

Abstract
Antagonistic coevolution between hosts and parasites has been proposed as a mechanism maintaining genetic diversity in both host and parasite populations. In particular, the high level of genetic diversity usually observed at the major histocompatibility complex (MHC) is generally thought to be maintained by parasite-driven selection. Among the possible ways through which parasites can maintain MHC diversity, diversifying selection has received relatively less attention. This hypothesis is based on the idea that parasites exert spatially variable selection pressures because of heterogeneity in parasite genetic structure, abundance or virulence. Variable selection pressures should select for different host allelic lineages resulting in population-specific associations between MHC alleles and risk of infection. In this study, we took advantage of a large survey of avian malaria in 13 populations of the house sparrow (Passer domesticus) to test this hypothesis. We found that (i) several MHC alleles were either associated with increased or decreased risk to be infected with Plasmodium relictum, (ii) the effects were population specific, and (iii) some alleles had antagonistic effects across populations. Overall, these results support the hypothesis that diversifying selection in space can maintain MHC variation and suggest a pattern of local adaptation where MHC alleles are selected at the local host population level.
Keywords: avian malaria, diversifying selection, Passer domesticus, Plasmodium relictum, resistance, susceptibility
1. Introduction
The outcome of the interaction between hosts and parasites has been shown to depend on the genetic makeup of the two partners as well as on the environment where the interaction takes place [1–3]. Resistance genes have been extensively searched for, in particular in animals and plants of economic interest [4–6]. In vertebrates, genes of the major histocompatibility complex (MHC) have become good candidates for resistance genes, for several reasons. MHC genes code for surface glycoprotein that bind to antigenic peptides and present them to the cells of the immune system (T-cells). MHC class I and II molecules present intra- and extracellular-derived peptides, respectively [7]. Antigen presentation is an essential component of the activation of the acquired immune response of vertebrates; therefore, individuals that fail to present specific pathogen-derived epitopes can be highly susceptible to the infection. To confirm the prominent role of MHC genes in the process of parasite resistance, a number of epidemiological studies have reported associations between MHC alleles and disease occurrence or severity. For instance, in humans, the human leukocyte antigen (HLA) allele BRB1*5301 is associated with a protective effect towards severe cerebral malaria in Gambia [8], the HLA alleles B*27 and B*57 are associated with a slower progression of disease in HIV-infected patients [9,10] and the HLA allele DRw53 is associated with a lower risk to contract leprosy in India [11]. In addition to these epidemiological data, investigations on model and non-model organisms have strengthened the idea that individuals with certain MHC haplotypes enjoy a selective advantage in the face of certain parasitic strains [12–18].
Although there is, now, little doubt that selection acts on MHC genes, the nature of the selection is open to debate [19,20]. MHC genes are usually considered as the most polymorphic of the vertebrate genome. Non-mutually exclusive hypotheses have been put forward to explain the maintenance of high MHC diversity (i.e. heterozygote advantage, negative frequency-dependent selection). These hypotheses are generally based on the idea that hosts and parasites are involved in coevolutionary cycles [21] and that parasites exert balancing selection on their hosts [22–24]. Investigations on MHC diversity have been conducted previously at different but complementary time scales: (i) at a macroevolutionary time scale, as shown by molecular analysis of the pattern of non-synonymous versus synonymous substitutions at the peptide-binding region [25,26], and (ii) at a microevolutionary timescale, based on the comparison of the pattern of population differentiation at MHC and microsatellite neutral loci [27,28].
Recently, theoretical work has suggested that variable, parasite-exerted, selection pressures in space and time can substantially contribute to maintain variation at the global scale [19]. This hypothesis of so-called diversifying selection is particularly appealing because we know that parasite abundance and diversity (both species and genetic diversity within species) vary in space and time, and are therefore probably to inflict variable selection pressures on hosts [17,29]. Diversifying selection has been investigated using a population genetics approach in fish and bird species [30,31]. Recently, we found that neutral processes are not sufficient to explain spatial variation in MHC class I among 13 house sparrow (Passer domesticus) populations [32]. In agreement with the prediction of diversifying selection [33], we found a stronger pattern of isolation by distance for MHC class I than for neutral markers (microsatellites). This result may arise because diversifying selection will retain population-specific lineages of alleles. This phenomenon should, therefore, contribute to the generation of a pattern of local adaptation where certain alleles are positively selected in some populations but not in others. Thus, at a contemporary timescale, we may predict a population-specific pattern of associations between MHC alleles and risk of disease.
The aim of the present work was to test this last prediction, using the house sparrow and a malaria parasite (Plasmodium relictum) as a study model. Haemosporidia (Plasmodium sp. and Haemoproteus sp.) are common avian blood parasites that have attracted considerable attention in the last years. The cost of infection with haemosporidian parasites has been measured recently by treating birds with antimalarial drugs [34–36] or by experimentally infecting them with Plasmodium strains [37–39]. These studies have confirmed that infected hosts can pay a substantial cost in terms of reproductive success and survival [40]. We might, therefore, expect that mechanisms of resistance towards avian haemosporidian parasites should be selected for. In agreement with this view, Westerdahl et al. [17] have reported evidence suggesting the involvement of MHC genes in the process of resistance towards Plasmodium ashfordi in great reed warblers (Acrocephalus arundinaceus). Similarly, in a previous study, Bonneaud et al. [41] have reported an association between MHC alleles and the risk of infection with a Plasmodium lineage in the house sparrow. This study was, however, only based on two populations, and we therefore wished to investigate the pattern of MHC alleles—P. relictum resistance at a larger geographical scale, involving a larger number of house sparrow populations.
2. Material and methods
(a). Sampling
The house sparrow is a common, sedentary bird [42], occurring in both rural and urban areas of Europe. Since 2004, a monitoring programme was started in 13 sites in France (figure 1 and table 1). Birds were caught with mist nets, ringed with a numbered metal ring and a blood sample was collected for each individual. Blood samples were collected between April 2004 and October 2005 and stored in lysis buffer (10 mM Tris-HCL pH 8, 100 mM EDTA, 2% SDS).
Figure 1.

Geographical localization of the 13 house sparrow populations used in this study. Scale bar, 100 km.
Table 1.
