCM, and TN subset was derived (% subset × number of CD3+CD8+/μL blood/100). Finally, we calculated the absolute number of CMV-specific CD8+ T cells/μL in each subset (% IFN-γ+ cells × absolute number of CD3+CD8+ TCM+TN/μL or CD3+CD8+ TEM/μL blood/100). (D, E) CFC assay of macaque CMV-specific T-cell lines detects IFN-γ+CD8+ CMV-specific T cells in sort-purified CD8+CD62L+ cells containing TCM (upper panels) and CD8+CD62L− TEM subsets (lower panels). Sort-purified subsets from 2 representative macaques were stimulated with autologous CMV peptide-pulsed antigen-presenting cells and assayed by CFC for production of IFN-γ after stimulation with medium alone (left panels), or CMV peptide antigen (right panels). Data are gated on CD3+CD8+ cells.
We used immunomagnetic bead-selection or cell-sorting to separate fresh PBMC into a CD62L+CD8+ fraction containing TCM and CD62L−CD8+ TEM. The purity of the CD62L+CD8+ and CD62L−CD8+ fractions was 97.5% (94.4-99.8%) and 98.3 % (95.9%-99.4%), respectively, and T cells specific for individual CMV-epitopes were detectable by CFC in both subsets [7]. The absolute number of CMV-specific TEM in the blood exceeded that of TCM by 10–40-fold (Fig. 2C). We then stimulated the sort-purified TEM and TCM for each 7 days with autologous CMV-peptide pulsed monocytes and IL-2 to enrich CMV-specific cells. The frequency of CMV-specific T cells by CFC analysis after stimulation was 15.3%-20.9% in CD62L− TEM-derived lines and 50.2%-69.4% in lines derived from CD62L+ T cells (Fig. 2D, E). CMV-specific CD8+ TE were cloned from polyclonal cultures derived from each TM subset. CMV-specific T-cell clones were identified 12-14 days after plating by screening an aliquot of each well for lysis of autologous peptide-pulsed but not unpulsed target cells. CMV-specific CD8+ TE clones were reliably derived from TCM in seven of seven animals and from TEM in three of three animals. The cloning efficiency (number of CMV+ wells×100/number of plated CMV+CD8+ T cells) was similarly achieved for TCM-derived cultures (34%-37%) and TEM-derived cultures (39%-63.8%). Thus, despite the fact that the CD8+ TCM are rare in the peripheral blood, CMV-specific TE clones could be reliably derived in vitro from both TCM and TEM subsets with comparable efficiency.
CMV-specific CD8+ TE clones from each of the TM subsets were then propagated using a rapid expansion method developed for growing human T cells for adoptive therapy [35, 37, 49, 51]. We stimulated the contents of a cloning well, typically containing 5×104-2×105 T cells, with anti-CD3/28 mAbs, irradiated feeder cells, and IL-2, over a 14-day cycle in vitro. A mean of 14 (range: 7-22) TCM-derived TE clones were expanded from individual micro-wells of each of seven animals and a mean of 11 (5-19) proliferated to a mean of 122.5×106 TE (65.8×106-178×106). A mean of eight (8-9) TEM-derived TE clones were expanded from each of three animals, and a mean of four (2-6) proliferated over 14 days to 120.2×106 TE (39.6×106-187.3×106). As reported previously, all TCM or TEM-derived CD8+ TE clones retained CMV-specific reactivity and displayed a CD28−CD62L−CCR7−CD127−granzyme B+ phenotype, consistent with their differentiation to TE cells as a consequence of T-cell receptor (TCR)-stimulation and growth in IL-2 [7]. Thus, CMV-specific TE clones can be reliably derived from both TCM and TEM subsets and a single in vitro expansion resulted reproducibly in the generation of sufficient numbers of functional TE cells that could be used to establish a cell-bank for subsequent expansion and adoptive transfer experiments.
Gene-marking of TM-derived CD8+ TE cells for in vivo tracking
Analysis of the fate and migration of adoptively transferred T cells in vivo is facilitated by the introduction of a marker gene to distinguish infused and endogenous T cells. Because foreign protein expression in T cells can be immunogenic [5, 6, 35], we developed retroviral vectors encoding macaque B-cell lineage surface molecules, either ΔCD19 or full-length CD20, to transduce TEM or TCM-derived TE [7]. The mean transduction efficiency of macaque TE as measured by surface-expression of the introduced marker was 30.7% (22.7%-45.3%; n=7) for the ΔCD19 retrovirus and 20.4% (10.3%-38.1%; n=3) for CD20 (Fig. 3A). The gene-marked TE cells were enriched by immunomagnetic selection to a purity of 95.8% (91.5%-98%) for ΔCD19 and 91.8% (86.9%-97.8%) for CD20 (Fig. 3A). To ensure genetic modification did not impact growth or function, we restimulated aliquots of the parental, ΔCD19+ or CD20+ CMV-specific CD8+ TE and examined proliferation and retention of CMV-specific cytotoxicity. Both ΔCD19+ and CD20+ TE clones proliferated comparable to the unmodified clones and mediated equivalent CMV-specific cytotoxicity (Fig. 3B, C).
Fig. 3. Gene-marking of macaque CD8+ TE with non-immunogenic B-cell lineage marker.

