

Abstract
Many tumor cells produce nitric oxide (NO) as an anti-apoptotic/pro-growth molecule which also promotes antiogenesis and tumor expansion. This study was designed to examine possible antagonistic effects of endogenous NO on tumor eradication by photodynamic therapy (PDT). Using COH-BR1 breast cancer cells sensitized in mitochondria with 5-aminolevulinic acid (ALA)-generated protoporphyrin IX as a model for ALA-based PDT, we found that caspase-9 activation and apoptotic death following irradiation were strongly enhanced by 1400W, an inhibitor of inducible nitric oxide synthase (iNOS). RT-PCR and Western analyses revealed a substantial upregulation of both iNOS mRNA and protein, beginning ~4 h after irradiation and persisting at least 20 h. Accompanying this was a strong 1400W-inhibitable increase in intracellular NO, as detected with the NO probe, DAF-2-DA. Short hairpin RNA-based iNOS knockdown in COH-BR1 cells dramatically reduced NO production under photostress while enhancing caspase-9 activation and apoptosis. These findings suggest that cytoprotective iNOS/NO induction in PDT-treated tumor cells could reduce treatment efficacy, and point to pharmacologic intervention with iNOS inhibitors for counteracting this.
INTRODUCTION
Photodynamic action is a unique type of oxidative process requiring the following components: (i) molecular oxygen; (ii) a light-absorbing dye or pigment acting as a sensitizing agent; and (iii) sensitizer-exciting light, typically in the visible wavelength range, but in some cases extending to the near ultraviolet or near infrared [1,2]. While this process often results in pathologic effects such as UVA-induced skin aging and cancer, it can also be exploited for beneficial effects, a prime example being antitumor photodynamic therapy (PDT) [3,4]. Clinical PDT typically involves systemic or topical administration of a tumor-localizing sensitizer or metabolic precursor, followed by laser source irradiation, which is restricted to the tumor area [4]. Tumor targeting of both sensitizer and exciting light make this approach highly site-specific. In addition, the sensitizer is usually innocuous until photoactivated, whereupon it gives rise to signaling and cytotoxic reactive oxygen species (ROS), singlet molecular oxygen (1O2) being one of the most prominent [3,4]. In 5-aminolevulinic acid (ALA)-based PDT, administered ALA or an ester thereof enters tumor cells and is metabolized to the active sensitizer, protoporphyrin IX (PpIX), via the heme biosynthetic pathway [5,6]. PpIX accumulates initially in mitochondria, reaching a level that is inversely proportional to the concentration of available iron used in its ferrochelatase-catalyzed conversion to heme, a non-sensitizer [7,8]. Although ALA or esterified ALA can be introduced systemically for PDT, topical application for skin cancers such as T-cell lymphoma and squamous cell carcinoma is both convenient and highly effective [6,9].
Nitric oxide produced naturally by nitric oxide synthase (NOS) enzymes plays a role in many different physiological processes, including vasodilation, neurotransmission, and antimicrobial action [10]. When generated at high rates, e.g. by activated neutrophils, NO can have prooxidant cytotoxic effects, whereas in low fluxes it can be cytoprotective, in some instances by acting as a free radical-scavenging antioxidant [11]. Many tumors produce low constitutive levels of NO, which has been reported to promote angiogenesis and tumor growth while inhibiting stress-induced apoptosis, e.g. that caused by ionizing radiation or chemotherapeutic agents [12-15]. The question of how tumor NO might influence PDT efficacy was first addressed 10-12 years ago in studies involving Photofrin-based PDT for various mouse tumors [16-18]. It was found that tumor eradication could be substantially improved by administering NOS inhibitors (L-NAME, L-NNA) and that the extent of this correlated with constitutive NO production, tumors with relatively high output responding best to the inhibitors [18]. The effects were attributed to suppression of NO’s vasodilatory action working in opposition to PDT-induced vasoconstriction [18]. However, important issues such as the following were not addressed: (i) whether the NO derived from tumor cells per se, tumor vasculature cells, or both; (ii) whether the NOS/NO involved was constitutive or stress-induced; and (iii) which of the three known NOS isoforms (neuronal, inducible, or endothelial) played the most important role in any given tumor type. Little attention had been given to these and related questions until we showed recently, using two breast tumor lines as ALA-PDT models [19], that inducible NOS (iNOS) was rapidly and persistently upregulated in photostressed cells and that the resulting NO suppressed apoptotic killing in iNOS inhibitor-reversible fashion. In the present study, we have extended these findings and have confirmed the role of stress-induced iNOS using an iNOS knock-down approach. Our observations are novel in terms of tumor cell response to photodynamic action. They suggest that tumor cells subjected to PDT may upregulate endogenous NOS/NO as a cytoprotective strategy and that this could compromise treatment efficacy.
MATERIALS AND METHODS
General materials
Sigma-Aldrich (St. Louis, MO) supplied the ALA, Hoechst 33258 (Ho), propidium iodide (PI), rhodamine 123 (Rh123), Dulbecco’s modified Eagle’s/Ham’s Nutrient F12 (DME/F12) medium, and fetal bovine serum. Pierce Chemical Co. (Rockford, IL) supplied the reagents for the bicinchoninic (BCA) protein assay. OptiME medium and LipofectAMINE-2000 were obtained from Invitrogen (Carlsbad, CA). Reagents for the terminal deoxynucleotidyl transferase dUTP nick end-lebeling (TUNEL) assay were obtained from R&D Systems (Minneapolis, MN). The Complete-Mini mixture of protease inhibitors was from Boehringer Mannheim (Indianapolis, IN). Calbiochem (Gibbstown, NJ) supplied the N-acetyl-Leu-Glu-His-Asp-7-amido-4-trifluoromethyl-coumarin (Ac-LEHD-AFC), EMD Biosciences (San Diego, CA) the DAF-2DA, and Cayman Chemicals (Ann Arbor, MI) the spermine NONOate (SPNO) and 1400W. The polyclonal antibody against human iNOS and monoclonal antibody against human β-actin were from Santa Cruz Biotechnology (Santa Cruz, CA). Horseradish peroxidase-conjugated IgG secondary antibodies were from Cell Signaling Technology (Danvers, MA). SureSilencing™ plasmids encoded for neomycin resistance and short hairpin RNA (shRNA) directed against human iNOS, along with a plasmid bearing scrambled shRNA as a negative control, were obtained from SABiosciences (Frederick, MD). The plasmids were expanded in E. coli according to supplier recommendations.
