

Dynamics and allosteric potential of the AMPA receptor N-terminal domain
The crystal structure of the AMPA receptor subunit GluA3 N-terminal domain (NTD) reveals unexpected structural flexibility of the NTD that might affect ion channel activity.
Keywords: AMPA receptor allostery, AMPA receptor function, GluA3 N-terminal domain structure, NTD ligand
Abstract
Glutamate-gated ion channels (ionotropic glutamate receptors, iGluRs) sense the extracellular milieu via an extensive extracellular portion, comprised of two clamshell-shaped segments. The distal, N-terminal domain (NTD) has allosteric potential in NMDA-type iGluRs, which has not been ascribed to the analogous domain in AMPA receptors (AMPARs). In this study, we present new structural data uncovering dynamic properties of the GluA2 and GluA3 AMPAR NTDs. GluA3 features a zipped-open dimer interface with unconstrained lower clamshell lobes, reminiscent of metabotropic GluRs (mGluRs). The resulting labile interface supports interprotomer rotations, which can be transmitted to downstream receptor segments. Normal mode analysis reveals two dominant mechanisms of AMPAR NTD motion: intraprotomer clamshell motions and interprotomer counter-rotations, as well as accessible interconversion between AMPAR and mGluR conformations. In addition, we detect electron density for a potential ligand in the GluA2 interlobe cleft, which may trigger lobe motions. Together, these data support a dynamic role for the AMPAR NTDs, which widens the allosteric landscape of the receptor and could provide a novel target for ligand development.
Introduction
Binding of L-glutamate to ionotropic glutamate receptors (iGluRs) initiates excitatory neurotransmission in vertebrate central nervous systems. This process is mediated by a series of conformational transitions, ultimately resulting in opening of the ion channel and depolarization of the post-synaptic membrane (Traynelis et al, 2010).
Ionotropic GluRs are arranged as dimers of dimers into receptor tetramers (Sobolevsky et al, 2009). The extracellular portion of each subunit consists of two domains, the ligand-binding domain (LBD) and the N-terminal domain (NTD), which resemble bacterial periplasmic-binding proteins (PBPs), ancient bilobate structures evolutionarily selected to capture ligand (O'Hara et al, 1993; Quiocho and Ledvina, 1996; Madden, 2002). Within subunit dimers, these domains are arranged as two-fold symmetric pairs of protomers, each consisting of two lobes, the upper and lower lobes (UL and LL; Armstrong and Gouaux, 2000; Mayer, 2005; Clayton et al, 2009; Jin et al, 2009). L-Glutamate docks to the membrane proximal LBD, which triggers lobe closure and initiation of the gating cascade. In AMPA-type iGluRs (AMPARs), the number of glutamate molecules bound to the receptor (up to four) determines open/closed-channel states and gives rise to complex gating properties, which ultimately shape excitatory signalling. AMPAR gating kinetics are modulated further by alternative RNA processing within the LBD and by a variety of drugs targeting the LBD dimer interface and binding cleft, respectively (Traynelis et al, 2010) (Lomeli et al, 1994; Mosbacher et al, 1994; Jin et al, 2005). Through their capacity to strengthen AMPAR transmission, small-molecule LBD modulators have entered clinical trials as cognitive enhancers (Lynch, 2002; Bowie, 2008; Ward et al, 2010).
The second, membrane-distal extracellular portion, the NTD, is structurally related to bacterial leucine-binding protein (Trakhanov et al, 2005), and also closely resembles the ligand-binding cores (LBCs) of natriuretic peptide receptors (He et al, 2005) and type-C G-protein-coupled receptors (GPCRs), including the type-B γ-aminobutyric receptor (GABABR) and the metabotropic glutamate receptors (mGluR1–8; Pin et al, 2003). In mGluRs, glutamate binding within the interlobe cleft triggers a ∼30° interlobe closure motion and a rearrangement of the dimer interface, which initiates G-protein signalling (Kunishima et al, 2000; Tsuchiya et al, 2002). To date, ligand binding to this distal domain in iGluRs has been associated exclusively with the NMDA-type receptors (NMDARs). Zn2+ docking to NR-2 subunit NTDs results in a downregulation of channel activity, presumably via closure of the NTD clamshell (Karakas et al, 2009; Hansen et al, 2010). The opposite effect, an increase in NMDAR open probability, was achieved by wedging the cleft open, implying bi-directional control of channel activity via NTD clamshell motions (Gielen et al, 2009). An allosteric path, originating in the ligand-binding cleft, successively transmitted through the NTD–LBD linker region, the LBD dimer interface and down to the channel gate, has been suggested for NMDARs (Gielen et al, 2008, 2009). Ifenprodil and related NTD-targeting drugs modulate NMDARs and thereby further enrich the functional spectrum of these ion channels (Mony et al, 2009; Hansen et al, 2010).
By contrast, AMPAR NTD ligands have not been described, and recent structural data of GluA2 and GluK2 NTDs in the nonNMDAR subfamily have been interpreted to rule out a signalling capacity for this domain (Jin et al, 2009; Kumar et al, 2009). Also, whereas in mGluRs, the LLs of the clamshell are free to rotate upward in response to ligand binding (Kunishima et al, 2000), the LLs in GluA2 and GluK2 NTDs appear constrained due to dimeric packing. As a result, the NTD has been suggested to function purely as a rigid subunit assembly device in nonNMDA receptors (Jin et al, 2009; Kumar et al, 2009).
In this study, we present new high-resolution AMPAR NTD structures and the first analysis of their structure-encoded dynamics, which reveal (i) electron density within the GluA2-binding pocket and thus potential ligand-binding capacity for AMPAR NTDs, (ii) a structurally labile GluA3 dimer interface, which facilitates interprotomer rearrangements; and (iii) an intrinsic ability of the protomers themselves to undergo clamshell-like motions, similar to other PBPs. Normal mode analysis (NMA) based on the anisotropic network model (ANM) (Atilgan et al, 2001; Bahar et al, 2010a, 2010b) suggests that classic clamshell motions are more prominent in GluA3 due to unconstrained LLs (similar to mGluR LBCs), but can also be discerned in the more tightly packed GluA2 NTD. NMA further demonstrates that iGluR NTD global motions resemble those of mGluR LBCs. In sum, AMPAR NTDs may have mGluR-like signalling capacity. Our data uncover an allosteric potential for AMPAR NTDs. Modulation via the NTD would widen the functional spectrum of AMPARs and potentially opens a currently unexplored target for ligand development.
Results
The GluA3 NTD features unconstrained LLs, resembling mGluRs
The iGluR NTD comprises the most distal portion of the receptor (Figure 1A and B) and is believed to interact with presynaptic components and secreted factors, including pentraxins in AMPARs (Hansen et al, 2010; see also Figure 6). Contrary to its well-established allosteric potential in NMDARs, this domain has been suggested to merely act as a rigid subunit assembly module in nonNMDARs (based on GluA2 and GluK2 structures). However, the assembly characteristics of the AMPAR NTDs show unexpected diversity with GluA2 and GluA3 lying at functional extremes (Rossmann et al, 2011; Sukumaran et al, 2011). The GluA3 NTD features the weakest homodimeric affinity in solution and harbours conspicuous sequence variations in the LL interface (Figure 1D). To better understand the biology of this elusive domain, we targeted GluA3 for X-ray crystallographic studies.
Figure 1.