(A) Efficient transduction and selection of macaque CD8+ TE with the ΔCD19 or CD20 marker. Macaque CD8+ TE were stimulated with anti-CD3/CD28 mAbs, transduced with ΔCD19 or CD20, and enriched by immunomagnetic selection. Aliquots of the unselected (□) or selected ( ) T cells were examined by flow cytometry after staining with mAbs against CD3, CD8, and CD19 or CD20 mAbs. Shown are mean and range of the results with the ΔCD19 (n=7) or CD20 (n=3) marker. (B) In vitro growth of gene-modified TE clones. Left panel: Representative TE clone either unmodified (◇) or ΔCD19+ (◆). Right panel: Representative TE clone either unmodified (◇) or CD20+ (●). Aliquots of T cells unmodified or transduced with ΔCD19+ (left panel) or CD20+ (right panel) were restimulated with anti-CD3/CD28 mAbs, irradiated feeder cells and IL-2, and numeric expansion was measured by counting viable cells on the indicated days. Data are representative of results with ΔCD19+ or CD20+ T cells obtained from each three macaques. (C) Gene-marked CMV-specific CD8+ TE clones retain CMV-specific reactivity. Aliquots of CMV-specific CD8+ TE either unmodified or either ΔCD19+ (left panel) and CD20+ (right panel) were restimulated in vitro and examined in a chromium release assay for recognition of autologous target cells, either unpulsed (□) or pulsed with the CMV cognate peptide at an effector-to-target (E/T) ratio of 10:1 (■), 5:1(
) T cells were examined by flow cytometry after staining with mAbs against CD3, CD8, and CD19 or CD20 mAbs. Shown are mean and range of the results with the ΔCD19 (n=7) or CD20 (n=3) marker. (B) In vitro growth of gene-modified TE clones. Left panel: Representative TE clone either unmodified (◇) or ΔCD19+ (◆). Right panel: Representative TE clone either unmodified (◇) or CD20+ (●). Aliquots of T cells unmodified or transduced with ΔCD19+ (left panel) or CD20+ (right panel) were restimulated with anti-CD3/CD28 mAbs, irradiated feeder cells and IL-2, and numeric expansion was measured by counting viable cells on the indicated days. Data are representative of results with ΔCD19+ or CD20+ T cells obtained from each three macaques. (C) Gene-marked CMV-specific CD8+ TE clones retain CMV-specific reactivity. Aliquots of CMV-specific CD8+ TE either unmodified or either ΔCD19+ (left panel) and CD20+ (right panel) were restimulated in vitro and examined in a chromium release assay for recognition of autologous target cells, either unpulsed (□) or pulsed with the CMV cognate peptide at an effector-to-target (E/T) ratio of 10:1 (■), 5:1( ), 2.5:1 (), or 1.25:1 (
), 2.5:1 (), or 1.25:1 ( ). Data are representative of results with ΔCD19+ or CD20+ T cells from each three macaques. (D, E) Stability of the marker-gene expression in macaque CD8+ T cells. The ΔCD19+ (D) or CD20+ (E) T cells were stimulated with anti-CD3/CD28 mAbs and examined by flow cytometry on day 6 (ii) and 14 (iii) of the stimulation cycle for ΔCD19 or CD20 expression. Unmodified T cells served as negative control (i). Aliquots of T cells were also rested and the ΔCD19 or CD20 expression was assessed by flow cytometry after 4 weeks of rest (iv) or 6 days after restimulation with anti-CD3/CD28 mAbs (v). Inset values show the % of CD3+CD8+ T cells positive for ΔCD19 or CD20 and the MFI is indicated for each time point. Data showing the difference in the stability of expression of ΔCD19 and CD20 at the cell surface is representative for experiments with ΔCD19 and CD20-modified T cells from 3 animals, and was observed in eight ΔCD19+ and CD20+ TE clones.
). Data are representative of results with ΔCD19+ or CD20+ T cells from each three macaques. (D, E) Stability of the marker-gene expression in macaque CD8+ T cells. The ΔCD19+ (D) or CD20+ (E) T cells were stimulated with anti-CD3/CD28 mAbs and examined by flow cytometry on day 6 (ii) and 14 (iii) of the stimulation cycle for ΔCD19 or CD20 expression. Unmodified T cells served as negative control (i). Aliquots of T cells were also rested and the ΔCD19 or CD20 expression was assessed by flow cytometry after 4 weeks of rest (iv) or 6 days after restimulation with anti-CD3/CD28 mAbs (v). Inset values show the % of CD3+CD8+ T cells positive for ΔCD19 or CD20 and the MFI is indicated for each time point. Data showing the difference in the stability of expression of ΔCD19 and CD20 at the cell surface is representative for experiments with ΔCD19 and CD20-modified T cells from 3 animals, and was observed in eight ΔCD19+ and CD20+ TE clones.
