

Abstract
Toward development of smoking cessation aids superior to bupropion (2), we describe synthesis of 2-(substituted phenyl)-3,5,5-trimethylmorpholine analogues 5a–5h and their effects on inhibition of dopamine, norepinephrine, and serotonin uptake, nicotinic acetylcholine receptor (nAChR) function, acute actions of nicotine, and nicotine-conditioned place preference (CPP). Several analogues encompassing aryl substitutions, N-alkylation, and alkyl extensions of the morpholine ring 3-methyl group provided analogues more potent in vitro than (S,S)-hydroxybupropion (4a) as inhibitors of dopamine or norepinephrine uptake and antagonists of nAChR function. All of the new (S,S)-5 analogues had better potency than (S,S)-4a as blockers of acute nicotine analgesia in the tail-flick test. Two analogues with highest potency at α3β4*-nAChR and among the most potent transporter inhibitors have better potency than (S,S)-4a in blocking nicotine-CPP. Collectively, these findings illuminate mechanisms of action of 2 analogues and identify deshydroxybupropion analogues 5a–5h as possibly superior candidates as aids to smoking cessation.
Keywords: Nicotine, bupropion, hydroxybupropion, structure activity relationship, dopamine uptake, norepinephrine uptake, nAChR antagonism, antinociception, locomotor activity, hypothermia
Introduction
Tobacco product use, principally via cigarette smoking, is the number one cause of premature death in the United States (Centers for Disease Control and Prevention, 2002). There are more than 440,000 deaths due to cigarette smoking, and more than $75 billion in annual medical costs is attributed to smoking (NIDA, 2006; Centers for Disease Control and Prevention, 2008; Centers for Disease Control and Prevention, 2005). It is now commonly accepted that smoking behavior is maintained to a large extent by the reinforcing effects of nicotine (1) and aversive effects of nicotine withdrawal.1–4 Both non-pharmacological and pharmacological interventions have demonstrated efficacy in smoking cessation.5 At present, first-line pharmaceutical treatments include nicotine replacement therapy or use of bupropion (2) or varenicline (3).6,7 While these treatments are useful in helping about 20% of smokers abstain long-term, new pharmacotherapies are needed that are either more effective or can impact those individuals not helped by existing treatments.
While nicotine replacement therapy and 3 act on nicotinic receptors as their primary targets, 2 seems to engage additional targets. We have hypothesized that 2, or more specifically its active (S,S)-hydroxymetabolite (S,S)-4a (see Carroll et al., 2010)8 fits the multiple molecular target model of drug action. This model postulates that the combination of effects of 2 or active metabolites on dopamine (DA) transporter (DAT), norepinephrine (NE) transporter (NET), and nicotinic acetylcholine receptor (nAChR) function is important to its therapeutic efficacy as a smoking cessation agent.9,10 The model also suggests that fine tuning of effects on DA and NE availability and on nAChR function could lead to superior aids to smoking cessation. We have chosen 2 as a template for such work.
Our earlier work toward a goal of developing superior aids to smoking cessation concerned analogues of 2, its (S,S)- or (R,R)-hydroxymetabolites (S,S)-4a or (R,R)-4a, respectively, or 3-phenyltropane-related compounds.8,11–13 Several of these agents have superior activities relative to 2 or its active metabolite (S,S)-4a as inhibitors of DA or NE uptake or of nAChR function and have very promising in vivo profiles as inhibitors of nicotine-induced dependence behaviors.8,11–13
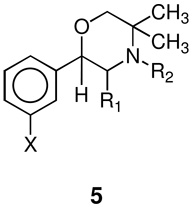
To explore potential utility as smoking cessation aids of compounds having related structural topology, and to define possible mechanism(s) of action of these compounds, we now describe the synthesis and in vitro and in vivo effects of 2-(substituted phenyl)-3,5,5-trimethylmorpholine analogues 5a–5h. Compounds (S,S)- and (R,R)-5a are analogues of the hydroxybupropion metabolites (S,S)- and (R,R)-4a, where the hydroxy group has been replaced by a hydrogen. We also synthesized and studied analogues (S,S)-5b–5h. In this study we report the identification of ligands with superior in vitro and preliminary in vivo activity profiles than those for (S,S)-4a.
Chemistry
Analogues 5a–5c, 5g, and 5h were synthesized in a fashion similar to that reported in the literature for optically active phenmetrazine (Scheme 1).14 The keto forms of the previously reported hydroxymorpholines 4a–4c, 4g, and 4h were reduced with sodium borohydride to afford mixtures of diastereomeric diols 6a–6c, 6g, and 6h, varying at the benzylic hydroxyl position. The morpholine ring structure could then be formed by cyclization using sulfuric acid in methylene chloride to form the optically active phenylmorpholines 5a–5c, 5g, and 5h. The diastereomic benzyl alcohol is presumably removed forming a benzylic cation, which is then trapped by the primary alcohol. Cyclization afforded the thermodynamically and kinetically more stable trans isomer. The optical activity is thus controlled by the methyl group alpha to the nitrogen. N-Methylation of (S,S)-5a to form 5d was done using methyl iodide in dimethylformamide at 70 °C. N-Alkylation of (S,S)-5a to form 5e and 5f was accomplished by standard reductive alkylation using acetaldehyde and propionaldehyde respectively and sodium triacetoxyborohydride in methylene chloride.
Scheme 1a.

a Reagents: a. NaBH4, CH3OH; b. CH2Cl2; H2SO4; c. MeI, DMF, 70°C for 5d; d. Na(OAc)3BH; acetaldehyde for 5e or propionaldehyde for 5f.
Analogue Characterization in Vitro
Compound (S,S)-4a has IC50 values of 630 and 180 nM for DA and NE uptake inhibition and is inactive for 5HT uptake inhibition (Table 1). The (R,R)-isomer (R,R)-4a is inactive as a DA and 5HT uptake inhibitor with much lower potency for NE uptake inhibition (IC50 = 9900 nM) than (S,S)-4a. Compound (S,S)-5a with IC50 values of 220, 100, and 390 nM for DA, NE, and 5HT uptake inhibition, respectively, is a more potent inhibitor of uptake of all three neurotransmitters than (S,S)-4a. The (R,R)-5a isomer is less potent at all three transporters than (S,S)-5a, as was seen for the pair of hydroxyl analogues, but was more potent than (R,R)-4a for DA and NE uptake inhibition.
Table 1.