Cell culture conditions
Wild-type human COH-BR1 cells, a breast tumor epithelial sub-line originally obtained from Dr. J. Doroshow (City of Hope Cancer Center, Duarte, CA) [20], were grown under standard culture conditions, using DME/F12 medium containing 10% serum, penicillin (100 units/ml), and streptomycin (100 μg/ml). All experiments were carried out with cells that had been passaged fewer than 10-times. Other details were as described previously [19].
iNOS knockdown procedure
COH-BR1 cells at 30–35% confluency in OptiME medium were transfected with 2.5 μg of iNOS shRNA-bearing plasmid or negative control plasmid, using LipofectAMINE-2000 as the permeabilizing agent. Of the four SABiosciences test plasmids with iNOS shRNA inserts, one (T2) with sequence: AGGCTCAAATCTCGGCAGAAT was found to produce the greatest diminution of iNOS mRNA. Thus, all of the knockdown experiments described were carried out with a clone generated using this vector. The control shRNA insert had the following scrambled sequence: TTCGGGAACTGGAGCTAAAGT. After an incubation period of 24 h, transfected cells were subcultured into 10-cm dishes. Forty-eight hours later, they were given fresh medium containing Geneticin (G418, 0.4 mg/ml) and maintained in this until colonies had formed. After single colony selection and proliferation, cells were subjected to RT-PCR and Western analysis for identifying positive knockdown clones and determining the extent of knockdown.
Cell sensitization, treatment with NOS inhibitors, and irradiation
COH-BR1 cells were metabolically sensitized with PpIX by treating with ALA (6); an empirical procedure was used [21,22] whereby most of the PpIX was located in mitochondria (where it originates [6]) when cells were irradiated. Briefly, cells at 60-65% confluence on coverslips in 35-mm culture dishes were incubated with 1.0 mM ALA in serum-free DME/F12 medium for 45 min in the dark. Since serum can act as a PpIX “sink”, omitting it assured that most of the porphyrin remained intracellular. Where indicated, a NOS inhibitor was introduced 15 min before ALA. After sensitization, cells were switched to fresh medium without ALA that either lacked or contained a NOS inhibitor, or inhibitor plus exogenous NO donor. Immediately thereafter, cell dishes were irradiated on a translucent plastic platform, using broad-band visible light at a fluence rate of ~11 W/m2; an irradiation time of 15 min corresponded to a delivered light fluence of ~1 J/cm2. The medium was then replaced with 1% serum-containing DME/F12 (without or with NOS inhibitor or NO donor, as specified) and cells were returned to the incubator for various time periods, after which parameters such as caspase activity, NOS mRNA or protein level, intracellular NO level, and apoptotic vs. necrotic cell death were determined.
Confocal microscopy
Subcellular location of PpIX prior to irradiation was assessed by confocal fluorescence microscopy. Immediately after incubation with ALA, cells were treated with the mitochondrial probe rhodamine 123 (Rh123), ~1 μg/ml for 10 min. After washing with PBS, the cells were overlaid with a dilute solution of Na2S2O4 in nitrogen-sparged PBS (to prevent porphyrin degradation during viewing) and analyzed by confocal microscopy using a Leica TCS/SCP2 instrument (Leica Microsystems, Knowlhill, UK). PpIX and Rh123 were photoexcited at 488 nm, the former being detected by its fluorescence emission in the 620-650 nm range, and the latter by its emission in the 520-580 nm range. Fluorescence intensities were quantified using a Metamorph software program.
Quantitative RT-PCR analysis
For determining iNOS mRNA expression level, total RNA from ALA/light treated cells was isolated at various post-irradiation times up to 20 h, using an RNeasy kit from Quigen Inc. (Valencia, CA) according to supplier instructions. Three microgram of total RNA was employed for the synthesis of cDNA, using an RT2 First Strand Kit (SABiosciences Corp., Frederick, MD). Real-time PCR was performed using the SABiosciences RT2 qPCR Master Mix (SABiosciences Corp.) following a protocol provided by the supplier. All quantitative gene expression by real-time RT-PCR was evaluated using the comparative threshold cycle (CT) method [23] and was normalized against endogenous glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The PCR primer sequences used were as follows: iNOS: 5′-AGCCAGAAGCGCTATCACGAAGAT-3′ and 5′-AATGCAGAGCTGGCTCCATCCTTA-3′; GAPDH: 5′-TCCTCACAGTTGCCATGTAGACCC-3′ and 5′-GGTTGAGCACAGGGTACTTTATTG-ATGG-3′.
Detection of NO formation in photostressed cells
Intracellular NO levels were monitored using the diaminofluorescein probe DAF-2DA [24,25], which enters cells and is trapped via hydrolysis to DAF-2. In aerobic systems, NO-derived nitrogen trioxide (N2O3) can nitrosate DAF-2 to give highly fluorescent DAF-2 triazole, which is detected [26,27]. A stock solution of 10 μM DAF-2DA in dimethyl sulfoxide was prepared immediately before experimental use and kept in the dark. COH-BR1 cells grown on cover slips in 30-mm dishes were exposed to a given light fluence, washed with PBS, and overlaid with phenol red- and serum-free DME/F12 medium containing 20 nM DAF-2DA. After 30 min of dark incubation at 37 °C, the cells were washed twice with PBS and affixed to the slide using one drop of Fluoromount G. Immediately thereafter, cells were examined for DAF-2 triazole accumulation, using a Nikon Eclipse 80i fluorescence microscope with FITC filter; excitation was set at 488 nm and emission at 610 nm. Total fluorescence intensity over a field of at least 50 cells was determined using Metamorph software.
Immunoblotting procedure
Western immunoblotting was used for assessing the effects of shRNA treatment or ALA/light stress on iNOS protein level in COH-BR1 cells. Treated cells, along with controls, were recovered by trypsinization, centrifuged, and washed with ice-cold PBS. The cells were suspended in cold pH 7.5 lysis buffer (25 mM HEPES/10% glycerol/0.3 M NaCl/1.5 mM MgCl2/2 mM EDTA/2 mM EGTA/1 mM dithiothreitol/20 mM β-glycerophosphate/1 mM Na3VO4/1% Triton X-100 plus Complete-Mini mixture of protease inhibitors) and homogenized by 5-6 passages through a 22-gauge needle. After centrifugation (8000g, 10 min), the supernatant fraction was analyzed for protein content by BCA assay. For immunoblotting, samples of equal protein content (typically ~120 μg) were separated by SDS-PAGE using 10% acrylamide/bis-acrylamide. Separated proteins were transferred to a polyvinylidene difluoride membrane, followed by exposure to primary antibody (anti-iNOS, anti-β-actin) and secondary antibody (peroxidase-conjugated IgG). A SuperSignal West Pico chemiluminescence detection kit (Thermo Scientific, Rockford, IL) was used for protein visualization. Integration of protein bands was accomplished using LabWorks image analysis software from UVP Systems (Upland, CA).
