

Abstract
Caspase-8 is an apical caspase which initiates programmed cell death following death receptor ligation. This central role in apoptosis has prompted significant clinical interest in regulating caspase-8 expression and proteolytic activity. However, caspase-8 has also been found to play a number of non-apoptotic roles in cells, such as promoting activation NFκB signaling, regulating autophagy and altering endosomal trafficking, and enhancing cellular adhesion and migration. Therefore, depending upon the specific cellular context, caspase-8 may either potentiate or suppress tumor malignancy. Accordingly, a marked heterogeneity exists in the expression patterns of caspase-8 among different tumor types. Therapeutics have been developed which can increase caspase-8 expression, yet it remains unclear whether this approach will be beneficial in all cases. Care is warranted, and the role of caspase-8 should be addressed on a case by case basis.
1. Caspase-8 is an Apical Caspase
Caspases are evolutionarily conserved cysteine endoproteinases, named for their ability to cleave peptide bonds after aspartic acid residues. For each caspase, a preferred recognition sequence of amino acid residues amino-terminal to the aspartic acid has been described, and in the case of caspase-8, an ‘ideal’ consensus recognition motif has been determined to be isoleucine-glutamic acid-threonine-aspartic acid [1]. However, absolute conservation of this sequence is not required, and caspase-8 can bind and cleave other tetrapeptide sequences, including a ‘classical’ aspartic acid - glutamic acid – valine - aspartic acid (DEVD) sequence that is the preferred target sequence for executioner caspases, such as caspase-3. The recognition of these motifs has lead to the development of caspase-selective inhibitory peptides, facilitating more widespread investigations of caspase biology. An unfortunate side-effect has been the assumption of caspase-specificity of these peptides, which continues to cause some concern in interpretation of the literature. In fact, significant off-target effects on other proteins can be associated with these peptides [2].
Like all caspases, the caspase-8 protein is translated as a monomeric, procaspase zymogen that is later activated by dimerization [3], typically accompanied by the maturation cleavage of the caspase itself [4; 5]. In the case of caspase-8 this yields large and small ‘subunits’ of approximately 18 and 12 kilodaltons. However, these two fragments are not actually disparate domains, but rather non-covalently associated parts of the same protease domain, consisting of a twisted β-sheet (6 stranded, one anti-parallel) bordered by 5 α helices [6]. Rather, the purpose of the observed cleavage is to accommodate the conformational rearrangements required for stable formation of the dimer and the catalytic sites [5], which are otherwise absent in the solved structure of the monomeric procaspase [7]. Nonetheless, the lack of cleavage of the caspase catalytic subunit, at least in the case of caspase-8, should not serve as an indication of an absolute absence of caspase-8 catalytic activity. Full length caspase-8 has been shown to retain low levels of catalytic activity [8] and accordingly, can be isolated using biotinylated probes (such as b-VAD) that covalently interact with the active site cysteine [9]. Recent evidence also supports an active form with a single cleavage between the small and large ‘subunits,’ with no loss of prodomains, particularly during non-apoptotic activation of the caspase. The mechanism by which the procaspase may transiently form a catalytic site is unclear, but given the requirement for dimerization to form a stable enzyme, it appears that this may represent an unfavored, but semi-stable conformer of the procaspase, or alternatively may reflect rearrangements of capsase-8 with other binding partners, such as c-FLIP [10]. Indeed, while dimerization of caspase-8 can be achieved in vitro with chaotropic salts, dimerization of caspase-8 in vivo appears to require maturation cleavage as an essential event [5].
The CASP8 gene on human chromosome 2q33.1 encodes an alternatively spliced message that is translated to several different protein isoforms. The most common forms, originally designated procaspase-8a and procaspase-8b by Scaffidi et al., [11], share two death effector domains (DEDs) and a catalytic domain, and differ only in an alternatively spliced sequence linking the DEDs to the catalytic domain. This ‘twin DEDs plus catalytic domain’ topology is shared by caspase-10 and caspase-18. Caspase-18 is absent in most mammals [12], while rodents also lack caspase-10. In man, these two DED-caspases have overlapping but not redundant functions. Mutations in either caspase have been linked to autoimmune lymphoproliferative syndrome. In addition, severe missense mutations in caspase-8 have been linked to immunodeficiency, likely due to a selective role for caspase-8 in promoting proximal signaling events among cells of the immune compartment [13].
Notably, at least four transcripts of the CASP8 gene encode ‘caspase’ isoforms that do not include the catalytic domain at all, but rather are composed solely of the prodomain DEDs followed by a peptide sequence to which no domain structure has been assigned [11]. Although the functions of such isoforms are not yet precisely known, the DEDs of caspase-8 are required for recruitment to the death inducing signaling complex (DISC), and it has been reported that the these DED-only proteins can act as competitive inhibitors to caspase-8 DISC recruitment [14] [15], as observed for twin DED-proteins found in viruses such as molluscum contagiosum [16]. This possible function is intriguing, as it also suggests that for each procaspase-8 activation event, one would actually result in generation of a caspase-8 inhibitor (ie., the mature prodomains). Interestingly, the prodomains of caspase-8 and/or the DED-only isoforms can be detected in a number of different cell types [17], while the activated catalytic domains cannot (possibly due to the presence of RING protein target sequences in the catalytic domain)[18]. However, it is not yet clear if physiological levels of expression of these cell-encoded proteins are sufficient for meaningful competition and subsequent inhibition of caspase-8 recruitment to the death complex.
In this regard, the inhibition of killing by expression of DEDs at physiologically relevant levels is not a consistent observation in response to expression of the DEDs. In contrast to a putative protective role, it has been shown that the over-expression of the DEDs of caspase-8 can form filamentous bodies in the cell which can recruit the procaspase holoprotein and promote apoptosis in caspase-8 expressing cells [19]. These ‘filaments’ absolutely require an intact microtubule structure – and associate best to apparently ‘stable’ microtubules- but collapse to simple aggregates in the presence of microtubule disrupting agents such as nocodozole [17]. Thus, the presence of elevated levels of endogenous prodomain DEDs, or alternatively spliced DED-isoforms, may actually decrease the proapoptotic threshold required via stimulation of cell surface death receptors (DR) by providing an amplification loop (Figure 1). Some DED-isoforms appear to bind tubulin at the centrosome, with a long term consequence of promoting mitotic crisis and senescence [20]. Given the evidence for both pro- and anti-apoptotic roles for DED-only proteins, the final impact of DED-expression is likely to be dependent upon the specific cellular context of expression, the relative accumulation of the DED protein, the precise splicing pattern of the DEDs, post-translational modifications and the associated carboxyl-peptide sequence.
Figure 1. Different levels of caspase-8 proteolytic activity correspond to different roles in the cell.