Analogue Inhibition of Monoamine Uptake and Nicotinic Acetylcholine Receptor (nAChR) Function
 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compd | R1 | R2 | X | Monoamine Uptake Inhibitiona IC50 (nM) |
nAChR Inhibitionb IC50 (µM) |
|||||
| [3H]DA | [3H]NE | [3H]5HT | α3β4*- | α4β2- | α4β4- | α1*- | ||||
| 2c | 660 ± 180 | 1900 ± 300 | IA | 1.8 (1.15) | 12 (1.15) | 15 (1.07) | 7.9 (1.12) | |||
| (S,S)-4ac | 630 ± 50 | 180 ± 4 | IA | 11 (1.48) | 3.3 (1.07) | 30 (1.10) | 28 (1.45) | |||
| (R,R)-4ac | IA | 9900 ± 1400 | IA | 6.5 (1.20) | 31 (1.12) | 41 (1.07) | 7.5 (1.10) | |||
| (S,S)-5a | CH3 | H | Cl | 220 ± 60 | 100 ± 30 | 387 ± 140 | 3.3 ± | 20 (0.06) | 30 (1.12) | NT |
| (R,R)-5a | CH3 | H | Cl | 1600 ± 270 | 1200 ± 300 | IA | 1.6 (1.07) | 17 (1.06) | 12 (1.06) | 9.4 (1.05) |
| (S,S)-5b | CH3 | H | F | 61 ± 20 | 32 ± 3 | 4600 ± 430 | 5.6 (1.04) | 23 (1.05) | 55 (1.10) | 34 (1.07) |
| (S,S)-5c | CH3 | H | Br | 44 ± 3 | 150 ± 20 | 390 ± 30 | 1.4 (1.12) | 12 (1.06) | 11 (1.07) | 7.3 (1.07) |
| (S,S)-5d | CH3 | CH3 | Cl | 230 ± 60 | 170 ± 20 | 540 ± 130 | 2.8 (1.09) | 16 (1.14) | 23 (1.09) | 21 (1.05) |
| (S,S)-5e | CH3 | C2H5 | Cl | 44 ± 9 | 24 ± 8 | 1500 ± 300 | 0.79 (1.06) | 7.2 (1.06) | 6.4 (1.04) | 14 (1.05) |
| (S,S)-5f | CH3 | C3H7 | Cl | 61 ± 20 | 13 ± 3.6 | 2900 ± 400 | 0.98 (1.06) | 12 (1.06) | 5.8 (1.04) | 5.5 (1.06) |
| (S,S)-5g | C2H5 | H | Cl | 23 ± 5 | 19 ± 3 | 1800 ± 30 | 5.6 (1.16) | 14 (1.05) | 15 (1.10) | 13 (1.07) |
| (S,S)-5h | C3H7 | H | Cl | 6.0 ± 1 | 9 ± 2 | 300 ± 100 | 3.1 (1.15) | 9.5 (1.04) | 8.0 (1.12) | 5.0 (1.08) |
Values for mean ± standard error of three independent experiments, each conducted with triplicate determination.
Mean micromolar IC50 values (to two significant digits) for (S,S)- and (R,R)-hydroxybupropions (4a and 4b) and the indicated 2-(substituted phenyl)-3,5,5-trimethylmorpholine analogues from three independent experiments for inhibition of functional responses to an EC80–EC90 concentration of carbamylcholine mediated by nAChR subtypes composed of the indicated subunits (where * indicates that additional subunits are or may be additional assembly partners with the subunits specified; see Methods and Materials). Numbers in parentheses indicate S.E.M. as a multiplication/division factor of the mean micromolar IC50 values shown [i.e., the value 1.8 (1.15) reflects a mean IC50 value of 1.8 µM with an S.E.M. range of 1.8 × 1.15 µM to 1.8/1.15 µM or 1.6–2.1 µM]. IA: IC50 >100 µM. NT: not determined.
Taken from reference 13.
Replacement of the chloro group in (S,S)-5a with a fluoro group to give (S,S)-5b results in a 3.7- and 3.2-fold increase in the potency for inhibition of DA and NE uptake but a 12-fold decrease in potency for inhibition of 5HT uptake (Table 1). Compared to (S,S)-4a, 5b is 10- and 5.6-fold more potent as a DA and NE uptake inhibitor. The arylbromo analogue 5c with an IC50 value of 44 nM for inhibition of DA uptake is 5-fold more potent than 5a at DAT but has essentially the same potency as 5a for inhibition of NE and 5HT uptake.
The addition of an N-methyl group to (S,S)-5a to give 5d had little effect on monoamine uptake inhibition potency (Table 1). In contrast, the addition of an N-ethyl or N-propyl group to 5a to give 5e and 5f, respectively, resulted in a 4- to 7.8-fold increase in potency [relative to (S,S)-5a] for DA and NE uptake inhibition and a 4- to 7-fold decrease in 5HT uptake inhibition potency.
Compounds 5g and 5h, which have 3-ethyl and 3-propyl groups in place of the 3-methyl group in (S,S)-5a, have IC50 values of 23 and 6.0 nM for DA uptake inhibition, 19 and 9 nM for NE uptake inhibition, and 1800 and 300 nM for 5HT uptake inhibition (Table 1). Thus, they are the most potent of the analogues tested as DA uptake inhibitors and share with the N-ethyl and N-propyl analogues highest potency as NE uptake inhibitors.
The effects of 3,5,5-trimethylmorpholine analogues (S,S)- and (R,R)-4a, (S,S)- and (R,R)-5a, and 5b–5h on function of diverse human nAChR subtypes naturally or heterologously expressed by human cell lines were assessed using 86Rb+ efflux assays that are specific only for nAChR function in the cells used. None of the analogues has activity as agonists at α1*-, α3β4*-, α4β2-, or α4β4-nAChR, because 86Rb+ efflux in the presence of these ligands alone at concentrations from ~5 nM to 100 µM (data not shown here) was indistinguishable from responses in cells exposed only to efflux buffer. 86Rb+ efflux assays also were used to assess whether ligands had activity as antagonists at human nAChR. Representative concentration-response curves for selected ligands illustrate their nAChR in vitro inhibitory profiles (Fig. 1; see also Table 1). Other studies (not shown here) indicate that each of the ligands acts via non-competitive inhibition of nAChR function.
Figure 1.

Specific 86Rb+ efflux (ordinate; percentage of control) was determined for functional, human muscle-type α1β1γδ-nAChR (■), ganglionic α3β4*-nAChR (○), α4β2-nAChR (▲), or α4β4-nAChR (∇) naturally or heterologously expressed in human cell lines in the presence of a receptor subtype-specific, EC80–EC90 concentration of the full agonist, carbamylcholine, either alone or in the presence of the indicated concentrations (abscissa, log molar) of (S,S)-5a, (R,R)-5a, 5e, or 5f as indicated. Mean micromolar IC50 values and SEM as a multiplication/division factor of the mean micromolar IC50 value are provided in Table 1.
Compound (S,S)-4a has IC50 values of 11, 3.3, 30 and 28 µM for functional antagonism of α3β4*-, α4β2-, α4β4-, and α1β1*-nAChRs, respectively, meaning that it has 3–10-fold selectivity for α4β2-nAChR over other subtypes. Its potency as a functional antagonist of α4β2-nAChR, which are strongly implicated in nicotine dependence, is ~10-fold higher than that of (R,R)-4a, which is slightly more potent than (S,S)-4a as an antagonist of α3β4*- and α1*-nAChR. The (S,S)-deshydroxy analogue, (S,S)-5a, with IC50 values of 3.3 µM at α3β4*-nAChR and 20 µM at α4β2-nAChR, is three times more potent at α3β4*-nAChR and seven times less potent at α4β2-nAChR than (S,S)-4a. Importantly, (S,S)-5a has selectivity for α3β4*-nAChR over α4β2-nAChR as does (R,R)-4a, but opposite to the α4β2-nAChR selectivity of the sister isomer, (S,S)-4a. Both (S,S)-4a and (S,S)-5a have lower potencies at α4β4- and α1β1*-nAChR than at α3β4*- or α4β2-nAChR. Interestingly, (R,R)-5a has an IC50 value of 1.6 µM at α3β4*-nAChR, making it the most potent at this nAChR subtype of the 4a or 5a ligands, and is 11-, 7.4-, or 5.8-fold more selective for α3β4*-nAChR relative to α4β2-, α4β4-, or α1*-nAChR subtypes. It is also 6.5- and 4-times more potent as an α3β4*-nAChR antagonist than (S,S)- and (R,R)-4a.