Assessment of caspase activation and cell death
Activation of caspase-9 in photodynamically stressed cells was monitored using the fluorogenic substrate Ac-LEHD-AFC. At the indicated postirradiation time(s), cells were washed once with ice-cold PBS and lysed by incubating for 30 min in ice-cold 130 mM sodium chloride/10 mM Tris-HCl/10 mM sodium pyrophosphate/10 mM sodium phosphate/10 mM dithiothreitol/1% Triton X-100 (pH 7.5). After centrifugation to remove any undissolved material, each lysate was analyzed for total protein content and caspase-9 activity. For the latter, an aliquot of lysate was incubated in the dark for 2 h at 37 °C with 75 μM Ac-LEHD-AFC in PBS containing 10% glycerol, 2 mM dithiothreitol, and 0.1 mM EDTA (pH 7.5). The fluorescence of liberated AFC was measured with a PTI QM-7SE spectrofluorimeter (London, ON, Canada), using 400 nm excitation and 505 nm emission.
The effects of photodynamic stress on overall cell viability were determined by thiazolyl blue (MTT) assay, as described previously [21]. The extent of apoptotic vs. necrotic cell death was evaluated using the nuclear fluorophores Ho and PI, the former to detect genuine sustained apoptosis and the latter to detect any necrosis, which is not always discernable with Ho alone [28]. At a given time of dark incubation after light exposure (typically 20 h), cells were treated with 5 μM Ho and 50 μM PI for 30 min and then examined, using a Nikon Diaphot-200 inverted fluorescence microscope with appropriate excitation and emission wavelength settings [19,22,28]. For each sample, the nuclei in 4-5 viewing fields of approximately 100 cells each were evaluated. The apoptotic fraction represented the number of cells with shrunken, brightly Ho-stained nuclei divided by total number of Ho-stained cells. For some experiments, the extent of apoptotic cell death was also determined by TUNEL assay, using conditions recommended by the reagent supplier.
Statistical analysis
The two-tailed Student’s t-test was used for determining the significance of perceived differences between experimental values, P <0.05 being considered statistically significant.
RESULTS
PpIX localization in ALA-treated cells
As shown by the confocal micrographs in Fig. 1A, wild type COH-BR1 cells treated with 1 mM ALA for 45 min in serum-free medium exhibited a perinuclear zone of PpIX fluorescence which clearly overlapped the fluorescence zone exhibited by the mitochondrial marker Rh123. Thus, most of the intracellular PpIX generated from ALA under these conditions was localized in mitochondria, and this is where it resided when cells were irradiated.
Figure 1.

Localization of PpIX in ALA-treated cells. Subconfluent wild type (A) and iNOS knockdown (B) COH-BR1 cells were dark-incubated with 1 mM ALA for 45 min in serum-free DME/F12 medium. The cells were then washed, treated with Rh123 (1 μg/ml) for 10 min, and examined by confocal fluorescence microscopy, using 488 nm excitation with 620-650 nm emission for PpIX, and 488 nm excitation with 520-580 nm emission for Rh123. Total PpIX fluorescence relative to Rh123 fluoresence for 5 randomly picked cells in 3 different viewing fields was 619 ± 10 for wild type and 618 ± 15 for iNOS-kd cells; means ± SD (n = 3). Scale bar: 50 μm.
Viability loss of ALA/light-treated cells in the absence vs. presence of NOS inhibitors
Using an MTT assay to monitor the viability status of ALA/light-treated COH-BR1 cells, we found that with increasing light fluence (J/cm2), there was a progressive loss of viability relative to a non-ALA/light control, as assessed 20 h after irradiation (Fig. 2). No significant loss was observed in a light-only or ALA-only (not shown) control, indicating that photoactivation of ALA-derived sensitizer (PpIX) was necessary for photokilling. When ALA/light treatment was carried out in the presence of 1.0 mM L-NAME, a non-specific inhibitor of NOS activity, loss of cell viability was substantially increased, the fluence for 50% loss decreasing from ~2.1 J/cm2 to ~1.4 J/cm2 (Fig. 2). An even greater rate of photokilling was observed when 0.1 mM 1400W, a highly specific iNOS inhibitor, was used, 50% viability loss in this case occurring at ~0.9 J/cm2 (Fig. 2). From these results, we deduced that endogenous NOS-derived NO made a significant contribution to cytoprotection against photokilling.
Figure 2.

Effects of NOS inhibitors on photosensitized loss of cell viability. COH-BR1 cells at ~60% confluency in phenol red- and serum-free DME/F12 medium were sensitized with PpIX by incubation with 1 mM ALA for 45 min in the dark. This was carried out in the absence or presence of L-NAME (1 mM) or 1400W (10 μM), introduced 15 min before ALA. Following sensitization, cells were exposed to increasing light fluences up to 2 J/cm2, and immediately thereafter switched to 1% serum-containing DME/F12 medium with or without L-NAME or 1400W at the indicated concentration. Twenty hours later, viability was determined by MTT assay. A light-only control (no ALA) was analyzed alongside. Plotted data are relative to those for non-treated cells (no ALA, light, or inhibitors); means ± SD of values from three separate experiments are shown; (○) no inhibitor; (▽) L-NAME; (Δ) 1400W; (x) light control.
Effects of NOS inhibitors on apoptotic killing of ALA/light-treated cells
In subsequent experiments, we examined the mechanism of cell killing by photodynamic stress and how this might be affected by NOS inhibitors. As shown in Fig. 3A and 3B, ALA/light-stressed cells died mainly by apoptosis, as assessed by Ho vs. PI staining and TUNEL assay 20 h after light exposure, a fluence of ~1 J/cm2 in this case. A dark control (20 h after ALA only) or light control (20 h after irradiation only) exhibited <5 % apoptosis compared with ~20% and 15% apoptosis (by Ho/PI and TUNEL assay, respectively) in cells exposed to both ALA and light (Fig. 3B). When cells were ALA/light-challenged in the presence of L-NAME, the apoptotic count increased to ~35% by Ho/PI and 25% by TUNEL. The count was even greater when 1400W was used, reaching ~60% (Ho/PI) and 45% (TUNEL) (Fig. 3A and 3B). Neither L-NAME nor 1400W at the concentrations used was cytotoxic in the dark or light. As shown in Fig. 3C, caspase-9 activity of ALA/light-treated cells 2 h after irradiation was ~1.7-fold greater than that of a dark control. This was far in advance of any measurable increases in apoptotic cell count and is consistent with activation of the intrinsic apoptotic pathway [29]. Irradiation in the presence of L-NAME or 1400W resulted in caspase-9 activity that was 3.2-fold and 4.3-fold greater, respectively, than that of the control (Fig. 3C). Thus, both caspase-9 activation and apoptotic count were substantially increased by the NOS inhibitors, 1400W having the greater effect on both parameters. These results further suggested that active iNOS played an important cytoprotective role against photodynamic stress, presumably through the generation of NO.