Total sample size (N), sample size by season (S: winter/spring), number of MHC alleles (NMHC) and parasite prevalence for the seven lineages of Plasmodium (SGS1, GRW11, COLL1, PADOM1, PADOM2, PADOM5, TURDUS1) and the one of Haemoproteus (PADOM3).
| population | N | S | NMHC | prevalence |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| all | SGS1 | GRW11 | COLL1 | PADOM1 | PADOM2 | PADOM3 | PADOM5 | TURDUS1 | ||||
| 1. Paris | 52 | 12/40 | 29 | 30.8 | 26.9 | 3.8 | 0 | 0 | 0 | 0 | 0 | 0 |
| 2. Cachan | 20 | 0/20 | 21 | 30.0 | 30.0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 3. Wissous | 39 | 12/27 | 27 | 48.7 | 41.0 | 7.7 | 0 | 7.7 | 2.5 | 0 | 0 | 0 |
| 4. Crégy | 85 | 67/18 | 31 | 49.4 | 31.8 | 3.5 | 0 | 0 | 0 | 14.1 | 0 | 0 |
| 5. Thieux | 45 | 0/45 | 23 | 33.3 | 26.7 | 6.7 | 0 | 0 | 0 | 0 | 0 | 0 |
| 6. Anglus | 58 | 58/0 | 26 | 38.0 | 31.0 | 3.4 | 3.4 | 0 | 0 | 0 | 0 | 0 |
| 7. Cours | 61 | 61/0 | 34 | 53.2 | 37.7 | 9.8 | 0 | 0 | 0 | 6.5 | 0 | 0 |
| 8. Crennes | 55 | 55/0 | 36 | 36.4 | 29.1 | 1.8 | 7.3 | 0 | 0 | 1.8 | 0 | 0 |
| 9. Rully | 58 | 18/40 | 29 | 39.6 | 29.3 | 6.9 | 0 | 0 | 0 | 3.4 | 0 | 0 |
| 10. Arles | 49 | 34/15 | 32 | 81.6 | 63.3 | 4.1 | 8.2 | 0 | 0 | 2.0 | 6.1 | 0 |
| 11. Chizé | 54 | 0/54 | 35 | 59.3 | 42.6 | 14.8 | 3.7 | 0 | 0 | 0 | 0 | 0 |
| 12. Hoedic | 44 | 0/44 | 22 | 6.8 | 6.8 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 13. Ouessant | 44 | 0/44 | 16 | 4.5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4.5 |
(b). Screening of MHC class I variation
Genomic DNA was extracted from blood samples using the Qiaquick 96 Purification Kit (QIAGEN) according to the manufacturer's instructions. We screened individuals to assess allelic diversity at the most variable MHC class I gene family using the polymerase chain reaction (PCR)-based denaturant gradient gel electrophoresis (DGGE) method [43]. This method allowed us to examine single-nucleotide polymorphism at MHC class I exon 3, corresponding to the highly variable peptide-binding site of the protein (α2 domain). We used motif-specific PCR to amplify a substantial proportion of the class I alleles (2–10 alleles per individual) from as many as five class I loci. Although not exhaustive, this gives a reliable estimate of the class I variation. In order to preferentially amplify transcribed alleles, we designed primers based on cDNA sequences: GCA21M 5′-CGTACAGCGGCTTGTTGGCTGTGA-3′ and fA23M 5′-GCGCTCCAGCTCCTTCTGCCCATA-3′ [44].
Amplifications were run in a final volume of 50 µl including 15–50 ng of DNA, 0.25 µM of each primer, 200 µM of dNTPs, 5 µl of 10× buffer and 0.5 U of Taq DNA polymerase. The thermal profile started with 90 s of denaturation at 94°C, followed by 35 cycles at 94°C, 65°C and 72°C for 30 s each and ended with an elongation step at 72°C for 10 min. The PCR products were separated using a DGGE. The DGGE gel contained 7 per cent 19 : 1 acrylamide/bisacrylamide, 1xTAE, formamide and a 40–65 per cent denaturating gradient of urea [44]. The gels were run at 60°C in 1xTAE buffer for 20 h at 180 V. Gels were stained with ethidium bromide and visualized under ultraviolet illumination. All gels always included two copies of a same marker (i.e. PCR fragments made from genomic DNA from three house sparrow individuals) to enable comparisons between gels. The migration distance of the bands on the DGGE gels were identified relative to these marker bands with a high repeatability. Only one person read all the DGGE gels to limit errors. In addition, 12 individuals were double-checked in at least two gels and in five cases in three DGGE gels. The same number and the same identity of DGGE bands were always found.
DGGE bands found in association with the infection status were excised from DGGE gels, and the bands were dissolved in 150 ml of ddH2O. This solution was frozen (−80°C) and melted (41°C), and re-amplified with the original primers and then directly sequenced. Sequences of these DGGE bands were deposed in GenBank (pado83: EU715815; pado109: EU715816; pado123: EF429132; pado133: EU715817).
(c). Screening for haemosporidian infections
We used a highly efficient nested PCR to amplify a 524 bp long fragment of the cytochrome b of both Plasmodium sp. and Haemoproteus sp. parasites from infected birds [45]. This method is highly repeatable and has a detection limit identifying parasitaemia as low as one infected red blood cell per 100 000.
Positive and negative controls were used: i.e. positive controls were from birds with known infections, and the negative controls used were purified water in place of DNA template, or else samples that were consistently void of parasites as confirmed by PCR. We identified lineages by sequencing the fragments on an ABI3730XL, Applied Biosystems.
(d). Statistical analyses
The association between the infection status and specific MHC alleles was tested using generalized linear models (hereafter GLM) with binomial distribution of errors and logit link function. Alleles with a frequency greater than 5 per cent were included in the analysis (n = 16 alleles).
In the first step, we investigated the relationship between the infection status (dependent, binary variable) and explanatory variables using all individuals in the same model (n = 658 individuals). Explanatory variables were the presence/absence of alleles (binary variables), season (binary variable; with the two following periods: autumn/winter (October–March) and spring/summer (April–September), site (factor) and first-order allele × site interactions. We also ran 16 independent models (one for each allele) and compared our qualitative results with the model including all 16 alleles. The p-values of each of these 16 models were adjusted using the Bonferroni correction for multiple tests.
In the second step, local effects of alleles were tested by considering each site separately. In each case, the best model was selected by starting from a full model with all explanatory variables and sequentially removing variables according to the Akaike information criterion. All analyses were conducted using the R software package [46].
In addition to the GLM models, ad hoc randomization tests were performed to compare the actual frequencies of infected individuals carrying each allele with the frequency distribution expected from a null model. This method allowed us to confirm that (i) observed site-specific associations between some alleles and infection status did not arise by chance and (ii) the effects of some alleles on infection status were different, and even antagonistic, among sites. The procedure used for the randomization models is fully described in the electronic supplementary material.
