

Abstract
Purpose
Sorafenib, a multikinase inhibitor targeting Raf and VEGFR has shown activity in unselected patients with NSCLC. At present there are no validated biomarkers indicative of sorafenib activity.
Experimental Design
Patients received sorafenib 400mg bid daily to determine activity and tolerability and to measure its biological effects. KRAS mutation status (N=34), angiogenesis markers (VEGF, bFGF, FLT-1, PLGF-1) and imaging with DCE-MRI to determine early changes in tumor vascular characteristics were evaluated. Three parameters Ktrans, Kep and ve, were measured by DCE-MRI at baseline and day 14 of cycle 1. Cytokine analysis was performed on days 0, 14, 28 and 54.
Results
37 pts with previously treated stage IV NSCLC were enrolled in this single center phase II trial. In 34 evaluable patients, 2 had partial responses, and 20 had stable disease for 3-17 months, a disease control rate of 65%. The median progression-free survival (PFS) was 3.4 months and median overall survival (OS) was 11.6 months. Toxicity was consistent with the known side effects of sorafenib. KRAS (32%) and EGFR mutations (22%), showed no correlation with response, PFS or OS. Kep, was significant in predicting an improvement in OS (p=0.035) and PFS (p=0.029). Cytokine analysis demonstrated an improved OS for bFGF day 0 <6 vs >6 pg/ml (p=0.042) whereas a PFS benefit was seen with bFGF at day 28 <6 vs >6 (p=0.028).
Conclusions
KRAS and EGFR mutational status showed no correlation with response, PFS or OS. Radiological and cytokine changes may act as biomarkers indicative of early angiogenesis inhibition.
Introduction
Treatment outcomes for advanced non small cell lung cancer (NSCLC) have been limited by the empiric administration of cytotoxic chemotherapy (1). Small molecule tyrosine kinase inhibitors have demonstrated single agent activity in a wide variety of solid tumors including NSCLC (2), however, the lack of validated predictive factors for many of these targeted treatments remains problematic. Recent evidence has highlighted the importance of individualizing therapy based on certain molecular characteristics (EGFR mutations and EML4-ALK translocations) (3-5). KRAS mutations are present in approximately 30% of NSCLC and are responsible for the proliferation signaling of the RAS/RAF/MEK/ERK pathway and indicate a poor prognosis and poor response to EGFR inhibitors (6, 7). Therapeutic targeting of the Ras pathway has so far been unsuccessful. RAF serine-threonine kinases are the principal effectors of RAS and are considered an important target for cancer therapy. Sorafenib is a multikinase inhibitor that inhibits C-RAF and B-RAF; VEGFRs 1, 2 and 3, and PDGF β; Flt3; RET; and c-KIT(8-11). Sorafenib has shown activity in preclinical models of NSCLC and several phase I (9, 12-15) and phase II trials (16, 17). To date there is limited data with regards to sorafenib sensitivity amongst NSCLC patients with different KRAS mutational status.
Vascular endothelial growth factor (VEGF) is up-regulated in many tumors(18). Unfortunately, there are no validated biomarkers in clinical practice available to predict those patients who will gain clinical benefit, or that can be used to monitor therapeutic response. Increases in VEGF and decreases in soluble VEGF receptors have been reported in phase I and phase II trials involving different VEGFR tyrosine kinase inhibitors (TKIs) and may represent a class effect (16, 19-23). Correlating these angiogenic cytokine changes with clinical benefit has however been difficult. The goal of this phase II trial was to determine if sorafenib is active in NSCLC and to determine the impact of K-Ras mutational status. Several correlative studies were performed during treatment to measure biological and clinical effects of sorafenib.
Patients and Methods
Patients
Thirty seven patients were enrolled between 2005 – 2009 onto the trial, which had been reviewed and approved by the National Cancer Institute's Institutional Review Board. Verbal and written consent was obtained from all patients. Enrollment criteria included age older than 18 years, an Eastern Cooperative Oncology Group (ECOG) performance status of 0-1, a life expectancy greater than 3 months and histologic or cytologic confirmation of recurrent or progressive advanced NSCLC, and only one line of prior chemotherapy. A protocol amendment in February 2008 allowed enrollment of patients who had received greater than one standard or investigational treatment regimen. Suitable candidates required adequate bone marrow, liver and renal function, measurable disease (RECIST 1.0) and to be at least 28 days since any prior radiation or major surgery. Patients with symptomatic brain metastases were excluded unless they had treatment for their brain metastases and had stable disease for at least 3 months without steroids. Originally patients with squamous cell carcinoma were allowed on study but a second protocol amendment excluded these patients due to side-effects seen in the ESCAPE trial(24). The study followed the current guidelines of the International Conference on Harmonization for good clinical practice and the Declaration of Helsinki.
Study Design
The primary objective of this single arm study was to determine the response rate (RR) and toxicity of sorafenib administered at a dose of 400 po mg BID daily in a 28 day cycle, in relapsed or recurrent NSCLC patients. Secondary objectives included a correlation of the patient's response to treatment with KRAS mutation status and with changes in angiogenic cytokines, an evaluation of the application of DCE-MRI to determine early changes in tumor vascularity during treatment, and an evaluation of time to progression and overall survival. Treatment continued until objective evidence of tumor progression or intolerable drug-related side effects. Adverse events were defined by the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) version 3.0. Two dose reductions were allowed for clinically significant toxicities attributed to sorafenib while on study: dose level -1 was 200mg BID and dose level -2 was 200mg OD. Doses were not re-escalated once toxicities had recovered. Drug could be held a maximum of 3 weeks prior to discontinuation from study.