Aryl halogen substitution of the chloro group in (S,S)-5a to a fluoro group to give (S,S)-5b slightly decreased whereas change to a bromo group in 5c produced an ~2-fold increase in antagonist potency at α3β4*-nAChR but also had similar effects on the other nAChR subtypes tested, thus not markedly altering nAChR selectivity.
Compound 5d, which is the N-methyl analogue of (S,S)-5a, has an nAChR profile similar to that of (S,S)-5a, with similar selectivity for α3β4*-nAChR over other subtypes but with slightly higher potency at α3β4*-nAChR. The N-ethyl and N-propyl analogues 5e and 5f have IC50 values of 0.79 and 0.98 µM at the α3β4*-nAChR, which makes them 4.2- and 3.4-fold more potent than (S,S)-5a and about two times more potent than (R,R)-5a at α3β4*-nAChRs. Compounds 5g and 5h have α3β4*-nAChR profiles similar to that of (S,S)-5a, although the propyl analogue 5h has slighty higher potency across all nAChR subtypes.
Behavioral Effects of Analogues
Compound (S,S)-4a blocks nicotine-induced antinociception in the tail-flick, hot-plate, locomotor depression, and hypothermia measures with AD50 values of 0.2, 1.0, 0.9, and 1.5 mg/kg, respectively (Table 2). None of the analogues was more potent than (S,S)-4a in the hot-plate and hypothermia tests, and only 5h with an AD50 of 0.49 mg/kg was more potent than (S,S)-4a in the locomotor test. However, eight analogues were more potent (AD50 values ranged from 0.006 to 0.13 mg/kg) in the tail-flick test than (S,S)-4a with four of the analogues also being substantially more potent than (S,S)-5a. Compound (S,S)-5a with an AD50 of 0.036 mg/kg in the tail-flick test is 5.5-times more potent than (S,S)-4a despite being inactive in the other three tests of acute nicotine action in mice. The arylbromo analogue 5c is six times more potent in the tail-flick test than (S,S)-5a and ~33-times more potent than (S,S)-4a and has an AD50 of 2.1 mg/kg in the locomotor test. The arylfluoro analog 5b has slightly lower potency than (S,S)-5c in the tail-flick and locomotor tasks. Moreover, each of the N-substituted and the alkyl extended analogues had higher potency than (S,S)-4a in the tail-flick assay.
Table 2.
Pharmacological evaluation of 3,5,5-trimethylmorpholine analogs as non-competitive nicotinic antagonistsa,b
| Compd | AD50 (mg/kg) | ||||
|---|---|---|---|---|---|
| Tail-flick | Hot-plate | Locomotion | Hypothermia | CPP | |
| 2c | 1.2 (1–1.18) | 15 (6–19) | 4.9 (0.9–46) | 9.2 (4–23) | 0.35 |
| (S,S)-4ac | 0.2 (0.06–0.7) | 1.0 (0.2–2.2) | 0.9 (0.2–5.7) | 1.5 (0.15–2.6) | 0.1 |
| (R,R)-4ac | 2.5 (1.2–3.6) | 10.3 (5.7–17.1) | IA | IA | NT |
| (S,S)-5a | 0.036 (0.012–0.1) | IA | IA | IA | NT |
| (R,R)-5a | 1.26 (0.39–4.1) | 3.9 (0.8–19) | IA | IA | NT |
| (S,S)-5b | 0.02 (0.008–0.03) | IA | 4.7 (1.2–19) | IA | NT |
| (S,S)-5c | 0.006 (0.003–0.01) | IA | 2.1 (0.9–4.8) | IA | NT |
| (S,S)-5d | 0.13 (0.03–0.6) | IA | 3.8 (1.2–12) | IA | NT |
| (S,S)-5e | 0.029 (0.004–0.23) | IA | IA | IA | 0.025 |
| (S,S)-5f | 0.056 (0.018–0.17) | IA | IA | IA | 0.03 |
| (S,S)-5g | 0.018 (0.009–0.03) | 9.6 (1.2–77) | 2.7 (0.7–10.5) | 2.5 (1.6–3.9) | NT |
| (S,S)-5h | 0.017 (0.002–0.15) | IA | 0.49(0.3–6.6) | IA | NT |
Results were expressed as AD50 (mg/kg) ± confidence limits (CL) or % effect at the highest dose tested. Dose-response curves were determined using a minimum of four different doses of test compound, and at least eight mice were used per dose group. IA = AD50 > 20 mg/kg.
NT = not tested.
Taken from reference 13.
In part because their in vitro potency as antagonists of α3β4*-nAChR and as inhibitors of DA and NE uptake were nearly the highest of the analogues tested, 5e and 5f were tested in mice for the ability to block nicotine rewarding effects as measured in the CPP test. Both compounds dose-dependently blocked the development of nicotine-induced CPP and they were 4- and 3-fold more potent in that assay than (S,S)-4a (Table 2).
Discussion
We have generated the 3,5,5-trimethylmorpholine analogues 5a–5h of the hydroxybupropion isomer (S,S)-4a where the 2-hydroxyl group has been replaced with a hydrogen and assessed the abilities of these analogues to affect DA, NE, and 5HT uptake, function of four nAChR subtypes, and the acute effects of nicotine and in CPP, which measures reward-related phenomena. The (S,S) analogues of 5a to 5h have greater potency than the reference compounds 2 and (S-S)-4a as inhibitors of DA or NE uptake. All the compounds have higher potency as antagonists of α3β4*-nAChR function than (S-S)-4a, and four of the compounds, 5a, 5c, 5e, and 5f, are more potent than 2. A comparison of the effects of aromatic substituents on 5a–5h shows a rank order potency at α3β4*-nAChR of alkyl-bromo > -chloro > -fluoro (5c > 5a > 5b). N-Alkylation of (S,S)-5a provided the N-methyl, N-ethyl, and N-propyl analogues 5d–5f. Whereas the N-methyl analogue 5d had about equal potencies at all the in vitro assays tested, the N-ethyl and N-propyl analogues 5e and 5f, respectively, had significantly higher potency for DA and NE uptake inhibition as well as antagonism of the α3β4*-nAChR. The carbon-3 extended 3-ethyl and 3-propyl analogues 5g and 5h, respectively, both had significantly higher DA and NE uptake inhibition relative to (S,S)-5a. Analogue 5g had slightly lower α3β4*-nAChR antagonist potency relative to (S,S)-5e, whereas 5h had about the same α3β4*-nAChR antagonist potency as (S,S)-5a. With AD50 values of 0.017 to 0.13 mg/kg, all N-substituted and carbon-3 extended chain analogues 5d–5h are potent antagonists of nicotine-induced antinociception in the tail-flick test. With AD50 values of 0.018 and 0.017 mg/kg, the extended chain analogues 5g and 5h have the highest potency in the tail-flick test. Importantly, the two analogues selected for further study based in part on in vitro profiling results, N-ethyl and N-propyl derivatives 5e and 5f with AD50 values of 0.025 and 0.03 mg/kg, respectively, have better potency as antagonists of nicotine-induced CPP than 2 and (S,S)-4a, which have AD50 values of 0.35 and 0.1 mg/kg, respectively. The antagonist activity of 2 in this assay is consistent with its ability to promote smoking cessation, probably via its hydroxymetabolite.