3. Results
(a). MHC variability
The diversity of MHC class I was assessed in a total of 658 house sparrows from the 13 populations (table 1 and figure 1; [32]). The number of MHC alleles per individual varied between two and 10; hence, we amplified at least five loci. Sixty different alleles were found. Only five alleles were found in all populations, whereas seven alleles were specific of one population. Because most alleles had relatively low frequencies (44 out of 60 were found in less than 5% of individuals), we focused on the alleles with a frequency higher than 5 per cent, for obvious statistical reasons (electronic supplementary material, table S1).
(b). Parasite prevalence and diversity
Overall (13 populations, 658 individuals screened), we found eight distinct mitochondrial haemosporidian parasite lineages (seven Plasmodium spp. and one Haemoproteus spp.; see the work of Bensch et al. [47] for a discussion on the biological relevance of these lineages). One Plasmodium lineage largely dominated the parasite community (SGS1, 31.31% of total infections), whereas in only 5.17, 3.04, 1.82, 0.45, 0.45, 0.30 and 0.15 per cent the infections were caused by the lineages Plasmodium GRW11, Haemoproteus PADOM3, Plasmodium COLL1, Plasmodium PADOM1, Plasmodium PADOM5, Plasmodium TURDUS1 and Plasmodium PADOM2 (table 1).
Based on morphological studies, the lineages SGS1 and GRW11 have been described as belonging to the species P. relictum [48,49]. In addition, these two lineages differ by only one synonymous substitution in the analysed cytochrome b sequence (0.2% divergence) and might represent a case of intra-specific mitochondrial polymorphism [47]. We decided, therefore, to analyse SGS1 and GRW11 together as P. relictum. Because of the low frequency of all other parasite lineages, we restricted our analyses to P. relictum.
(c). Association between MHC allele diversity and P. relictum
As a first step, we compared two models. The first model contained the site, season, allele and all the two-way interactions between site and allele. The second model only included site, season and allele (all the two-way interactions between site and allele having been removed). The model including the site × allele interactions had a statistically significant better fit than the model without interactions (χ2 = 230.5, d.f. = 162, p = 0.0003). This result is not due to overdispersion of binomial variances and still holds when the two models were re-implemented with a quasi-binomial error structure ([50]; see details in the electronic supplementary material).
When considering all individuals and all alleles in the same model, the analysis revealed that the probability of infection differed among sites (LR: χ2 = 92.9, d.f. = 12, p < 10−4) and seasons (LR: χ2 = 6.18, d.f. = 1, p = 0.013). No allele had a significant effect on the infection status at the level of the whole sample. Most importantly, the GLM model revealed that there were significant site × allele interaction terms for three alleles (pado83: LR χ2 = 26.44, d.f. = 8, p < 10−3; pado109: LR χ2 = 18.4, d.f. = 8, p = 0.018; pado133: LR χ2 = 29.3, d.f. = 12, p = 0.0035) suggesting that the effects of these alleles on the infection probability varied among sites.
Since prevalence varied between seasons, we performed a number of supplementary analyses to ensure that the observed site × allele interactions did not arise because of sampling at different seasons in different populations. We therefore (i) reduced the dataset to those sites that were sampled in both seasons and (ii) analysed separately data gathered in winter and spring. The results of these models were similar to those provided by the overall analysis, in spite of a reduced statistical power (see the electronic supplementary material). In particular, we found statistically significant site × allele interactions for the pado83, pado133 and pado123 in winter (see electronic supplementary material).
The lineages SGS1 and GRW11 show morphological and molecular similarities, however very little is known about their virulence. Therefore, considering the two lineages together in the same model might provide misleading results. To take into account this possible confounding factor, we reran the model focusing on the SGS1-infected individuals (the low prevalence of GRW11 prevented us from analysing this lineage alone). Again the results were similar to those provided by the overall model (see electronic supplementary material).
As a complement to these models, we performed 16 different correlative analyses between malaria prevalence and each MHC allele. These analyses provided the same general results as the model including all alleles: (i) the site effect was significant in all 16 models (p < 10−4 in all cases after Bonferroni correction for multiple testing), (ii) no allele had a significant effect on infection status at the scale of the whole dataset, and (iii) in the absence of correction for multiple testing, pado83, pado109 and pado133 showed significant interactions with site (p = 0.0028, 0.025 and 0.0032, respectively; analyses of deviance showed that the interaction term significantly improved model fit in all three cases). After Bonferroni correction, site × allele interactions remained significant at the 5 per cent level for pado83 and pado133 (figure 2).
Figure 2.

Difference between the percentage of infected individuals carrying the MHC allele and the percentage of infected individuals not carrying the allele, for the five and 10 populations of house sparrows (Passer domesticus) which, respectively, harbour pado83 and pado133. Positive values represent increased risk of infection linked to the presence of the MHC allele, whereas negative values indicate decreased risk associated with the allele. Stars indicate significant association between MHC allele and infection status.
The randomization model provided similar results: (i) certain MHC alleles are associated with P. relictum prevalence (table 2) and (ii) the effect of pado83, pado109 and pado133 is population specific (table 2).
Table 2.
Association between MHC class I allele and Plasmodium relictum prevalence by population. Increased protection is coded by ‘+’ and increased risk of infection by ‘−’.
| population | MHC allele | effect | 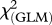 |
d.f. | p-value(GLM) | p-value(resampling) |
|---|---|---|---|---|---|---|
| 1. Paris | none | |||||
| 2. Cachan | none | |||||
| 3. Wissous | none | |||||
| 4. Crégy | pado109 | ‘−’ | 4.52 | 1 | 0.03 | 0.05 |
| pado123 | ‘−’ | 11.61 | 1 | 0.0007 | 0.001 | |
| 5. Thieux | pado83 | ‘−’ | 7.90 | 1 | 0.005 | 0.03 |
| pado133 | ‘−’ | 5.02 | 1 | 0.025 | 0.05 | |
| 6. Anglus | pado133 | ‘+’ | 4.51 | 1 | 0.03 | 0.047 |
| 7. Cours | pado83 | ‘+’ | 9.51 | 1 | 0.002 | 0.03 |
| pado133 | ‘−’ | 6.46 | 1 | 0.01 | 0.037 | |
| 8. Crennes | none | |||||
| 9. Rully | none | |||||
| 10. Arles | pado133 | ‘+’ | 9.13 | 1 | 0.0025 | 0.035 |
| 11. Chizé | none | |||||
| 12. Hoedic | none | |||||
| 13. Ouessant | none |
When considering each site separately, we found strong evidence for statistical associations between alleles and infection status in five out of 13 sites, with again a strong consistency between the GLM and the resampling models (table 2). In three cases, the presence of an allele was associated with a decreased risk to harbour the infection, whereas alleles were associated with a higher likelihood to be infected with P. relictum in five cases. Importantly, the three alleles associated with a significant site × allele interaction term in the GLM model were significantly correlated with the infection status in some local sites. Among them, pado83 and pado133 had opposing effects (increased and decreased probability of infection) in different sites (figure 2).