Figure 3.

Effects of NOS inhibitors on photosensitized caspase-9 activation and apoptotic cell death. COH-BR1 cells were sensitized with ALA-induced PpIX and irradiated (1 J/cm2) in the absence or presence of 1 mM L-NAME or 10 μM 1400W, introduced 1 h before light. (A) Representative fluorescence micrographs of cells treated with Ho/PI or TUNEL reagents 20 h after irradiation under the indicated conditions (N: L-NAME; W: 1400W); scale bar: 200 μm. (B) Integrated fluorescence levels for micrographs such as shown in panel A. Means ± SD of values from three replicate experiments are plotted; gray bars, Ho/PI assay; white bars, TUNEL assay. *P<0.01 compared with corresponding ALA/hν; #P<0.001 compared with corresponding ALA/hν. (C) Caspase-9 activity measured 2 h after irradiation using Ac-LEHD-AFC as substrate. Plotted values are relative to the ALA-only control; *P<0.001 compared with ALA/hν.
Transcriptional and translational expression of iNOS in ALA/light-stressed cells
Using Western analysis, we showed previously that exposure of ALA-treated COH-BR1 cells to a 2 J/cm2 light fluence resulted in a strong, sustained upregulation of iNOS, which became apparent after 2 h of post-irradiation incubation [19]. No neuronal-type enzyme (nNOS, NOS-1) was detected in these cells, either before or after a photochallenge. However, a low constitutive level of endothelial-type enzyme (eNOS, NOS-3) was observed, but this was not upregulated by ALA/light treatment [19]. In the present study, we examined iNOS expression at the transcriptional as well as translational level, using COH-BR1 cells sensitized as described and exposed to a 1 J/cm2 light fluence. As shown in Fig. 4A, there was no significant change in steady state iNOS mRNA level for as long as 2 h after irradiation, but by 4 h it reached ~1.8-times the control level and remained similarly elevated for at least 20 h. Western analysis revealed that there was a corresponding post-irradiation increase in iNOS protein, which was nearly doubled by 4 h and remained so out to 20 h (Fig. 4B). Thus, iNOS mRNA as well as protein was induced in photodynamically stressed cells.
Figure 4.

Transcriptional and translational expression of iNOS in ALA/light-treated cells. ALA-treated COH-BR1 cells were exposed to a 1 J/cm2 light fluence. After the indicated periods of post-irradiation dark incubation up to 20 h, the cells were sampled for (A) iNOS mRNA determination by RT-PCR analysis and (B) iNOS protein determination by immunoblot analysis. An ALA-only dark control (DC) at 20 h was analyzed alongside. (A) Means ± SD of values from three separate experiments are shown; *P<0.01 compared with DC. (B) Densitometrically-determined iNOS band intensity, normalized to β-actin and relative to DC, is shown for each time point; results are from one experiment representative of three.
DAF-2DA-based detection of NO accumulation in ALA/light-treated cells
The effect of photostress on steady state level of intracellular NO was examined using DAF-2DA, which, as DAF-2, is nitrosated to give the fluorescent product, DAF-2 triazole [24-27]. As shown in Fig. 5A, COH-BR1 cells treated with ALA and kept in the dark for 20 h exhibited relatively little fluorescence with DAF-2DA, whereas ALA/light-challenged cells exhibited very strong fluorescence after 20 h, the integrated intensity being ~8-times greater than that of the dark control. There was a striking 77% reduction in ALA/light-elicited fluorescence when irradiation and subsequent dark incubation were carried out in the presence of 1400W (Fig. 5A). This is consistent with a causal relationship between upregulated iNOS and the large increase in steady state NO observed in photostressed cells. Using the same cell line, we showed previously [19] that ALA/light-provoked caspase-3/7 activation and apoptotic cell count were strongly enhanced by the highly specific NO scavenger cPTIO. This suggests that the elevated NO observed with ALA/light (Fig. 4A) played a prominent role in cellular resistance to photokilling.
Figure 5.

Imaging of NO formation in photostressed cells: effects of iNOS inhibition and iNOS knockdown. (A) Wild-type COH-BR1 cells were preincubated with ALA and either kept in the dark (ALA) or irradiated (1 J/cm2) in the absence (ALA/hν) or presence of 10 μM 1400W (ALA/W/hν). After 20 h of dark incubation, the cells were treated with 20 nM DAF-2DA for 30 min, then examined by fluorescence microscopy, using 488 nm excitation and 610 nm emission. (B) Cells from an iNOS-knockdown clone (kd) of COH-BR1 cells, along with those from a negative control (c-kd), were treated as described for wild type cells. The numbers below the image frames in panels (A) and (B) represent integrated fluorescence intensities; means ± SD of values from 3 different viewing fields of 50 cells each are shown. Scale bar: 200 μm.
Effects of shRNA-induced knockdown on photostress-induced iNOS and NO
To further substantiate the involvement of iNOS induction in NO-mediated hyperresistance to photokilling, we examined the effects of knocking down the iNOS transcript, using RNA interference methodology. The shRNA approach employed [30] allowed us to generate and select stable clones of COH-BR1 cells exhibiting greatly diminished iNOS expression. As shown by the Western blot in Fig. 6A, the selected clone exhibited an iNOS knockdown of ~80% relative to protein expressed in wild type cells or in a scrambled shRNA control. When iNOS-knockdown (kd) cells were pre-incubated with ALA as described for wild type cells and exposed to a 1 J/cm2 light fluence, iNOS induction over a 20 h post-irradiation period was greatly diminished (Fig. 6B) compared with that of wild type cells (Fig. 4B). The small effect seen in Fig. 6B is attributed to the residual constitutive iNOS observed after knockdown. In contrast, a control prepared with scrambled shRNA exhibited the same timing and extent of iNOS induction after photostress as observed in wild type cells (results not shown).
Figure 6.