Introduction of genes encoding a unique surface marker allows quantifiable measurement of T-cell persistence in vivo by both PCR for vector sequences and flow cytometry. However, flow cytometric methods require stable cell-surface marker expression, even after T cells enter a resting state. We examined the stability of ΔCD19 and CD20 on transduced T cells in vitro six and 14 days after anti-CD3/28-restimulation, and again after four weeks of rest in IL-15 (0.1 ng/mL) in the absence of TCR-stimulation. This experiment was performed with TCM-derived TE cells because TEM-derived TE cells survive poorly upon rest under these conditions [7]. Aliquots were removed from the cultures at intervals during the growth and resting phase, and were analyzed by flow cytometry for expression of ΔCD19 or CD20. The mean fluorescence intensity (MFI) of ΔCD19-expression on ΔCD19+ T cells declined modestly from 7759 to 6160 between day six and 14 of the 14-day stimulation cycle and was only slightly lower (MFI: 6014) at the end of the resting phase (Fig. 3D). By contrast, the level of CD20-expression decreased profoundly from an MFI of 5558 to 1200 between day six and 14 of the culture, and declined further after the 28-day rest period such that only 56.9% of the T cells expressed detectable cell-surface CD20 (Fig. 3E). To examine whether the lower CD20-expression reflected activation-dependence or the selective survival of non-transduced T cells, we restimulated aliquots of rested ΔCD19+ and CD20+ TE with anti-CD3/CD28 mAbs and analyzed the cell-surface expression of ΔCD19 and CD20. Six days after restimulation, the CD20-transduced TE upregulated CD20-expression and 99.9% of the cells expressed high levels of CD20 (MFI: 8758; Fig. 3E). The rested ΔCD19+ T cells also showed an increased level of ΔCD19 upon activation (MFI: 7896) although ΔCD19-expression was not as dependent on T-cell activation as CD20 (Fig. 3D). Similar results were observed in additional ΔCD19+ and CD20+ TE cells. This differential stability of ΔCD19 and CD20 was surprising since the expression of both transgenes is driven by the same retroviral promoter and the high transduction efficiency would suggest multiple retroviral integration sites, minimizing the potential that transgene-expression would vary as a result of the genome insertion site. This data suggests the CD20 molecule may undergo more rapid turnover, reducing expression at the cell-surface when promoter activity is diminished during periods of cell quiescence. Thus, while both the ΔCD19 or CD20-marker allow detection of transferred, gene-marked T cells by PCR, the greater stability of ΔCD19-expression indicates the ΔCD19 vector would be superior for in vivo tracking of transferred T cells by flow cytometry.
T-cell infusions and monitoring of toxicity
The infusion of large numbers of antigen-specific TE requires clinical monitoring for potential toxicities [10, 35, 49, 51]. We propagated autologous ΔCD19+ or CD20+ CMV-specific CD8+ TE clones over 12-14 days to large numbers (3.5-5×109) and transferred four TCM-derived TE clones at a dose of 3-5×108/kg to four macaques (Supplementary Fig. 1A). For each infusion, animals were closely monitored for adverse effects. We did not observe any clinical toxicity during or after the infusions. Laboratory monitoring of CBC and serum chemistry identified a transient decrease in the lymphocyte count in all animals one day after the infusion as the only significant alteration (Supplementary Fig. 1B).
Monitoring T-cell persistence by real-time qPCR and flow cytometry
We evaluated both qPCR and flow cytometry for tracking transferred ΔCD19+ and CD20+ T cells in vivo [7]. Sensitivity of the PCR assay using primer pairs amplifying unique sequences within each of the retroviral vectors (Fig. 4A) was determined by spiking serial 1:10 dilutions of aliquots of ΔCD19+ or CD20+ TE into aliquots of pre-infusion PBMC as described in the Method section. The titration experiments showed that the qPCR assay detected both ΔCD19+ and CD20+ T cells with comparable sensitivity of approximately one transduced T cell per reaction, each of which contained DNA from 105 pre-infusion PBMC (Fig. 4B).
Fig. 4. Tracking of ΔCD19+ or CD20+ CMV-specific CD8+ TE clones following adoptive transfer by qPCR.

(A) Schematic design of retroviral vector constructs encoding for macaque B-cell lineage marker genes and location of primer and fluorescent probe (red bar) used for the qPCR assay. Abbreviations: MPSV-LTR, myeloproliferative sarcoma virus retroviral long terminal repeat; PRE, woodchuck hepatitis virus post-transcriptional regulatory element. (B) Detection of ΔCD19 or CD20-marked T cells within PBMC by qPCR. Samples of titrated numbers of ΔCD19+ () or CD20+ T cells (■) were spiked into aliquots of pre-infusion PBMC (each 105 PBMC/reaction) and examined by real-time qPCR for detection of marker-positive T cells. Data are representative of each 3 assays with ΔCD19+ or CD20+ T cells. (C) Enumeration of transferred ΔCD19+ or CD20+ T cells determined by real-time qPCR for vector sequences. Autologous ΔCD19+ () or CD20+ (■) TCM-derived TE clones were expanded in vitro and transferred to each one of the macaques at a dose of 5×108/kg. DNA was isolated from samples of PBMC obtained before and at indicated time-points after the T-cell infusion and examined by real-time qPCR for detection of marker-positive T cells.
We have shown previously that transferred TCM-derived CD8+ TE cells survive long-term in vivo, but those derived from TEM did not persist [7]. Thus, to examine the relative utility of the ΔCD19 and CD20-marker for tracking T cells in vivo with qPCR and flow cytometry, we studied two animals that received TCM-derived TE cells (5×108/kg). Analysis of PBMC (∼50,000–150,000 cell equivalents per reaction) obtained before and one day after infusion of TCM-derived TE marked with ΔCD19 or CD20 by qPCR showed that both the transferred ΔCD19+ and CD20+ TE cells were easily detected at a level corresponding to 60,687 copies/106 PBMC and 37,767 copies/106 PBMC, respectively (Fig. 4C). The frequency of the transferred ΔCD19+ or CD20+ T cells gradually declined over 8 weeks after infusion, but both ΔCD19+ and CD20+ T cells remained detectable by PCR at all time points (Fig. 4C) and were also identified in samples of BM and LN obtained 14 days after the infusion [7].