Imaging Studies - DCE-MRI
Dynamic contrast enhanced magnetic resonance imaging (DCE MRI) was used to evaluate changes in vascularity at baseline and at day 14 (+/- 3 days) of cycle 1. DCE MRI was performed with a 1.5-T MR system (Philips Achieva, The Best, The Netherlands) using a dedicated receive-only six-channel phased array coil (see supplemental text). Data from DCE MRI were analyzed using a 2-compartment (Kety) model, also known as the general kinetic model (GKM)(25, 26). The GKM model analysis was performed with an IDL-based (Interactive Data Language; Research Systems Inc., Boulder, CO, USA) research tool (Cine Tool, GE Healthcare, USA). Manual region-of-interest measurements were obtained from each slice of the target lesion. The GKM model produces three parameters: kep the reverse contrast transfer rate, Ktrans, the forward contrast transfer rate and ve the extravascular fraction. Baseline and follow up after treatment cycles kep, Ktrans, and ve values were obtained.
Cytokine analysis
Serial plasma samples were collected from all patients in an ETDA-containing vacutainer at pretreatment (baseline – day 0), and on day 14, 28 and 54. After centrifugation, the samples were aliquoted, immediately frozen, and stored at -80°C. Plasma analysis performed included evaluation of VEGF, placental-derived growth factor (PLGF), basic fibroblast growth factor (bFGF), and VEGF receptor 1 (sVEGFR1 or FLT-1) using multiplex array plates from Meso-Scale Discovery (Gaithersburg, MD). The concentrations of the cytokines were determined with recombinant standards and expressed as pg/ml.
KRAS/EGFR/BRAF Mutation Identification
Five to ten 5-micron sections from formalin-fixed, paraffin-embedded (FFPE) tissue sections were de-paraffinized by standard methods. Macro-dissection was performed on selected specimens to obtain at least 10% tumor cell content for DNA isolation, as necessary. DNA isolation was carried out with the QIAmp FFPE DNA Kit on an automated QIAcube instrument (Qiagen, Germantown, MD). Targeted analysis of KRAS codons 12, 13, and 61 was performed using pyrosequencing technology on a PyroMark Q24 instrument (Qiagen) and the PyroMark Q24 KRAS v2.0 kit (Qiagen), as described by Ogino et al(27). Targeted analysis for EGFR mutations involving exon 20 codon 790, exon 21 codons 858/861/863 was performed using pyrosequencing, while exon 19 deletions were assessed by capillary electrophoresis using a Genetic Analyzer 3130xl (Applied Biosystems), as described by Pan et al(28). Primers for the EGFR pyrosequencing reactions were designed in our laboratory and are available on request. For BRAF V600 mutation detection, the extracted DNA was subjected to an initial PCR using a single primer set (sequence available on request) encompassing codon V600. Pyrosquencing was carried out on a Qiagen PyroMark Q24 system (Valencia, CA).
Statistical Considerations
The study was conducted using a phase II optimal design in order to determine if sorafenib was able to be associated with a response rate (PR + CR) which could rule out 5% (p0=0.05) in favor of a more desirable 20% response rate (p1=0.20). Using alpha=0.10 (probability of accepting a poor drug) and beta=0.10 (probability of rejecting a good drug), initially 12 patients were to be enrolled onto the trial. If zero of the 12 patients responded, then accrual would end while if 1 or more of the first 12 patients had a CR or a PR, then accrual would continue until a total of 37 patients with measurable disease had been enrolled. If 4 or more of 37 had a response, then this would warrant further investigation in a subsequent trial. The associations between KRAS and EGFR mutations and response (PR+SD vs. PD) were determined by a Fisher's exact test. Comparisons of continuous parameters were made using an exact Wilcoxon rank sum test. The difference from baseline to days 14, 28, and 54 was determined by a Wilcoxon signed rank test. Survival and progression free survival analyses were initially performed using univariate Cox models to screen for the association between outcome and values of a continuous parameter less than or greater than the median value. For the cases in which a trend was identified, the p-value was confirmed by a log-rank test p-value. In view of the exploratory nature of these analyses, any p-values reported have not been adjusted for multiple comparisons, and the results of these analyses are considered hypothesis generating.
Results
Patient Characteristics
Baseline demographic data and disease characteristics are listed in table 1. All patients had received prior therapy with 21 patients (57%) having had at least 2 prior regimens. Seventeen patients (46%) had received only one previous regimen prior to study enrollment. In total 43% had received prior EGFR tyrosine kinase inhibitors and 40% had prior bevacizumab.
Table 1. Clinical Characteristics of Patients (N=37).