GluA2 and GluA3 NTDs differ structurally. (A) Left: Topology of an iGluR subunit. The NTD segment is denoted as a green curve and the transmembrane segments as grey columns. Right: Structure of the bipartite GluA2 NTD dimer (PDB 3HSY). The two chains/protomers are coloured green and cyan. Upper and lower lobes (UL, LL) are denoted and their respective interprotomer interfaces are circled. Secondary structural elements contributing to the LL interface are labelled. (B) Structure of the GluA3 NTD (dimer I), with the two protomers coloured red and blue. The UL dimer interface analogous to GluA2 is circled, and the LL interface is shown by a box and an arrow indicating the increased space between the LLs, compared with GluA2. Segments homologous to the GluA2 LL interface segments (from A) are labelled. (C) Lower lobe packing markedly differs between GluA2 and GluA3 NTDs. The lower lobe interface of GluA2 (green) and GluA3 (red) are shown after aligning common secondary structure segments. Note the significantly closer packing of the GluA2 LL interface. Also shown are arginines from GluA3 that project into the interface; this unfavourable electrostatic interaction may contribute to the increased interlobe distance. (D) Sequence conservation in the NTD LL of the AMPA and kainate subfamilies. Different background colours indicate different conservation patterns; for example, conserved sites (columns) within a subfamily are coloured red. Residues that project across the interface are denoted with asterisks (*). Note the markedly higher conservation of the LL interface within the kainate subfamily. See also Supplementary Figures S1 and S2.
GluA3 NTD crystals diffracted to 2.2 Å. The structure was solved by molecular replacement using GluA2 (PDB 3HSY; Greger et al, 2009) as a search probe; two dimers (I and II) are present in the asymmetric unit (Supplementary Table I). Overall, the architecture of the bilobed protomer and packing across the UL interface in dimer I was highly similar to GluA2 (root-mean-square displacement (RMSD) 0.6 Å; Figure 1B; Supplementary Figure S1). The most striking difference is a repositioning of the LLs, in which GluA3 dimer I are widely separated, up to 8 Å relative to the spacing between the GluA2 LLs (Figure 1B; Supplementary Figure S2C).
The LL arrangement observed in the GluA3 NTD bears a striking resemblance to mGluR1 and the natriuretic peptide receptor LBCs, where signalling via flexible LLs is well established (Kunishima et al, 2000; He et al, 2001; Tsuchiya et al, 2002). In fact, GluA3 and mGluR1 show a very similar degree of LL separation (Supplementary Figure S2). Thus, unlike GluA2, in GluA3 the LLs are not constrained by dimeric packing, but have greater freedom to move and may thus propagate signal. It is worth pointing out that ligand-independent clamshell motions have been deduced from experimental data in NMDARs (Gielen et al, 2009); a related scenario mGluA3 (see below). The structure also provides an immediate explanation for the relatively low GluA3 NTD dimer affinity measured in solution (Rossmann et al, 2011).
A closer examination of the LL interface reveals that, contrary to GluA2, the GluA3 LL interface is largely polar in dimer I, which was not anticipated previously from sequence alignments (Jin et al., 2009). In particular, Arg163 and Arg184 project towards the interface (Figure 1C) generating positive electrostatic potential (Supplementary Figure S3A); charge repulsion presumably contributes to the increased lobe separation seen in dimer I. Arg163 is replaced by hydrophobic residues in the other AMPAR subunits—in GluA2 Ile157 takes its place and engages Ala148 of the opposite protomer in hydrophobic contacts (Figure 1D). GluK1–3 kainate receptors also harbour Arg at this position (Figure 1D); however, the positive charge is shielded effectively by Glu186 and Glu192 (Kumar et al, 2009). Interestingly, Figure 1D also shows that in kainate receptors the LLs are well conserved, in apparent contrast to AMPARs. We conclude that the previously described ‘locked' GluA2 dimer, which is also seen in the GluK2 kainate receptor (Jin et al, 2009; Kumar et al, 2009), is not universally found in all nonNMDARs.
Interprotomer rearrangements in the GluA3 NTD
NTD-driven allostery involves the dimer interface in NMDARs (Hansen et al, 2010) and in the analogous mGluRs, where a large-scale reorientation of dimeric contacts were observed crystallographically (Kunishima et al, 2000). A similar picture is seen with the ANP receptor (He et al, 2001). Similarly, interfacial rearrangements of the membrane-proximal LBD in AMPARs couple between active and non-active, desensitized states (Mayer and Armstrong, 2004; Mayer, 2005; Armstrong et al, 2006). Comparison of two GluA3 quaternary conformations observed in the crystal structures, dimer I and dimer II, show large differences, suggesting that GluA3 protomers also possess the ability to adopt alternative quaternary forms. Dimer II features a counter-rotation along an axis perpendicular to the dimer interface, relative to dimer I, resulting in rearrangements across both the UL and LL dimer interfaces (Figure 2B). Indeed, in a different crystal form of the GluA3 NTD, we find an additional dimer form, dimer III (PDB 3P3W), alongside the original dimer I configuration (Figure 2A; Supplementary Table I), underscoring the fact that the GluA3 NTD can adopt multiple quaternary structures. As dimer I is found in both crystal forms, it seems to be energetically favoured. Although new contacts between the LLs are formed in dimers II and III, contacts within the UL interface are diminished, which may result in an overall less stable and thus more heterogeneous interface (Supplementary Table II and Figure S4A). The relevance of GluA3 dimer II could be assessed in solution: side chains of M150 in the apposing LLs come into close proximity to one another (Supplementary Figure S4B). Conservative mutation of this position to Cys, M150C, resulted in a greater proportion of crosslinked dimer on non-reducing SDS–PAGE (Supplementary Figure S4B), suggesting that dimer II is accessible in solvent, even under more dilute, non-crystallographic conditions.
Figure 2.

GluA3 NTD crystal structures exhibit different protomerprotomer packings and interfacial contacts. (A) GluA3 crystallizes in three distinct dimeric forms. The dimeric arrangements of each form are shown from above and from the side, with the molecular surfaces of one protomer from dimers I, II and III coloured red, brown and yellow, respectively. (B) Dimers I, II and III are related by rigid-body motions of their protomers. The grey protomers from panel A have been aligned to within 0.5 Å RMSD of each other, whereas the second (coloured) protomer in each structure is left free. The resulting superposition is shown in the inset (the side view from panel A) and turned ∼90°, looking onto the packing surface of each dimer in the main panel. Translational and rotational shifts between interface helices (UL: B and C, LL: E and F) are shown. Note there is a large (∼16 Å) difference between the packing of LL helices in dimer I (red) and II (brown), whereas dimer III (yellow) assumes an intermediate position. The shifts in the UL are indicated for rotation between dimers I and II for helices B and C. The structural difference is more accentuated in the LL, due to intraprotomer structural variabilities. (C) Superposition from B, viewed from the bottom. The two-fold symmetry axis and the plane of the interface are shown as a circle and a dashed line, respectively.
Overall, the three alternative quaternary conformations I, II and III provide examples of dimeric rearrangements that are energetically accessible to the GluA3 NTD. As shown in Figure 2B, interprotomer translational and rotational displacements up to 16 Å and ∼11° are observed, whereas no significant intraprotomer changes are apparent (RMSDs 0.45–0.52 Å, when superimposing main chain Cα atoms). As shown below, NMA reveals that the repositioning of the two protomers with respect to each other is enabled by the top-ranking, or softest, normal mode intrinsically accessible to the GluA3 dimeric architecture, and that the observed structures could represent snapshots along this readily accessible mode of motion.