4. Discussion
The nature of the selection acting on MHC genes has been and still is widely debated. Although often mentioned as the most important force maintaining MHC polymorphism, theoretical and empirical work suggests that heterozygote advantage alone is unlikely to explain the observed pattern of MHC diversity [20,51]. Hedrick [19] has suggested that variation in space and time of parasite abundance and diversity, which probably results in heterogeneous selection pressures, can substantially contribute to maintain MHC diversity at the global scale. Incidentally, this diversifying selection should generate a pattern of local adaptation where MHC alleles are positively selected in some populations but not in others. Identifying the selective agents responsible for this spatially heterogeneous selection is a tantalizing task. We have been studying the association between MHC and P. relictum in 13 populations of house sparrows and found a pattern in agreement with the predictions of the diversifying selection hypothesis. Namely, we found that a few MHC class I alleles were associated with malaria prevalence in five populations. Moreover, for three alleles, there was a statistical support for a population-specific effect, as shown by a significant interaction between the presence/absence of the allele and the population on the infection status. The evidence in support of the hypothesis of diversifying selection in the house sparrow reported here adds to the findings of another study where we compared the pattern of genetic differentiation based on the MHC and neutral markers in the same 13 sparrow populations [32]. In agreement with the hypothesis of diversifying selection, we found that population differentiation was stronger for the MHC than for microsatellites, especially so for geographically distant populations.
Although the role of MHC genes in the process of parasite resistance is now relatively well established, population-specific associations between MHC alleles and infectious diseases have been seldom reported. In a seminal study on human populations, Hill et al. [8,52] reported that the class I HLA allele BRB1*5301 reduced the risk of severe malaria in Gambia (West Africa), whereas in Kenya (East Africa) another allele (BRB1*0101) had a protective effect towards the risk to develop severe malaria. This result, once again, is suggestive of a spatial structure where antigenic variants differ between sites and select for different HLA alleles. In agreement with this view, Gilbert et al. [53] have further reported that HLA type can affect the distribution of parasite alleles and that geographical variation in parasite allele frequencies can affect the strength of the association between host HLA alleles and infectious disease.
In contrast to the MHC of chickens, the MHC of passerines is usually characterized by gene duplication and deletion which produce a variable number of loci even within species and a higher polymorphism both for class I and II genes [54–56]. In agreement with these previous results, we amplified at least five class I loci with a number of MHC alleles per individual varying between two and 10.
As already mentioned above, the aim of the present study was to test the idea that the association between MHC alleles and risk of malaria infection varies in space. This idea rests on two main assumptions: (i) infection with malaria parasites is costly for the host and (ii) hosts are exposed to genetically variable lineages of parasites resulting in variable selection in space. Although, by definition, parasites are costly for their hosts because they divert resources that are no longer available for the vital functions of the host, the magnitude of the cost can be quite variable [38,57,58]. In some circumstances, hosts can cope with the infection [37] and the pathogen can enter a phase of latency with no apparent cost for the host [59,60]. In other case, pathogens can rapidly induce damage leading to host death [38,39,61,62]. For years, evidence for the cost paid by hosts of avian malaria has been scant. Correlative studies where the prevalence of infection was associated with fitness components (survival and/or reproductive success) do not provide evidence for a causal link between infection and fitness. However, experimental work conducted on both natural populations and poultry has shown that infection with Haemoproteus and Plasmodium incurs a cost for the host [38,62–64]. In particular, experimental infection of captive house sparrows with the SGS1 lineage has shown that infected birds pay a cost in terms of reduction in body mass [37] and haematocrit (E. Guivier, S. Garnier, E. Arnoux, B. Faivre & G. Sorci 2007, unpublished data) compared with control non-infected birds. Overall, these results support the idea that avian malaria exerts selection pressures on their hosts.
The second assumption underlying the hypothesis of population-specific associations between MHC alleles and risk of infection is that parasites and the selection pressures they exert vary in space. Genetic variation of malaria among populations has been reported for human parasites (P. falciparum; [65]). We are not aware of similar studies conducted on avian malaria, but it seems reasonable to assume that P. relictum does genetically vary in space. Similarly, we do not have strong evidence showing that selection intensity varies in space. However, recent studies have reported extensive spatial variation in the prevalence of avian haemosporidian parasites, even at very small scales [66–70]. For instance, Wood et al. [70] have shown that the prevalence of infection with the Plasmodium lineages (SGS1, TURDUS1 and BT7) in the blue tit (Cyanistes caeruleus) can vary between 10 and 60 per cent even within the same population, depending on landscape heterogeneities. At a larger spatial scale, we have also found large among-population variation in prevalence, with values ranging from 4.5 to 81.6 per cent. There are several reasons that can explain this variability. Haemosporidian parasites are transmitted by arthropod vectors [71,72] and, therefore, ecological factors associated with vector abundance and diversity may explain differences in parasite prevalence [67,73,74].
Besides the population-specific effect of MHC alleles on risk of malaria infection, we also found that some alleles had antagonistic effects across populations. In particular, pado83 and pado133 were associated with both increased protection and increased risk of infection in different sites. Although the presence of susceptibility alleles might appear puzzling from an evolutionary point of view, MHC alleles that are associated with an increased risk of infection have been reported in several host–parasite systems [9,16,41,75–77]. Given the cost of infection, at a first glance, one might expect that natural selection would rapidly clear such susceptibility alleles from the population [78,79]. However, several mechanisms might explain the persistence of susceptibility alleles in spite of the cost of infection [75,80,81]. First, MHC alleles might have a pleiotropic effect on different parasite species or lineages. Second, the observed increase in prevalence associated with the presence of a given allele can be the result of MHC-altered competition between two parasite species/lineages [82].
To conclude, our results support the hypothesis that diversifying selection in space can (i) maintain MHC variation and (ii) generate a pattern of local adaptation. Our study also highlights the importance of the spatial scale for the understanding of the forces maintaining the polymorphism of immune genes. Whereas local selection pressures might tend to reduce diversity at the site level, variable selection in space can contribute to maintain high levels of polymorphism at a larger scale, here at the country level. However, dispersal among sites and temporal variation in selection pressures can also intervene to maintain diversity within local populations. To go further in the investigation of the diversifying selection hypothesis, we believe that more effort should be devoted to estimate (i) the spatial variability in the cost of infection, (ii) the temporal change in allele frequencies, and (iii) the genetic variation of the parasite. Finally, experimental infections of hosts of known MHC with sympatric and allopatric parasites would probably provide conclusive evidence in support of the hypothesis of diversifying selection.