In our previous studies,8,11–13 we succeeded in generating analogues with reasonably higher inhibitory potency than 2 or either of its hydroxymetabolite isomers (R,R)-4a and (S,S)-4a in both the in vitro and in vivo assays used in this study, whether on the 2, hydroxybupropion, or 3-phenyltropane backbones.8,12,13 The current studies show that several 3,5,5-morpholino analogues 5a–5h also have higher potency than 2, (R,R)-4a and (S,S)-4a in the same test.
Interestingly, (S,S)-5a, which has a hydrogen in place of the 2-hydroxy group in (S,S)-4a, has a higher potency than (S,S)-4a at the targets of interest, DAT, NET, and α3β4*-nAChR, but not at α4β2-nAChR, a subtype that has been implicated in nicotine dependence. Since DA in the nucleus accumbens undoubtedly plays a role in reinforcement and reward, and NE input from the locus coeruleus can gate activity in dopaminergic nuclei and enhance attention, inhibition of reuptake at either could contribute to nicotine’s dependence-related behavioral effects.
Recent genetic studies suggest that α3β4*-nAChR subtypes play an important role in nicotine dependence. Indeed, genetic variants in the nAChR α3/β3/α5 subunit gene cluster are associated with susceptibility to nicotine dependence in humans, perhaps consistent with the presence of α3β4*-nAChR at key portions in the extended reward circuit.15–17
Our earlier studies indicated that conversion of 2 to its (S,S)-hydroxymetabolite (S,S)-4a was associated with an increase in compound potency in inhibition of acute nicotine effects to the same degree that the latter compound also displayed an increase in potency as an inhibitor, and selectivity for α4β2-nAChR.9 This suggested that efforts to increase potency at α4β2-nAChR might lead to discovery of better bupropion-related aids to smoking cessation. It was not clear whether the hydroxy moiety itself, the specific atomic topography of the (S,S)-hydroxymetabolite, or some combination of both, contributed to the increased behavioral and α4β2-nAChR potency of the compound relative to 2.
The current studies indicate that the hydroxy moiety could contribute to enhanced selectivity of bupropion-related compounds for α4β2-nAChR, as its removal from (S,S)-4a in (S,S)-5a correlates with a decline in antagonist potency at α4β2-nAChR. Interestingly, as opposed to the preference of (R,R)-4a for α3β4*-nAChR and of (S,S)-4a for α4β2-nAChR, both isomers (R,R)-and (S,S)-5a are slightly selective for α3β4*- over α4β2-nAChR and have indistinguishable antagonists potencies at α4β2-nAChR, although (S,S)-4a or -5a are both less potent than their (R,R)-equivalents at α3β4*-nAChR. Moreover, all of the other (S,S)-5 analogues have higher potency at α3β4*- than at α4β2-nAChR.
With regard to activity as monoamine uptake inhibitors, higher potency for (S,S) as opposed to (R,R)-4a and -5a is evident at DAT and NET, and there is a substantial increase in potency when either isomeric form is changed from the hydroxyl- to the hydrogen form. Moreover, each of the (S,S)-5a variants has higher DA and NE uptake inhibitory potency except for the N-methyl analogue 5d and the bromo-substituted analogue 5c acting at the NET. The alkyl extension analogues 5g and 5h also have higher potency for 5HT uptake inhibition.
It is possible that good activity in DA and/or NE uptake inhibition and slight adjustments in activity at nAChR may be key to developing a compound with desired, increased efficacy as a smoking cessation aid. However, strong activity in either DA or NE uptake inhibition also might be adequate to decrease nicotine dependence measures. In no case is potency at nAChR greater than that for DA or NE uptake inhibition for analogues 5b–5h. Submicromolar IC50 values for inhibition of DA uptake is a characteristic of the most potent inhibitors of nicotine effects in the tail-flick assay. Comparative differences in activity in the tail-flick assay do not match with comparative differences in inhibitory potency at any single molecular target. The similarities in the abilities of 5e and 5f to block nicotine-induced CPP (AD50 = 0.025 and 0.03 mg/kg, respectively) can be reconciled with the similar activities of those agents at DA and NE uptake inhibition and/or α3β4*-nAChR antagonism, as their 3–4-fold better activity in nicotine-CPP blockade relative to (S,S)-4a, could be attributed to their ~10-fold higher potency at these molecular targets.
In summary, replacement of 2-hydroxyl groups in the (S,S)-hydroxybupropion (4a) with a hydrogen to give (S,S)-5a resulted in increased potency for inhibition of DA and NE uptake, antagonism of α3β4*-nAChR and increased potency for antagonizing nicotine-induced antinociception in the tail-flick test. The N-ethyl and N-propyl analogues, 5e and 5f, respectively, of (S,S)-5a were more potent DA and NE uptake inhibitors as well as antagonists of α3β4*-nAChRs than (S,S)-4a and (S,S)-5a. The aryl fluoro and bromo analogues 5b and 5c, respectively, and carbon-3 extended chain analogues 5g and 5h all have higher DA uptake inhibition potency than (S,S)-4a and (S,S)-5a. All (S,S)-5 analogues are more potent in the tail-flick test than (S,S)-4a. The N-ethyl and N-propyl analogues 5e and 5f, respectively, which have the highest antagonist potency at α3β4*-nAChR as well as high potency for DA and NE uptake inhibition also had better potency than (S,S)-4a in the nicotine-CPP test. Thus, 2 and (S,S)-4a analogues 5a–5h, particularly 5e and 5f represent exciting new lead structures for the development of new pharmacotherapies to treat nicotine addiction (smokers).
Experimental
Nuclear magnetic resonance (1H NMR and 13C NMR) spectra were recorded on a 300 MHz (Bruker AVANCE 300) unless otherwise noted. Chemical shift data for the proton resonances were reported in parts per million (δ) relative to internal (CH3)4Si (δ 0.0). Optical rotations were measured on an AutoPol III polarimeter, purchased from Rudolf Research. Elemental analyses were performed by Atlantic Microlab, Norcross, GA. Purity of compounds (>95%) was established by elemental analyses. Analytical thin-layer chromatography (TLC) was carried out on plates precoated with silica gel GHLF (250 µM thickness). TLC visualization was accomplished with a UV lamp or in an iodine chamber. All moisture-sensitive reactions were performed under a positive pressure of nitrogen maintained by a direct line from a nitrogen source. Anhydrous solvents were purchased from Aldrich Chemical Co.