| Characteristic | Number of Patients (%) | |
|---|---|---|
| Sex | Female | 18 (49%) |
| Male | 19 (51%) | |
| Histology | Adenocarcinoma | 22 (60%) |
| Adenocarinoma with BAC features | 9 (24%) | |
| Squamous cell carcinoma | 3 (8%) | |
| Poorly differentiated carcinoma | 2 (5%) | |
| Large cell neuroendocrine | 1 (3%) | |
| Ethnicity | White | 25 (68%) |
| African American | 5 (13%) | |
| Asian | 4 (11%) | |
| Hispanic/Latino | 3 (8%) | |
| ECOG performance status | PS 1 | 5 (14%) |
| PS 0 | 32 (86%) | |
| Age | Median age | 61yrs |
| range | 30-85 yrs | |
| No. of prior chemotherapy and targeted regimens | 0 | 0 (0%) |
| 1 | 17 (46%) | |
| 2 | 4 (11%) | |
| 3 | 8 (22%) | |
| 4 | 3 (8%) | |
| 5 | 3 (8%) | |
| 6 | 2 (5%) | |
| Previous therapy | platinum (Cisplatin/Carboplatin) | 33 (89%) |
| Taxane (Docetaxel/Paclitaxel) | 29 (78%) | |
| Erlotinib/Gefitinib | 16 (43%) | |
| Bevacizumab | 15 (40%) | |
| Pemetrexed | 13 (35%) | |
Efficacy
All patients had progressive disease at the time of enrollment. The median duration of treatment was approximately 3 months (97 days, range, 12 to 517 days). Three patients were considered not evaluable for response, one patient withdrew from study after 4 weeks and the 2 others due to a lack of target lesions. In total, two partial responses (6%) were seen (Figure 1). The first patient had metastatic squamous cell carcinoma and had received only one previous regimen of carboplatin and paclitaxel for 6 cycles. His tumor was wildtype for KRAS, EGFR and BRAF. The second patient had metastatic adenocarcinoma to the brain and adrenal gland. He had received one previous chemotherapy regimen of carboplatin, gemcitabine and bevacizumab prior to commencing sorafenib. His tumor was wildtype for KRAS and BRAF and not enough tissue was available for EGFR analysis.
Figure 1. Maximum percent reduction or best response of target lesions in patients (N=34) with available post baseline tumor measurements.
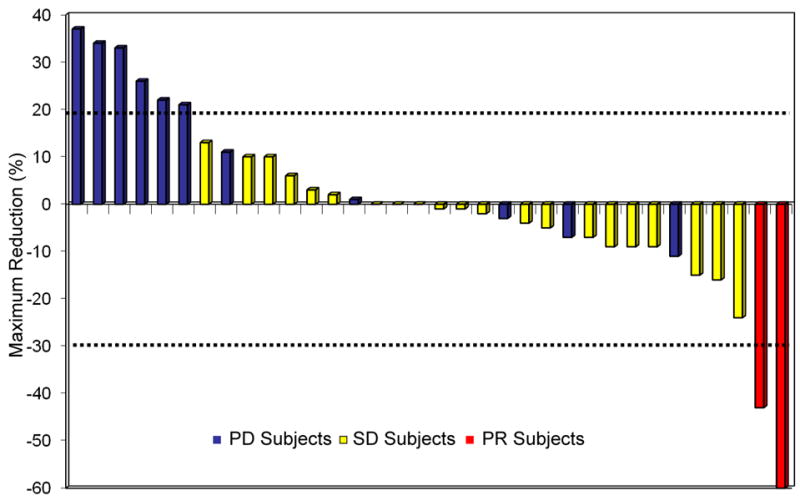
PD, progressive disease; SD, stable disease; PR, partial responses. Two PRs measuring 60% and 43% and 20 patients (59%) with stable disease with a median duration of 5.4 months. Tumor shrinkage (1% to 24%) was seen in 12 patients with stable disease and tumor growth (2% to 16%) was seen in the remaining 8 patients. In total 8 patients (24%) had stable disease ≥ 6 months, with 3 patients on study for 6 months, 1 for 7 months, 2 for 8 months, 1 for 10 months and 1 patient for 17 months respectively. Progressive disease as best response was seen in 12 patients (35%), with 3 patients considered not evaluable.
Median progression free survival (PFS) for evaluable patients was 3.4 months and median overall survival (OS) was 11.6 months (Figure 2). The disease control rate was 65% with 2 PRs (6%) and 20 SD (59%). In patients who had received only one previous therapy 2 PRs, and 7 SD were seen. In an evaluation of all 37 patients, stable disease greater than 3 months (3 cycles) ranging from 3 to 17 months was documented in 20 patients (54%) with a median duration of 5.4 months. One patient had clinical disease progression and died 3 weeks after sorafenib was discontinued. There were no differences in terms of RR, PFS or OS depending on whether patients had one or multiple lines of prior therapy or between squamous cell and adenocarcinomas.
Figure 2. Overall survival (OS) and Progression free survival (PFS).
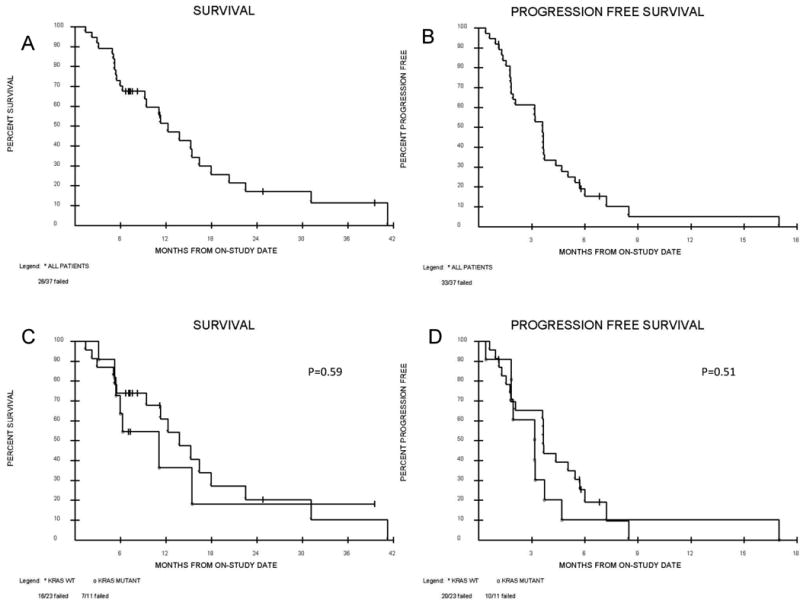
Kaplan meier curves for (A) OS (B) PFS (C) OS according to KRAS wildtype or mutation (D) PFS according to KRAS wildtype or mutation. Median OS for all patients on study is 11.6 months. Median OS for KRAS wildtype is 13.2 months and 7.2 months for KRAS mutant (p=0.59). Median PFS for all patients on study is 3.4 months. Median PFS for KRAS wildtype is 3.6 months and 2.6 months for KRAS mutant.