The GluA2 LLs are not tightly packed and exhibit structural variabilities
The structural variabilities observed in GluA3 prompted us to analyse and compare the GluA2 NTD dimer interface, where the LLs appear constrained by dimeric packing (Figure 1). Protein interfaces have been classified into functionally relevant and those generated by crystal packing, on the basis of the physicochemical properties intrinsic to the interface (Bahadur et al, 2004; Bordner and Abagyan, 2005). One property that reliably discriminates between stable biological interactions versus nonspecific and comparatively weak crystal-packing interfaces is the local atomic contact density (LD). Nonspecific interfaces have been shown to have an LD below 40, whereas values above indicate stable, biologically relevant packing interfaces (Bahadur et al, 2004). Using this method, we investigated the highest resolution structures of GluA2 and GluA3 currently available (PDB 3HSY, 3O21). As expected from the crystal structures, the GluA3 dimer interfaces showed extensive variability. For example, we find that the LD is 42.7 in the UL interface of dimer I, but is <5 in the LL (Figure 3A and Supplementary Figure S4A), indicating that the dimer I UL represents a biologically relevant interface. In contrast, dimer II showed reduced LDs for both UL and LL (approximately 31–32; Supplementary Figure 4A), suggesting that both interfaces in dimer II are less stable than the dimer I UL. Because of lower resolution (4.2 Å), dimer III was not subjected to atomic level analysis, but due to its similar arrangement to dimer II, we expect that dimer III will also be less stable than dimer I. We examined other interface parameters studied by Bahadur et al, (2004), including solvent-accessible surface area, hydrophobicity and evolutionary conservation, which also indicated that the UL and LL interfaces of dimer II are less stable than the UL of dimer I (Supplementary Table II).
Figure 3.

The LLs in GluA2 and GluA3 NTDs exhibit fewer interfacial contacts and larger structural variabilities compared with the ULs. (A) View onto the dimer interface. Atoms making contacts across the dimer interface within 4.5 Å are depicted as spheres on the molecular surface of the respective monomer. Spheres are coloured by number of contacts from blue (1 contact) to red (⩾7 contacts). Local contact density (LD) is also noted for each interface. (B) Structural variations from known GluA2 NTD structures. All GluA2 NTD crystal structures at resolution of 2.5 Å or better (PDB 3HSY, 2WJW, 3H5V) were analysed for different backbone conformations. UL and LL cores from each chain were aligned to a reference chain (3HSY chain B) to within 0.5 Å RMSD and deviations were measured for backbone atoms; these backbone atom deviations are mapped back onto the 3HSY chain B structure and coloured on a logarithmic scale from 0.1 Å (blue) to 10 Å (red). Whereas loops in the UL were largely invariant, specific loops in the LL showed deviations of up to 0.1 Å. Inset: Using the above alignment and superposition algorithm, segments from 3H5V (αF and αG) and 2WJW (αI) exhibiting the most extreme displacements from 3HSY chain B are highlighted in grey. See also Supplementary Figure S4.
Interestingly, when extended to GluA2, we find that these parameters similarly point to relatively weak GluA2 LL contacts, whereas the UL interface classifies as stronger and biologically relevant (Supplementary Table II). The atomic packing densities in the UL are ∼2-fold larger than those computed for the LL, with an LD of 43.5 versus 21.5 (Figure 3A). In addition, hydrophobicity and evolutionary conservation are markedly reduced between LLs (Supplementary Table II), whereas the solvent content is increased (two- to three-fold), signifying more polar, less stable contacts between the LLs compared with the UL interface (Supplementary Figure S3A, B; Dey et al, 2010). In sum, this analysis concludes that the LL interface in GluA2 makes weaker contacts compared with the UL interface. Therefore, the GluA2 LLs could accommodate rearrangements.
In order to examine potential flexibility in the GluA2 LL, we compiled published GluA2 NTD data sets (7 chains ⩽2.5 Å resolution) and quantified relative displacements of the main chains (Materials and methods). While the cores of each structure could be fit to within 0.5 Å RMSD and rigid-body motions of the two lobes could be disqualified, isolated backbone segments in the LL showed displacements up to ∼10 Å (coloured red in Figure 3B). Those ‘flexible' portions include αF, αG, αI plus attached loops: αF and αG showed rotational motions of up to 47° and 21°, respectively, and alternate loop conformations encompassed RMSDs of up to 3.6 Å, whereas segments in the UL were mostly invariant (RMSD 0.2–0.3 Å; Figure 3B; inset). Together, this analysis suggests dynamics for GluA2 LL segments, in addition to the interprotomer dynamic potential suggested above. To characterize further the hierarchy of motions intrinsically accessible to GluA2 and GluA3, we next performed an NMA of these structures.
Two hierarchical scales of global oscillation in AMPAR NTDs—interlobe cleft closure and interprotomer counter-rotation
The dynamic potential of AMPAR NTDs was analysed by NMA using the Gaussian Network Model (GNM; Bahar et al, 1997; Haliloglu et al, 1997) and the ANM (Atilgan et al, 2001). Motions (or global modes) near the native state, resulting from structure-encoded residue fluctuations, can be simulated with elastic network models (Materials and methods). Normal mode analysis performed on these elastic networks assesses the magnitude and direction of residue fluctuations and predicts the most probable collective motions. This approach allowed us to extract the global modes of motions robustly encoded by each biomolecular architectures (Bahar et al, 2010b). We focused on the most probable, top-ranking or (‘softest') modes of motion (modes 1–3) accessible to AMPAR NTDs. The softest modes lie at the lowest frequency end of the mode spectrum; they are usually distinguished by their high degree of collectivity and provide insights into the cooperative mechanisms relevant to biological function (Bahar et al, 2010a, 2010b). Calculations were performed for both monomeric and dimeric arrangements of GluA2, GluA3 and mGluR1.
The structural differences between GluA2 and GluA3 NTDs can be described at two hierarchical levels as a first approximation: interprotomer packing rearrangements and interlobe (intraprotomer) clamshell motions originating from the high mobility of the LLs. As shown below, the observed structures do not just represent static snapshots, but undergo cooperative fluctuations (or interconversions) between each other. The predisposition of the AMPAR NTDs to undergo these two levels of movements (intra- and interprotomer) renders them competent to transmit conformational changes to downstream portions of the receptor.
Regarding intraprotomer motions, PBP-like opening/closing of the two clamshell lobes, which is well established for mGluR1 (Kunishima et al, 2000) and other PBPs (Quiocho and Ledvina, 1996), is also readily accessed by the GluA2- and GluA3 NTD monomers in slowmode 2, as shown for GluA3 in Figure 4A (and for GluA2 and mGluR1 in Supplementary Figure S5A). This common trait is robustly defined by the bilobate structure of each protomer. The occurrence probabilities (reflected by the reciprocal eigenvalues/frequencies associated with these eigenmodes; see Materials and methods) are comparable in the three GluRs, except for a higher predisposition of mGluR1 to undergo these motions.
Figure 4.

Global dynamics of GluA2 and GluA3 NTDs. (A) ANM predicts GluA2 and GluA3 NTDs to adopt classical clamshell motions. One of the dominant modes of motion predicted by ANM simulations for the GluA3 NTD monomer is a classical clamshell opening/closing with a large range of motion. The major deformation is an opening of the cleft (cleft opening angle shown as bars) along the ‘hinge' axis; open (green) and closed (pink) states of GluA3 are shown. (B) Deformations within the dimer assembly are a bit different. The dominant mode is now an anti-correlated motion between the monomers along the axis denoted by the dashed red line, and manifests as a counter-rotation when viewed from the direction indicated by the black arrow. (C) Mobility profile of GluA2, GluA3 and GluK2 NTDs. Residue-specific fluctuations in mode 1 are shown for GluA2 (red), GluA3 (blue) and GluK2 (green; PDB 3H6G) NTDs. The correlation coefficients between the mobility distributions are as follows: 0.82 between GluA2 and GluA3, 0.86 between GluA2 and GluK2 and 0.94 between GluA3 and GluK2. Secondary structural segments that exhibit large fluctuations in the lower lobe (αF, αG and αI) are labelled. Upper and lower lobes are identified as brown and green bars below the x axis. (D) Dimer assemblies also show mobility. A GluA2 NTD dimer is shown with residues coloured by magnitude of the fluctuations from the first 10 modes of GNM, from least (blue) to most mobile (red). Note that the lower lobe is more mobile than the upper lobe, with the putative output region contacting the LBD exhibiting the most mobility. See also Supplementary Figure S5.