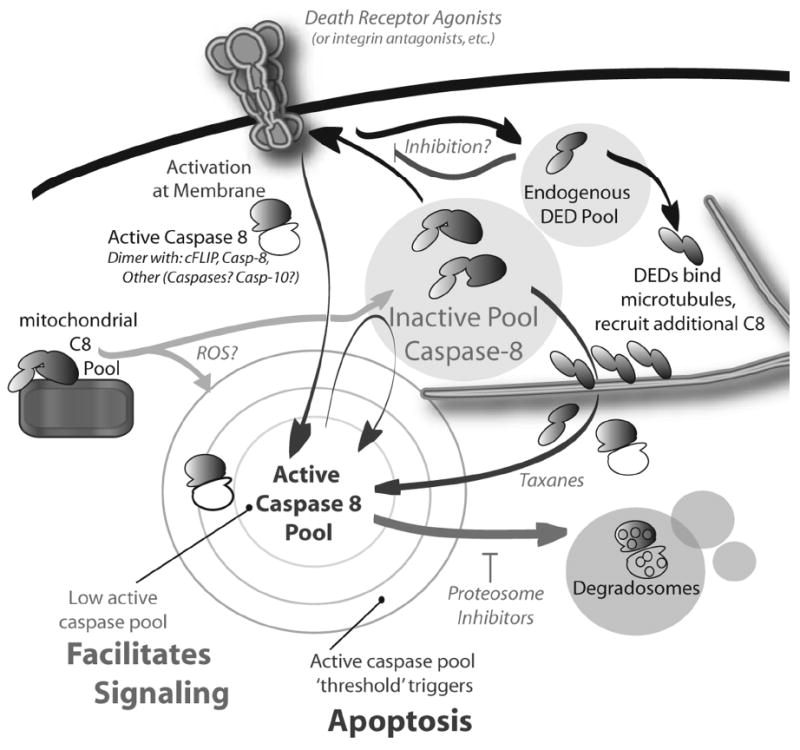
Activation of caspase-8 by death receptors (or other receptors such as integrins or toll-like receptors) may be sufficient to activate apoptosis, if threshold levels of active caspase-8 are achieved. However, insufficient activation of caspase-8 can occur due to proteosomal degradation of the caspase (inhibited by protoesomal inhibitors), competition for access to the DISC due to inhibitors such as cFLIP, or phosphorylation of the catalytic domain, which blocks maturation cleavage and/or dimerization. In this case, insufficient caspase-3 is activated to trigger cell death. Low or negligible levels of caspase-8 activity can, however, support most (if not all) of the nonapoptotic functions of caspase-8. In some cases, caspase-8 activation could potentially be enhanced by cooperativity between receptors systems or by secondary amplification of mitochondrial or microtubule-associated caspase-8 (enhanced by reactive oxygen species, ROS, and taxanes, respectively). This would be expected to assist cells in achieving threshold levels of caspase-8 activity necessary to trigger apoptosis.
2. Caspase-8 Activation via Death Receptors
Caspase-8 was identified as a key component of the DISC which forms following ligation of CD95/FAS [21]. Ligation of the CD95/FAS with Fas Ligand (FASL) promotes conformational changes in the receptor permitting interaction of a cytosolic ‘Death Domain’ (DD) from FAS/CD95 with a DD in the adaptor protein FADD (Fas-Associated, Death Domain containing). DDs are part of the structural superfamily composed of DEDs as well as CARD (caspase recruitment domain) and pyrin domains [22]. FADD itself is composed simply of a DD and a DED; binding to CD95/Fas via its DD is permissive for subsequent DED:DED mediated recruitment of caspase-8. In turn, this facilitates caspase-8 dimerization and maturation processing. The characterization of this minimal DISC cemented the designation of CD95/FAS as a DR. Structurally, DRs are defined by homology to the tumor necrosis factor α (TNFα) receptor in the extracellular region, permitting homotrimerization of the receptor, as well as a cytosolic DD that can bind FADD and promote cell death. A number of members of the TNFα receptor family have been characterized, and at least 9 appear to have a cytosolic DD [23].
This has had important implications for the development of DR-targeted pro-apoptotic therapies. Although CD95/Fas is present in most tissues and is frequently present in tumors, and although agonistic antibodies that trigger CD95/Fas mediated apoptosis have been described, the capacity for caspase-8 to elicit programmed cell death following CD95/Fas ligation can nonetheless vary dramatically from cell type to cell type. This implies that there are additional factors that regulate signals originating from this pathway. For example, hepatocytes are sensitive to apoptosis induced via Fas ligand, presenting a potential complication and limitation to the development of therapeutic CD95/FAS agonists [24], while expression of K-ras inhibits DR-mediated killing in a Raf1 dependent manner in colon carcinoma [25]. Similarly, the systemic effects of the potent pro-inflammatory mediator TNFα prevents its general use as proapoptotic ligands, although some clinical approaches using ‘high dose’ TNFα by enforcing ‘compartmentalization’ of the therapy have met with promise [26].
To date, the greatest clinical success among the DR ligands is related to exploiting the tumor necrosis factor-related apoptosis inducing ligand (Apo 2 ligand, commonly known as TRAIL). In this case, the apoptosis-transducing signaling receptors for TRAIL, DR4 and DR5 are not present on most tissue cells, but instead are frequently upregulated during malignancy. This selective upregulation is significant, as it allows avoidance of general systemic toxic effects of other DRs, and TRAIL based therapies have been approved for clinical use [27]. Nonetheless, TRAIL-based therapies are not issue-free, and variation in tumor susceptibility is seen, again illustrating the complexity of achieving ‘therapeutic’ programmed cell death. The travails of TRAIL mediated therapy are addressed by other authors within this volume.
3. Evaluating Caspase-8 Expression in Tumors
Targeting a programmed cell death pathway in a therapeutic setting depends upon continued expression of the minimal necessary functional determinants of that death cascade. Disregulation of a single component of a multiprotein cascade can be sufficient to compromise apoptosis and permit tumor ‘escape.’ In the case of DR-mediated killing, the loss of the DR, an adaptor protein (eg., FADD) or a caspase can compromise the death cascade. Moreover, DRs such as CD95/Fas and TNFα RI are also associated with non-apoptotic signaling events [28; 29]. Therefore, in a worst-case scenario, the disregulation of programmed cell death components could result in a switch to a non-apoptotic signaling pathway, thus aiding tumor growth or survival. For example, expression of the DR CD95/Fas has been linked to enhanced tumor growth [30], intimating that this receptor is usurped to nonapoptotic functions during tumor progression.
In the case of caspase-8, there are different answers as to whether the caspase is actually ‘lost’ during tumor progression. Notably, an overt loss of caspase-8 expression is not commonly observed in most of the epithelial-derived cancers [31; 32; 33; 34]. Among tumors, such as colon carcinoma, that progress through multiple stages, the loss of caspase-8 is nonetheless a rare event. Inactivating mutations in the gene occur relatively rarely, though this still accounts for a substantial number of cases due to the prevalence of colon cancer. Many developing carcinoma express oncogenic activities, such as supraphysiologic expression of caspase inhibitors, or activation of growth factor receptors or Src kinases which can inhibit caspase-8 activation [35; 36; 37]. It is likely that the multiplicity of lesions present in these tumors permits maintenance of caspase-8 expression, and it seems reasonable that, in the absence of its pro-apoptotic activity, caspase-8 expression could be selected for in this tumor environment.