KRAS and EGFR mutations
Table 2 summarizes the characteristics of patients with either wildtype or KRAS/EGFR. Three patients out of 37 did not have tissue available for KRAS analysis. KRAS mutations occurred in 11/34 patients (32%). Fourteen patients (38%) did not have evaluable tissue to perform EGFR mutation analysis. Of the remaining 23 patients there were 5 (22%) subjects with an activating EGFR mutation. There was reciprocal exclusion of EGFR and KRAS mutations, as reported by others (29). Interestingly 3 (8%) never smoking patients with adenocarcinomas demonstrated KRAS mutations. There was no correlation between the mutational status of KRAS or EGFR and RR, PFS or OS. The DCR observed in KRAS mutant and KRAS wildtype patients was 60% and 71% respectively (p=0.69). The DCR observed in EGFR mutant and EGFR wildtype patients was 40% and 69% respectively (p=0.33).
Table 2. KRAS and EGFR mutation status.
| Patient | Sex (M/F) | Ethnicity | Smoking Status | Histology | KRAS | EGFR |
|---|---|---|---|---|---|---|
| 1 | F | White | Never | Adenocarcinoma | N/A | N/A |
| 2 | M | White | Smoker | Adeno-BAC | WT | WT |
| 3 | F | Asian | Never | Adenocarcinoma | G12C | WT |
| 4 | M | White | Smoker | Squamous cell | WT | WT |
| 5 | M | White | Smoker | Squamous cell | WT | N/A |
| 6 | M | Hispanic | Smoker | Adenocarcinoma | WT | WT |
| 7 | M | White | Smoker | Adenocarcinoma | G13D | WT |
| 8 | M | White | Smoker | Adeno-BAC | G12C | WT |
| 9 | F | Hispanic | Smoker | NSCLC | WT | N/A |
| 10 | M | White | Smoker | Large cell | WT | WT |
| 11 | M | White | Never | Adenocarcinoma | WT | N/A |
| 12 | F | Black | Never | Adenocarcinoma | WT | L858R |
| 13 | F | White | Smoker | Squamous cell | WT | WT |
| 14 | M | White | Smoker | Adenocarcinoma | WT | N/A |
| 15 | M | Asian | Smoker | Adenocarcinoma | N/A | N/A |
| 16 | F | White | Smoker | Adeno-BAC | WT | WT |
| 17 | M | Black | Smoker | Poorly differentiated | WT | N/A |
| 18 | F | White | Never | Adenocarcinoma | G12D | N/A |
| 19 | F | White | Smoker | Adeno-BAC | G12V | WT |
| 20 | M | White | Smoker | Adeno-BAC | WT | WT |
| 21 | M | Black | Smoker | Adeno-BAC | WT | N/A |
| 22 | M | White | Smoker | Adenocarcinoma | Q61H | WT |
| 23 | F | Asian | Never | Adenocarcinoma | WT | Ex19del15 |
| 24 | F | White | Smoker | Adenocarcinoma | WT | N/A |
| 25 | M | White | Never | Adeno-BAC | G12C | WT |
| 26 | M | White | Smoker | Adenocarcinoma | N/A | N/A |
| 27 | F | Asian | Never | Adenocarcinoma | WT | Ex19del15 |
| 28 | F | White | Smoker | Adeno-BAC | G12D | WT |
| 29 | F | White | Smoker | Adenocarcinoma | G12C | WT |
| 30 | F | Hispanic | Never | Adenocarcinoma | WT | N/A |
| 31 | M | White | Smoker | Adenocarcinoma | WT | L858R |
| 32 | M | Black | Never | Adenocarcinoma | WT | Ex19del18 |
| 33 | F | Black | Never | Adenocarcinoma | WT | N/A |
| 34 | F | White | Smoker | Adenocarcinoma | WT | WT |
| 35 | F | White | Smoker | Adenocarcinoma | G12C | N/A |
| 36 | F | White | Smoker | Poorly differentiated | WT | WT |
| 37 | F | White | Smoker | Adeno-BAC | G12C | WT |
Adeno-BAC: Adenocarinoma with bronchioloaveolar features; WT, wildtype; N/A, not available
Plasma Biomarker Analysis
All 37 patients are evaluable for analysis. Thirty two patients (86%) had plasma drawn at baseline whereas only 14 samples (38%) were available for day 54 analysis. Increases in VEGF and decreases in VEGR1 from baseline to day 54 were detected. Increases in plasma PLGF levels were also seen at these time-points (supplemental table 1). The 4 cytokines were each divided at their respective median values and evaluated for their association with overall survival and progression free survival. Four of the parameters evaluated demonstrated an association with improved OS and PFS by having univariate two-tailed p-values <0.10 (supplemental table 2 and 3). Of these parameters, bFGF at baseline <6 vs. >6 was significant for OS (p=0.042) and similar levels on day 28 were significant for PFS (p=0.028) (Figure 3). The difference between day 28-day 0 PLGF was significant for OS (<11 v >12), p=0.0027 (supplemental Figure 2). Cytokine analysis did not predict response to sorafenib.
Figure 3. Cytokine and DCE-MRI analysis.