We also used flow cytometry to visualize the transferred T cells by co-staining samples of PBMC with CD3, CD8, and CD19 or CD20. One and three days after infusion of ΔCD19+ TE, we identified ΔCD19+ T cells at a frequency of 26.3% of CD3+ cells or 45.1% of CD3+CD8+ T cells, respectively. A distinct ΔCD19+ population of CD3+CD8+ T cells remained detectable at a frequency of 1.7% in the blood at week three after the T cells had become quiescent (Fig. 5A) and migrated to LN and BM [7]. Flow cytometry also detected transferred CD20+ TE in the peripheral blood one and three days after transfer at a frequency of 20.2% of CD3+CD8+ T cells or 12.3% of CD3+CD8+ T cells, respectively (Fig. 5B). However, accurate quantification of the CD20+ T cells in the blood by flow cytometry was difficult for two reasons. First, there was a small subset of endogenous CD8+ T cells that stained positive for CD20 in the pre-infusion sample in some donors consistent with previous reports [19] (Fig. 5B). Secondly, by three weeks post-infusion the transferred CD20+ TE did not segregate as a distinct subset, perhaps due to a lower expression level of CD20 in rested T cells as predicted by our in vitro experiments (Fig. 5B). We stimulated PBMC obtained before and six days after infusion with anti-CD3/CD28 mAbs and examined the CD20 cell-surface expression by flow cytometry after four days to determine if CD20 was upregulated. We found the absolute frequency of CD20+ T cells did not change substantially, but the MFI of CD20-expression on the CD20+ subset increased upon activation (Fig. 5C). Of interest, CD20-expression level of the endogenous CD20+CD8+ T-cell subset was also activation-dependent (Fig. 5C). Thus, the ΔCD19-marker is superior to the CD20-marker for tracking transferred T cells in vivo using flow cytometry, both because the expression level is less activation-dependent and because there is no endogenous T-cell subset that expresses ΔCD19.
Fig. 5. Tracking of transferred ΔCD19+ or CD20+ CD8+ TE clones by flow cytometry.

(A) An autologous CMV-specific TE clone modified to express ΔCD19 was expanded in vitro and transferred back to a macaque at a cell dose of 5×108/kg. PBMC were collected before (pre) and on day 1 and 3, and at week 3 after infusion and examined by flow cytometry after staining with mAbs to CD3, CD8, and CD19. Data are gated on CD3+CD8+ T cells and are representative for results in three animals. Inset values show the frequency (%) of CD3+CD8+ TE positive for ΔCD19. (B) An autologous CD20+ CMV-specific TE clone was given to a macaque at a dose of 5×108/kg. PBMC were collected at the indicated times and analyzed by flow cytometry for the frequency (%) of CD20+ T cells within the CD3+CD8+ T-cell subset. (C) Activation-dependence of the CD20-expression in persisting CD20+ T cells. Flow cytometry analysis of PBMC obtained before (left panel) and 6 days after the CD20+ T-cell infusion (second right panel) after staining with mAbs to CD3, CD8, and CD20. After 4 days of activation with anti-CD3/CD28 mAbs, aliquots of the activated pre-infusion PBMC (second left panel) and ‘Day 6’ PBMC (right panel) were examined by flow cytometry for cell-surface expression of CD20 after gating on CD3+CD8+ T cells. Inset values show the frequency (%) of CD3+CD8+ TE positive for CD20. The MFI of the CD20+ TE (R1) is shown.
Phenotype and quality of T-cell memory established by adoptive T-cell transfer
In prior work, we showed that CMV-specific TE derived and expanded by in vitro culture from CD8+ TCM persisted in vivo and a subset of the transferred cells reacquired phenotypic markers of TCM including either CD127, CD28, CCR7, or CD62L [7]. Here, we used the ΔCD19-marker to distinguish transferred TE and employed for the first time polychromatic flow cytometry to simultaneously assess co-expression of several of these TM markers on the T cells that persisted in vivo. We obtained PBMC four weeks after the infusion of a ΔCD19+ TCM-derived TE clone and stained aliquots for CD3, CD8, and CD19 to identify the transferred ΔCD19+ T cells and then examined this subset for the co-expression of CD95, CD62L, CCR7, CD127, or CD28 (Fig. 6A, B). A subset of the infused ΔCD19+ TE clone differentiated to CD62L+ cells while others maintained a CD62L− phenotype in vivo (Fig. 6B, upper row). ΔCD19+ T cells that acquired CD62L also co-expressed CCR7 (Fig. 6B, upper row) and the majority were positive for CD28 and CD127 (Fig. 6B, upper and lower row), markers the cells did not express at the time of infusion but which are characteristic of TCM (Fig. 6B). The fraction of ΔCD19+ T cells that did not re-acquire CD62L was CD28+/−CD127− CCR7−, consistent with a TEM phenotype.
Fig. 6. Analysis of phenotype and function of adoptively transferred ΔCD19+ TCM-derived TE clones.

(A) Multiparameter flow cytometry of macaque PBMC obtained 4 weeks after a ΔCD19+ TE infusion. Transferred ΔCD19+ T cells were identified in PBMC after staining with mAbs to CD3, CD8, and CD19. (B) The expression of TM marker on CD3+CD8+ΔCD19+ T cells was determined by flow cytometry after co-staining with CD62L, CCR7, CD28, CD127 or CD95. (C) Aliquots of macaque PBMC were obtained 8 weeks after infusion of a TCM-derived ΔCD19+CD8+ TE clone. CCR7+CD95+ TCM and CCR7−CD95+ TEM subsets either in the CD3+CD8+ΔCD19− or CD3+CD8+ΔCD19+ cells were identified as described in (A) and (B). Cells were then stained for intracellular expression of Ki-67. Inset values show the frequency (%) of Ki-67+ T cells. (D) Analysis of intracellular Ki-67 in PBMC obtained from 2 macaques 6-8 weeks after infusion of a TCM-derived ΔCD19+CD8+ TE clone, and in samples of BM and LN obtained from 3 macaques 2 weeks after the infusion. PBMC: mean; BM and LN: mean ± SD. (E) Function of re-isolated ΔCD19+ TE obtained from TCM or TEM subsets. Aliquots of post-infusion PBMC were sort-purified in CD8+ΔCD19+CD62L+ and CD8+ΔCD19+CD62L− subsets, restimulated in vitro using anti-CD3/CD28 mAbs, and examined in a chromium release assay for recognition of unpulsed (□) or peptide-pulsed target cells. E/T (■) 2.5:1, () 1.25:1. The transferred ΔCD19+ TE clone served as control. (F) Aliquots of the infused ΔCD19+ TE clone (■) and re-isolated ΔCD19+ TE either obtained from TCM () or TEM (□) were stimulated with medium or CMV peptide-pulsed antigen-presenting cells, and examined by CFC for production of IFN-γ, TNF-α, and IL-2 after gating on CD3+CD8+ T cells.