By contrast, the loss of caspase-8 expression occurs very frequently in malignant neuroendocrine tumors, such as neuroblastoma [38; 39], primitive neuroectoderm tumors [40] and small cell lung carcinoma [41], as well as in brain tumors such as medulloblastoma [42] and relapsing glioblastoma [43]. Many of these tumors can arise rapidly, and/or during development, and it is possible that without a long term oncogenic niche (ie., an appropriate microenvironment) to develop in, the expression of caspase-8 is simply not tolerated well in these tumors. In this regard, it may be worth noting that many of the tumors which lose caspase-8 arise from cells that do not form extensive integrin-extracellular matrix (ECM) contacts, and thus are lacking in a relatively potent survival mechanism. Similarly, among lymphoid cells, which tend towards (a) susceptibility to DR-mediated killing and (b) exhibit high levels of caspase-8 expression, there is a tendency towards decreased (but not lost) expression of caspase-8 that is associated with malignant transformation. In this case, it is likely that the maintenance of low levels of caspase-8 expression reflects a balance between a requirement for caspase-8 signaling in the immune compartment [28; 44], while in turn attenuating apoptotic function. Frequently, the depression in caspase-8 expression occurs via methylation of the caspase-8 promoter (with deletion of the gene occasionally observed). In contrast to epithelial cancers, where a link between caspase-8 and tumor malignancy is less established, the loss of caspase-8 expression is linked to poor outcome in neuroendocrine tumors.
The expression of caspase-8 proteins in normal and tumor tissue has been documented as part of the Human Protein Atlas (HPA) project [45] (Figure 2). These studies are particularly useful, because they demonstrate the actual presence and persistence of the protein antigen, rather than simply measuring whether an mRNA transcript is present. Indeed, in many cases its not clear which isotypes are identified. For caspase-8, HPA uses a panel of four different antibodies. Of these, the most relevant, based on specificity and strength of interaction, appear to be HPA001302, which is raised against the p10 peptide of the catalytic domain, (excluding a penultimate conserved sequence also found in the related caspase-10) and HPA005688, which is raised against the second DED/linker/early catalytic domain (this antisera may recognize a number of differently spliced isoforms, but will vary in its capacity to recognize any given isoform). These immunohistological studies provide validation for the assertion that caspase-8 expression is not ‘generally lost’ in the majority of cancers, and that the down-regulation seen in neuroectoderm-derived tumors represents a special, and interesting, case. It is noteworthy that the conclusions of the HPA pathologists evaluating the immunohistological distribution of caspase-8 actually support a caspase-8 ‘up-regulation’ in many malignancies, particularly hepatocarcinoma, as well as pancreatic and cervical cancers. In contrast to these epithelial cancers, detection of caspase-8 was decreased in prostate cancer. Reasons for decreased detection are not yet clear, but could relate to the relatively specialized niche in which the prostate tumor evolves.
Figure 2. Caspase-8 Expression in Cancer.
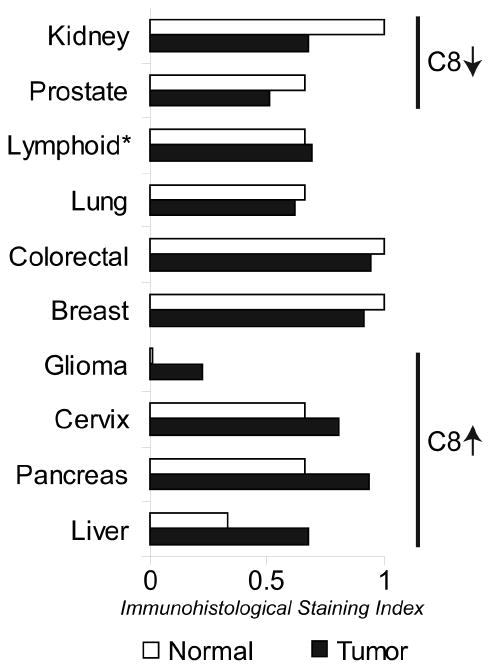
The graph shows the relative expression of caspase-8 in normal and neoplastic tissues. Data are derived as a measure of the pathologist-scored immunohistology using antisera to the p10 region of the catalytic domain (HPA001302; staining for full length caspase-8 isoforms). *Lymphoid compares lymphoma to activated lymphoid cells in lymph node reactive centers. Naïve lymphoid cells, outside these regions, do express higher levels of caspase-8 (~1).
Glioma also represents a special case. Although caspase-8 has been documented to be absent in many glioblastoma, its not clear that glial cells themselves actually express detectable full length caspase-8. In contrast to the glial cells, it has been documented that neighboring neurons do express caspase-8 [46]. Thus, it appears that glioma that express caspase-8 may actually express it de novo, and may upregulate caspase-8 expression, apparently as a mechanism that contributes to malignancy [47]. The up-regulation of a pro-death enzyme such as caspase-8 might, at first, appear to present a paradox, but is very likely to be explained by recent studies that firmly establish non-apoptotic roles for caspase-8.
4. Known Anti-tumor roles for Caspase-8
Most investigations of caspase-8 as an anti-tumor protein have focused on its role as an effector of DR-mediated killing, and its seems cleat that cell surface death ligands such as TRAIL, FASL or TNFα might be used to enforce tissue integrity or aid immune surveillance. Other roles are possible, however. Studies in our lab have supported a causal relationship between expression of caspase-8 and neuroblastoma aggression [48]. Genetic manipulation of caspase-8 expression, either via shRNA-mediated knockdown or by reconstitution to ‘physiological’ expression levels, can directly influence the metastatic potential of neuroblastoma in vivo. Perhaps not surprisingly, caspase-8 expression suppressed metastasis independent of the cellular capacity to undergo DR-mediated apoptosis. We observed that, in tumors where death ligands (TRAIL, TNFα, FASL) did not induce cell death, a caspase-8 dependent killing was still observed among cells in an inappropriate ECM microenvironment. We speculated that this could be due to Integrin-mediated death, a pathway associated with anoikis which we defined some time ago [9]. Alternatively, it might also relate to changes in cell susceptibility due to decreases in the level of caspase-8 activation required to initiate apoptosis.