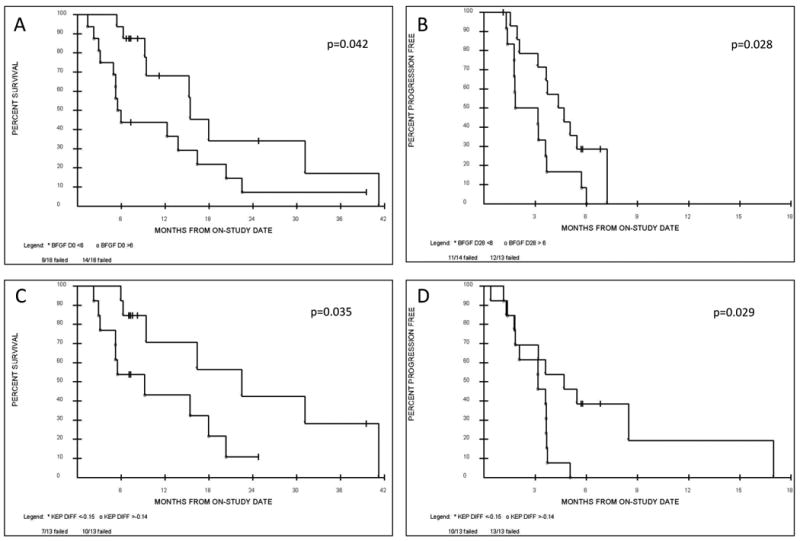
Exploratory analysis demonstrating potential trends towards an association with OS and PFS by having univariate two-tailed p-values <0.10 included basic fibroblast growth factor (bFGF) and the reverse constant transfer rate (Kep) as measured via DCE-MRI. (A) Patients with baseline (day 0) bFGF levels <6 pg/ml had an improved OS compared to patients with higher baseline bFGF levels >6 pg/ml. (B) Patients with lower bFGF levels <6 pg/ml as measured post cycle 1 (day 28) had an improved PFS than patients with higher day 28 bFGF levels >6 pg/ml, indicating a potential prognostic and predictive role for bFGF. Similarly Kep as measured by DCE-MRI may act as a radiological biomarker. Patients demonstrating a Kep difference of < - 0.15 (the difference between the day 14 and the day 0 measurements i.e. day14-day0) compared to patients with a Kep difference of > - 0.14 showed (C) an overall survival benefit and (D) a progression free survival benefit.
DCE MRI
Twenty-six patients (70%) had DCE-MRI scans at baseline and on day 14 (+/- 3 days) of cycle 1. Target lesions were localized in the lung (20 patients), adrenal gland (2 patients), liver (1 patient), mediastinum (1 patient), chest wall (1 patient) and neck (1 patient). Decreases in either Kep or Ktrans were seen in twenty-one patients (81%), whereas an increase was observed in five patients (19%) (Supplemental Figure 1). Kep, Ktrans and ve measurements at day 0, day 14 and the difference between the day 14 and the day 0 measurements (day14-day0) were each divided at their respective median values and evaluated for their association with RR, PFS and OS. In a univariate exploration the Kep difference of < - 0.15 vs. > - 0.14 was statistically significant, with an improved OS (p=0.035) and PFS (p=0.029) for those patients demonstrating a Kep difference of < - 0.15 compared to patients with a Kep difference of > - 0.14 (Figure 3). Non significant differences were associated with Ktrans and ve. DCE-MRI did not help predict response to sorafenib.
Safety
The toxicities of sorafenib have been well documented and similar side effects to those previously described were seen in this study (Table 3). In total, 16 patients tolerated the full dose of 400mg po bid but 21 patients needed a dose reduction (57%). Of these, 11 patients required one dose reduction and 10 required a second reduction to 200mg po OD. All patients who were reduced to dose level -2 tolerated sorafenib at this dosage. Twelve patients (32%) required a dose reduction during cycle one. The most frequent grade 3 drug-related AEs were: hypertension (16%), hand/foot syndrome (14%), dyspnea (14%), and hypophosphatemia (14%). One patient developed a squamous cell carcinoma of the skin while on study which was treated by excision and discontinuation of drug. A second patient developed a keratoacanthoma, which was removed and observed while continuing on drug. The most common reason for treatment discontinuation was disease progression (n = 33; 94%). To date 26 patients have died, 7 (27%) died within 60 days from coming off study. There was one death while on study secondary to clinical progression.
Table 3. Toxicity.
| Any Grade | Grade 3-4 | |||
|---|---|---|---|---|
| Adverse Event | Number | % | Number | % |
| Palmar-plantar erythrodysesthesia | 22 | 59 | 6 | 16 |
| Rash/desquamation | 20 | 54 | 2 | 5 |
| Hypertension | 16 | 43 | 6 | 16 |
| Diarrhea | 14 | 38 | 1 | 3 |
| Fatigue | 11 | 30 | 3 | 8 |
| Anorexia | 9 | 24 | 0 | 0 |
| Nausea | 8 | 22 | 1 | 3 |
| Dry skin | 7 | 19 | 0 | 0 |
| Mucositis | 7 | 19 | 1 | 3 |
| Pruritus | 7 | 19 | 1 | 3 |
Discussion
In this trial sorafenib demonstrated activity with a response rate of 6%, a DCR of 65%, a PFS of 3.4 months, and an OS of 11.6 months. Two other phase II clinical trials have reported activity of sorafenib in heavily pretreated patients with NSCLC with an OS of 6.7 months, median PFS of 2.7 months and stable disease in 59%(16) and in ECOG 2501 a DCR of 47% and a median PFS of 3.6 months (17). There is little known of the possible differences in the sensitivity of NSCLC to sorafenib according to KRAS mutational status. Furthermore the identification of biomarkers predictive of treatment outcomes for anti-VEGF therapies has so far met with limited success. In the BATTLE trial the overall DCR for patients treated with sorafenib was 58%. Subset analyses showed DCR of 61% in patients with KRAS mutations vs 56% KRAS wildtype but only a 23% DCR in patients with an EGFR mutation compared to 64% in patients without an EGFR mutation (p=0.048)(30). Here we report a DCR of 60% in patients with KRAS mutations vs 71% in KRAS wildtype (p=0.69) and a DCR of 40% in EGFR mutations vs 69% in patients without an EGFR mutation (p=0.33). Smit et al observed 3 PRs and a median PFS of 3 months (95% CI: 2.2-3.8 months) in 10 patients with previously treated advanced NSCLC all harboring KRAS mutations(31).