(S,S)-2-(3'-Chlorophenyl)-3,5,5-trimethylmorpholine [(S,S)-5a] Hemi-d-tartrate
A solution of (S,S)-2-(3'-chlorophenyl)-3,5,5-trimethylmorpholine-2-ol [(S,S)-4a] hemi-d-tartrate (990 mg, 3.00 mmol) in 12 mL of 50% aqueous ethanol was cooled at 0 °C and treated with NaBH4 (450 mg, 12 mmol). The reaction mixture was stirred overnight at room temperature. The reaction mixture was quenched at 0 °C by the slow addition of 4.5 mL of concentrated HCl. The clear solution was basified with saturated aqueous solution of NaCO3 and extracted twice with ethyl acetate. The combined extracts were dried (Na2SO4), filtered, and concentrated under reduced pressure affording 470 mg of crude mixture of diols. The crude reaction mixture was dissolved in 5 mL of CH2Cl2, cooled to 0 °C and treated dropwise with 4 mL of concentrated H2SO4. The mixture was stirred overnight with warming to room temperature. The reaction mixture was added to crushed ice, basified with aqueous solution of sodium carbonate and extracted with ether (twice). The combined extracts were dried (Na2SO4), filtered, and concentrated under reduced pressure. The resulting oil was purified by flash chromatography using CH2Cl2-methanol (10:1) plus 1% ammonium hydroxide as the eluent to afford 300 mg (42%) of (S,S)-5a. 1H NMR (CDCl3) δ 7.38–7.35 (m, 1H), 7.28–7.20 (m, 3H), 3.78 (d, 1H, J = 9.3 Hz), 3.68 (d, 1H, J = 11.0 Hz), 3.34 (d, 1H, J = 11.0 Hz), 3.12–3.01 (m, 1H), 1.39 (s, 3H), 1.07 (s, 3H), 0.82 (d, 3H, J = 6.3 Hz); 13C NMR (CDCl3) δ 142.2, 134.5, 129.8, 128.24, 127.7, 125.9, 86.2, 77.4, 51.1, 49.9, 27.4, 23.6, 18.5.
A sample of the (S,S)-5a was converted to the hemi-d-tartrate salt: mp 209–210 °C; [α]20D +7.6° (c 0.7, CH3OH). 1H NMR (methanol-d4) δ 7.48–7.45 (m, 1H), 7.41–7.35 (m, 3H), 4.37 (s, 1H), 4.29 (d, 1H, J = 10.0 Hz), 3.76 (dd, 2H, J = 30.5, J = 12.3 Hz), 3.58–3.50 (m, 1H), 1.58 (s, 3H), 1.35 (s, 3H), 1.04 (d, 3H, J = 6.5 Hz); 13C NMR (methanol-d4) δ 177.6, 140.8, 135.6, 131.3, 130.2, 128.7, 127.3, 83.5, 74.9, 74.4, 55.1, 52.2, 23.8, 21.2, 15.4; MS (ESI) m/z 240.2 [(M−tartrate)+; M = C13H18ClNO • 0.5 C4H6O6]. Anal. (C15H21ClNO4 • 0.25 H2O) C, H, N.
(R,R)-2-(3'-Chlorophenyl)-3,5,5-trimethylmorpholine [(R,R)-5a] Hemi-l-tartrate
A procedure similar to the one reported for (S,S)-2-(3'-chlorophenyl)-3,5,5-trimethylmorpholine (S,S)-5a was used. A sample of (R,R)-2-(3'-chlorophenyl)-3,5,5-trimethylmorpholine-2-ol [(R,R)-4a] hemi-d-tartrate (660 mg, 2.00 mmol) in 8 mL of 50% aqueous ethanol was treated with NaBH4 (300 mg, 8.00 mmol) to give 540 mg of a crude mixture of diols 6a. A solution of the crude sample in CH2Cl2 (6 mL) was treated with 3 mL of concentrated H2SO4 to afford 364 mg (76% yield) of (2R,3R)-5a. 1H NMR (CDCl3) δ 7.38–7.36 (m, 1H), 7.29–7.20 (m, 3H), 3.77 (d, 1H, J = 9.3 Hz), 3.69 (d, 1H, J = 9.0 Hz), 3.34 (d, 1H, J = 12.0 Hz), 3.11–3.02 (m, 1H), 1.43 (s, 3H), 1.07 (s, 3H), 0.81 (d, 3H, J = 6.0 Hz); 13C NMR (CDCl3) δ 142.2, 134.4, 129.7, 128.3, 127.7, 126.0, 86.2, 77.5, 51.1, 49.7, 27.4, 23.6, 18.5.
A sample of the (R,R)-5a was converted to the hemi-l-tartrate salt: mp 210–211 °C; [α]20D - 10.2° (c 0.5, CH3OH). 1H NMR (methanol-d4) δ 7.47–7.43 (m, 1H), 7.40–7.31 (m, 3H), 4.35 (s, 1H), 4.26 (d, 1H, J = 10.2 Hz), 3.74 (dd, 2H, J = 32.1, J = 12.2 Hz), 3.57–3.41 (m, 1H), 1.56 (s, 3H), 1.32 (s, 3H), 1.02 (d, 3H, J = 6.6 Hz); 13C NMR (methanol-d4) δ 178.1, 141.2, 135.6, 131.2, 130.1, 128.7, 127.3, 83.8, 75.2, 74.7, 54.6, 52.2, 24.1, 21.4, 15.6; MS (ESI) m/z 240.1 [(M−tartrate)+; M = C13H18ClNO • 0.5 C4H6O6]. Anal. (C15H21ClNO4) C, H, N.
(S,S)-2-(3’-Fluorophenyl)-3,5,5-trimethylmorpholine (5b) Hemi-d-tartrate
A solution of (S,S)-2-(3'-fluorophenyl)-3,5,5-trimethylmorpholine (4b) hemi-d-tartrate (220 mg, 0.700 mmol) in 4 mL EtOH/H2O (1:1) was cooled at 0 °C and treated with NaBH4 (106 mg, 2.80 mmol). The reaction mixture was stirred at room temperature overnight. After cooling the reaction mixture at 0 °C, 1 mL of HCl 1.6 M solution in EtOH was added slowly to the reaction vessel, and the mixture was allowed to warm to room temperature. Ether and NaHCO3 saturated aqueous solution were added to the reaction vessel, and the organic layer was separated. The aqueous phase was extracted with ether (three times). The combined organic extracts were washed (water, brine), dried (Na2SO4), and concentrated to give 6b as a white solid 124 mg (74% yield). 1H NMR (CDCl3) δ 7.33–7.27 (m, 1H), 7.11–7.04 (m, 2H), 6.98–6.89 (m, 1H), 4.58 (d, 1H, J = 4.0 Hz), 3.37 (dd, 2H, J = 26.2, J = 10.7 Hz), 3.13–3.02 (m, 1H), 1.12 (s, 3H), 1.10 (s, 3H), 0.85 (d, 3H, J = 6.7 Hz); 13C NMR (CDCl3) δ 129.4 (d), 121.9 (d), 114.1, 113.8, 113.5, 113.2, 75.5, 69.7, 54.3, 51.5, 25.2, 24.6, 18.2; MS (ESI) m/z 242.3 [(M + H)+, M = C13H18FNO2].