In the dimeric context, a new interprotomer mode of motion dominates: an anticorrelated movement of the protomers, which is essentially imparted by the softest (i.e., the highest probability or lowest frequency) mode 1 in both GluA2 and GluA3. In this mode, the two protomers undergo almost rigid-body counter-rotations about the central axis (Figure 4B). It is this mode that allows GluA3 to sample the different quaternary conformations (dimers I, II and III), in which the protein was crystallized (Figure 2B), and allows mGluR1 to transition between its resting and active states (Kunishima et al, 2000). Interestingly, this mode also allows the dimer assemblies of both GluA2 and GluA3 to access the conformational landscape of mGluR1 (discussed below). Moreover, we find that the intraprotomer clamshell-type motions described for the monomers above (Figure 4A and Supplementary Figure S5A) are also accessible in the dimer, in mode 3. The associated eigenvalues are 0.47 (GluA2), 0.35 (GluA3) and 0.23 (mGluR1), revealing that GluA2 is the stiffest and mGluR1 the most mobile for this clamshell-like motion. Therefore, the NTD dimer maintains, and presumably exploits, the intrinsic propensities of the individual monomers.
GluA3 can transit into mGluR1 conformations
Motivated by the similarity of the intrinsic mobility encoded by the dimeric structures of GluA2 and GluA3, we examined if these motions could allow access to the mGluR1 apo form (PDB 1EWT). The apo form of mGluR1 features a large (∼70°) rotation about the dimeric interface and is the mGluR structure structurally most different to the iGluR NTDs (RMSD of ∼14 Å); furthermore, the apo form exhibits the functionally relevant dimeric rearrangement. The overlap (or correlation cosine) between mode 1 of mGluR1 and GluA3 is 0.83 (see Materials and methods). This suggests that there is a direct path connecting even the most extreme conformation of mGluR1 to GluA3 in conformational space (Supplementary Figure S5B). Interestingly, this path is accessible via mGluR1's most dominant modes, allowing a reconfiguration of mGluR1 into GluA3 and matching the allosteric rearrangement of the mGluR LBC upon glutamate binding. The reverse passage, from GluA3 to mGluR1 conformations, can also be achieved on displacement along a small subset of soft GluA3 modes (Supplementary Figure S5B), although the contribution of the softest mode (mode 1) in this case is slightly smaller (∼0.55).
The results obtained for all transitions between the three structures indicate that the experimentally resolved GluA2, 3 and mGluR1 structures essentially represent conformers in a subspace readily accessed via modes that are naturally favoured by the shared overall architecture. We note that the passage between GluA2 and GluA3 structures requires higher modes, as the structural difference between these two conformers is relatively small and involves more localized (as opposed to global) changes (Supplementary Figure S5B). In summary, normal mode analysis underscores intrinsic similarities between the dynamics of AMPAR NTDs and the mGluR LBC dimer assemblies; the closer ‘functional' relationship between mGluR1 and GluA3 can be explained by their similar LL arrangements (Figure 1 and Supplementary Figure S2A).
Quantifying NTD dynamics
Mobility revealed by normal mode analysis can be quantified and mapped back to the primary or tertiary structure to dissect specific contributions from critical residues or regions. Figure 4C gives such an analysis, in which the distributions of square mobilities of residues, as driven by mode 1, are plotted as a function of residue index for GluA2, GluA3, and the GluK2 kainate receptor NTD (PDB 3H6G) as a reference. Two features are immediately apparent—the shapes of the curves are very similar (correlation coefficients range from 0.82 to 0.94; Figure 4C), with peaks (most mobile residues) and minima (most constrained residues) located at the same positions. Interestingly, in the GluK2 kainate receptor NTD, which similar to GluA2 features a ‘closed' LL interface (Kumar et al, 2009), the sizes of motions are very similar in magnitude to GluA2 (with an identical eigenvalue, 0.19). Second, the size of motions is generally higher in GluA3, indicating a higher propensity to undergo en bloc counter-rotations, consistent with fewer LL interface contacts in GluA3. In fact, the ratio of eigenvalues of GluA3 versus GluA2 is 0.11:0.19, suggesting that GluA3 can experience ∼1.7 times larger square displacements along this particular mode (en bloc counter-rotations) than GluA2 or GluK2. Despite the differences in ‘stiffness', the distributions of motions (among residues) are very similar between all three nonNMDAR NTDs, as can be extrapolated from the high correlation for all pairs.
Regarding critical regions in the tertiary structure, Figure 4D summarizes overall dynamics most accessible to the GluA2 dimer through an ‘average' depiction of the top 10 modes, determined via the GNM (Bahar et al, 1997; Haliloglu et al, 1997). The most mobile regions (in red) locate to the front and bottom of the LL, which presumably functions as an output region down to the receptor's LBD. Notably, flexibility in this region is even greater in GluA3; the most mobile segments in this region (helices F, G and I) are indicated in Figures 4C and D. ‘Mobility' in this region has been identified independently by analysis of available GluA2 NTD structures (see above; Figure 3B), and by single-molecule experiments (H Neuweiler, in preparation).
The minima in Figure 4B correspond to residues predicted to be critical for mediating the global intraprotomer/clamshell and interprotomer/counter-rotation modes of motion. Interestingly, these residues, shown as spheres in Figure 5A, fell into two groups (green ellipsoids): the interfacial residues between the ULs, and those acting as hinge sites for the clamshell motion between the UL and LL in each protomer. These residues, which are mostly conserved between GluA2 and GluA3, are expected to have a critical role, not only in mediating the global, collective movements of mGluR and GluA NTDs, but also in allosteric signal propagation emanating from the NTD interlobe cleft.
Figure 5.

High-resolution crystal structure of a GluA2 NTD shows ligand density in the canonical substrate-binding cleft. (A) Residues in GluA2 and GluA3 NTDs critical for collective dynamics. Residues that coordinate hinging motions of the GluA2 and GluA3 NTD dimers are shown as spheres, mapped on the GluA3 dimer I structure. The residues comprise two major groups, based on the two dominant mechanisms of motions: those that coordinate counter-rotations of protomers (UL interfacial residues) and those that coordinate clamshell motions of individual protomers (interlobe hinge residues, i.e., minima from Figure 4B). (B) Fo–Fc electron density maps contoured at 3.0 σ (magenta) and 2.0 σ (blue) show non-protein, non-water molecules in the cleft. Omit difference maps were generated by stripping the cleft of heteroatoms and waters. Putative ligand-coordinating residues are indicated as sticks, with those at analogous positions to ligand-coordinating residues in mGluR1 indicated by red asterisks. Residues identified from normal mode analysis that coordinate collective motions are indicated in blue with analogous positions from GluA3 (from panel B) given in parentheses. All density-coordinating residues are conserved among all AMPA receptors (see Supplementary Figure S6A). The chemical nature of bound ligand is under investigation. See also Supplementary Figure S6.
Electron density in the GluA2 NTD binding cleft
In PBPs, lobe motions are triggered by small-molecule ligands, docking to the interlobe cleft (Quiocho and Ledvina, 1996; Figure 5A). We observed electron density deep within the cleft of the GluA2 NTD, at 1.75 Å resolution, which could not be attributed to GluA2 side chains or structured waters due to location, size and geometry. This density was detected independently in different high-resolution GluA2 data sets, illustrated by the Fo–Fc omit map in Figure 5B. At present, a sulphate ion (present in the crystallization buffer) was placed into parts of the density (PDB 3HSY). In accord with previous findings (Jin et al, 2009), no cleft density was observed at lower resolution (>2 Å; MR, MS and IHG, unpublished observations).