Currently, no direct RAS inhibitor has proven clinically effective and agents such as sorafenib that bypass RAS and inhibit effector molecules downstream of the mutant GTPase (e.g. RAF) are being evaluated. Preclinical data have suggested that sorafenib inhibits cell growth by inducing G1 arrest in NSCLC cell lines independent of KRAS genotype(32). Here we report that sorafenib inhibited the growth of NSCLC in a manner independent of KRAS mutational status with no differences in RR, PFS or OS being detected between patients with KRAS wildtype or mutant tumors. The inhibitory effect of sorafenib in KRAS wildtype tumors was also independent of whether there was a mutant EGFR gene present or not. Preclinical studies have indicated that sorafenib blocks the ERK signaling pathway only in wild type KRAS tumors and it inhibits NSCLC cell growth by targeting B-RAF in cells with wild-type KRAS and C-RAF in cells with mutant KRAS(32, 33). These results need to be validated in clinical trials and were not assessed in this study.
It has been suggested that for each class of drug it may be possible to identify certain cytokine changes that occur during treatment that may serve as pharmacodynamic or efficacy markers. In this trial we noted an increase in baseline VEGF levels and a decrease in sVEGFR-1 which have been reported in other studies and likely represents a class effect. The predictive role of pretreatment VEGF levels in patients with NSCLC and who are treated with antiangiogenic therapy remains controversial with some suggestion that bioavailable, rather than circulating VEGF may provide the most predictive value(34-37). Germline polymorphisms in VEGF are also being evaluated as a means to help predict patients likely to respond to sorafenib(38). In an exploratory analysis of 4 plasma cytokines, we found distinct patterns of cytokine changes that may act as predictors of response to sorafenib. BFGF levels at day 0 and day 28 showed significance in terms of an OS (p=0.042) and PFS benefit (p=0.028) and may act as prognostic and predictive biomarkers (Figure 3). These correlative studies are considered hypothesis generating and may form the basis for future trials.
Functional and molecular imaging may also be used for pre-therapy molecular phenotyping and may prove effective as pharmacodynamic predictive biomarkers. DCE-MRI is non-invasive and is sensitive to tumor perfusion parameters such as vascular volume, vascular permeability and flow. Typically, aggressive tumors are characterized by a rapid enhancement followed by a subsequent rapid wash-out period(26, 39). In this study, Kep demonstrated a significant predictive value for OS (p=0.035) and PFS (p=0.029). Kep is considered more robust than the other parameters since it is not as dependent on the T1 values of the tissue or Ve(26). Although Ktrans and Kep are correlated with each other, it is not surprising that one parameter might better predict OS and PFS than another. For instance, Ktrans can be influenced by changes in ve values after treatment, variability in image noise, T1 measurement and is very sensitive to patient motion. In this study most of the target lesions were localized to the lungs, adrenal glands and liver all of which are subject to significant motion.
In conclusion, this trial demonstrates that sorafenib provides clinical benefit for patients with heavily pretreated advanced NSCLC irrespective of their KRAS mutational status. Although this study is limited by the relatively small sample size and varied population it is indicative of the population most often seen in thoracic oncology clinics whereby a wide array of histologies and prior therapies are encountered. Establishing predictive biomarkers for anti-angiogenics remains a significant challenge as discovery and validation will have to be tailored to the known mechanisms of action of a certain agent in a certain disease, and will require standardization of biomarker assays amongst protocols. Challenges to overcome include establishing adequate criteria to measure response and the need for spatially resolved dynamic biomarkers to meet the heterogeneous and dynamic nature of cancer. Preliminary biomarker data are emerging but mostly from single arm trials making it difficult to ascertain whether the marker is prognostic or predictive. A combination of vascular permeability imaging and circulating factors measured at various time points may yield a ‘composite biomarker’ to make robust predictions. Ultimately these data will have to be tested and validated in large, well-designed, prospective clinical trials.
Statement of Translational Relevance.
KRAS mutations are present in approximately 30% of NSCLC and indicate a poor prognosis and a poor response to EGFR inhibitors. Currently, no direct RAS inhibitor has proven clinically effective and agents such as sorafenib that bypass RAS and inhibit effector molecules downstream of the mutant GTPase (e.g. RAF) are being evaluated. To our knowledge this paper is the first completed study of the direct impact of RAS mutations in NSCLC patients treated with sorafenib. The most striking observation from this report is that sorafenib inhibited the growth of NSCLC in a manner independent of KRAS mutational status with no differences in RR, PFS or OS being detected between patients with KRAS wildtype or mutant tumors. The inhibitory effect of sorafenib in KRAS wildtype tumors was also independent of whether there was a mutant EGFR gene present or not.