A solution of crude diol 6b (110 mg, 0.455 mmol) in CH2Cl2 (2 mL) was cooled at 0 °C and treated with 1 mL concentrated H2SO4. The reaction mixture was stirred at room temperature overnight, then poured into a flask with crushed ice. The mixture was neutralized with NaHCO3 saturated aqueous solution, followed by extraction with ether (three times). The organic layers were separated, combined, washed (water, brine), separated, dried (Na2SO4), and concentrated to a white solid 58 mg (57% yield). 1H NMR (CDCl3) δ 7.34–7.28 (m, 1H), 7.14–6.97 (m, 3H), 3.78 (d, 1H, J = 9.2 Hz), 3.70 (d, 1H, J = 11.0 Hz), 3.34 (d, 1H, J = 11.0 Hz), 3.10–3.01 (m, 1H), 1.39 (s, 3H), 1.08 (s, 3H), 0.82 (d, 3H, J = 6.3 Hz); 13C NMR (CDCl3) δ 164.6, 142.7, 129.9 (d), 123.3 (d), 115.1 (d), 114.3 (d), 86.2, 77.4, 51.1, 49.2, 27.4, 23.5, 18.5; MS (ESI) m/z 222.4 [(M − H)+ M = C13H18FNO].
A sample of free base (54 mg, 0.24 mmol) in 2 mL ether was treated with a solution of d-tartaric acid (18 mg, 0.12 mmol) in MeOH (1 mL) to give 61 mg (85% yield) of 5b • tartrate as a white solid: mp 167–168 °C; [α]20D +9.1° (c 0.9, CH3OH). 1H NMR (methanol-d4) δ 7.45–7.36 (m, 1H), 7.25–7.09 (m, 3H), 4.36 (s, 1H), 4.28 (d, 1H, J = 10.0 Hz), 3.74 (dd, 2H, J = 30.4, J = 12.0 Hz), 3.52–3.43 (m, 1H), 1.56 (s, 3H), 1.32 (s, 3H), 1.02 (d, 3H, J = 6.5 Hz); 13C NMR (CD3OD) δ 165.9, 141.6, 131.5 (d), 124.7 (d), 116.7 (d) 115.4 (d), 83.8, 75.2, 74.7, 54.7, 52.2, 24.1, 21.4, 15.6; MS (ESI) m/z 224.3 [(M − tartrate)+, M = C13H18FNO • 0.5 C4H6O6]. Anal. (C15H21FNO4 • 0.25 H2O) C, H, N.
(S,S)-2-(3'-Bromophenyl)-3,5,5-trimethylmorpholine (5c) Hemi-d-tartrate
A procedure similar to the one reported for (S,S)-2-(3'-chlorophenyl)-3,5,5-trimethylmorpholine (S,S)-5a was used to synthesize 5c. A solution of (S,S)-2-(3'-bromophenyl)-3,5,5-trimethylmorpholine-2-ol (4c) d-tartrate (265 mg, 0.710 mmol) in 4 mL of 50% aqueous ethanol was treated with NaBH4 (107 mg, 2.83 mmol) to give 215 mg of a crude mixture of diols. The crude reaction mixture was dissolved in CH2Cl2 (4 mL) and treated with 2 mL of concentrated H2SO4 to afford 150 mg (74%) of (S,S)-5c. 1H NMR (CDCl3) δ 7.53–7.50 (m, 1H), 7.44–7.40 (m, 1H), 7.25–7.17 (m, 2H), 3.75 (d, 1H, J = 9.3 Hz), 3.69 (d, 1H, J = 11.1 Hz), 3.33 (d, 1H, J = 11.4 Hz), 3.11–3.01 (m, 1H), 1.39 (s, 3H), 1.07 (s, 3H), 0.81 (d, 3H, J = 6.3 Hz); 13C NMR (CDCl3) δ 142.5, 131.3, 130.5, 130.0, 126.4, 122.7, 86.1, 77.4, 50.8, 49.8, 27.4, 23.6, 18.6.
A sample of the free base was converted to the title compound: mp 212–213 °C; [α]20D +7.6° (c 0.63, CH3OH). 1H NMR (methanol-d4) δ 7.59–7.51 (m, 1H), 7.39–7.25 (m, 3H), 4.35 (s, 1H), 4.26 (d, 1H, J = 10.0 Hz), 3.79 (d, 1H, J = 12.2 Hz), 3.68 (d, 1H, J = 12.2 Hz), 3.51–3.45 (m, 1H), 1.56 (s, 3H), 1.32 (s, 3H), 1,02 (d, 3H, J = 6.6 Hz); 13C NMR (methanol-d4) δ 177.8, 141.5, 133.0, 131.5 (d), 127.7, 123.5, 84.0, 75.4, 74.6, 54.3, 52.2, 24.3, 21.5, 15.8; MS (ESI) m/z 284.7 [(M − tartrate)+; M = C13H18BrNO • 0.5 C4H6O6]. Anal. (C15H21BrNO4) C, H, N.
(S,S)-2-(3'-Chlorophenyl)-3,4,5,5-tetramethylmorpholine (5d) Hydrochloride
A sample of (S,S)-2-(3'-chlorophenyl)-3,5,5-trimethylmorpholine (5a) (60 mg, 0.25 mmol) and potassium carbonate (104 mg, 0.750 mmol) in 1.5 mL of DMF were charged in a sealed flask apparatus and treated with CH3I (19 µL, 0.30 mmol). The reaction vessel was sealed and stirred overnight at 70 °C. The reaction mixture was cooled to room temperature, diluted with water, and extracted twice with ether. The combined extracts were dried (Na2SO4), filtered, and concentrated under reduced pressure. The resulting oil was purified by column chromatography using CH2Cl2-methanol (30:1) as eluent, afforded 45 mg (71%) of 5d. 1H NMR (CDCl3) δ 7.38–7.36 (m, 1H), 7.25–7.21 (m, 3H), 4.04 (d, 1H, J = 9.6 Hz), 3.54 (q, 1H, J = 11.1 Hz), 2.62–2.53 (m, 1H), 2.25 (s, 3H), 1.19 (s, 3H), 1.07 (s, 3H), 0.83 (d, 3H, J = 6.3 Hz); 13C NMR (CDCl3) δ 142.6, 134.3, 129.5, 128.29, 128.04, 126.4, 85.5, 78.1, 57.3, 34.2, 25.0, 15.7, 14.1.
A sample of 5d was converted to the hydrochloride salt: mp 212–213 °C; [α]20D +51.9° (c 0.75, CH3OH); MS (ESI) m/z 254.6 [(M − HCl)+; M = C14H20ClNO • HCl]. Anal. (C14H21Cl2NO) C, H, N.