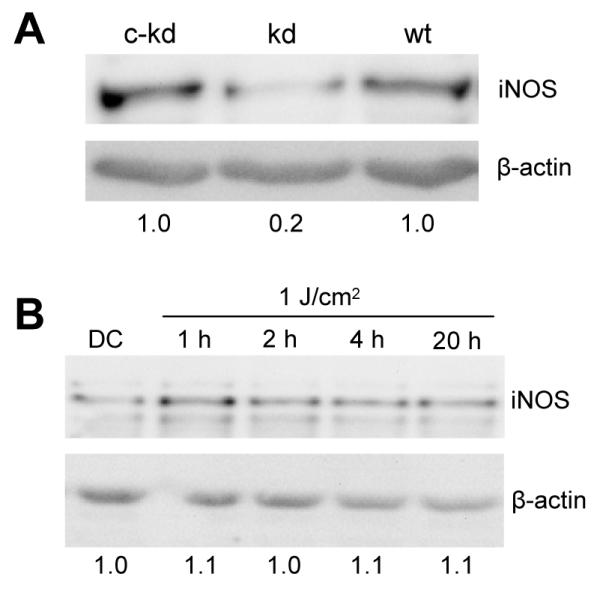
iNOS knockdown: immunodetectable protein before and after ALA/light treatment. (A) Immunoblot of iNOS in wild type COH-BR1 cells (wt), an iNOS-knockdown clone (kd), and its negativecontrol (c-kd). Protein load: 125 μg per lane. Densitometrically-assessed iNOS band intensity, normalized to β-actin and expressed relative to c-kd, is indicated below each lane. (B) Immunoblot of iNOS-knockdown COH-BR1 cells at various times after exposure to ALA and a 1 J/cm2 light fluence. Protein load: 125 μg per lane. Number below each lane is iNOS band intensity, normalized to β-actin and relative to the dark control (DC).
As shown by the confocal images in Fig. 1B, the pre-irradiation location of PpIX in ALA-treated iNOS-kd cells was indistinguishable from that in wild type cells (Fig. 1A). Moreover, integrated PpIX fluorescence intensity relative to that of Rh123 over a field of 4 or 5 cells was essentially the same for ALA-treated wild type and iNOS-kd cells (Fig. 1A,B), implying that PpIX content was the same for both. Therefore, altered porphyrin synthesis or localization was ruled out as a possible reason for the much diminished iNOS stress response in Fig. 6B compared with Fig. 4B.
As anticipated, DAF-2-monitored NO was greatly diminished in ALA/light-treated iNOS-kd cells (Fig. 5B) compared with wild type counterparts (Fig. 5A). However, the NO level in ALA/light-treated iNOS-kd control cells (Fig. 5B) was similar to that in treated wild type (Fig. 5A), consistent with the Western blot data in Fig. 6A. Taken together, the results in Figs. 5 and 6 confirm that NO was overproduced in photodynamically stressed cells and that upregulated iNOS was the source of this NO.
Effect of iNOS knockdown on ALA/light-induced apoptosis
We examined the effects of iNOS knockdown on cell sensitivity to photokilling. As shown in Fig. 7A, the number of COH-BR1 cells succumbing to Ho/PI- or TUNEL-assessed apoptosis was significantly greater in the iNOS-knockdown clone than in a negative control generated with scrambled shRNA. Approximately 5% of the knockdown cells were apoptotic in an ALA-treated dark control, and this increased to ~40% (TUNEL) and 53% (Ho/PI) after ALA/light treatment, whereas only ~15-20% of the ALA/light-treated scrambled control cells were apoptotic (Fig. 7B). This clearly indicates that stress-induced iNOS, and by implication iNOS-derived NO, played a prominant role in cell resistance to apoptotic photokilling.
Figure 7.

Effect of iNOS knockdown on photostress-induced apoptosis. iNOS-knockdown and negative control cells were treated with ALA and irradiated as described in Fig. 3. After 20 h of dark incubation, extent of apoptosis was evaluated using Ho/PI or TUNEL nuclear staining with fluorescence microscopy. (A) Representative fluorescence micrographs for photostressed knockdown (kd/ALA/hν), dark control (kd/ALA), and photostressed control knockdown (c-kd/ALA/hν) cells; scale bar: 200 μm. (B) Integrated fluorescence intensities for micrographs such as shown in panel A. For each condition, the average of 4 image fields, each with ~100 cells, is represented. Means ± SD of values from three separate experiments are plotted; gray bars, Ho/PI; white bars, TUNEL; *P<0.001 compared with corresponding c-kd/ALA/hν.
Rescue of iNOS-knockdown cells by exogenous NO
We reasoned that if endogenous NO insufficiency due to iNOS knockdown was responsible for the diminished resistance to photokilling observed in Fig. 7, then supplying NO from an exogenous source should reverse this effect. To address this question, we used spermine NONOate (SPNO), a chemical donor which releases 2 molar equivalents of NO with a rate constant of ~1.0 h−1 at 37 °C [31]. As shown in Fig. 8A, caspase-9 activity in iNOS-kd COH-BR1 cells was increased ~4-fold by ALA/light stress, whereas a much smaller effect was seen in the negative control (c-kd). Added to iNOS-kd cells after ALA and shortly before irradiation, SPNO in non toxic concentration (100 μM) decreased caspase-9 activation by more than 60%, whereas fully decomposed SPNO (used as a control) had little, if any, effect (Fig. 8A). As shown in Fig. 8B, ALA/light treatment of iNOS-kd cells resulted in an apoptotic count of ~60%, which was ~3-times greater than that observed with identically treated negative control or wild type cells. Consistent with its effects on caspase-9 activation, active SPNO abolished the large increase in stress-induced apoptosis observed with positive iNOS knockdown, and this effect was eliminated when decomposed SPNO was used (Fig. 8B). Thus, exogenous NO was able to “rescue” cells from the deleterious effects of iNOS depletion, mimicking the effects of endogenous NO in induced resistance to photodynamic stress.
Figure 8.

Reversal of iNOS knockdown effects by exogenous NO. Wild type (wt), iNOS-knockdown (kd), and control knockdown (c-kd) cells were preincubated with ALA and irradiated (1 J/cm2) in the absence or presence of 100 μM active SPNO (S) or decomposed SPNO (dS), added 10 min before irradiation. (A) Caspase-9 activity measured 2 h after irradiation. (B) Apoptosis assessed with Ho/PI 20 h after irradiation. Means ± SD of values from three replicate experiments are plotted in (A) and (B). *P<0.001 compared with c-kd/ALA/hν; **P<0.001 compared with kd/ALA/hν; #P<0.01 compared with kd/S/ALA/hν.
DISCUSSION
Many tumors exhibit a dual response to endogenous or exogenous NO. In low (nanomolar range) concentrations produced by tumor cells themselves or vascular endothelial cells, NO tends to promote tumor growth [15, 32-35], whereas in relatively high (micromolar range) concentrations, typically produced by activated resident macrophages, it tends to be tumoricidal [36,37]. In addition to promoting tumor cell proliferation, low level endogenous NO is known to oppose the antitumor effects of ionizing radiation and certain chemotherapeutic agents [15,35]. A similar negative effect of NO has been reported for animal tumor PDT models. Using Photofrin-based PDT with various implanted mouse tumors, two independent groups [16-18] found that NOS inhibitors such as L-NAME and L-NNA, introduced immediately after irradiation, markedly improved tumor loss and animal survival, the tumors with relatively high constitutive NO production responding best [18]. Moreover high-NO producing tumors exhibited substantially less intrinsic sensitivity to PDT than low-NO counterparts. These findings indicated that endogenous NO was somehow antagonizing the salutary effects of PDT. Since the NOS inhibitors were found to decrease tumor blood flow [18], the observed effects were attributed to NO’s well known ability to cause vasodilation. These effects would work in opposition to tumor ablation by PDT, given that this is attributed at least in part to vasoconstriction and diminished oxygen supply [17,38]. However, very little biochemical or cellular characterization of these NO effects has been carried out since these original reports [16-18], leaving several questions unsettled, e.g. (i) whether NOS upregulation occurs under PDT stress and if so, which isoform(s) and for what duration, (ii) the relative importance of tumor cell vs. vascular cell NOS/NO in anti-PDT activity, and (iii) the signaling pathways that are engaged or disengaged by the induced NO.