It is known that endogenous CD8+ TCM and TEM subsets undergo slow cell division in response to homeostatic cytokines in vivo [41]. To assess whether the TCM-derived ΔCD19+ TE persisting in vivo as memory cells returned to quiescence comparable to endogenous TM, we examined the transferred ΔCD19+ T cells for expression Ki-67, which identifies cycling T cells [15]. The majority of the ΔCD19+ TE clone was Ki-67+ at the time of transfer (data not shown). However, the ΔCD19+ T cells that persisted for >6 weeks and differentiated to TCM and TEM in vivo reverted to quiescence and the fraction that expressed Ki-67+ was comparable to endogenous CD8+ TCM and TEM (Fig. 6C, D). Similarly, only a small fraction of ΔCD19+ TCM or ΔCD19+ TEM cells that were present in samples of BM or LN obtained 14 days after transfer expressed Ki-67+ (Fig. 6D), illustrating that the transferred TCM derived TE cells established reservoirs of quiescent TM cells in memory niches.
We next investigated the functional attributes of the transferred virus-specific TM cells. We sort-purified ΔCD19+ TEM and TCM subsets from pooled samples of PBMC obtained 28-56 days after adoptive transfer and stimulated the cells in vitro with anti-CD3/CD28 mAbs. After 14 days of culture, we examined aliquots of the cells for lysis of peptide-pulsed target cells and for cytokine production by CFC. We found the ΔCD19+ T cells re-isolated from CD8+CD62L+ TCM or CD8+CD62L− TEM subsets efficiently differentiated to cytolytic TE in vitro (Fig. 6E), and produced IFN-γ and TNF-α upon antigen encounter. A subset of the TE re-isolated from TCM also produced IL-2 in response to viral antigen stimulation, but only a small fraction of the TE re-isolated from TEM produced IL-2 (Fig. 6F). Thus, the adoptive transfer of a CMV-specific TE clone derived from TCM precursors can establish heterogeneous TM cells that return to the quiescent memory pool, respond to TCR-stimulation, and produce effector cytokines in addition to IL-2 after re-isolation in vitro.
Discussion
Establishing durable and functional antigen-specific T-cell responses is a goal of clinical adoptive T-cell therapy for both malignant and infectious disease [8, 14, 20, 40]. Our prior work in macaques showed that CMV-specific TCM, but not TEM-derived TE clones exhibit the capacity to survive after transfer as long-lived and functional TM [7]. To enable future studies that might evaluate strategies to enhance the persistence of TE cells from each TM subset, we describe detailed methods developed for deriving CMV-specific TE clones from distinct TM subsets in M. nemestrina. An important aspect was the use of culture conditions and cell doses that are comparable to human T-cell therapy regimens [35, 49, 51]. In our study in 23 immunocompetent animals, we used a peptide panel encompassing the CMV IE-1 protein and selected CMV-IE-2 and pp65B peptides and detected responses in the majority (78%) of M. nemestrina. The complete IE-1 panel was selected because it is a major target antigen in humans, and including full peptide panels to additional CMV proteins would likely increase the fraction of animals from which T cells could be derived [25, 44]. Numerous CMV-specific CD8+ TE clones were readily derived from both TM subsets in seven out of seven CMV-immune animals with comparable efficiency and the clones retained CMV-specific function during transduction, selection, and propagation. Thus, this model provides a feasible platform to systematically examine potential approaches to improve cell transfer efficiency including homeostatic cytokines or pharmacologic modulation [1, 2, 13, 31, 47].
For the adoptive transfer experiments to examine the utility of non-immunogenic marker genes for longitudinal analysis of the in vivo survival and phenotype of transferred CMV-specific TE clones, we used exclusively TCM derived TE cells since they have been proven to be capable of surviving long-term in vivo [7]. Our data show that both the ΔCD19 and CD20 B-cell lineage markers were safe in vivo and permitted tracking of the transferred TE by qPCR. An unexpected finding was the differential stability of the cell-surface expression of ΔCD19 and CD20 despite the fact that they were driven off the identical retroviral promoter. Suboptimal retroviral expression of full-length CD20 in human T cells has been described previously [16, 43, 46] and may in part reflect the structural complexity of the full-length CD20 protein or its rapid turnover [45]. The presence of endogenous CD20+ T cells in a subset of donors [19] is another factor rendering the ΔCD19-marker superior for tracking studies by flow cytometry and suggests ΔCD19 is a potential candidate marker gene for human translation.