(S,S)-2-(3'-Chlorophenyl)-4-ethyl-3,5,5-trimethylmorpholine (5e) Di-p-Toluoyl-l-tartrate
A sample of (S,S)-2-(3'-chlorophenyl)-3,5,5-trimethylmorpholine (5a) (320 mg, 1.33 mmol) was dissolved in 5 mL of dichloroethane and treated with NaBH(OAc)3 (117 mg, 2.66 mmol) and an excess amount of acetaldehyde. The reaction mixture was stirred at room temperature overnight. The reaction was quenched with aqueous solution of sodium carbonate and extracted with ether. The combined organic layers were dried (Na2SO4), filtered, and concentrated. The crude product was purified by column chromatography on silica gel using cyclohexane-ethyl acetate (5:1) with 1% NH4OH as the eluent to give 150 mg (42%) of (S,S)-5e as colorless oil. 1H-NMR (CDCl3) δ 7.36 (s, 1H), 7.25 (m, 3H), 4.00 (d, 1H), 3.50 (dd, 2H), 2.72 (m, 2H), 2.27 (m, 1H), 1.22 (s, 3H), 1.05 (t, 3H), 1.04 (s, 3H), 0.82 (d, 3H). 13C-NMR (CDCl3) δ 142.9, 134.5, 129.8, 128.6, 128.4, 126.7, 86.2, 78.5, 57.5, 54.6, 42.6, 25.4, 19.3, 17.1, 16.4. m/z 268.0 [(M+H)+. M = C15H22ClNO]
A sample of the 5e was converted to the di-p-toluoyl-L-tartrate salt: mp 165–166 °C; [α]20D -81.4° (c 0.56, CH3OH). Anal. (C35H40ClNO9) C, H, N.
(S,S)-2-(3'-Chlorophenyl)-3,5,5-trimethyl-4-propylmorpholine (5f) Di-p-toluoyl-L-tartrate
Compound 5f was prepared in the same fashion as 5e, using (S,S)-2-(3'-chlorophenyl)-3,5,5-trimethylmorpholine (5a) (320 mg, 1.33 mmol) in 5 mL of dichloroethane and was treated with NaBH(OAc)3 (790 mg, 3.74 mmol) and an excess amount of propionaldehyde to afford 220 mg (75%) of 5f. 1H-NMR (CDCl3) δ 7.36 (s, 1H), 7.23 (m, 3H), 4.00 (d, 1H), 3.50 (dd, 2H), 2.72 (m, 1H), 2.55 (m, 1H), 2.10 (m, 1H), 1.45 (m, 2H), 1.22 (s, 3H), 1.02 (s, 3H), 0.82 (t, 3H), 0.79 (d, 3H); 13C-NMR (CDCl3) δ 142.9, 134.5, 129.8, 128.5, 128.4, 126.7, 86.1, 78.3, 57.7, 54.4, 51.2, 27.2, 25.5, 17.2, 16.1, 11.9. m/z 282.6 [(M+H)+, M = C16H24ClNO].
A sample of the 5f was converted to the di-p-toluoyl-L-tartrate salt: mp 144–145 °C; [α]20D - 67.2° (c 0.6, CH3OH). Anal. (C36H42ClNO9) C, H, N.
(S,S)-2-(3'-Chlorophenyl)-3-ethyl-5,5-dimethylmorpholine (5g) Hemi-d-tartrate
A procedure similar to the one described for (S,S)-2-(3'-chlorophenyl)-3,5,5-trimethylmorpholine (S,S)-5a was used to synthesize (S,S)-5g. A sample of (S,S)-2-(3'-chlorophenyl)-3-ethyl-5,5-dimethylmorpholine-2-ol d-tartrate (4g) (100 mg, 0.230 mmol) in 2 mL of 50% aqueous ethanol was treated with NaBH4 (45 mg, 1.2 mmol) to give 74 mg of a crude mixture of diols 6g. A solution of the crude sample in 2 mL of CH2Cl2 was treated with 1 mL of concentrated sulfuric acid to afford 54 mg (98%) of (S,S)-5g. The free base was converted to its hemi-d-tartrate salt by dissolving the free base on methanol and adding 16.9 mg (0.5 equivalent) of d-tartaric acid dissolved in methanol: mp 203–204 °C; [α]20D-4.2° (c 0.5, CH3OH). 1H-NMR (CD3OD) δ 7.47 (s, 1H), 7.39 (s, 3H), 4.36 (s, 1H), 4.27 (d, 1H), 3.76 (d, 1H), 3.66 (d, 1H), 1.56 (s, 3H), 1.40 (m, 2H), 1.32 (s, 3H), 0.75 (t, 3H); 13C NMR (CD3OD) δ 178.2, 141.8, 136.0, 131.7, 130.5, 129.3, 127.9, 83.7, 75.7, 74.9, 58.2, 55.0, 24.5, 21.9, 10.7. m/z 254.0 [(M−tartrate)+. M = C16H23ClNO4] Anal. (C16H23ClNO4 • 0.25 H2O) C, H, N.
(S,S)-2-(3'-Chlorophenyl)-5,5-dimethyl-3-propylmorpholine (5h) Hemi-d-tartrate
A procedure similar to the one reported for (S,S)-2-(3'-chlorophenyl)-3,5,5-trimethylmorpholine (S,S)-5a was used to synthesize 5h. Treatment of (S,S)-2-(3'-chlorophenyl)-5,5-dimethyl-3-propylmorpholine-2-ol (4h) hemi-d-tartrate (360 mg,1.00 mmol) in 6 mL of 50% aqueous ethanol with NaBH4 (151 mg, 4.00 mmol) afforded 295 mg of a crude mixture of diols. The crude sample was dissolved in 4 mL of CH2Cl2 and treated with 2 mL of concentrated H2SO4 to give 250 mg (98%) of 5h. Compound 5h was converted to its hemi-d-tartrate salt: mp 232–233 °C; [α]20D-19.0° (c 1.1, CH3OH). 1H NMR (CD3OD) δ 7.47 (s, 1H), 7.39 (m, 3H), 4.36 (s, 1H), 4.30 (d, 1H), 3.78–3.66 (m, 2H), 3.37–3.29 (m, 2H), 1.57 (s, 3H), 1.33 (s, 3H), 1.26–1.40 (m, 1H), 0.94–0.92 (m, 1H), 0.74 (t, 3H); 13C NMR (CD3OD) δ 178.4, 141.8, 136.0, 131.7, 130.5, 129.3, 83.7, 75.5, 75.1, 56.6, 55.1, 33.6, 24.4, 21.8, 20.1, 14.4; m/z 268.0 [(M−tartrate)+. M = C17H25ClNO4)] Anal. (C17H25ClNO4) C, H, N.
Cell lines and culture
Human embryonic kidney (HEK-293) cells stably expressing human DAT, NET, or SERT were maintained as previously described.18 Several human cell lines that naturally or heterologously express specific, functional, human nAChR subtypes also were used as described earlier.8,12
Transporter Assays
The abilities of compounds to inhibit uptake of [3H]DA, [3H]5HT, or [3H]NE by the respective, human transporters were evaluated using the appropriate HEK-293 cell line as previously reported.18
nAChR Functional Assays
86Rubidium ion efflux assays were used as previously described8,12 to characterize functional effects of analogues.