Supplementary Material
Acknowledgments
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health. This work was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organization imply endorsement by the U.S. Government.
References
- 1.Schiller JH, Harrington D, Belani CP, et al. Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. N Engl J Med. 2002;346:92–8. doi: 10.1056/NEJMoa011954. [DOI] [PubMed] [Google Scholar]
- 2.Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353:123–32. doi: 10.1056/NEJMoa050753. [DOI] [PubMed] [Google Scholar]
- 3.Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–57. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 4.Rosell R, Moran T, Queralt C, et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med. 2009;361:958–67. doi: 10.1056/NEJMoa0904554. [DOI] [PubMed] [Google Scholar]
- 5.Shaw AT, Yeap BY, Mino-Kenudson M, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol. 2009;27:4247–53. doi: 10.1200/JCO.2009.22.6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eberhard DA, Johnson BE, Amler LC, et al. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J Clin Oncol. 2005;23:5900–9. doi: 10.1200/JCO.2005.02.857. [DOI] [PubMed] [Google Scholar]
- 7.O'Byrne KJ, Bondarenko I, Barrios C, et al. Molecular and clinical predictors of outcome for cetuximab in non-small cell lung cancer (NSCLC): Data from the FLEX study. J Clin Oncol (Meeting Abstracts) 2009;27:8007. [Google Scholar]
- 8.Wilhelm S, Chien DS. BAY 43-9006: preclinical data. Curr Pharm Des. 2002;8:2255–7. doi: 10.2174/1381612023393026. [DOI] [PubMed] [Google Scholar]
- 9.Wilhelm SM, Carter C, Tang L, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099–109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- 10.Wilhelm S, Carter C, Lynch M, et al. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov. 2006;5:835–44. doi: 10.1038/nrd2130. [DOI] [PubMed] [Google Scholar]
- 11.Plaza-Menacho I, Mologni L, Sala E, et al. Sorafenib functions to potently suppress RET tyrosine kinase activity by direct enzymatic inhibition and promoting RET lysosomal degradation independent of proteasomal targeting. J Biol Chem. 2007;282:29230–40. doi: 10.1074/jbc.M703461200. [DOI] [PubMed] [Google Scholar]
- 12.Strumberg D, Richly H, Hilger RA, et al. Phase I clinical and pharmacokinetic study of the Novel Raf kinase and vascular endothelial growth factor receptor inhibitor BAY 43-9006 in patients with advanced refractory solid tumors. J Clin Oncol. 2005;23:965–72. doi: 10.1200/JCO.2005.06.124. [DOI] [PubMed] [Google Scholar]
- 13.Clark JW, Eder JP, Ryan D, Lathia C, Lenz HJ. Safety and pharmacokinetics of the dual action Raf kinase and vascular endothelial growth factor receptor inhibitor, BAY 43-9006, in patients with advanced, refractory solid tumors. Clin Cancer Res. 2005;11:5472–80. doi: 10.1158/1078-0432.CCR-04-2658. [DOI] [PubMed] [Google Scholar]
- 14.Moore M, Hirte HW, Siu L, et al. Phase I study to determine the safety and pharmacokinetics of the novel Raf kinase and VEGFR inhibitor BAY 43-9006, administered for 28 days on/7 days off in patients with advanced, refractory solid tumors. Ann Oncol. 2005;16:1688–94. doi: 10.1093/annonc/mdi310. [DOI] [PubMed] [Google Scholar]
- 15.Flaherty KT, Schiller J, Schuchter LM, et al. A phase I trial of the oral, multikinase inhibitor sorafenib in combination with carboplatin and paclitaxel. Clin Cancer Res. 2008;14:4836–42. doi: 10.1158/1078-0432.CCR-07-4123. [DOI] [PubMed] [Google Scholar]
- 16.Blumenschein GR, Jr, Gatzemeier U, Fossella F, et al. Phase II, multicenter, uncontrolled trial of single-agent sorafenib in patients with relapsed or refractory, advanced non-small-cell lung cancer. J Clin Oncol. 2009;27:4274–80. doi: 10.1200/JCO.2009.22.0541. [DOI] [PubMed] [Google Scholar]
- 17.Schiller JH, Lee JW, Hanna NH, Traynor AM, Carbone DP. A randomized discontinuation phase II study of sorafenib versus placebo in patients with non-small cell lung cancer who have failed at least two prior chemotherapy regimens: E2501. J Clin Oncol (Meeting Abstracts) 2008;26:8014. [Google Scholar]
- 18.Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol. 2005;23:1011–27. doi: 10.1200/JCO.2005.06.081. [DOI] [PubMed] [Google Scholar]
- 19.Ebos JM, Lee CR, Christensen JG, Mutsaers AJ, Kerbel RS. Multiple circulating proangiogenic factors induced by sunitinib malate are tumor-independent and correlate with antitumor efficacy. Proc Natl Acad Sci U S A. 2007;104:17069–74. doi: 10.1073/pnas.0708148104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nikolinakos PG, Altorki N, Yankelevitz D, et al. Plasma cytokine and angiogenic factor profiling identifies markers associated with tumor shrinkage in early-stage non-small cell lung cancer patients treated with pazopanib. Cancer Res. 70:2171–9. doi: 10.1158/0008-5472.CAN-09-2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Longo R, Gasparini G. Challenges for patient selection with VEGF inhibitors. Cancer Chemother Pharmacol. 2007;60:151–70. doi: 10.1007/s00280-006-0403-6. [DOI] [PubMed] [Google Scholar]