Residues projecting towards the density include F95, R108, R135, N218 and Y274 (Figure 5B and C). Analogous side chains coordinate L-glutamate in mGluR1, 3 and 7 (red stars in Figure 5B; Muto et al, 2007) as well as in prokaryotic LIV-BP (Trakhanov et al, 2005) and sugar-binding proteins (Vyas et al, 1991), and a subset were independently identified from normal mode analysis as critical residues mediating collective motions (Figure 5A and B). All five positions are conserved across GluA1–4 AMPARs in higher vertebrates, but not in the related kainate receptor family (Supplementary Figure S6A; see also Clayton et al, 2009). Whether this density indeed represents a bona fide AMPAR NTD ligand is unclear; its chemical nature is currently under investigation. Thus, in analogy to the NMDAR2 NTDs and similar to other PBPs, AMPAR NTDs may also have the capacity to coordinate small molecules in the binding cleft. A ligand could similarly trigger interlobe motions (Figure 4A) and transmit allosteric changes between the extracellular portion and the ion channel, and in turn extend the functional repertoire of AMPARs.
Discussion
Electron density in the AMPAR NTD cleft
Apart from its role in subunit-selective assembly (Ayalon and Stern-Bach, 2001; Greger et al, 2007; Hansen et al, 2010; Rossmann et al, 2011), the function of the most distal AMPAR domain, the NTD, is not understood. The NTD forms a zinc sensor in NMDARs, capable of modulating channel activity in a subunit-dependent manner (Gielen et al, 2009; Yuan et al, 2009). Zn2+, as well as an increasing list of clinically relevant synthetic compounds, binds within the interlobe cleft (Paoletti et al, 2000; Karakas et al, 2009) and propagates allosteric modulation (Mony et al, 2009; Traynelis et al, 2010). This mode of ligand-triggered clamshell motion is comparable with a multitude of other PBPs (Tam and Saier, 1993; Quiocho and Ledvina, 1996). We detect density in the GluA2 cleft <2 Å (but not at lower resolution). The shape of the electron density varied, which can be explained either by ligand promiscuity, a characteristic of PBPs (Quiocho and Ledvina, 1996), differing crystallization pH and solvent conditions, or by low occupancy. Notably, the cleft opening-angle between the current and published structures was indistinguishable. This is analogous to the NR2B NTD, in which both the Zn2+-bound and apo structures feature similar conformations, with an RMSD of ∼ 0.56 Å (Karakas et al, 2009). In NR2B, an explanation may be provided by the fact that a Na+ and three Cl− ions are present in the apo, Zn2+-free form, which could stabilize the ‘closed-cleft' conformation (Karakas et al, 2009). It should also be noted that both in GluK2 and GluA2 unassigned density is found in the cleft (Clayton et al, 2009; Kumar et al, 2009). In GluK2 a tartrate ion, present in the crystallization buffer, has been modelled (PDB 3H6G), in GluA2, as yet unidentified density is observed (PDB 2WJW), which is overlaying the density depicted in Figures 5B. In fact, in that study the authors did not exclude a signalling capacity for the NTD in AMPARs (Clayton et al, 2009).
No density was observed in GluA3 at 2.2 Å. It is currently unclear whether AMPAR NTDs exhibit subunit-selective ligand-binding activity, which is seen in NR2A and -2B NMDARs; NMDAR2 subunits have vastly different Zn2+ sensitivities (Paoletti et al, 1997), and ifenprodil binds more selectively to NR2B-containing receptors. Interestingly, whereas binding-site residues are conserved between AMPAR paralogs, the electrostatic potential in the substrate-binding cleft varies substantially between GluA2 and GluA3. The GluA2 cavity exhibits positive potential, whereas in GluA3 the cavity is more neutral but features a negatively charged cleft entry (Supplementary Figure S6B, C). Whether these features facilitate docking of subunit-selective AMPAR NTD ligands requires further study.
The NTD dimer interface—stability versus sliding
Whereas in monomeric, prokaryotic PBPs interlobe motions are central to downstream function (Tam and Saier, 1993; Quiocho and Ledvina, 1996), eukaryotic PBPs are fused to a variety of multimeric receptors (Felder et al, 1999). Here, lobe motions can be transmitted via interface rearrangements, which have been described in the dimeric natriuretic peptide receptor and in mGluRs (Kunishima et al, 2000; He et al, 2005). Similar signalling mechanisms are likely to operate in other members of the vast group of type-C GPCRs, in which PBPs in dimeric arrangement generally function as ligand sensors (Pin et al, 2003). In mGluR1, ligand binding induces a 70° reorientation of the dimer interface from the ‘resting' to the ‘active' states. However, the ‘active' conformation is also seen in an apo crystal structure, suggesting a dynamic equilibrium between the two conformations that is shifted towards ‘active' by ligand (Kunishima et al, 2000).
We also observe dimer reorientations in GluA3; the most frequently encountered conformation, dimer I, is very similar to the ‘active' conformation of mGluR1 with the characteristic LL separation (Supplementary Figure S2). We envisage that collisions between the free lobes may form transient LL interfaces (Supplementary Figure S3A), which facilitate rearrangement of the much tighter UL interface; alternatively, an extrinsic signal (e.g., pentraxin binding or a cleft ligand) may deform the UL interface, which might induce compensatory LL interface rearrangements (Figure 6). If these alternative conformations are sampled in the context of the full receptor, they would be transmitted to the ∼16 amino-acid linker between the NTD and LBD, a functional output region in the NMDAR NTD (Gielen et al, 2009), and may affect the conformational dynamics of the closely packed LBDs. It is currently unclear whether these motions have functional consequences on the ion channel, although dimeric rearrangements generally and ‘sliding' interface motions specifically have been shown to be relevant in other contexts, including haemoglobin oligomers (Mueser et al, 2000).
Figure 6.

Allosteric potential of the AMPA receptor NTD. Potential extrinsic NTD modulators may engage different modes of motion. iGluR NTDs have been shown to interact with a variety of extracellular presynaptic, secreted and small-molecule factors. These modulators potentially bind the NTD at multiple sites that can affect the overall conformation in different ways: (1) perturb interprotomer dimer-packing via the sliding interface (purple); (2) regulate intraprotomer conformation via cleft motions (red); or (3) pry apart of pack together the labile lower-lobe interface (blue). Finally, extrinsic factors can affect interdomain conformations by binding to the mobile interdomain linker (4), for example, auto-antibodies in the case of Rasmussen's encephalitis and autoimmune epilepsy (inset, partial epitope shown in brown). These protein–protein interactions, by modulating NTD conformations, have the potential to allosterically control channel function.
Contrasting with GluA3, GluA2 NTD dimers pack very similarly in all currently available crystal structures. Here both lobes participate in packing; however, different physicochemical properties are apparent: in GluA2 the LL interface is more polar, is less conserved and has reduced atomic packing density (Supplementary Table II and Figure S3). Moreover, we find that UL contacts are well preserved between different data sets, whereas LL contact densities vary (data not shown). In sum, this points to a relatively weaker LL interface, which may rearrange, particularly in the context of the receptor where the lobes are connected to a dynamic gating device, the LBD. The difference in GluA2 and GluA3 interface flexibility most likely resides in the LL interface, where in GluA3 repelling like-charges (R163, R184) are likely the driving force for alternative dimer conformations along the bipartite NTD interface. The LL interface in low-affinity kainate receptors is strikingly well conserved (Figure 1D); it will be revealing to elucidate structural information for these domains and to conduct simulations of the type described herein to compare their dynamic properties.