Acknowledgements
We thank Véronique Morin, Anne Oudin and Thomas Ruby for their assistance during the screening of MHC alleles. We thank all the volunteers from the CRBPO (National Museum of Natural History of Paris) for their help on the field and for providing samples: F. Baroteau, J. Birard, L. Brucy, G. Chaussi, J. Marion, F. Martavan, G. Massez, P. Ollivier, V. Ternois. We also thank A. Z. Lendvai and M. Giraudeau for captures on the field (Chizé) and B. Faivre for his help on Ouessant and Hoedic. Financial support was provided by the Région Ile de France to C.L., R.J. and G.S., the Région Bourgogne to G.S. and the Agence Nationale de la Recherche (EVO-INF-ECOL, NT05-2_44272) to G.S.
References
- 1.Forde S. E., Thompson J. N., Holt R. D., Bohannan B. J. M. 2008. Coevolution drives temporal changes in fitness and diversity across environments in a bacteria–bacteriophage interaction. Evolution 62, 1830–1839 [DOI] [PubMed] [Google Scholar]
- 2.Laine A. L., Tellier A. 2008. Heterogeneous selection promotes maintenance of polymorphism in host–parasite interactions. Oikos 117, 1281–1288 10.1111/j.0030-1299.2008.16563.x (doi:10.1111/j.0030-1299.2008.16563.x) [DOI] [Google Scholar]
- 3.Vale P. F., Stjernman M., Little T. J. 2008. Temperature-dependent costs of parasitism and maintenance of polymorphism under genotype-by-environment interactions. J. Evol. Biol. 21, 1418–1427 10.1111/j.1420-9101.2008.01555.x (doi:10.1111/j.1420-9101.2008.01555.x) [DOI] [PubMed] [Google Scholar]
- 4.Kaufman J. 2000. The simple chicken major histocompatibility complex: life and death in the face of pathogens and vaccines. Phil. Trans. R. Soc. Lond. B 355, 1077–1084 10.1098/rstb.2000.0645 (doi:10.1098/rstb.2000.0645) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jann O. C., Werling D., Chang J. S., Haig D., Glass E. J. 2008. Molecular evolution of bovine Toll-like receptor 2 suggests substitutions of functional relevance. BMC Evol. Biol. 8, 288. 10.1186/1471-2148-8-288 (doi:10.1186/1471-2148-8-288) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Manosalva P. M., Davidson R. M., Liu B., Zhu X. Y., Hulbert S. H., Leung H., Leach J. E. 2009. A Germin-like protein gene family functions as a complex quantitative trait locus conferring broad-spectrum disease resistance in rice. Plant Physiol. 149, 286–296 10.1104/pp.108.128348 (doi:10.1104/pp.108.128348) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klein J. 1986. The natural history of the major histocompatibility complex. New York, NY: John Wiley & Sons [Google Scholar]
- 8.Hill A. V. S., et al. 1991. Common west African Hla antigens are associated with protection from severe malaria. Nature 352, 595–600 10.1038/352595a0 (doi:10.1038/352595a0) [DOI] [PubMed] [Google Scholar]
- 9.Hendel H., et al. 1999. New class I and II HLA alleles strongly associated with opposite patterns of progression to AIDS. J. Immunol. 162, 6942–6946 [PubMed] [Google Scholar]
- 10.Carrington M., O'Brien S. J. 2003. The influence of HLA genotype on AIDS. Annu. Rev. Med. 54, 535–551 10.1146/annurev.med.54.101601.152346 (doi:10.1146/annurev.med.54.101601.152346) [DOI] [PubMed] [Google Scholar]
- 11.Meyer C. G., May J., Stark K. 1998. Human leukocyte antigens in tuberculosis and leprosy. Trends Microbiol. 6, 148–154 10.1016/S0966-842X(98)01240-2 (doi:10.1016/S0966-842X(98)01240-2) [DOI] [PubMed] [Google Scholar]
- 12.Lakshmanan N., Gavora J. S., Lamont S. J. 1997. Major histocompatibility complex class II DNA polymorphisms in chicken strains selected for Marek's disease resistance and egg production or for egg production alone. Poultry Sci. 76, 1517–1523 [DOI] [PubMed] [Google Scholar]
- 13.Kurtz J., Kalbe M., Aeschlimann P. B., Haberli M. A., Wegner K. M., Reusch T. B. H., Milinski M. 2004. Major histocompatibility complex diversity influences parasite resistance and innate immunity in sticklebacks. Proc. R. Soc. Lond. B 271, 197–204 10.1098/rspb.2003.2567 (doi:10.1098/rspb.2003.2567) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harf R., Sommer S. 2005. Association between major histocompatibility complex class II DRB alleles and parasite load in the hairy-footed gerbil, Gerbillurus paeba, in the southern Kalahari. Mol. Ecol. 14, 85–91 10.1111/j.1365-294X.2004.02402.x (doi:10.1111/j.1365-294X.2004.02402.x) [DOI] [PubMed] [Google Scholar]
- 15.Meyer-Lucht Y., Sommer S. 2005. MHC diversity and the association to nematode parasitism in the yellow-necked mouse (Apodemus flavicollis). Mol. Ecol. 14, 2233–2243 10.1111/j.1365-294X.2005.02557.x (doi:10.1111/j.1365-294X.2005.02557.x) [DOI] [PubMed] [Google Scholar]
- 16.Schad J., Ganzhorn J. U., Sommer S. 2005. Parasite burden and constitution of major histocompatibility complex in the malagasy mouse lemur, Microcebus murinus. Evolution 59, 439–450 [PubMed] [Google Scholar]
- 17.Westerdahl H., Waldenstrom J., Hansson B., Hasselquist D., von Schantz T., Bensch S. 2005. Associations between malaria and MHC genes in a migratory songbird. Proc. R. Soc. B 272, 1511–1518 10.1098/rspb.2005.3113 (doi:10.1098/rspb.2005.3113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kjoglum S., Larsen S., Bakke H. G., Grimholt U. 2008. The effect of specific MHC class I and class II combinations on resistance to furunculosis in Atlantic salmon (Salmo salar). Scand. J. Immunol. 2, 160–168 [DOI] [PubMed] [Google Scholar]