In the present study, which advances from our recently published work [19], we have focused mainly on questions (i) and (ii) above. Initial experiments showed that MTT-assessed photokilling of ALA-primed COH-BR1 cells was enhanced by L-NAME and more so by 1400W at a far lower concentration, suggesting a cytoprotective role for iNOS specifically under photostress. Follow-up experiments using Ho/PI, TUNEL, and caspase-9 activation assays revealed that photokilling occurred mainly by apoptosis, most likely originating in mitochondria (intrinsic pathway). Under the sensitization conditions used, ALA-derived PpIX was localized in the mitochondrial compartment and highly reactive 1O2 generated by PpIX photoactivation would have reacted primarily in this compartment [6], consistent with activation of the intrinsic pathway. As seen in MTT-detected photokilling, caspase-9 activation and apoptosis were both strongly stimulated by 1400W, suggesting iNOS-modulated phototoxicity. A key finding relating to iNOS involvement was the rapid and prolonged transcriptional and translational upregulation of iNOS in response to ALA/light-induced stress. Post-irradiation elevation of iNOS protein began more slowly than observed previously [19], most likely because the light fluence used in most of this study (~1 J/cm2) was one-half of that used earlier. The extent of normalized protein overexpression was about one-half of that observed with 2 J/cm2 [19], but again it remained at the maximal level for at least 20 h after stress was imposed. Using DAF-2DA as an NO-scavenging fluorescent probe, we found that there was a strong sustained increase in 1400W-inhibitable NO production in ALA/light-treated cells, supporting the idea that hyperresistance to apoptotic photokilling was elicited by this NO, which derived mainly from induced iNOS. These NO imaging data (Fig. 5) confirm our earlier evidence based on indirect measurement of NO (as nitrite), using a chemiluminescence-based NO analyzer [19]. Our argument about upregulated iNOS/NO involvement in cytoprotection was further strengthened by showing that iNOS knockdown strongly promoted photokilling, in agreement with the effects of 1400W and the NO scavenger cPTIO described previously [19]. These results make it highly unlikely that the observed stress response was due to constitutive iNOS somehow undergoing a gain of activity. The knockdown results also confirm previous immunoblot data showing that iNOS was the only NOS isoform in COH-BR1 cells to be upregulated by ALA/light treatment [19]. We showed earlier, moreover, that apoptotic photokilling of breast adenocarcinoma MDA-MB-231 cells was also stimulated by iNOS inhibition [19], suggesting that photostress-elevated resistance mediated by NO may be a general phenomenon in iNOS-expressing tumor cells.
The mechanism of iNOS/NO induction in photostressed cells has not been examined in this study, but based on evidence obtained with other stress systems, activation of transcription factor NF-κB might be involved [39,40]. In recent preliminary experiments, we observed that the pro-survival kinase Akt (PKB) is rapidly phosphorylation-activated in ALA/light-stressed COH-BR1 cells (results not shown), and well in advance of iNOS induction (Fig. 4B). One of the downstream events in Akt signaling is NF-κB activation [41,42], leading to expression of several stress response genes, including iNOS [39]. Although this pathway could explain iNOS upregulation in our system, additional supporting evidence is needed, and we are actively pursuing this in ongoing studies.
The mechanism by which iNOS-generated NO inhibits apoptotic photokilling is also not yet clear. Since NO does not scavenge 1O2 (43), this can be ruled out as a possible upstream explanation. Other plausible possibilities include (i) inhibition of pro-apoptotic mitogen-activated kinases such as ASK1 and JNK via S-nitrosation [44,45]; (ii) S-nitrosation inhibition of caspase activation or activity [46]; (iii) downregulation of pro-apoptotic Bax or upregulation of anti-apoptotic Bcl-xL [22]; and (iv) activation of cyclic GMP (cGMP)-generating soluble guanylyl cyclase (sGC). A previous study (47) focusing on the latter possibility for aluminum phthalocyanine-sensitized lymphoblastoid cells showed that photokilling was suppressed by chemical donor-derived NO and that this could be antagonized by an inhibitor of sGC or protein kinase G (PKG), suggesting involvement of cGMP-dependent PKG in the NO effect. In contrast, we have found that ALA/light-induced apoptosis of COH-BR1 cells is not enhanced by the same sGC inhibitor at higher concentration (results not shown), tentatively ruling out cGMP involvement in the cytoprotective effects of endogenous stress-generated NO which we describe.
In the context of ALA-PDT or other modes of PDT in the clinical setting, the present findings provide new insights into NO-based resistance of tumor cells to eradication by this treatment and how such resistance might be countered by the rational use of NOS inhibitors. One can postulate that the efficacy of such interventions might depend on the aggressiveness of the PDT challenge in terms of sensitizer concentration at key target sites, applied light dose, and ambient O2 level. Factors such as these can determine the importance of tumor cells per se vs. tumor vascular (e.g. endothelial) cells as PDT targets [3,4]. Considering direct effects on tumor cells, if the applied PDT pressure (e.g. sensitizer and/or light fluence level) is relatively low, this might elicit pro-survival/growth upregulation of iNOS/NO (as we have observed in vitro), resulting in a reduced overall cell kill. In this case, a suitable inhibitor known to be highly selective for iNOS would be expected to improve treatment outcome, assuming that iNOS/NO induction is not overshadowed by other resistance mechanisms. If, on the other hand, the applied PDT oxidative pressure is high, inflammatory processes may play a greater role in tumor cell death [48], in which case more copious NO (along with superoxide) generated by activated neutrophils and macrophages might enhance the kill rather than suppress it. For this situation, one would expect an iNOS inhibitor to reduce overall cell kill, in contrast to its effect under relatively low photodynamic pressure. In published PDT studies with animal models [17,18], NOS inhibitors only improved the cure rate of NO-producing tumors, suggesting that gross inflammation was not a major contributor to tumor cell death. Apoptosis resulting from relatively modest photostress is the preferred mode of tumor cell eradication in clinical PDT [49]. Under these conditions, there is a good possibility that administration of an appropriate NOS inhibitor will improve the treatment outcome, a prospect that remains to be tested in clinical trial format.