AMPAR NTD dynamics
Normal mode analysis further suggests that the GluA2 and GluA3 NTDs exhibit dynamics capable of allosteric communication. In both NTD monomers, PBP-like clamshell motions were apparent in one of the dominant modes of motion, indicating that lobe dynamics are a common trait robustly defined by the bilobate structure, irrespective of the particular sequence (GluA2, GluA3 or mGluR1). This observation is in line with previous studies undertaken for proteins sharing common, PBP-like architectures (Keskin et al, 2000). In the dimeric assemblies, clamshell motions were also observed, but were less dominant. The fact that the dimeric form maintains and presumably exploits the intrinsic propensities of the protomers and allows access to hinging motions of the individual protomers suggests that these motions allow for potential ligand-mediated conformational modulation (similar to mGluRs).
The dominant mode in the dimeric assemblies of GluA2, GluA3 and mGluR1 was an interprotomer counter-rotation motion. In GluA3, this mode of motion underlies the interface rearrangements observed crystallographically in dimers I, II and III (Figure 2), and is the main contributor in the transition to the mGluR1 conformation. These findings agree with the structural analysis of the dimeric interfaces, which indicate that the UL and LL interfaces in GluA3 and the LL interface in GluA2 are labile, as well as the mobility of specific LL segments in GluA2 (compare the mobility distributions in the LL computed crystallographically in Figure 3B and via NMA in Figure 4C). Furthermore, these results also suggest that GluA2 may potentially access intradimer mobility similar to GluA3 and that iGluR NTDs possess the ability to undergo conformational changes similar to those seen in the mGluRs, which trigger allosteric communication with downstream segments of the receptor and transmembrane signal transduction (Muto et al, 2007).
Therefore, it is perhaps not surprising that a quantitative analysis of the different dimeric conformations of GluA2, GluA3 and mGluR1 reveals a facile path of interconversion between the three crystal structures (PDB 3HSY, 3O21 and 1EWT, respectively, Supplementary Figure S5B). The striking structural similarity of the GluA3 NTD with the mGluR1 LBC, the fact that mGluR1-like collective dynamics is accessible by both GluA2 and GluA3, even in their dimeric forms, and the reported modulation of channel activity by some autoimmune anti-GluA3 NTD antibodies associated with Rasmussen's encephalitis and epilepsy (Rogers et al, 1994; Cohen-Kashi Malina et al, 2006) imputes as-yet unrecognized allosteric potential to the NTD of AMPA-type iGluRs and suggests an unappreciated role of this domain on channel function.
Of note, the recently solved structure of the GluK5 NTD dimer reveals a close resemblance to the LL orientation/packing to the GluA3 dimer II reported in our study (Kumar and Mayer, 2010).
Moreover, Farina et al (in press) provide the crystal structure of the NR1a NMDAR NTD dimer. This structure reveals yet another lower lobe arrangement distinct to what is known from nonNMDAR NTDs and from the mGluR LBCs, underlying the versatile nature of LL packing in iGluR NTDs.
Materials and methods
Protein crystallography
GluA3 NTD constructs were designed and expressed as described in Rossmann et al (2011). Crystallization was performed using the vapour diffusion method (Benvenuti and Mangani, 2007); GluA3 NTD crystals grew as thin rods in 16–20% PEG 3350, 200–250 mM NH4H2PO4, pH 4.6. Crystals were cryo-protected in the mother liquor supplemented with 30% glycerol and 200 mM ammonium sulphate.
Diffraction data were collected from beamline I03 at the Diamond Light Source (Oxford, UK) and a Rigaku FR-E Super Bright rotating anode laboratory X-ray source (Supplementary Table II). Data were processed using the IMOSFLM and XDS packages (Leslie, 2006; Kabsch, 1993). The structure was solved by molecular replacement with PHASER (McCoy et al, 2005), using the GluA2 NTD monomer (PDB 3HSY) as a search probe. The model was initially refined using REFMAC (Murshudov et al, 1997) and then alternately refined using PHENIX (Adams et al, 2002) and manually rebuilt in COOT (Emsley and Cowtan, 2004). MOLPROBITY (Davis et al, 2004) and PROCHECK (Laskowski et al, 1993) were used to validate model stereochemistry.
Structural and evolutionary analysis
Each chain from high-resolution GluA2 structures (PDBs 3HSY, 2WJW, 3H5V) was aligned to the reference structure 3HSY chain B with the program PINQ (Lesk, 1986) to an overall RMSD of 0.5 Å or better, and displacements for individual backbone atoms were calculated. Accessible surface areas were calculated using the Lee and Richards algorithm (Lee and Richards, 1971) with the NACCESS program (Hubbard and Thornton, 1993). The approach of Bahadur et al. (2004) was used to compute values for residue propensity, fraction of buried hydrophobic surface (fnp), LD and hydrophobic interaction (detailed in Supplemental Methods). Evolutionary sequence analysis was conducted on 334 annotated sequences identified as AMPAR homologues and collected from publicly available databases (NCBI, ENSEMBLE). Multiple sequence alignments were generated first in PROMALS3D (Pei et al, 2008) and manually adjusted. Patterns of conservation were investigated using the ConSurf package (Landau et al, 2005).
ANM and GNM
Extensive reviews of these methods have been given in our previous work, for example, Bahar and Rader (2005) and Bahar et al, (2010a); the reader is referred to these studies for details. In the ANM, the collective dynamics is controlled by the Hessian matrix H, which for a network of N nodes is a 3N × 3N matrix composed of N × N super elements of the form (Atilgan et al, 2001)
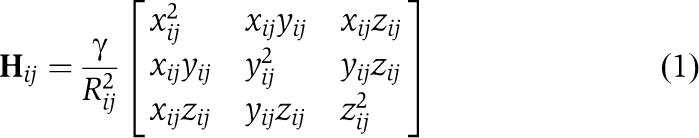 |
for i≠j, and Hii=−Σj, j≠i Hij. Here xij, yij and zij are the components of the distance vector Rij between residues i and j, Rij is its magnitude, γ is a uniform force constant for all springs in the network. In the GNM, the N × N Kirchhoff matrix Γ replaces H, with elements given by Γij=−1, if Rij<Rcut and 0 otherwise, and Γii=−Σj, j≠iΓij. The movement along a given mode k is described by the kth eigenvector (uk) and eigenvalue (λk) of H (or Γ), such that the cross-correlations between residue motions may be expressed a summation over all (or a subset of dominant/subset) modes as
 |
Here C is the 3N × 3N (or N × N) covariance matrix for ANM (or GNM), the diagonal elements of which provide information on mean-square fluctuations of individual residues, and the off diagonal elements reflect the correlations 〈ΔRi.ΔRj〉 in the GNM, or their 3 × 3 decomposition in terms of x-, y- and z-components in the ANM. The contributions to these properties made by individual modes may be computed by selecting the particular elements in the summation over modes (Equation 2).
Overlap between ANM modes and experimentally observed structural changes
Each structure can be characterized by a 3N-dimensional configuration (or state) vector R, the elements of which are the coordinates of the N sites (e.g., α-carbons in ANM/GNM). The 3N-6 ANM modes form a complete orthonormal basis set, such that the transition from state RA to state RB can be achieved by moving along these 3N-6 directions (e.g., eigenvectors/modes). It has been shown in previous work (Valadie et al, 2003; Bahar et al, 2010b) that a few low frequency modes account for ∼65% of the conformation change observed between closed and open states. The contribution of a given mode k to the transition RA → RB may be assessed by examining the overlap between the difference vector d=RB–RA and the ANM eigenvectors uk predicted for RA. Thus, the overlap or correlation cosine is given by I(k)=d.uk/∣d∣, where ∣d∣ designates the magnitude of d, and the cumulative overlap over a subset of modes is CO=(Σk [I(k)]2)½. A high overlap achieved by soft modes (e.g., modes 1–3) indicates that the transition is ‘easily' accessible.