Interestingly, the role of caspase-8 in suppressing tumor progression was limited to suppressing metastasis, but not primary tumor growth, in our models. In contrast, it has been reported that caspase-8 expression can directly suppress the ability of normal cells to undergo oncogenic transformation [49], however, this may be a semantic interpretation of the two model systems. A common feature of either system is that when a ‘correct’ ECM is present, ie., one which ligates a quorum of cell surface integrins as a rigid scaffold, the expression of caspase-8 is less relevant. However, when a cell finds itself within a ‘transit’ or foreign environment, such as an agarose-filled tissue culture dish or an inappropriate tissue, selection against caspase-8 expressing cells is facilitated. In this regard, it is interesting to note that tumors which arise in a niche where ligation of integrins by a robust ECM occurs appear to have a lesser requirement for suppression of caspase-8. Sites with rich integrin contacts promote signaling events, such as the activation of oncogenic tyrosine kinases [37] and the upregulation of bcl-2 proteins [50] that allow cells to tolerate caspase-8 expression. Small cell tumors, hematologic tumors and tumors which arise in the brain (which has a largely proteoglycan-based ECM) have a greater tendency to lack caspase-8 expression.
5. Targeting Caspase-8 Regulation via Small Molecules and Therapeutics
The expression of caspase-8 also predisposes tumors to apoptosis induced by chemotherapeutic agents other than DR-antagonists [51]. Microtubule-stabilizing agents, such as taxanes, promote caspase-8 mediated apoptosis, possibly via upregulation of components of the TRAIL pathway [51; 52] or by amplification of caspase-8 activation via microtubule-anchored DED ‘filaments’ [17]. In contrast, protection against FAS-mediated killing is observed in vivo with microtubule disrupting agents such as colchicine [24]. Thus, caspase-8 mediated killing may be influenced by the status of cellular tubulin, which would appear to have implications for the design of rationale combinatorial therapies.
Similarly, the capacity for caspase-8 to promote apoptosis is linked to the production of reactive oxygen species (ROS) in the cell. At least in the case of DRs, the presence of ROS favors a proapoptotic over NF-κB signaling mode [53], though it is possible that it is death ligand independent [54], and requires only the DR and/or caspase. It is not clear exactly how the ROS act. It is possible that it mobilizes sequestered pools of existing caspase-8 from sites such as the mitochondria, a key site for ROS generation. Caspase-8 can be bound to the mitochondrial surface via Bifunctional Apoptosis Regulator (BAR) [55; 56]. This makes it tempting to speculate that ROS production at the mitochondria (regulated positively by JNK and negatively by NF-κb) might mobilize this sequestered pool of zymogen. The production of ROS does interact with known caspase-8 signaling pathways. Mechanistically, the production of specific ROS such as nitric oxide can result in nitrosylation of both the p50 and p65 subunits of NF-κB [57]. This inhibits transcription of NF-κB target genes, including mitochondrial superoxide dismutase [58], a key enzyme limiting ROS accumulation at mitochondria. Thus, levels of ROS in a target cell may act as one of the switches that toggle between the apoptotic and nonapoptotic functions of caspase-8.
Increased caspase-8 ‘expression’ has also been reported after treatment with proteosomal inhibitors such as Bortezomib and NPI-0052. These small molecules exhibit increased potency when used against tumor cell populations that express caspase-8, likely via multiple mechanisms. Proteosomal inhibition can increase total cellular caspase-8 levels [59] presumably by blocking degradation [18]. Upregulation of DRs [60], and blockade of NF-κB-mediated signaling [61] also occur, thus promoting ROS production and mobilizing other death-promoting factors. However, these treatments do not result in de facto decreases in prosurvival factors such as Bcl-2 proteins, suggesting that this may not be absolutely necessary to induce cell death.
Clinically, it is possible to up-regulate the transcription of caspase-8 in some cellular contexts via treatment with therapeutic agents, cytokines and agents of differentiation. In the case of neuroblastoma, retinoic acid, 5-aza-cytidine [38] and interferon-γ all promote caspase-8 expression [62], though via different mechanisms. While retinoic acids such as fenretinide appear to impact both transcription (presumed to be indirect, via the induction of secondary differentiation factors) [63] as well as activation (via the coordinated induction of DR signaling components) [64], Vidaza and 5-aza-cytidines act by preventing methylation in the caspase-8 promoter. Interferon-γ can promote caspase-8 expression completely independent of methylation status [65], acting via transcription factors of the STAT family. Clinically, fenretinide and interferon-γ would appear most promising, as they fulfill two key roles; increasing caspase-8 expression and favoring the activation of cell death pathways.
6. Nonapoptotic Functions of Caspase-8
Promoting caspase-8 expression and activity to initiate programmed cell death presents a therapeutic opportunity in cancer. Nonetheless, caspase-8 expression is heterogenous and sometimes even elevated in tumors. Therefore, some doubt remains as to whether it is always desirable to promote caspase-8 expression. Certainly, a number of malignant tumors tolerate very high levels of procaspase-8. Given the intense selection pressure present in a tumor’s microenvironment, it seems likely that the apparent upregulation of procaspase-8 in these tumors represents exploitation of one or more nonapoptotic roles of caspase-8.
In mice, the loss of caspase-8 expression during development is lethal due to profound vascular and neural tube defects independent of its proapoptotic activity [28]. Within the immune compartment, caspase-8 plays a key role in signaling via the NF-κB pathway downstream of the antigen receptor [44], possibly via interactions with RIP and NIK [66] but independent of FADD [67]. However, FADD plays a role in promoting caspase-8 recruitment and survival during autophagic signaling in activated T-cells [68]. In tumors, limited autophagy is protective, and can promote resistance to DR-mediated killing [69], providing a precedent for caspase-8-mediated tumor survival.
Caspase-8 expression can also directly promote tumor aggression. Some of the peripheral pool of caspase-8 can localize to focal contacts formed by integrin binding to the ECM [70], as well as to ruffles at the leading edge of migrating cells. At these sites, caspase-8 is phosphorylated by nonreceptor tyrosine kinases, such as Src. This may impact this critical periperhal pool of caspase-8 in several ways. First, phosphorylation of caspase-8 on tyrosine 380 inhibits its proteolytic activation [36; 37; 71], (although this may conversely be regulated by the actions of phosphatases) [71]. Secondly, it promotes binding of caspase-8 by several Src homology domain 2 (SH2) proteins, including Src-family kinases themselves [72; 73] and the p85 subunit of phosphoinositide 3’ kinase [74], which in turn promotes cell migration and invasion via the activation of calpains [72; 75] and by alterations to small G-protein signaling and to integrin recycling to the cell surface [76]. Thus, interaction between the ECM may be cell-protective via a capacity to toggle caspase-8 from apoptotic to non-apoptotic roles.
Among cells with a functional cell death pathway, the potentially malignant effects of caspase-8 expression, such as enhanced cell migration, may be balanced by the ongoing death of cells leaving the primary tumor site. However, among cells with deficiencies in the caspase cascade [70] or among cells with constitutive (presumably oncogenic) phosphorylation of caspase-8 [37], caspase-8 function may be selectively usurped to non-apoptotic functions. In such cases, the enhanced expression of caspase-8 in a tumor would be undesirable.