Acknowledgements
This work was supported by USPHS Grant CA70823 from the National Cancer Institute. We are grateful to Jared Schmitt for expert technical assistance. Helpful discussions with Neil Hogg and Witold Korytowski are also appreciated.
REFERENCES
- 1.Foote CS. Mechanisms of photosensitized oxidation. Science. 1968;162:963–970. doi: 10.1126/science.162.3857.963. [DOI] [PubMed] [Google Scholar]
- 2.Girotti AW. Mechanisms of photosensitization. Photochem. Photobiol. 1983;38:745–751. doi: 10.1111/j.1751-1097.1983.tb03610.x. [DOI] [PubMed] [Google Scholar]
- 3.Henderson BW, Dougherty TJ. How does photodynamic therapy work? Photochem. Photobiol. 1992;55:145–157. doi: 10.1111/j.1751-1097.1992.tb04222.x. [DOI] [PubMed] [Google Scholar]
- 4.Dougherty TJ, Gomer CJ, Henderson BW, Jori G, Kessel D, Korbelik M, Moan J, Peng Q. Photodynamic therapy. J. Natl. Cancer Inst. 1998;90:889–905. doi: 10.1093/jnci/90.12.889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Malik Z, Lugaci H. Destruction of erythroleukemic cells by photoactivation of endogenous porphyrins. Br. J. Cancer. 1987;56:589–595. doi: 10.1038/bjc.1987.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peng Q, Berg K, Moan J, Kongshaug M, Nesland JM. 5-Aminolevulinic acid-based photodynamic therapy: principles and experimental research. Photochem. Photobiol. 1997;65:235–251. doi: 10.1111/j.1751-1097.1997.tb08549.x. [DOI] [PubMed] [Google Scholar]
- 7.Hanania J, Malik Z. The effect of EDTA and serum on endogenous porphyrin accumulation and photodynamic sensitization of human K562 cells. Cancer Lett. 1992;65:127–131. doi: 10.1016/0304-3835(92)90156-p. [DOI] [PubMed] [Google Scholar]
- 8.Berg K, Anhold H, Bech O, Moan J. The influence of iron chelators on the accumulation of protoporphyrin IX in 5-aminolevulinic acid-treated cells. Br. J. Cancer. 1996;74:688–697. doi: 10.1038/bjc.1996.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krammer B, Plaetzer K. ALA and its clinical impact: from bench to bedside. Photochem. Photobiol. Sci. 2008;7:283–289. doi: 10.1039/b712847a. [DOI] [PubMed] [Google Scholar]
- 10.Moncada SR, Palmer RMJ, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol. Rev. 1991;43:109–143. [PubMed] [Google Scholar]
- 11.Wink DA, Mitchell JB. Chemical biology of nitric oxide: insights into the regulatory, cytotoxic, and cytoprotective mechanisms of nitric oxide. Free Radic. Biol. Med. 1998;25:434–456. doi: 10.1016/s0891-5849(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 12.Jenkins DC, Charles IG, Thomsen LL, Moss DW, Holmes LS, Baylis SA, Rhodes P, Westmore K, Emson PC, Moncada S. Roles of nitric oxide in tumor growth. Proc. Natl. Acad. Sci. USA. 1995;92:4392–4396. doi: 10.1073/pnas.92.10.4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fukumura D, Jain RK. Role of nitric oxide in angiogenesis and microcirculation in tumors. Cancer Metastasis Rev. 1998;17:77–89. doi: 10.1023/a:1005908805527. [DOI] [PubMed] [Google Scholar]
- 14.Kolb JP. Mechanisms involved in the pro- and anti-apoptotic role of NO in human leukemia. Leukemia (Baltimore) 2000;14:1685–1694. doi: 10.1038/sj.leu.2401896. [DOI] [PubMed] [Google Scholar]
- 15.Crowell JA, Steele VE, Sigman CC, Fay JR. Is inducible nitric oxide synthase a target for chemoprevention? Mol. Cancer Ther. 2003;2:815–823. [PubMed] [Google Scholar]
- 16.Korbelik M, Shibuya H, Cecic I. Relevance of nitric oxide to the response of tumors to photodynamic therapy. Proc. SPIE. 1998;3247:98–105. [Google Scholar]
- 17.Henderson BW, Sitnik-Busch TM, Vaughan LA. Potentiation of photodynamic therapy antitumor activity in mice by nitric oxide synthase inhibition is fluence rate dependent. Photochem. Photobiol. 1999;70:64–71. [PubMed] [Google Scholar]
- 18.Korbelik M, Parkins CS, Shibuya H, Cecic I, Stratford MRL, Chaplin DJ. Nitric oxide production by tumor tissue: impact on the response to photodynamic therapy. Br. J. Cancer. 2000;82:1835–1843. doi: 10.1054/bjoc.2000.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bhowmick R, Girotti AW. Cytoprotective induction of nitric oxide in a cellular model of 5-aminolevulinic acid-based photodynamic therapy. Free Radic. Biol. Med. 2010;48:1296–1301. doi: 10.1016/j.freeradbiomed.2010.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Esworthy RS, Baker MA, Chu F-F. Expression of selenium-dependent glutathione peroxidase in human breast tumor cell lines. Cancer Res. 1995;55:957–962. [PubMed] [Google Scholar]
- 21.Kriska T, Korytowski W, Girotti AW. Hyperresistance to photosensitized lipid peroxidation and apoptotic killing in 5-aminolevulinate-treated tumor cells overexpressing mitochondrial GPx4. Free Radic. Biol. Med. 2002;33:1389–1402. doi: 10.1016/s0891-5849(02)01078-x. [DOI] [PubMed] [Google Scholar]
- 22.Bhowmick R, Girotti AW. Signaling events in apoptotic photokilling of 5-aminolevulinic acid-treated tumor cells: inhibitory effects of nitric oxide. Free Radic. Biol. Med. 2009;47:731–740. doi: 10.1016/j.freeradbiomed.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nolan T, Hands RE, Bustin SA. Quantification of mRNA using real-time RT-PCR. Nature Protocols. 2006;1:1559–1582. doi: 10.1038/nprot.2006.236. [DOI] [PubMed] [Google Scholar]
- 24.Suzuki N, Kojima H, Urano Y, Kikucki K, Hirata Y, Nagano T. Orthogonality of calcium concentration and ability of 4,5-diaminofluorescein to detect NO. J. Biol. Chem. 2002;277:47–49. doi: 10.1074/jbc.M108195200. [DOI] [PubMed] [Google Scholar]