- 19.Hedrick P. W. 2002. Pathogen resistance and genetic variation at MHC loci. Evolution 56, 1902–1908 [DOI] [PubMed] [Google Scholar]
- 20.De Boer R. J., Borghans J. A. M., van Boven M., Kesmir C., Weissing F. J. 2004. Heterozygote advantage fails to explain the high degree of polymorphism of the MHC. Immunogenetics 55, 725–731 10.1007/s00251-003-0629-y (doi:10.1007/s00251-003-0629-y) [DOI] [PubMed] [Google Scholar]
- 21.Anderson R. M., May R. M. 1982. Coevolution of hosts and parasites. Parasitology 85, 411–426 [DOI] [PubMed] [Google Scholar]
- 22.Nielsen R. 2005. Molecular signatures of natural selection. Annu. Rev. Genet. 39, 197–218 10.1146/annurev.genet.39.073003.112420 (doi:10.1146/annurev.genet.39.073003.112420) [DOI] [PubMed] [Google Scholar]
- 23.Prugnolle F., Manica A., Balloux F. 2005. Geography predicts neutral genetic diversity of human populations. Curr. Biol. 15, R159–R160 10.1016/j.cub.2005.02.038 (doi:10.1016/j.cub.2005.02.038) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Piertney S. B., Oliver M. K. 2006. The evolutionary ecology of the major histocompatibility complex. Heredity 96, 7–20 [DOI] [PubMed] [Google Scholar]
- 25.Hughes A. L., Hughes M. K. 1995. Natural selection on the peptide-binding regions of major histocompatibility complex. Immunogenetics 42, 233–243 10.1007/BF00176440 (doi:10.1007/BF00176440) [DOI] [PubMed] [Google Scholar]
- 26.Takahata N., Satta Y., Klein J. 1992. Polymorphism and balancing selection at major histocompatibility complex loci. Genetics 130, 925–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Landry C., Bernatchez L. 2001. Comparative analysis of population structure across environments and geographical scales at major histocompatibility complex and microsatellite loci in Atlantic salmon (Salmo salar). Mol. Ecol. 10, 2525–2539 10.1046/j.1365-294X.2001.01383.x (doi:10.1046/j.1365-294X.2001.01383.x) [DOI] [PubMed] [Google Scholar]
- 28.Aguilar A., Garza J. C. 2006. A comparison of variability and population structure for major histocompatibility complex and microsatellite loci in California costal steelhead (Oncorhunchus mykiss Walbaum). Mol. Ecol. 15, 923–937 10.1111/j.1365-294X.2006.02843.x (doi:10.1111/j.1365-294X.2006.02843.x) [DOI] [PubMed] [Google Scholar]
- 29.Westerdahl H., Hansson B., Bensch S., Hasselquist D. 2004. Between-year variation of MHC allele frequencies in great reed warblers: selection or drift? J. Evol. Biol. 17, 485–492 10.1111/j.1420-9101.2004.00711.x (doi:10.1111/j.1420-9101.2004.00711.x) [DOI] [PubMed] [Google Scholar]
- 30.Piertney S. B. 2003. Major histocompatibility complex B-LB gene variation in red grouse Lagopus lagopus scoticus. Wildlife Biol. 9, 251–259 [Google Scholar]
- 31.Ekblom R., Saether S. A., Grahn M., Fiske P., Kalas J. A., Hoglund J. 2007. Major histocompatibility complex variation and mate choice in a lekking bird, the great snipe (Gallinago media). Mol. Ecol. 13, 3821–3828 10.1111/j.1365-294X.2004.02361.x (doi:10.1111/j.1365-294X.2004.02361.x) [DOI] [PubMed] [Google Scholar]
- 32.Loiseau C., Richard M., Garnier S., Chastel O., Julliard R., Zoorob R., Sorci G. 2009. Diversifying selection on MHC class I in the house sparrow (Passer domesticus). Mol. Ecol. 18, 1331–1340 10.1111/j.1365-294X.2009.04105.x (doi:10.1111/j.1365-294X.2009.04105.x) [DOI] [PubMed] [Google Scholar]
- 33.Garrigan D., Hedrick P. W. 2003. Detecting adaptive molecular polymorphism: lessons from the MHC. Evolution 57, 1707–1722 [DOI] [PubMed] [Google Scholar]
- 34.Atkinson C. T., Lease J. K., Drake B. M., Shema N. P. 2001. Pathogenicity, serological responses, and diagnosis of experimental and natural malarial infections in native Hawaiian thrushes. Condor 103, 209–218 10.1650/0010-5422(2001)103[0209:PSRADO]2.0.CO;2 (doi:10.1650/0010-5422(2001)103[0209:PSRADO]2.0.CO;2) [DOI] [Google Scholar]
- 35.Martinez-De La Puente J., Merino S., Tomas G., Moreno J., Morales J., Lobato E., Garcia-Fraile S. 2007. Can the host immune system promote multiple invasions of erythrocytes in vivo? Differential effects of medication and host sex in a wild malaria-like model. Parasitology 134, 651–655 [DOI] [PubMed] [Google Scholar]
- 36.Knowles S. C. L., Palinauskas V., Sheldon B. C. 2010. Chronic malaria infections increase family inequalities and reduce parental fitness: experimental evidence from a wild bird population. J. Evol. Biol. 23, 557–569 10.1111/j.1420-9101.2009.01920.x (doi:10.1111/j.1420-9101.2009.01920.x) [DOI] [PubMed] [Google Scholar]
- 37.Palinauskas V., Valkiūnas G., Bolshakov C. V., Bensch S. 2008. Plasmodium relictum (lineage P-SGS1): effects on experimentally infected passerine birds. Exp. Parasitology 120, 372–380 10.1016/j.exppara.2008.09.001 (doi:10.1016/j.exppara.2008.09.001) [DOI] [PubMed] [Google Scholar]
- 38.Zehtindjiev P., Ilieva M., Westerdahl H., Hansson B., Valkiūnas G., Bensch S. 2008. Dynamics of parasitemia of malaria parasites in a naturally and experimentally infected migratory songbird, the great reed warbler Acrocephalus. Exp. Parasitol. 119, 99–110 10.1016/j.exppara.2007.12.018 (doi:10.1016/j.exppara.2007.12.018) [DOI] [PubMed] [Google Scholar]
- 39.Cellier-Holzem E., Esparza-Salas R., Garnier S., Sorci G. 2010. Effect of repeated exposure to Plasmodium relictum (lineage SGS1) on infection dynamics in domestic canaries. Int. J. Parasitol. 40, 1447–1453 10.1016/j.ijpara.2010.04.014 (doi:10.1016/j.ijpara.2010.04.014) [DOI] [PubMed] [Google Scholar]