Behaviorial Assays
All animal experiments were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals and Institutional Animal Care and Use Committee guidelines. Male Institute of Cancer Research (ICR) mice (weighing 20–25 g) obtained from Harlan (Indianapolis, IN) were used to test effects of analogues on acute actions of nicotine (tail-flick, hot-plate, locomotor and body temperature studies) as previously described.8,12 Conditioned place preference (CPP) assays also were conducted as specified earlier respectively) or 3-phenyltropanes-related compounds.8,11
Supplementary Material

Acknowledgment
This work was supported by National Institutes of Health National Cooperative Drug Discovery Group grant U19 DA019377.
Abbreviations
- NRT
nicotine replacement therapy
- DA
dopamine
- 5HT
serotonin
- NE
norepinephrine
- HEK
human embryonic kidney
- DAT
dopamine transporter
- SERT
serotonin transporter
- NET
norepinephrine transporter
- nAChR
nicotine acetylcholine receptor(s)
- VTA
ventral tegmental area
- MPE
maximum possible effect
- CPP
conditioned place preference
Footnotes
Supporting Information Available: Elemental analysis data. This material is available free of charge via the Internet at http://pubs.acs.org.
Contributor Information
Bruce E. Blough, Email: beb@rti.org.
Hernán A. Navarro, Email: han@rti.org.
S. Wayne Mascarella, Email: swm@rti.org.
M. Imad Damaj, Email: mdamaj@vcu.edu.
Ronald J. Lukas, Email: Ron.Lukas@CHW.EDU.
References
- 1.Benowitz NL. Nicotine addiction. Prim. Care. 1999;26:611–631. doi: 10.1016/s0095-4543(05)70120-2. [DOI] [PubMed] [Google Scholar]
- 2.Benowitz NL. Clinical pharmacology of nicotine: implications for understanding, preventing, and treating tobacco addiction. Clin. Pharmacol. Ther. 2008;83:531–541. doi: 10.1038/clpt.2008.3. [DOI] [PubMed] [Google Scholar]
- 3.Hughes JR, Higgins ST, Bickel WK. Nicotine withdrawal versus other drug withdrawal syndromes: similarities and dissimilarities. Addiction. 1994;89:1461–1470. doi: 10.1111/j.1360-0443.1994.tb03744.x. [DOI] [PubMed] [Google Scholar]
- 4.Tutka P, Mosiewicz J, Wielosz M. Pharmacokinetics and metabolism of nicotine. Pharmacol. Rep. 2005;57:143–153. [PubMed] [Google Scholar]
- 5.West R, McNeill A, Raw M. Smoking cessation guidelines for health professionals: an update. Health Education Authority. Thorax. 2000;55:987–999. doi: 10.1136/thorax.55.12.987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tutka P. Nicotinic receptor partial agonists as novel compounds for the treatment of smoking cessation. Expert Opin. Investig. Drugs. 2008;17:1473–1485. doi: 10.1517/13543784.17.10.1473. [DOI] [PubMed] [Google Scholar]
- 7.Rollema H, Coe JW, Chambers LK, Hurst RS, Stahl SM, Williams KE. Rationale, pharmacology and clinical efficacy of partial agonists of alpha4beta2 nACh receptors for smoking cessation. Trends Pharmacol. Sci. 2007;28:316–325. doi: 10.1016/j.tips.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 8.Carroll FI, Blough BE, Mascarella SW, Navarro HA, Eaton JB, Lukas RJ, Damaj MI. Synthesis and Biological Evaluation of Bupropion Analogues as Potential Pharmacotherapies for Smoking Cessation. J. Med. Chem. 2010;53:2204–2214. doi: 10.1021/jm9017465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Damaj MI, Carroll FI, Eaton JB, Navarro HA, Blough BE, Mirza S, Lukas RJ, Martin BR. Enantioselective effects of hydroxy metabolites of bupropion on behavior and on function of monoamine transporters and nicotinic receptors. Mol. Pharmacol. 2004;66:675–682. doi: 10.1124/mol.104.001313. [DOI] [PubMed] [Google Scholar]
- 10.Slemmer JE, Martin BR, Damaj MI. Bupropion is a nicotinic antagonist. J. Pharmacol. Exp. Ther. 2000;295:321–327. [PubMed] [Google Scholar]
- 11.Damaj MI, Grabus SD, Navarro HA, Vann RE, Warner JA, King LS, Wiley JL, Blough BE, Lukas RJ, Carroll FI. Effects of hydroxymetabolites of bupropion on nicotine dependence behavior in mice. J. Pharmacol. Exp. Ther. 2010 doi: 10.1124/jpet.110.166850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lukas RJ, Muresan AZ, Damaj MI, Blough BE, Huang X, Navarro HA, Mascarella SW, Eaton JB, Marxer-Miller SK, Carroll FI. Synthesis and Characterization of In Vitro and In Vivo Profiles of Hydroxybupropion Analogues: Aids to Smoking Cessation. J. Med. Chem. 2010;53:4731–4748. doi: 10.1021/jm1003232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carroll FI, Blough BE, Mascarella SW, Navarro HA, Eaton JB, Lukas RJ, Damaj I. Nicotinic Acetylcholine Receptor Efficacy and Pharmacological Properties of 3-(Substituted phenyl)-2β-substituted Tropanes. J. Med. Chem. 2010 doi: 10.1021/jm100994w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clarke FH. cis- and trans-3-Methyl-2-phenylmorpholine. J. Org. Chem. 1962;27:3251–3253. [Google Scholar]
- 15.Bierut LJ, Stitzel JA, Wang JC, Hinrichs AL, Grucza RA, Xuei X, Saccone NL, Saccone SF, Bertelsen S, Fox L, Horton WJ, Breslau N, Budde J, Cloninger CR, Dick DM, Foroud T, Hatsukami D, Hesselbrock V, Johnson EO, Kramer J, Kuperman S, Madden PA, Mayo K, Nurnberger J, Jr, Pomerleau O, Porjesz B, Reyes O, Schuckit M, Swan G, Tischfield JA, Edenberg HJ, Rice JP, Goate AM. Variants in nicotinic receptors and risk for nicotine dependence. Am. J. Psychiatry. 2008;165:1163–1171. doi: 10.1176/appi.ajp.2008.07111711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saccone SF, Hinrichs AL, Saccone NL, Chase GA, Konvicka K, Madden PA, Breslau N, Johnson EO, Hatsukami D, Pomerleau O, Swan GE, Goate AM, Rutter J, Bertelsen S, Fox L, Fugman D, Martin NG, Montgomery GW, Wang JC, Ballinger DG, Rice JP, Bierut LJ. Cholinergic nicotinic receptor genes implicated in a nicotine dependence association study targeting 348 candidate genes with 3713 SNPs. Hum Mol Genet. 2007;16:36–49. doi: 10.1093/hmg/ddl438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Whitten L. Studies link family of genes to nicotine addiction. NIDA Notes. 2009;22:10–14. [Google Scholar]
- 18.Eshleman AJ, Carmolli M, Cumbay M, Martens CR, Neve KA, Janowsky A. Characteristics of drug interactions with recombinant biogenic amine transporters expressed in the same cell type. J. Pharmacol. Exp. Ther. 1999;289:877–885. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.