Our work illustrates the use of the macaque model to evaluate the qualities and fate of transferred TCM-derived CD8+ TE clones [7]. We used multicolor flow cytometry to extend our prior work and show that transferred TE cells derived from a single TCM precursor persist in vivo, differentiate to heterogeneous TCM and TEM phenotypes, and return to cell quiescence comparable to endogenous TCM and TEM. A recent study in two rhesus macaques could not detect consistent survival of transferred TCM-derived TE [28]. In this study, TE clones derived from TCM or TEM were induced by SIV DNA vaccination. The TE clones were cultured for prolonged times without costimulation, labeled with a fluorescent-dye for in vivo tracking, and infused three days after intravenous SIV infection followed by a 10-day course of low-dose IL-2. The results reported in this study showed that both the TEM and TCM-derived TE clone could be detected in the blood only briefly, and persistence in bronchioalveolar-lavage at 6 weeks did not segregate with the derivation of the clone [28]. Thus, under the experimental conditions used in the SIV study, undefined clonal variation rather than intrinsic programming of TM subsets appeared to account for the differential migration and survival in hosts where antigen is present at the time of transfer. We are currently analyzing gene-expression profiles and epigenetic alterations in TE cells that we derived, which may provide further insights into signaling pathways responsible for differences in the fate of transferred TCM-derived TE cells in vivo and elucidate the molecular basis and plasticity of the TCM-derived TE [21, 42]. Utilizing a nonhuman primate model to improve selection, persistence, and function of T cells should significantly enhance the potential to obtain clinically-relevant information for future adoptive therapy trials.
Supplementary Material
Acknowledgments
We would like to thank staff of the Washington National Primate Research Center, especially Mike Gough, Carole Elliott, and Jaclyn Bogue, for their expert handling of the animals and technical assistance. We thank W. Uckert, Max-Delbrück-Center, Germany, for the plasmid pMP71pre, and L. Picker and A. Sylvester (Oregon Health & Science University) for the CMV peptide and discussions. This work was supported by National Institutes of Health grants CA114536, AI053193, P51-RR000166-47S1, and RR00166.
Footnotes
Competing Interest Statement: The authors declare that they have no competing financial interest.
References
- 1.Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, Larsen CP, Ahmed R. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460:108–112. doi: 10.1038/nature08155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berger C, Berger M, Hackman RC, Gough M, Jensen MC, Riddell SR. Safety and immunological effects of IL-15 administration in nonhuman primates. Blood. 2009;114:2417–2426. doi: 10.1182/blood-2008-12-189266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berger C, Blau CA, Clackson T, Riddell SR, Heimfeld S. CD28 costimulation and immunoaffinity-based selection efficiently generate primary gene-modified T cells for adoptive immunotherapy. Blood. 2003;101:476–484. doi: 10.1182/blood-2002-07-2142. [DOI] [PubMed] [Google Scholar]
- 4.Berger C, Blau CA, Huang ML, Iuliucci JD, Dalgarno DC, Gaschet J, Heimfeld S, Clackson T, Riddell SR. Pharmacologically regulated Fas-mediated death of adoptively transferred T cells in a nonhuman primate model. Blood. 2004;103:1261–1269. doi: 10.1182/blood-2003-08-2908. [DOI] [PubMed] [Google Scholar]
- 5.Berger C, Flowers ME, Warren EH, Riddell SR. Analysis of transgene-specific immune responses that limit the in vivo persistence of adoptively transferred HSV-TK-modified donor T cells after allogeneic hematopoietic cell transplantation. Blood. 2006;107:2294–2302. doi: 10.1182/blood-2005-08-3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berger C, Huang ML, Gough M, Greenberg PD, Riddell SR, Kiem HP. Nonmyeloablative immunosuppressive regimen prolongs in vivo persistence of gene-modified autologous T cells in a nonhuman primate model. J Virol. 2001;75:799–808. doi: 10.1128/JVI.75.2.799-808.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest. 2008;118:294–305. doi: 10.1172/JCI32103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berger C, Turtle CJ, Jensen MC, Riddell SR. Adoptive transfer of virus-specific and tumor-specific T cell immunity. Curr Opin Immunol. 2009;21:224–232. doi: 10.1016/j.coi.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davis MM. A prescription for human immunology. Immunity. 2008;29:835–838. doi: 10.1016/j.immuni.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartentruber DJ, Topalian SL, Sherry R, Restifo NP, Hubicki AM, Robinson MR, Raffeld M, Duray P, Seipp CA, Rogers-Freezer L, Morton KE, Mavroukakis SA, White DE, Rosenberg SA. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, Robbins PF, Huang J, Citrin DE, Leitman SF, Wunderlich J, Restifo NP, Thomasian A, Downey SG, Smith FO, Klapper J, Morton K, Laurencot C, White DE, Rosenberg SA. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol. 2008;26:5233–5239. doi: 10.1200/JCO.2008.16.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Engels B, Cam H, Schüler T, Indraccolo S, Gladow M, Baum C, Blankenstein T, Uckert W. Retroviral vectors for high-level transgene expression in T lymphocytes. Hum Gene Ther. 2003;14:1155–1168. doi: 10.1089/104303403322167993. [DOI] [PubMed] [Google Scholar]