- 25.Nagano T. Bioimaging probes for reactive oxygen species and reactive nitrogen species. J. Clin Biochem. Nutr. 2009;45:111–124. doi: 10.3164/jcbn.R09-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wardman P. Fluorescent and luminescent probes for measurement of oxidative and nitrosative species in cells and tissues: progress, pitfalls, and prospects. Free Radic. Biol. Med. 2007;43:995–1022. doi: 10.1016/j.freeradbiomed.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 27.Lancaster JR. The use of diaminofluorescein for nitric oxide detection; conceptual and methodological distinction between NO and nitrosation. Free Radic. Biol. Med. 2010;49:1145. doi: 10.1016/j.freeradbiomed.2010.06.015. [DOI] [PubMed] [Google Scholar]
- 28.Spector DL, Goldman RD, Leinwald LA. Cells: A Laboratory Manual. Vol. 1. Cold Spring Harbor Laboratory Press; 1997. Ch. 15. [Google Scholar]
- 29.Ow Y-LP, Green DR, Hao Z, Mak TW. Cytochrome c: functions beyond respiration. Nature Reviews. 2008;9:532–542. doi: 10.1038/nrm2434. [DOI] [PubMed] [Google Scholar]
- 30.Sandy P, Ventura A, Jacks T. Mammalian RNAi: a practical guide. BioTechniques. 2005;39:215–224. doi: 10.2144/05392RV01. [DOI] [PubMed] [Google Scholar]
- 31.Keefer LK, Nims RW, Davies KM, Wink DA. “NONOates” (1-substituted diazen-1-ium-1,2-diolates) as nitric oxide donors: convenient nitric oxide dosage forms. Methods Enzymol. 1996;268:281–293. doi: 10.1016/s0076-6879(96)68030-6. [DOI] [PubMed] [Google Scholar]
- 32.Stepnik M. Roles of nitric oxide in carcinogenesis: protumorigenic effects. Int. J. Occup. Med. Environ. Health. 2002;15:219–227. [PubMed] [Google Scholar]
- 33.Siegert A, Rosenberg C, Schmitt WD, Denkert C, Hauptmann S. Nitric oxide of human colorectal adenocarcinoma cell lines promotes tumor cell invasion. Br. J. Cancer. 2002;87:1310–1315. doi: 10.1038/sj.bjc.6600224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pervin S, Singh R, Hernandez E, Wu G, Chaudhuri G. Nitric oxide in physiologic concentrations targets the translational machinery to increase the proliferation of human breast cancer cells: involvement of mammalian target of rapamycin/eIF4E pathway. Cancer Res. 2007;67:289–299. doi: 10.1158/0008-5472.CAN-05-4623. [DOI] [PubMed] [Google Scholar]
- 35.Sikora AG, Gelbard A, Davies MA, Sano D, Ekmekcioglu S, Kwon J, Hailemichael Y, Jayaraman P, Myers JN, Grimm EA, Overwijk WW. Targeted inhibition of inducible nitric oxide synthase inhibits growth of human melanoma in vivo, and synergizes with chemotherapy. Clin. Cancer Res. 2010;16:1834–1844. doi: 10.1158/1078-0432.CCR-09-3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Farias-Eisner RF, Sherman MP, Aeberhard E, Chaudhuri G. Nitric oxide is an important mediator for tumoricidal activity in vivo. Proc. Natl. Acad. Sci USA. 1994;91:9407–9411. doi: 10.1073/pnas.91.20.9407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Juang S-H, Xie K, Xu L, Shi Q, Wang Y, Yoneda J, Fidler IJ. Suppression of tumorigenicity and metastasis of human renal carcinoma cells by infection with retroviral vectors harboring the murine inducible nitric oxide synthase gene. Gene Ther. 1998;9:845–854. doi: 10.1089/hum.1998.9.6-845. [DOI] [PubMed] [Google Scholar]
- 38.Fingar VH, Henderson BW. Drug and light dose dependence of photodynamic therapy: a study of tumor and normal tissue response. Photochem. Photobiol. 1987;46:837–841. doi: 10.1111/j.1751-1097.1987.tb04856.x. [DOI] [PubMed] [Google Scholar]
- 39.Xie QW, Kashiwabara Y, Nathan C. Role of transcription factor NF-κB/Rel in induction of nitric oxide synthase. J. Biol. Chem. 1994;269:4705–4708. [PubMed] [Google Scholar]
- 40.Ahn KS, Aggarwal BB. Transcription factor NF-κB: a sensor for smoke and stress signals. Ann. N.Y. Acad. Sci. 2005;1056:218–233. doi: 10.1196/annals.1352.026. [DOI] [PubMed] [Google Scholar]
- 41.Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase Akt pathway in human cancer. Nature Rev. Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 42.Stephens LR, Williams, Hawkins P. Phosphoinositide 4-kinases as drug targets in cancer. Curr. Opin. Pharmacol. 2005;5:357–365. doi: 10.1016/j.coph.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 43.Niziolek M, Korytowski W, Girotti AW. Chain-breaking antioxidant and cytoprotective action of nitric oxide in photodynamically stressed tumor cells. Photochem. Photobiol. 2003;78:262–270. doi: 10.1562/0031-8655(2003)078<0262:caacao>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 44.Park HS, Wu J-W, Cho JH, Kim M-S, Huh S-H, Ryoo K, Choi E-J. Inhibition of apoptosis signal-regulating kinase 1 by nitric oxide through a thiol redox mechanism. J. Biol. Chem. 2004;279:7484–7490. doi: 10.1074/jbc.M304183200. [DOI] [PubMed] [Google Scholar]
- 45.Park HS, Huh S-H, Kim M-S, Lee S-H, Choi E-J. Nitric oxide negatively regulates c-Jun N-terminal kinase/stress activated protein by means of S-nitrosylation. Proc. Natl. Acad. Sci. USA. 2000;97:14382–14387. doi: 10.1073/pnas.97.26.14382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li C-Q, Wogan GN. Nitric oxide as a modulator of apoptosis. Cancer Lett. 2005;226:1–15. doi: 10.1016/j.canlet.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 47.Gomes ER, Almeida RD, Carvalho AP, Duarte CB. Nitric oxide modulates tumor cell death induced by photodynamic therapy through a cGMP-dependent mechanism. Photochem. Photobiol. 2002;76:423–430. doi: 10.1562/0031-8655(2002)076<0423:nomtcd>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 48.Castano AP, Mroz P, Hamblin MR. Photodynamic therapy and tumor immunity. Nature Rev. Cancer. 2006;6:535–545. doi: 10.1038/nrc1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oleinick NL, Morris RL, Belichenko I. The role of apoptosis in response to photodynamic therapy: what, where, why, and how. Photochem. Photobiol. Sci. 2000;1:1–21. doi: 10.1039/b108586g. [DOI] [PubMed] [Google Scholar]