7. Conclusions
Caspase-8 was first identified as a zymogenic protease that acted as a critical effector of DR-mediated killing, but has since been determined to be a highly versatile molecule that impacts many cellular signaling pathways on a day to day basis (Figure 3). Given the variety of roles possible for caspase-8, and the fact that it is not generally lost in tumors of breast, colon or lung (non small cell), caution should be taken in assigning it a general ‘anti-tumor’ role, and therefore also in adopting therapeutic strategies to elevate caspase-8 expression. In extreme cases, it might even be desirable to suppress caspase-8 expression. Nonetheless, as our understanding of caspase-8 function progresses, and molecular mechanisms toggling caspase-8 between apoptotic and nonapoptotic roles become clear, it should be possible to devise coordinated therapeutic strategies that elevate caspase-8 expression while enhancing its death-inducing functions.
Figure 3. Caspase-8 Plays Diverse Cellular Roles.
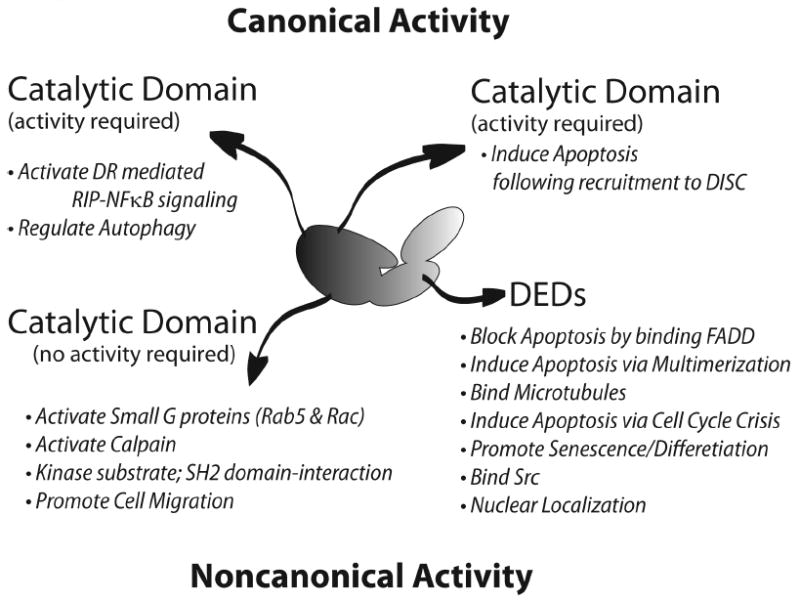
Many of the known roles of caspase-8 are summarized, coupled to the domains required to execute those activities.
Footnotes
Conflicts of interest Statement None Declared
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Garcia-Calvo M, Peterson EP, Leiting B, Ruel R, Nicholson DW, Thornberry NA. Inhibition of human caspases by peptide-based and macromolecular inhibitors. J Biol Chem. 1998;273:32608–32613. doi: 10.1074/jbc.273.49.32608. [DOI] [PubMed] [Google Scholar]
- 2.Rozman-Pungercar J, Kopitar-Jerala N, Bogyo M, Turk D, Vasiljeva O, Stefe I, Vandenabeele P, Bromme D, Puizdar V, Fonovic M, Trstenjak-Prebanda M, Dolenc I, Turk V, Turk B. Inhibition of papain-like cysteine proteases and legumain by caspase-specific inhibitors: when reaction mechanism is more important than specificity. Cell Death Differ. 2003;10:881–888. doi: 10.1038/sj.cdd.4401247. [DOI] [PubMed] [Google Scholar]
- 3.Boatright KM, Renatus M, Scott FL, Sperandio S, Shin H, Pedersen IM, Ricci JE, Edris WA, Sutherlin DP, Green DR, Salvesen GS. A unified model for apical caspase activation. Mol Cell. 2003;11:529–541. doi: 10.1016/s1097-2765(03)00051-0. [DOI] [PubMed] [Google Scholar]
- 4.Pop C, Fitzgerald P, Green DR, Salvesen GS. Role of proteolysis in caspase-8 activation and stabilization. Biochemistry. 2007;46:4398–4407. doi: 10.1021/bi602623b. [DOI] [PubMed] [Google Scholar]
- 5.Oberst A, Pop C, Tremblay AG, Blais V, Denault JB, Salvesen GS, Green DR. Inducible dimerization and inducible cleavage reveal a requirement for both processes in caspase-8 activation. J Biol Chem. 2010 doi: 10.1074/jbc.M109.095083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blanchard H, Donepudi M, Tschopp M, Kodandapani L, Wu JC, Grutter MG. Caspase-8 specificity probed at subsite S(4): crystal structure of the caspase-8-Z-DEVD-cho complex. J Mol Biol. 2000;302:9–16. doi: 10.1006/jmbi.2000.4041. [DOI] [PubMed] [Google Scholar]
- 7.Keller N, Grutter MG, Zerbe O. Studies of the molecular mechanism of caspase-8 activation by solution NMR. Cell Death Differ. 2009 doi: 10.1038/cdd.2009.155. [DOI] [PubMed] [Google Scholar]
- 8.Muzio M, Stockwell BR, Stennicke HR, Salvesen GS, Dixit VM. An induced proximity model for caspase-8 activation. J Biol Chem. 1998;273:2926–2930. doi: 10.1074/jbc.273.5.2926. [DOI] [PubMed] [Google Scholar]
- 9.Stupack DG, Puente XS, Boutsaboualoy S, Storgard CM, Cheresh DA. Apoptosis of adherent cells by recruitment of caspase-8 to unligated integrins. J Cell Biol. 2001;155:459–470. doi: 10.1083/jcb.200106070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boatright KM, Deis C, Denault JB, Sutherlin DP, Salvesen GS. Activation of caspases-8 and -10 by FLIP(L) Biochem J. 2004;382:651–657. doi: 10.1042/BJ20040809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scaffidi C, Medema JP, Krammer PH, Peter ME. FLICE is predominantly expressed as two functionally active isoforms, caspase-8/a and caspase-8/b. J Biol Chem. 1997;272:26953–26958. doi: 10.1074/jbc.272.43.26953. [DOI] [PubMed] [Google Scholar]
- 12.Eckhart L, Ballaun C, Hermann M, VandeBerg JL, Sipos W, Uthman A, Fischer H, Tschachler E. Identification of novel mammalian caspases reveals an important role of gene loss in shaping the human caspase repertoire. Mol Biol Evol. 2008;25:831–841. doi: 10.1093/molbev/msn012. [DOI] [PubMed] [Google Scholar]
- 13.Chun HJ, Zheng L, Ahmad M, Wang J, Speirs CK, Siegel RM, Dale JK, Puck J, Davis J, Hall CG, Skoda-Smith S, Atkinson TP, Straus SE, Lenardo MJ. Pleiotropic defects in lymphocyte activation caused by caspase-8 mutations lead to human immunodeficiency. Nature. 2002;419:395–399. doi: 10.1038/nature01063. [DOI] [PubMed] [Google Scholar]
- 14.Himeji D, Horiuchi T, Tsukamoto H, Hayashi K, Watanabe T, Harada M. Characterization of caspase-8L: a novel isoform of caspase-8 that behaves as an inhibitor of the caspase cascade. Blood. 2002;99:4070–4078. doi: 10.1182/blood.v99.11.4070. [DOI] [PubMed] [Google Scholar]
- 15.Miller MA, Karacay B, Zhu X, O’Dorisio MS, Sandler AD. Caspase 8L, a novel inhibitory isoform of caspase 8, is associated with undifferentiated neuroblastoma. Apoptosis. 2006;11:15–24. doi: 10.1007/s10495-005-3258-0. [DOI] [PubMed] [Google Scholar]
- 16.Tsukumo SI, Yonehara S. Requirement of cooperative functions of two repeated death effector domains in caspase-8 and in MC159 for induction and inhibition of apoptosis, respectively. Genes Cells. 1999;4:541–549. doi: 10.1046/j.1365-2443.1999.00280.x. [DOI] [PubMed] [Google Scholar]