- 13.Gattinoni L, Klebanoff CA, Restifo NP. Pharmacologic induction of CD8+ T cell memory: better living through chemistry. Sci Transl Med. 2009;1:11ps12. doi: 10.1126/scitranslmed.3000302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gattinoni L, Powell DJ, Jr, Rosenberg SA, Restifo NP. Adoptive immunotherapy for cancer: building on success. Nat Rev Immunol. 2006;6:383–393. doi: 10.1038/nri1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gerdes J, Lemke H, Baisch H, Wacker HH, Schwab U, Stein H. Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J Immunol. 1984;133:1710–1715. [PubMed] [Google Scholar]
- 16.Griffioen M, van Egmond EHM, Kester MGD, Willemze R, Falkenburg JHF, Heemskerk MHM. Retroviral transfer of human CD20 as a suicide gene for adoptive T-cell therapy. Haematologica. 2009;94:1316–1320. doi: 10.3324/haematol.2008.001677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heslop HE, Slobod KS, Pule MA, Hale GA, Rousseau A, Smith CA, Bollard CM, Liu H, Wu MF, Rochester RJ, Amrolia PJ, Hurwitz JL, Brenner MK, Rooney CM. Long term outcome of EBV specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood. 2010;115:925–935. doi: 10.1182/blood-2009-08-239186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horn PA, Topp MS, Morris JC, Riddell SR, Kiem HP. Highly efficient gene transfer into baboon marrow repopulating cells using GALV-pseudotype oncoretroviral vectors produced by human packaging cells. Blood. 2002;100:3960–3967. doi: 10.1182/blood-2002-05-1359. [DOI] [PubMed] [Google Scholar]
- 19.Hultin LE, Hausner MA, Hultin PM, Giorgi JV. CD20 (pan-B cell) antigen is expressed at a low level on a subpopulation of human T lymphocytes. Cytometry. 1993;14:196–204. doi: 10.1002/cyto.990140212. [DOI] [PubMed] [Google Scholar]
- 20.June CH, Blazar BR, Riley JL. Engineering lymphocyte subsets: tools, trials and tribulations. Nat Rev Immunol. 2009;9:704–716. doi: 10.1038/nri2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaech SM, Hemby S, Kersh E, Ahmed R. Molecular and functional profiling of memory CD8 T cell differentiation. Cell. 2002;111:837–851. doi: 10.1016/s0092-8674(02)01139-x. [DOI] [PubMed] [Google Scholar]
- 22.Kaur A, Daniel MD, Hempel D, Lee-Parritz D, Hirsch MS, Johnson RP. Cytotoxic T-lymphocyte responses to cytomegalovirus in normal and simian immunodeficiency virus-infected rhesus macaques. J Virol. 1996;70:7725–7733. doi: 10.1128/jvi.70.11.7725-7733.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klebanoff CA, Gattinoni L, Torabi-Parizi P, Kerstann K, Cardones AR, Finkelstein SE, Palmer DC, Antony PA, Hwang ST, Rosenberg SA, Waldmann TA, Restifo NP. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci USA. 2005;102:9571–9576. doi: 10.1073/pnas.0503726102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maecker HT, Dunn HS, Suni MA, Khatamzas E, Pitcher CJ, Bunde T, Persaud N, Trigona W, Fu TM, Sinclair E, Bredt BM, McCune JM, Maino VC, Kern F, Picker LJ. Use of overlapping peptide mixtures as antigens for cytokine flow cytometry. J Immunol Methods. 2001;255:27–40. doi: 10.1016/s0022-1759(01)00416-1. [DOI] [PubMed] [Google Scholar]
- 25.Manley TJ, Luy L, Jones T, Boeckh M, Mutimer H, Riddell SR. Immune evasion proteins of human cytomegalovirus do not prevent a diverse CD8+ cytotoxic T cell response in natural infection. Blood. 2004;104:1075–1082. doi: 10.1182/blood-2003-06-1937. [DOI] [PubMed] [Google Scholar]
- 26.Manuel ER, Charini WA, Sen P, Peyerl FW, Kuroda MJ, Schmitz JE, Autissier P, Sheeter DA, Torbett BE, Letvin NL. Contribution of TCR repertoire breadth to the dominance of epitope-specific CD8+ T lymphocyte responses. J Virol. 2006;80:12032–12040. doi: 10.1128/JVI.01479-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mestas J, Hughes CCW. Of mice and not men: differences between mouse and human immunology. J Immunol. 2004;172:2731–2738. doi: 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- 28.Minang JT, Trivett MT, Bolton DL, Trubey CM, Estes JD, Li Y, Smedley J, Pung R, Rosati M, Jalah R, Pavlakis GN, Felber BK, Piatak M, Jr, Roederer M, Lifson JD, Ott DE, Ohlen C. Distribution, persistence, and efficacy of adoptively transferred central and effector memory-derived autologous simian immunodeficiency virus-specific CD8+ T cell clones in rhesus macaques during acute infection. J Immunol. 2010;184:315–326. doi: 10.4049/jimmunol.0902410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nikolich-Zugich J. Non-human primate models of T-cell reconstitution. Semin Immunol. 2007;19:310–317. doi: 10.1016/j.smim.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Onlamoon N, Hudson K, Bryan P, Mayne AE, Bonyhadi M, Berenson R, Sundstrom BJ, Bostik P, Ansari AA, Villinger F. Optimization of in vitro expansion of macaque CD4+ T cells using anti-CD3 and co-stimulation for autotransfusion therapy. J Med Primatol. 2006;35:178–193. doi: 10.1111/j.1600-0684.2006.00182.x. [DOI] [PubMed] [Google Scholar]