- 40.Tomas G., Merino S., Moreno J., Morales J., Martinez-de la Puente J. 2007. Impact of blood parasites on immunoglobulin level and parental effort: a medication field experiment on a wild passerine. Funct. Ecol. 21, 125–133 [Google Scholar]
- 41.Bonneaud C., Pérez-Tris J., Federici P., Chastel O., Sorci G. 2006. Major histocompatibility alleles associated with local resistance to malaria in a passerine. Evolution 60, 383–389 [PubMed] [Google Scholar]
- 42.Summers-Smith J. D. 1998. The sparrows: a study of the genus Passer. Calton, UK: T. & A.D. Pyser Ltd [Google Scholar]
- 43.Myers R. M., Maniatis T., Lerman L. S. 1987. Detection and localization of single base changes by denaturant gradient gel electrophoresis. Methods Enzymol. 155, 501–527 10.1016/0076-6879(87)55033-9 (doi:10.1016/0076-6879(87)55033-9) [DOI] [PubMed] [Google Scholar]
- 44.Bonneaud C., Sorci G., Morin V., Westerdahl H., Zoorob R., Wittzell H. 2004. Diversity of MHC class I and IIB genes in house sparrows (Passer domesticus). Immunogentics 55, 855–865 10.1007/s00251-004-0648-3 (doi:10.1007/s00251-004-0648-3) [DOI] [PubMed] [Google Scholar]
- 45.Waldenström J., Bensch S., Hasselquist D., Ostman O. 2004. A new nested polymerase chain reaction method very efficient in detecting Plasmodium and Haemoproteus infections from avian blood. J. Parasitol. 90, 191–194 10.1645/GE-3221RN (doi:10.1645/GE-3221RN) [DOI] [PubMed] [Google Scholar]
- 46.R Development Core Team: R. 2004. A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing [Google Scholar]
- 47.Bensch S., Perez-Tris J., Waldenstrom J., Hellgren O. 2004. Linkage between nuclear and mitochondrial DNA sequences in avian malaria parasites: multiple cases of cryptic speciation? Evolution 58, 1617–1621 [DOI] [PubMed] [Google Scholar]
- 48.Palinauskas V., Kosarev V., Shapoval A., Bensch S., Valkiūnas G. 2007. Comparison of mitochondrial cytochrome b lineages and morphospecies of two avian malaria parasites of the subgenera Haemamoeba and Giovannolaia (Haemosporida: Plasmodiidae). Zootaxa 1626, 39–50 [Google Scholar]
- 49.Valkiūnas G., Zehtindjiev P., Dimitrov D., Krizanauskiene A., Iezhova T. A., Bensch S. 2008. Polymerase chain reaction-based identification of Plasmodium (Huffia) elongatum, with remarks on species identify of haemosporidian lineages deposited in GenBank. Parasitol. Res. 102, 1185–1193 10.1007/s00436-008-0892-9 (doi:10.1007/s00436-008-0892-9) [DOI] [PubMed] [Google Scholar]
- 50.Crawley M. J. 2007. The R book, pp. 569–590 Chichester, UK: John Wiley & Sons Ltd [Google Scholar]
- 51.Ilmonen P., Penn D. J., Damjanovich K., Morrison L., Ghotbi L., Potts W. K. 2007. Major histocompatibility complex heterozygosity reduces fitness in experimentally infected mice. Genetics 176, 2501–2508 10.1534/genetics.107.074815 (doi:10.1534/genetics.107.074815) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hill A. V. S. 1998. The immunogenetics of human infectious diseases. Annu. Rev. Immunol. 16, 593–617 10.1146/annurev.immunol.16.1.593 (doi:10.1146/annurev.immunol.16.1.593) [DOI] [PubMed] [Google Scholar]
- 53.Gilbert S. C., et al. 1998. Association of malaria parasite population structure, HLA, and immunological antagonism. Science 279, 1173–1177 10.1126/science.279.5354.1173 (doi:10.1126/science.279.5354.1173) [DOI] [PubMed] [Google Scholar]
- 54.Westerdahl H., Wittzell H., von Schantz T. 1999. Polymorphism and transcription of MHC class I genes in a passerine bird, the great reed warbler. Immunogenetics 49, 158–170 10.1007/s002510050477 (doi:10.1007/s002510050477) [DOI] [PubMed] [Google Scholar]
- 55.Balakrishnan C. N., et al. 2010. Gene duplication and fragmentation in the zebra finch major histocompatibility complex. BMC Biol. 8, 29. 10.1186/1741-7007-8-29 (doi:10.1186/1741-7007-8-29) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bollmer J. L., Dunn P. O., Whittingham L. A., Wimpee C. 2010. Extensive MHC class II B gene duplication in a passerine, the common yellowthroat (Geothlypis trichas). J. Hered. 101, 448–460 10.1093/jhered/esq018 (doi:10.1093/jhered/esq018) [DOI] [PubMed] [Google Scholar]
- 57.Long G. H., Chan B. H. K., Allen J. E., Read A. F., Graham A. L. 2008. Experimental manipulation of immune-mediated disease and its fitness costs for rodent malaria parasites. BMC Evol. Biol. 8, 128. 10.1186/1471-2148-8-128 (doi:10.1186/1471-2148-8-128) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Marzal A., Bensch S., Reviriego M., Balbontin J., de Lope F. 2008. Effects of malaria double infection in birds: one plus one is not two. J. Evol. Biol. 21, 979–987 10.1111/j.1420-9101.2008.01545.x (doi:10.1111/j.1420-9101.2008.01545.x) [DOI] [PubMed] [Google Scholar]
- 59.Atkinson C. T., Dusek R. J., Lease J. K. 2000. Serological responses and immunity to superinfection with avian malaria in experimentally-infected Hawaii Amakihi. J. Wildlife Dis. 37, 20–27 [DOI] [PubMed] [Google Scholar]
- 60.Perkins S. L., Kerwin A. S., Rothschild A. D. 2009. Patterns of infection of the lizard malaria parasite, Plasmodium floridense, in invasive brown anoles (Anolis sagrei) in Southwestern Florida. Parasitol. Res. 104, 1191–1196 10.1007/s00436-008-1310-z (doi:10.1007/s00436-008-1310-z) [DOI] [PubMed] [Google Scholar]