- 17.Mielgo A, Torres VA, Clair K, Barbero S, Stupack DG. Paclitaxel promotes a caspase 8-mediated apoptosis through death effector domain association with microtubules. Oncogene. 2009;28:3551–3562. doi: 10.1038/onc.2009.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McDonald ER, 3rd, El-Deiry WS. Suppression of caspase-8- and -10-associated RING proteins results in sensitization to death ligands and inhibition of tumor cell growth. Proc Natl Acad Sci U S A. 2004;101:6170–6175. doi: 10.1073/pnas.0307459101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Siegel RM, Martin DA, Zheng L, Ng SY, Bertin J, Cohen J, Lenardo MJ. Death-effector filaments: novel cytoplasmic structures that recruit caspases and trigger apoptosis. J Cell Biol. 1998;141:1243–1253. doi: 10.1083/jcb.141.5.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mielgo A, Torres VA, Schmid MC, Graf R, Zeitlin S, Lee P, Shields DJ, Barbero S, Jamora C, Stupack DG. The death effector domains of caspase-8 induce terminal differentiation. PLoS ONE. 2009 doi: 10.1371/journal.pone.0007879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Medema JP, Scaffidi C, Kischkel FC, Shevchenko A, Mann M, Krammer PH, Peter ME. FLICE is activated by association with the CD95 death-inducing signaling complex (DISC) Embo J. 1997;16:2794–2804. doi: 10.1093/emboj/16.10.2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park HH, Lo YC, Lin SC, Wang L, Yang JK, Wu H. The death domain superfamily in intracellular signaling of apoptosis and inflammation. Annu Rev Immunol. 2007;25:561–586. doi: 10.1146/annurev.immunol.25.022106.141656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mahmood Z, Shukla Y. Death receptors: targets for cancer therapy. Exp Cell Res. 2010;316:887–899. doi: 10.1016/j.yexcr.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 24.Feng G, Kaplowitz N. Colchicine protects mice from the lethal effect of an agonistic anti-Fas antibody. J Clin Invest. 2000;105:329–339. doi: 10.1172/JCI7398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoogwater FJH, et al. Oncogenic K-Ras Turns Death Receptors Into Metastasis-Promoting Receptors in Human and Mouse Colorectal Cancer Cells. Gastroenterology. 2010;138 doi: 10.1053/j.gastro.2010.02.046. In press. [DOI] [PubMed] [Google Scholar]
- 26.Lejeune FJ, Ruegg C. Recombinant human tumor necrosis factor: an efficient agent for cancer treatment. Bull Cancer. 2006;93:E90–100. [PubMed] [Google Scholar]
- 27.Bellail AC, Qi L, Mulligan P, Chhabra V, Hao C. TRAIL agonists on clinical trials for cancer therapy: the promises and the challenges. Rev Recent Clin Trials. 2009;4:34–41. doi: 10.2174/157488709787047530. [DOI] [PubMed] [Google Scholar]
- 28.Kang TB, Ben-Moshe T, Varfolomeev EE, Pewzner-Jung Y, Yogev N, Jurewicz A, Waisman A, Brenner O, Haffner R, Gustafsson E, Ramakrishnan P, Lapidot T, Wallach D. Caspase-8 serves both apoptotic and nonapoptotic roles. J Immunol. 2004;173:2976–2984. doi: 10.4049/jimmunol.173.5.2976. [DOI] [PubMed] [Google Scholar]
- 29.Barnhart BC, Legembre P, Pietras E, Bubici C, Franzoso G, Peter ME. CD95 ligand induces motility and invasiveness of apoptosis-resistant tumor cells. Embo J. 2004;23:3175–3185. doi: 10.1038/sj.emboj.7600325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen L, Park SM, Tumanov AV, Hau A, Sawada K, Feig C, Turner JR, Fu YX, Romero IL, Lengyel E, Peter ME. CD95 promotes tumour growth. Nature. 2010;465:492–496. doi: 10.1038/nature09075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim HS, Lee JW, Soung YH, Park WS, Kim SY, Lee JH, Park JY, Cho YG, Kim CJ, Jeong SW, Nam SW, Kim SH, Lee JY, Yoo NJ, Lee SH. Inactivating mutations of caspase-8 gene in colorectal carcinomas. Gastroenterology. 2003;125:708–715. doi: 10.1016/s0016-5085(03)01059-x. [DOI] [PubMed] [Google Scholar]
- 32.Soung YH, Lee JW, Kim SY, Jang J, Park YG, Park WS, Nam SW, Lee JY, Yoo NJ, Lee SH. CASPASE-8 gene is inactivated by somatic mutations in gastric carcinomas. Cancer Res. 2005;65:815–821. [PubMed] [Google Scholar]
- 33.Lubahn J, Berndt SI, Jin CH, Klim A, Luly J, Wu WS, Isaacs S, Wiley K, Isaacs WB, Suarez BK, Hayes RB, Kibel AS. Association of CASP8 D302H polymorphism with reduced risk of aggressive prostate carcinoma. Prostate. 2010;70:646–653. doi: 10.1002/pros.21098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.MacPherson G, Healey CS, Teare MD, Balasubramanian SP, Reed MW, Pharoah PD, Ponder BA, Meuth M, Bhattacharyya NP, Cox A. Association of a common variant of the CASP8 gene with reduced risk of breast cancer. J Natl Cancer Inst. 2004;96:1866–1869. doi: 10.1093/jnci/dji001. [DOI] [PubMed] [Google Scholar]
- 35.Iwase M, Takaoka S, Uchida M, Yoshiba S, Kondo G, Watanabe H, Ohashi M, Nagumo M. Epidermal growth factor receptor inhibitors enhance susceptibility to Fas-mediated apoptosis in oral squamous cell carcinoma cells. Oral Oncol. 2008;44:361–368. doi: 10.1016/j.oraloncology.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 36.Keller N, Mares J, Zerbe O, Grutter MG. Structural and biochemical studies on procaspase-8: new insights on initiator caspase activation. Structure. 2009;17:438–448. doi: 10.1016/j.str.2008.12.019. [DOI] [PubMed] [Google Scholar]