- 31.Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, Jones RG, Choi Y. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. 2009;460:103–107. doi: 10.1038/nature08097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peggs KS, Verfuerth S, Pizzey A, Khan N, Guiver M, Moss PA, Mackinnon S. Adoptive cellular therapy for early cytomegalovirus infection after allogeneic stem-cell transplantation with virus-specific T-cell lines. Lancet. 2003;362:1375–1377. doi: 10.1016/S0140-6736(03)14634-X. [DOI] [PubMed] [Google Scholar]
- 33.Pitcher CJ, Hagen SI, Walker JM, Lum R, Mitchell BL, Maino VC, Axthelm MK, Picker LJ. Development and homeostasis of T cell memory in rhesus macaque. J Immunol. 2002;168:29–43. doi: 10.4049/jimmunol.168.1.29. [DOI] [PubMed] [Google Scholar]
- 34.Powers C, Früh K. Rhesus CMV: an emerging animal model for human CMV. Med Microbiol Immunol. 2008;197:109–115. doi: 10.1007/s00430-007-0073-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Riddell SR, Elliott M, Lewinsohn DA, Gilbert MJ, Wilson L, Manley SA, Lupton SD, Overell RW, Reynolds TC, Corey L, Greenberg PD. T-cell mediated rejection of gene-modified HIV-specific cytotoxic T lymphocytes in HIV-infected patients. Nat Med. 1996;2:216–223. doi: 10.1038/nm0296-216. [DOI] [PubMed] [Google Scholar]
- 36.Riddell SR, Rabin M, Geballe AP, Britt WJ, Greenberg PD. Class I MHC-restricted cytotoxic T lymphocyte recognition of cells infected with human cytomegalovirus does not require endogenous viral gene expression. J Immunol. 1991;146:2795–2804. [PubMed] [Google Scholar]
- 37.Riddell SR, Watanabe KS, Goodrich JM, Li CR, Agha ME, Greenberg PD. Restoration of viral immunity in immunodeficient humans by the adoptive transfer of T cell clones. Science. 1992;257:238–241. doi: 10.1126/science.1352912. [DOI] [PubMed] [Google Scholar]
- 38.Robbins PF, Dudley ME, Wunderlich J, El-Gamil M, Li YF, Zhou J, Huang J, Powell DJ, Jr, Rosenberg SA. Cutting edge: persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. J Immunol. 2004;173:7125–7130. doi: 10.4049/jimmunol.173.12.7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rooney CM, Smith CA, Ng CYC, Loftin SK, Sixbey JW, Gan Y, Srivastava DK, Bowman LC, Krance RA, Brenner MK, Heslop HE. Infusion of cytotoxic T cells for the prevention and treatment of Epstein-Barr virus-induced lymphoma in allogeneic transplant recipients. Blood. 1998;92:1549–1555. [PubMed] [Google Scholar]
- 40.Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8:299–308. doi: 10.1038/nrc2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol. 2004;22:745–763. doi: 10.1146/annurev.immunol.22.012703.104702. [DOI] [PubMed] [Google Scholar]
- 42.Sarkar S, Kalia V, Haining WN, Konieczny BT, Subramaniam S, Ahmed R. Functional and genomic profiling of effector CD8 T cell subsets with distinct memory fates. J Exp Med. 2008;205:625–640. doi: 10.1084/jem.20071641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Serafini M, Bonamino M, Golay J, Introna M. Elongation factor (EF1α) promoter in a lentiviral backbone improves expression of the CD20 suicide gene in primary T lymphocytes allowing efficient rituximab-mediated lysis. Haematologica. 2004;89:86–95. [PubMed] [Google Scholar]
- 44.Sylwester AW, Mitchell BL, Edgar JB, Taormina C, Pelte C, Ruchti F, Sleath PR, Grabstein KH, Hosken NA, Kern F, Nelson JA, Picker LJ. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J Exp Med. 2005;202:673–685. doi: 10.1084/jem.20050882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tedder TF, Klejman G, Schlossman SF, Saito H. Structure of the gene encoding the human B lymphocyte differentiaiton antigen CD20 (B1) J Immunol. 1989;142:2560–2568. [PubMed] [Google Scholar]
- 46.van Meerten T, Claessen MJ, Hagenbeek A, Ebeling SB. The CD20/αCD20 ‘suicide’ system: novel vectors with improved safety and expression profiles and efficient elimination of CD20-transgenic T cells. Gene Ther. 2006;13:789–797. doi: 10.1038/sj.gt.3302705. [DOI] [PubMed] [Google Scholar]
- 47.Waldmann TA. The biology of interleukin-2 and interleukin-15: implications for cancer therapy and vaccine design. Nat Rev Immunol. 2006;6:595–601. doi: 10.1038/nri1901. [DOI] [PubMed] [Google Scholar]
- 48.Walker JM, Maecker HT, Maino VC, Picker LJ. Multicolor flow cytometric analysis in SIV-infected rhesus macaque. Methods Cell Biol. 2004;75:535–557. doi: 10.1016/s0091-679x(04)75022-0. [DOI] [PubMed] [Google Scholar]
- 49.Walter EA, Greenberg PD, Gilbert MJ, Finch RJ, Watanabe KS, Thomas ED, Riddell SR. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N Engl J Med. 1995;333:1038–1044. doi: 10.1056/NEJM199510193331603. [DOI] [PubMed] [Google Scholar]
- 50.Wiseman RW, Karl JA, Bimber BN, O'Leary CE, Lank SM, Tuscher JJ, Detmer AM, Bouffard P, Levenkova N, Turcotte CL, Szekeres E, Wright C, Harkins T, O'Connor DH. Major histocompatibility complex genotyping with massively parallel pyrosequencing. Nat Med. 2009;15:1322–1326. doi: 10.1038/nm.2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yee C, Thompson JA, Byrd D, Riddell SR, Roche P, Celis E, Greenberg PD. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: In vivo persistence, migration, and antitumor effect of transferred cells. Proc Natl Acad Sci USA. 2002;99:16168–16173. doi: 10.1073/pnas.242600099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yue Y, Barry PA. Rhesus cytomegalovirus: A nonhuman primate model for the study of human cytomegalovirus. Adv Virus Res. 2008;72:207–226. doi: 10.1016/S0065-3527(08)00405-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.