Accession codes
Coordinates for the GluA3 NTD dimers I and II (3O21) and dimers I and III (3P3W) were deposited in the Protein Data Bank.
Supplementary Material
Acknowledgments
We thank Alexej Murzin and Cyrus Chothia for helpful discussions, Nigel Unwin for critical reading of the manuscript and Mette Hogh Jensen for helping with GluA3 crystal trials. MS, MR and IHG were funded by the MRC; MS was also funded by the NIH and IHG by the Royal Society. IB gratefully acknowledges support from NIH awards 5R01GM086238-02 and U54GM087519.
Footnotes
The authors declare that they have no conflict of interest.
References
- Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC (2002) PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr 58(Part 11): 1948–1954 [DOI] [PubMed] [Google Scholar]
- Armstrong N, Gouaux E (2000) Mechanisms for activation and antagonism of an AMPA-sensitive glutamate receptor: crystal structures of the GluR2 ligand binding core. Neuron 28: 165–181 [DOI] [PubMed] [Google Scholar]
- Armstrong N, Jasti J, Beich-Frandsen M, Gouaux E (2006) Measurement of conformational changes accompanying desensitization in an ionotropic glutamate receptor. Cell 127: 85–97 [DOI] [PubMed] [Google Scholar]
- Atilgan AR, Durell SR, Jernigan RL, Demirel MC, Keskin O, Bahar I (2001) Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys J 80: 505–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayalon G, Stern-Bach Y (2001) Functional assembly of AMPA and kainate receptors is mediated by several discrete protein-protein interactions. Neuron 31: 103–113 [DOI] [PubMed] [Google Scholar]
- Bahadur RP, Chakrabarti P, Rodier F, Janin J (2004) A dissection of specific and non-specific protein-protein interfaces. J Mol Biol 336: 943–955 [DOI] [PubMed] [Google Scholar]
- Bahar I, Atilgan AR, Erman B (1997) Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential. Fold Des 2: 173–181 [DOI] [PubMed] [Google Scholar]
- Bahar I, Lezon TR, Bakan A, Shrivastava IH (2010a) Normal mode analysis of biomolecular structures: functional mechanisms of membrane proteins. Chem Rev 110: 1463–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahar I, Lezon TR, Yang LW, Eyal E (2010b) Global dynamics of proteins: bridging between structure and function. Annu Rev Biophys 39: 23–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahar I, Rader AJ (2005) Coarse-grained normal mode analysis in structural biology. Curr Opin Struct Biol 15: 586–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benvenuti M, Mangani S (2007) Crystallization of soluble proteins in vapor diffusion for x-ray crystallography. Nat Protoc 2: 1633–1651 [DOI] [PubMed] [Google Scholar]
- Bordner AJ, Abagyan R (2005) Statistical analysis and prediction of protein-protein interfaces. Proteins 60: 353–366 [DOI] [PubMed] [Google Scholar]
- Bowie D (2008) Ionotropic glutamate receptors & CNS disorders. CNS Neurol Disord Drug Targets 7: 129–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton A, Siebold C, Gilbert RJ, Sutton GC, Harlos K, McIlhinney RA, Jones EY, Aricescu AR (2009) Crystal structure of the GluR2 amino-terminal domain provides insights into the architecture and assembly of ionotropic glutamate receptors. J Mol Biol 392: 1125–1132 [DOI] [PubMed] [Google Scholar]
- Cohen-Kashi Malina K, Ganor Y, Levite M, Teichberg VI (2006) Autoantibodies against an extracellular peptide of the GluR3 subtype of AMPA receptors activate both homomeric and heteromeric AMPA receptor channels. Neurochem Res 31: 1181–1190 [DOI] [PubMed] [Google Scholar]
- Davis IW, Murray LW, Richardson JS, Richardson DC (2004) MOLPROBITY: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res 32 (Web Server issue): W615–W619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey S, Pal A, Chakrabarti P, Janin J (2010) The subunit interfaces of weakly associated homodimeric proteins. J Mol Biol 398: 146–160 [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60 (Part 12 Part 1): 2126–2132 [DOI] [PubMed] [Google Scholar]
- Farina AN, Blain KY, Maruo T, Kwiatkowski W, Choe S, Nakagawa T Separation of domain contacts is required for heterotetrameric assembly of functional NMDA receptors. J Neurosci (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felder CB, Graul RC, Lee AY, Merkle HP, Sadee W (1999) The Venus flytrap of periplasmic binding proteins: an ancient protein module present in multiple drug receptors. AAPS PharmSci 1: E2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gielen M, Le Goff A, Stroebel D, Johnson JW, Neyton J, Paoletti P (2008) Structural rearrangements of NR1/NR2A NMDA receptors during allosteric inhibition. Neuron 57: 80–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gielen M, Siegler Retchless B, Mony L, Johnson JW, Paoletti P (2009) Mechanism of differential control of NMDA receptor activity by NR2 subunits. Nature 459: 703–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greger IH, Rossmann M, Sukumaran M, Penn AC, Veprintsev DB (2009) Structure and function of the AMPA receptor N-terminal domain. Soc Neurosci Abstr 512: 4 [Google Scholar]
- Greger IH, Ziff EB, Penn AC (2007) Molecular determinants of AMPA receptor subunit assembly. Trends Neurosci 30: 407–416 [DOI] [PubMed] [Google Scholar]
- Haliloglu T, Bahar I, Erman B (1997) Gaussian dynamics of folded proteins. Phys Rev Lett 79: 3090–3093 [Google Scholar]
- Hansen KB, Furukawa H, Traynelis SF (2010) Control of assembly and function of glutamate receptors by the amino-terminal domain. Mol Pharmacol 78: 535–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Chow D, Martick MM, Garcia KC (2001) Allosteric activation of a spring-loaded natriuretic peptide receptor dimer by hormone. Science 293: 1657–1662 [DOI] [PubMed] [Google Scholar]
- He XL, Dukkipati A, Wang X, Garcia KC (2005) A new paradigm for hormone recognition and allosteric receptor activation revealed from structural studies of NPR-C. Peptides 26: 1035–1043 [DOI] [PubMed] [Google Scholar]
- Hubbard SJ, Thornton JM. (1993) Naccess. Computer Program. London: Department of Biochemistry and Molecular Biology, University College [Google Scholar]
- Jin R, Clark S, Weeks AM, Dudman JT, Gouaux E, Partin KM (2005) Mechanism of positive allosteric modulators acting on AMPA receptors. J Neurosci 25: 9027–9036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin R, Singh SK, Gu S, Furukawa H, Sobolevsky AI, Zhou J, Jin Y, Gouaux E (2009) Crystal structure and association behaviour of the GluR2 amino-terminal domain. EMBO J 28: 1812–1823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W (1993) Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J Appl Crystallogr 26: 795–800 [Google Scholar]
- Karakas E, Simorowski N, Furukawa H (2009) Structure of the zinc-bound amino-terminal domain of the NMDA receptor NR2B subunit. EMBO J 28: 3910–3920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keskin O, Jernigan RL, Bahar I (2000) Proteins with similar architecture exhibit similar large-scale dynamic behavior. Biophys J 78: 2093–2106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar J, Mayer ML (2010) Crystal structures of the glutamate receptor ion channel GluK3 and GluK5 amino-terminal domains. J Mol Biol 404: 680–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar J, Schuck P, Jin R, Mayer ML (2009) The N-terminal domain of GluR6-subtype glutamate receptor ion channels. Nat Struct Mol Biol 16: 631–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunishima N, Shimada Y, Tsuji Y, Sato T, Yamamoto M, Kumasaka T, Nakanishi S, Jingami H, Morikawa K (2000) Structural basis of glutamate recognition by a dimeric metabotropic glutamate receptor. Nature 407: 971–977 [DOI] [PubMed] [Google Scholar]