- 37.Cursi S, Rufini A, Stagni V, Condo I, Matafora V, Bachi A, Bonifazi AP, Coppola L, Superti-Furga G, Testi R, Barila D. Src kinase phosphorylates Caspase-8 on Tyr380: a novel mechanism of apoptosis suppression. Embo J. 2006;25:1895–1905. doi: 10.1038/sj.emboj.7601085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hopkins-Donaldson S, Bodmer JL, Bourloud KB, Brognara CB, Tschopp J, Gross N. Loss of caspase-8 expression in highly malignant human neuroblastoma cells correlates with resistance to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis. Cancer Res. 2000;60:4315–4319. [PubMed] [Google Scholar]
- 39.Teitz T, Lahti JM, Kidd VJ. Aggressive childhood neuroblastomas do not express caspase-8: an important component of programmed cell death. J Mol Med. 2001;79:428–436. doi: 10.1007/s001090100233. [DOI] [PubMed] [Google Scholar]
- 40.Grotzer MA, Eggert A, Zuzak TJ, Janss AJ, Marwaha S, Wiewrodt BR, Ikegaki N, Brodeur GM, Phillips PC. Resistance to TRAIL-induced apoptosis in primitive neuroectodermal brain tumor cells correlates with a loss of caspase-8 expression. Oncogene. 2000;19:4604–4610. doi: 10.1038/sj.onc.1203816. [DOI] [PubMed] [Google Scholar]
- 41.Shivapurkar N, Toyooka S, Eby MT, Huang CX, Sathyanarayana UG, Cunningham HT, Reddy JL, Brambilla E, Takahashi T, Minna JD, Chaudhary PM, Gazdar AF. Differential inactivation of caspase-8 in lung cancers. Cancer Biol Ther. 2002;1:65–69. doi: 10.4161/cbt.1.1.45. [DOI] [PubMed] [Google Scholar]
- 42.Ebinger M, Senf L, Wachowski O, Scheurlen W. Promoter methylation pattern of caspase-8, P16INK4A, MGMT, TIMP-3, and E-cadherin in medulloblastoma. Pathol Oncol Res. 2004;10:17–21. doi: 10.1007/BF02893403. [DOI] [PubMed] [Google Scholar]
- 43.Martinez R, Setien F, Voelter C, Casado S, Quesada MP, Schackert G, Esteller M. CpG island promoter hypermethylation of the pro-apoptotic gene caspase-8 is a common hallmark of relapsed glioblastoma multiforme. Carcinogenesis. 2007;28:1264–1268. doi: 10.1093/carcin/bgm014. [DOI] [PubMed] [Google Scholar]
- 44.Su H, Bidere N, Zheng L, Cubre A, Sakai K, Dale J, Salmena L, Hakem R, Straus S, Lenardo M. Requirement for caspase-8 in NF-kappaB activation by antigen receptor. Science. 2005;307:1465–1468. doi: 10.1126/science.1104765. [DOI] [PubMed] [Google Scholar]
- 45.Ponten F, Jirstrom K, Uhlen M. The Human Protein Atlas--a tool for pathology. J Pathol. 2008;216:387–393. doi: 10.1002/path.2440. [DOI] [PubMed] [Google Scholar]
- 46.Zhang X, Graham SH, Kochanek PM, Marion DW, Nathaniel PD, Watkins SC, Clark RS. Caspase-8 expression and proteolysis in human brain after severe head injury. Faseb J. 2003;17:1367–1369. doi: 10.1096/fj.02-1067fje. [DOI] [PubMed] [Google Scholar]
- 47.Gdynia G, Grund K, Eckert A, Bock BC, Funke B, Macher-Goeppinger S, Sieber S, Herold-Mende C, Wiestler B, Wiestler OD, Roth W. Basal caspase activity promotes migration and invasiveness in glioblastoma cells. Mol Cancer Res. 2007;5:1232–1240. doi: 10.1158/1541-7786.MCR-07-0343. [DOI] [PubMed] [Google Scholar]
- 48.Stupack DG, Teitz T, Potter MD, Mikolon D, Houghton PJ, Kidd VJ, Lahti JM, Cheresh DA. Potentiation of neuroblastoma metastasis by loss of caspase-8. Nature. 2006;439:95–99. doi: 10.1038/nature04323. [DOI] [PubMed] [Google Scholar]
- 49.Krelin Y, Zhang L, Kang TB, Appel E, Kovalenko A, Wallach D. Caspase-8 deficiency facilitates cellular transformation in vitro. Cell Death Differ. 2008;15:1350–1355. doi: 10.1038/cdd.2008.88. [DOI] [PubMed] [Google Scholar]
- 50.Reddig PJ, Juliano RL. Clinging to life: cell to matrix adhesion and cell survival. Cancer Metastasis Rev. 2005;24:425–439. doi: 10.1007/s10555-005-5134-3. [DOI] [PubMed] [Google Scholar]
- 51.Muhlethaler-Mottet A, Bourloud KB, Auderset K, Joseph JM, Gross N. Drug-mediated sensitization to TRAIL-induced apoptosis in caspase-8-complemented neuroblastoma cells proceeds via activation of intrinsic and extrinsic pathways and caspase-dependent cleavage of XIAP, Bcl-xL and RIP. Oncogene. 2004;23:5415–5425. doi: 10.1038/sj.onc.1207704. [DOI] [PubMed] [Google Scholar]
- 52.Nimmanapalli R, Perkins CL, Orlando M, O’Bryan E, Nguyen D, Bhalla KN. Pretreatment with paclitaxel enhances apo-2 ligand/tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis of prostate cancer cells by inducing death receptors 4 and 5 protein levels. Cancer Res. 2001;61:759–763. [PubMed] [Google Scholar]
- 53.Chen C, Liu Y, Zheng D. An agonistic monoclonal antibody against DR5 induces ROS production, sustained JNK activation and Endo G release in Jurkat leukemia cells. Cell Res. 2009;19:984–995. doi: 10.1038/cr.2009.60. [DOI] [PubMed] [Google Scholar]
- 54.Ling YH, Lin R, Perez-Soler R. Erlotinib induces mitochondrial-mediated apoptosis in human H3255 non-small-cell lung cancer cells with epidermal growth factor receptorL858R mutation through mitochondrial oxidative phosphorylation-dependent activation of BAX and BAK. Mol Pharmacol. 2008;74:793–806. doi: 10.1124/mol.107.044396. [DOI] [PubMed] [Google Scholar]
- 55.Zhang H, Xu Q, Krajewski S, Krajewska M, Xie Z, Fuess S, Kitada S, Pawlowski K, Godzik A, Reed JC. BAR: An apoptosis regulator at the intersection of caspases and Bcl-2 family proteins. Proc Natl Acad Sci U S A. 2000;97:2597–2602. doi: 10.1073/pnas.97.6.2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stegh AH, Barnhart BC, Volkland J, Algeciras-Schimnich A, Ke N, Reed JC, Peter ME. Inactivation of caspase-8 on mitochondria of Bcl-xL-expressing MCF7-Fas cells: role for the bifunctional apoptosis regulator protein. J Biol Chem. 2002;277:4351–4360. doi: 10.1074/jbc.M108947200. [DOI] [PubMed] [Google Scholar]