- 22.Deprimo SE, Bello CL, Smeraglia J, et al. Circulating protein biomarkers of pharmacodynamic activity of sunitinib in patients with metastatic renal cell carcinoma: modulation of VEGF and VEGF-related proteins. J Transl Med. 2007;5:32. doi: 10.1186/1479-5876-5-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Drevs J, Zirrgiebel U, Schmidt-Gersbach CI, et al. Soluble markers for the assessment of biological activity with PTK787/ZK 222584 (PTK/ZK), a vascular endothelial growth factor receptor (VEGFR) tyrosine kinase inhibitor in patients with advanced colorectal cancer from two phase I trials. Ann Oncol. 2005;16:558–65. doi: 10.1093/annonc/mdi118. [DOI] [PubMed] [Google Scholar]
- 24.Scagliotti G, Novello S, von Pawel J, et al. Phase III Study of Carboplatin and Paclitaxel Alone or With Sorafenib in Advanced Non-Small-Cell Lung Cancer. J Clin Oncol: JCO. 2009;26:1321. doi: 10.1200/JCO.2009.26.1321. [DOI] [PubMed] [Google Scholar]
- 25.Kety SS. The theory and applications of the exchange of inert gas at the lungs and tissues. Pharmacol Rev. 1951;3:1–41. [PubMed] [Google Scholar]
- 26.Choyke PL, Dwyer AJ, Knopp MV. Functional tumor imaging with dynamic contrast-enhanced magnetic resonance imaging. J Magn Reson Imaging. 2003;17:509–20. doi: 10.1002/jmri.10304. [DOI] [PubMed] [Google Scholar]
- 27.Ogino S, Kawasaki T, Brahmandam M, et al. Sensitive sequencing method for KRAS mutation detection by Pyrosequencing. J Mol Diagn. 2005;7:413–21. doi: 10.1016/S1525-1578(10)60571-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pan Q, Pao W, Ladanyi M. Rapid polymerase chain reaction-based detection of epidermal growth factor receptor gene mutations in lung adenocarcinomas. J Mol Diagn. 2005;7:396–403. doi: 10.1016/S1525-1578(10)60569-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shigematsu H, Lin L, Takahashi T, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst. 2005;97:339–46. doi: 10.1093/jnci/dji055. [DOI] [PubMed] [Google Scholar]
- 30.Herbst RS, Blumenschein GR, Jr, Kim ES, et al. Sorafenib treatment efficacy and KRAS biomarker status in the Biomarker-Integrated Approaches of Targeted Therapy for Lung Cancer Elimination (BATTLE) trial. J Clin Oncol (Meeting Abstracts) 28:7609. [Google Scholar]
- 31.Smit EF, Dingemans AM, Thunnissen FB, Hochstenbach MM, van Suylen RJ, Postmus PE. Sorafenib in patients with advanced non-small cell lung cancer that harbor K-ras mutations: a brief report. J Thorac Oncol. 5:719–20. doi: 10.1097/JTO.0b013e3181d86ebf. [DOI] [PubMed] [Google Scholar]
- 32.Takezawa K, Okamoto I, Yonesaka K, et al. Sorafenib inhibits non-small cell lung cancer cell growth by targeting B-RAF in KRAS wild-type cells and C-RAF in KRAS mutant cells. Cancer Res. 2009;69:6515–21. doi: 10.1158/0008-5472.CAN-09-1076. [DOI] [PubMed] [Google Scholar]
- 33.Cichowski K, Janne PA. Drug discovery: inhibitors that activate. Nature. 464:358–9. doi: 10.1038/464358a. [DOI] [PubMed] [Google Scholar]
- 34.Fontanini G, Boldrini L, Chine S, et al. Expression of vascular endothelial growth factor mRNA in non-small-cell lung carcinomas. Br J Cancer. 1999;79:363–9. doi: 10.1038/sj.bjc.6690058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davidoff AM, Ng CY, Zhang Y, et al. Careful decoy receptor titering is required to inhibit tumor angiogenesis while avoiding adversely altering VEGF bioavailability. Mol Ther. 2005;11:300–10. doi: 10.1016/j.ymthe.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 36.Dowlati A, Gray R, Sandler AB, Schiller JH, Johnson DH. Cell adhesion molecules, vascular endothelial growth factor, and basic fibroblast growth factor in patients with non-small cell lung cancer treated with chemotherapy with or without bevacizumab--an Eastern Cooperative Oncology Group Study. Clin Cancer Res. 2008;14:1407–12. doi: 10.1158/1078-0432.CCR-07-1154. [DOI] [PubMed] [Google Scholar]
- 37.Bonnesen B, Pappot H, Holmstav J, Skov BG. Vascular endothelial growth factor A and vascular endothelial growth factor receptor 2 expression in non-small cell lung cancer patients: relation to prognosis. Lung Cancer. 2009;66:314–8. doi: 10.1016/j.lungcan.2009.02.013. [DOI] [PubMed] [Google Scholar]
- 38.Zhang W, Lee J, Schiller JH, Carbone DP, Chung CH, Lenz H. Use of germline polymorphisms in VEGF to predict tumor response and progression-free survival in non-small cell lung cancer (NSCLC) patients treated with sorafenib: Subset pharmacogenetic analysis of Eastern Cooperative Oncology Group (ECOG) trial E2501. J Clin Oncol (Meeting Abstracts) 28:7607. [Google Scholar]
- 39.Knopp MV, Giesel FL, Marcos H, von Tengg-Kobligk H, Choyke P. Dynamic contrast-enhanced magnetic resonance imaging in oncology. Top Magn Reson Imaging. 2001;12:301–8. doi: 10.1097/00002142-200108000-00006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.