- Landau M, Mayrose I, Rosenberg Y, Glaser F, Martz E, Pupko T, Ben-Tal N (2005) ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res 33 (Web Server issue): W299–W302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, MacArthhur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26: 283–291 [Google Scholar]
- Lee B, Richards FM (1971) The interpretation of protein structures: estimation of static accessibility. J Mol Biol 55: 379–400 [DOI] [PubMed] [Google Scholar]
- Lesk AM (1986) Integrated access to sequence and structural data. In Biosequences: Perspectives and User Services in Europe, Saccone C (ed), pp 23–28. Belgium: Bruxelles [Google Scholar]
- Leslie AG (2006) The integration of macromolecular diffraction data. Acta Crystallogr D Biol Crystallogr 26: 48–57 [DOI] [PubMed] [Google Scholar]
- Lomeli H, Mosbacher J, Melcher T, Hoger T, Geiger JR, Kuner T, Monyer H, Higuchi M, Bach A, Seeburg PH (1994) Control of kinetic properties of AMPA receptor channels by nuclear RNA editing. Science 266: 1709–1713 [DOI] [PubMed] [Google Scholar]
- Lynch G (2002) AMPA receptor modulators as cognitive enhancers. Curr Opin Pharmacol 4: 4–11 [DOI] [PubMed] [Google Scholar]
- Madden DR (2002) The structure and function of glutamate receptor ion channels. Nat Rev Neurosci 3: 91–101 [DOI] [PubMed] [Google Scholar]
- Mayer ML (2005) Glutamate receptor ion channels. Curr Opin Neurobiol 15: 282–288 [DOI] [PubMed] [Google Scholar]
- Mayer ML, Armstrong N (2004) Structure and function of glutamate receptor ion channels. Annu Rev Physiol 66: 161–181 [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ (2005) Likelihood-enhanced fast translation functions. Acta Crystallogr D Biol Crystallogr 61(Part 4): 458–464 [DOI] [PubMed] [Google Scholar]
- Mony L, Kew JN, Gunthorpe MJ, Paoletti P (2009) Allosteric modulators of NR2B-containing NMDA receptors: molecular mechanisms and therapeutic potential. Br J Pharmacol 157: 1301–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosbacher J, Schoepfer R, Monyer H, Burnashev N, Seeburg PH, Ruppersberg JP (1994) A molecular determinant for submillisecond desensitization in glutamate receptors. Science 266: 1059–1062 [DOI] [PubMed] [Google Scholar]
- Mueser TC, Rogers PH, Arnone A (2000) Interface sliding as illustrated by the multiple quaternary structures of liganded hemoglobin. Biochemistry 39: 15353–15364 [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 53(Part 3): 240–255 [DOI] [PubMed] [Google Scholar]
- Muto T, Tsuchiya D, Morikawa K, Jingami H (2007) Structures of the extracellular regions of the group II/III metabotropic glutamate receptors. Proc Natl Acad Sci USA 104: 3759–3764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Hara PJ, Sheppard PO, Thogersen H, Venezia D, Haldeman BA, McGrane V, Houamed KM, Thomsen C, Gilbert TL, Mulvihill ER (1993) The ligand-binding domain in metabotropic glutamate receptors is related to bacterial periplasmic binding proteins. Neuron 11: 41–52 [DOI] [PubMed] [Google Scholar]
- Paoletti P, Ascher P, Neyton J (1997) High-affinity zinc inhibition of NMDA NR1-NR2A receptors. J Neurosci 17: 5711–5725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P, Perin-Dureau F, Fayyazuddin A, Le Goff A, Callebaut I, Neyton J (2000) Molecular organization of a zinc binding n-terminal modulatory domain in a NMDA receptor subunit. Neuron 28: 911–925 [DOI] [PubMed] [Google Scholar]
- Pei J, Kim BH, Grishin NV (2008) PROMALS3D: a tool for multiple protein sequence and structure alignments. Nucleic Acids Res 36: 2295–2300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pin JP, Galvez T, Prezeau L (2003) Evolution, structure, and activation mechanism of family 3/C G-protein-coupled receptors. Pharmacol Ther 98: 325–354 [DOI] [PubMed] [Google Scholar]
- Quiocho FA, Ledvina PS (1996) Atomic structure and specificity of bacterial periplasmic receptors for active transport and chemotaxis: variation of common themes. Mol Microbiol 20: 17–25 [DOI] [PubMed] [Google Scholar]
- Rogers SW, Andrews PI, Gahring LC, Whisenand T, Cauley K, Crain B, Hughes TE, Heinemann SF, McNamara JO (1994) Autoantibodies to glutamate receptor GluR3 in Rasmussen′s encephalitis. Science 265: 648–651 [DOI] [PubMed] [Google Scholar]
- Rossmann M, Sukumaran M, Penn AC, Veprintsev DB, Madan Babu M, Greger IH (2011) Subunit-selective N-terminal domain associations organize the formation of AMPA receptor heteromers. EMBO J 30: 959–971 [DOI] [PMC free article] [PubMed]
- Sobolevsky AI, Rosconi MP, Gouaux E (2009) X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature 462: 745–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumaran M, Penn AC, Greger IH (2011) AMPA receptor assembly—atomic determinants and built-in modulators. In Synaptic Plasticity—Dynamics, Development and Disease, Kreutz M, Sala C (eds). Wien, New York: Springer (in press) [DOI] [PubMed] [Google Scholar]
- Tam R, Saier MH Jr (1993) Structural, functional, and evolutionary relationships among extracellular solute-binding receptors of bacteria. Microbiol Rev 57: 320–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trakhanov S, Vyas NK, Luecke H, Kristensen DM, Ma J, Quiocho FA (2005) Ligand-free and -bound structures of the binding protein (LivJ) of the Escherichia coli ABC leucine/isoleucine/valine transport system: trajectory and dynamics of the interdomain rotation and ligand specificity. Biochemistry 44: 6597–6608 [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R, Sibley D (2010) Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev 62: 405–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya D, Kunishima N, Kamiya N, Jingami H, Morikawa K (2002) Structural views of the ligand-binding cores of a metabotropic glutamate receptor complexed with an antagonist and both glutamate and Gd3+. Proc Natl Acad Sci USA 99: 2660–2665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valadie H, Lacapcre JJ, Sanejouand YH, Etchebest C (2003) Dynamical properties of the MscL of Escherichia coli: a normal mode analysis. J Mol Biol 332: 657–674 [DOI] [PubMed] [Google Scholar]
- Vyas NK, Vyas MN, Quiocho FA (1991) Comparison of the periplasmic receptors for L-arabinose, D-glucose/D-galactose, and D-ribose. Structural and functional similarity. J Biol Chem 266: 5226–5237 [PubMed] [Google Scholar]
- Ward SE, Bax BD, Harries M (2010) Challenges for and current status of research into positive modulators of AMPA receptors. Br J Pharmacol 160: 181–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, Hansen KB, Vance KM, Ogden KK, Traynelis SF (2009) Control of NMDA receptor function by the NR2 subunit amino-terminal domain. J Neurosci 29: 12045–12058 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.