- 57.Kelleher ZT, Matsumoto A, Stamler JS, Marshall HE. NOS2 regulation of NF-kappaB by S-nitrosylation of p65. J Biol Chem. 2007;282:30667–30672. doi: 10.1074/jbc.M705929200. [DOI] [PubMed] [Google Scholar]
- 58.Bubici C, Papa S, Pham CG, Zazzeroni F, Franzoso G. NF-kappaB and JNK: an intricate affair. Cell Cycle. 2004;3:1524–1529. doi: 10.4161/cc.3.12.1321. [DOI] [PubMed] [Google Scholar]
- 59.Thorpe JA, Christian PA, Schwarze SR. Proteasome inhibition blocks caspase-8 degradation and sensitizes prostate cancer cells to death receptor-mediated apoptosis. Prostate. 2008;68:200–209. doi: 10.1002/pros.20706. [DOI] [PubMed] [Google Scholar]
- 60.Voortman J, Resende TP, Abou El Hassan MA, Giaccone G, Kruyt FA. TRAIL therapy in non-small cell lung cancer cells: sensitization to death receptor-mediated apoptosis by proteasome inhibitor bortezomib. Mol Cancer Ther. 2007;6:2103–2112. doi: 10.1158/1535-7163.MCT-07-0167. [DOI] [PubMed] [Google Scholar]
- 61.Chauhan D, Singh A, Brahmandam M, Podar K, Hideshima T, Richardson P, Munshi N, Palladino MA, Anderson KC. Combination of proteasome inhibitors bortezomib and NPI-0052 trigger in vivo synergistic cytotoxicity in multiple myeloma. Blood. 2008;111:1654–1664. doi: 10.1182/blood-2007-08-105601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Casciano I, Banelli B, Croce M, De Ambrosis A, di Vinci A, Gelvi I, Pagnan G, Brignole C, Allemanni G, Ferrini S, Ponzoni M, Romani M. Caspase-8 gene expression in neuroblastoma. Ann N Y Acad Sci. 2004;1028:157–167. doi: 10.1196/annals.1322.017. [DOI] [PubMed] [Google Scholar]
- 63.Raguenez G, Muhlethaler-Mottet A, Meier R, Duros C, Benard J, Gross N. Fenretinide-induced caspase-8 activation and apoptosis in an established model of metastatic neuroblastoma. BMC Cancer. 2009;9:97. doi: 10.1186/1471-2407-9-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kalli KR, Devine KE, Cabot MC, Arnt CR, Heldebrant MP, Svingen PA, Erlichman C, Hartmann LC, Conover CA, Kaufmann SH. Heterogeneous role of caspase-8 in fenretinide-induced apoptosis in epithelial ovarian carcinoma cell lines. Mol Pharmacol. 2003;64:1434–1443. doi: 10.1124/mol.64.6.1434. [DOI] [PubMed] [Google Scholar]
- 65.Fulda S, Debatin KM. IFNgamma sensitizes for apoptosis by upregulating caspase-8 expression through the Stat1 pathway. Oncogene. 2002;21:2295–2308. doi: 10.1038/sj.onc.1205255. [DOI] [PubMed] [Google Scholar]
- 66.Shikama Y, Yamada M, Miyashita T. Caspase-8 and caspase-10 activate NF-kappaB through RIP, NIK and IKKalpha kinases. Eur J Immunol. 2003;33:1998–2006. doi: 10.1002/eji.200324013. [DOI] [PubMed] [Google Scholar]
- 67.Arechiga AF, Bell BD, Solomon JC, Chu IH, Dubois CL, Hall BE, George TC, Coder DM, Walsh CM. Cutting edge: FADD is not required for antigen receptor-mediated NF-kappaB activation. J Immunol. 2005;175:7800–7804. doi: 10.4049/jimmunol.175.12.7800. [DOI] [PubMed] [Google Scholar]
- 68.Bell BD, Leverrier S, Weist BM, Newton RH, Arechiga AF, Luhrs KA, Morrissette NS, Walsh CM. FADD and caspase-8 control the outcome of autophagic signaling in proliferating T cells. Proc Natl Acad Sci U S A. 2008;105:16677–16682. doi: 10.1073/pnas.0808597105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Han J, Hou W, Goldstein LA, Lu C, Stolz DB, Yin XM, Rabinowich H. Involvement of protective autophagy in TRAIL resistance of apoptosis-defective tumor cells. J Biol Chem. 2008;283:19665–19677. doi: 10.1074/jbc.M710169200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Barbero S, Mielgo A, Torres V, Teitz T, Shields DJ, Mikolon D, Bogyo M, Barila D, Lahti JM, Schlaepfer D, Stupack DG. Caspase-8 association with the focal adhesion complex promotes tumor cell migration and metastasis. Cancer Res. 2009;69:3755–3763. doi: 10.1158/0008-5472.CAN-08-3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jia SH, Parodo J, Kapus A, Rotstein OD, Marshall JC. Dynamic Regulation of Neutrophil Survival through Tyrosine Phosphorylation or Dephosphorylation of Caspase-8. J Biol Chem. 2008;283:5402–5413. doi: 10.1074/jbc.M706462200. [DOI] [PubMed] [Google Scholar]
- 72.Barbero S, Barila D, Mielgo A, Stagni V, Clair K, Stupack D. Identification of a critical tyrosine residue in caspase 8 that promotes cell migration. J Biol Chem. 2008 doi: 10.1074/jbc.M800549200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Finlay D, Howes A, Vuori K. Critical role for caspase-8 in epidermal growth factor signaling. Cancer Res. 2009;69:5023–5029. doi: 10.1158/0008-5472.CAN-08-3731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Senft J, Helfer B, Frisch SM. Caspase-8 interacts with the p85 subunit of phosphatidylinositol 3-kinase to regulate cell adhesion and motility. Cancer Res. 2007;67:11505–11509. doi: 10.1158/0008-5472.CAN-07-5755. [DOI] [PubMed] [Google Scholar]
- 75.Helfer B, Boswell BC, Finlay D, Cipres A, Vuori K, Bong Kang T, Wallach D, Dorfleutner A, Lahti JM, Flynn DC, Frisch SM. Caspase-8 promotes cell motility and calpain activity under nonapoptotic conditions. Cancer Res. 2006;66:4273–4278. doi: 10.1158/0008-5472.CAN-05-4183. [DOI] [PubMed] [Google Scholar]
- 76.Torres VA, Mielgo A, Barbero S, Hsiao R, Wilkins JA, Stupack DG. Rab5 mediates caspase-8-promoted cell motility and metastasis. Mol Biol Cell. 2010;21:369–376. doi: 10.1091/mbc.E09-09-0769. [DOI] [PMC free article] [PubMed] [Google Scholar]