- 61.Atkinson C. T., Woods K. L., Dusek R. J., Sileo L. S., Iko W. M. 1995. Wildlife disease and conservation in Hawaii: pathogenicity of avian malaria (Plasmodium relictum) in experimentally infected Iiwi (Vestiaria coccinea). Parasitology 111, S59–S69 10.1017/S003118200007582X (doi:10.1017/S003118200007582X) [DOI] [PubMed] [Google Scholar]
- 62.Donovan T. A., Schrenzel M., Tucker T. A., Pessier A. P., Stalls I. H. 2008. Hepatic hemorrhage, hemocoelom, and sudden death due to Haemoproteus infection in passerine birds: eleven cases. J. Vet. Diagn. Invest. 20, 304–313 [DOI] [PubMed] [Google Scholar]
- 63.Yorinks N., Atkinson C. T. 2000. Effects of malaria on activity budgets of experimentally infected juvenile Apapane (Himatione sanguinea). Auk 117, 731–738 10.1642/0004-8038(2000)117[0731:EOMOAB]2.0.CO;2 (doi:10.1642/0004-8038(2000)117[0731:EOMOAB]2.0.CO;2) [DOI] [Google Scholar]
- 64.Permin A., Juhl J. 2002. The development of Plasmodium gallinaceum infections in chickens following single infections with three different dose levels. Vet. Parasitol. 105, 1–10 10.1016/S0304-4017(01)00645-8 (doi:10.1016/S0304-4017(01)00645-8) [DOI] [PubMed] [Google Scholar]
- 65.Prugnolle F., Durand P., Koella J., Razakandrainibe F., Arnathau C., Villarreal D., Rousset F., de Meeus T., Renaud F. 2008. A comparison of Anopheles gambiae and Plasmodium falciparum genetic structure over space and time. Microbes Infect. 10, 269–275 10.1016/j.micinf.2007.12.021 (doi:10.1016/j.micinf.2007.12.021) [DOI] [PubMed] [Google Scholar]
- 66.Merila J., Bjorklund M., Bennett G. F. 1995. Geographic and individual variation in haematozoan infections in the greenfinch, Carduelis chloris. Can. J. Zool. 73, 798–1804 [Google Scholar]
- 67.Sol D., Jovani R., Torres J. 2000. Geographical variation in blood parasites in feral pigeons: the role of vectors. Ecography 23, 307–314 10.1034/j.1600-0587.2000.d01-1639.x (doi:10.1034/j.1600-0587.2000.d01-1639.x) [DOI] [Google Scholar]
- 68.Bensch S., Akesson A. 2003. Temporal and spatial variation of hematozoans in Scandinavian willow warblers. J. Parasitol. 89, 388–391 10.1645/0022-3395(2003)089[0388:TASVOH]2.0.CO;2 (doi:10.1645/0022-3395(2003)089[0388:TASVOH]2.0.CO;2) [DOI] [PubMed] [Google Scholar]
- 69.Gibb C. E., Jones J., Girvan M. K., Barg J. J., Robertson R. J. 2005. Geographic variation in prevalence and parasitemia of Haemoproteus paruli in the cerulean warbler (Dendroica cerulea). Can. J. Zool. 83, 626–629 [Google Scholar]
- 70.Wood M. J., Cosgrove C. L., Wilkin T. A., Knowles S. C. L., Day K. P., Sheldon B. C. 2007. Within-population variation in prevalence and lineage distribution of avian malaria in blue tits, Cyanistes caeruleus. Mol. Ecol. 16, 3263–3273 10.1111/j.1365-294X.2007.03362.x (doi:10.1111/j.1365-294X.2007.03362.x) [DOI] [PubMed] [Google Scholar]
- 71.Atkinson C. T., van Riper C. 1991. Pathogenicity and epizootiology of avian haematozoa: Plasmodium, Leucocytozoon, and Haemoproteus. In Bird-parasite interactions, ecology, evolution, and behaviour (eds Loye J. E., Zuk M.), pp. 19–48 Oxford, UK: Oxford University Press [Google Scholar]
- 72.Valkiūnas G. 2005. Avian malaria parasites and other haemosporidia. Boca Raton, FL: CRC Press [Google Scholar]
- 73.Garvin M. C., Greiner E. C. 2003. Epizootiology of Haemoproteus danilewskyi (Haemosporina: Haemoproteidae) in blue jays (Cyanocitta cristata) in Southcentral Florida. J. Wildlife Dis. 39, 1–9 [DOI] [PubMed] [Google Scholar]
- 74.Malmqvist B., Strasevicius D., Hellgren O., Adler P. H., Bensch S. 2004. Vertebrate host specificity of wild-caught blackflies revealed by mitochondrial DNA in blood. Proc. R. Soc. Lond. B 271, S152–S155 10.1098/rsbl.2003.0120 (doi:10.1098/rsbl.2003.0120) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Apanius V., Penn D., Slev P. R., Ruff L. R., Potts W. K. 1997. The nature of selection on the major histocompatibility complex. Crit. Rev. Immunol. 17, 179–224 [DOI] [PubMed] [Google Scholar]
- 76.Segal S., Hill A. V. S. 2003. Genetic susceptibility to infectious disease. Trends Microbiol. 11, 445–448 10.1016/S0966-842X(03)00207-5 (doi:10.1016/S0966-842X(03)00207-5) [DOI] [PubMed] [Google Scholar]
- 77.Frodsham A. J., Hill A. V. S. 2004. Genetics of infectious diseases. Hum. Mol. Genet. 13, R187–R194 10.1093/hmg/ddh225 (doi:10.1093/hmg/ddh225) [DOI] [PubMed] [Google Scholar]
- 78.Carrington M., et al. 1999. HLA and HIV-1: heterozygote advantage and B*35-Cw*04 disadvantage. Science 283, 1748–1752 10.1126/science.283.5408.1748 (doi:10.1126/science.283.5408.1748) [DOI] [PubMed] [Google Scholar]
- 79.Wills C. 1999. Selection against susceptibility to HIV-1. Science 285, 11. 10.1126/science.285.5424.11a (doi:10.1126/science.285.5424.11a) [DOI] [Google Scholar]
- 80.Zhang X. S., Wang J. L., Hill W. G. 2002. Pleiotropic model of maintenance of quantitative genetic variation at mutation selection balance. Genetics 161, 419–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lorang J. M., Sweat T. A., Wolpert T. J. 2007. Plant disease susceptibility conferred by a resistance gene. Proc. Natl Acad. Sci. USA 11, 14 861–14 866 10.1073/pnas.0702572104 (doi:10.1073/pnas.0702572104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Loiseau C., Zoorob R., Garnier S., Birard J., Federici P., Julliard R., Sorci G. 2008. Antagonistic effects of a MHC class I allele on malaria-infected house sparrows. Ecol. Lett. 11, 258–265 10.1111/j.1461-0248.2007.01141.x (doi:10.1111/j.1461-0248.2007.01141.x) [DOI] [PubMed] [Google Scholar]