

Abstract
The low-molecular-weight compound JRC-II-191 inhibits infection of HIV-1 by blocking the binding of the HIV-1 envelope glycoprotein gp120 to the CD4 receptor and is therefore an important lead in the development of a potent viral entry inhibitor. Reported here is the use of two orthogonal screening methods, GOLD docking and ROCS shape-based similarity searching, to identify amine-building blocks that, when conjugated to the core scaffold, yield novel analogues that maintain similar affinity for gp120. Use of this computational approach to expand SAR produced analogues of equal inhibitory activity but with diverse capacity to enhance viral infection. The novel analogues provide additional lead scaffolds for the development of HIV-1 entry inhibitors that employ protein-ligand interactions in the vestibule of gp120 Phe 43 cavity.
Keywords: HIV, gp120, CD4, Entry inhibitor, Virtual Screening
1. Introduction
The acquired immunodeficiency syndrome (AIDS) is a consequence of the depletion of CD4+ lymphocytes caused by the human immunodeficiency virus (HIV-1) in an infected individual.1,2 The infection is initiated by a series of attachment events, mediated by the HIV-1 viral spike which is comprised of two glycoproteins, gp120 and gp41.3 The attachment of HIV to the target T-cell lymphocyte occurs via binding of gp120 to the host CD4 receptor.4 CD4 binding causes a large conformational structuring of gp1205,6 that forms and exposes the binding site for one of two chemokine receptors, either CCR5 or CXCR4.7 Chemokine receptor binding is the second obligatory event in viral entry,8,9 and is followed by insertion of the gp41 fusion peptide into the host cell membrane,10 promoting viral and cell membrane fusion and viral entry. 11
Several X-ray crystal structures of complexes of gp120 with CD4 and various human antibodies have been determined.12,13 CD4 residues identified by mutagenesis and the crystal structure to be critical for binding to gp120 are Phe 43 and Arg 59.14 Residue Phe 43, located on the CD4 β-turn that contacts a large cavity that is formed upon gp120-CD4 binding, is flanked by the inner, outer and bridging sheet domains of gp120 (Figure 1). Additionally, Arg59 forms an electrostatic interaction with Asp 368 on an adjacent α-helix.12,15 Studies carried out by isothermal titration calorimetry show that CD4 binds to gp120 with large favorable enthalpy and unfavorable entropy changes (ΔH = -63 kcal/mol, -TΔS = 52 kcal/mol), observed for the binding of core gp120 proteins, as well as for the full-length gp120, to CD4, that can be attributed to the large conformational structuring of the previously unstructured regions in gp120.16,17
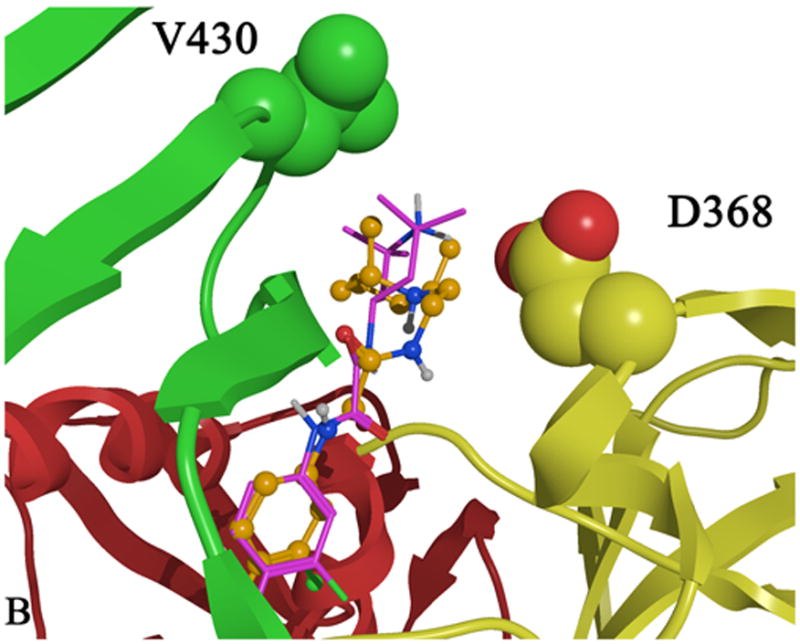
Figure 1.

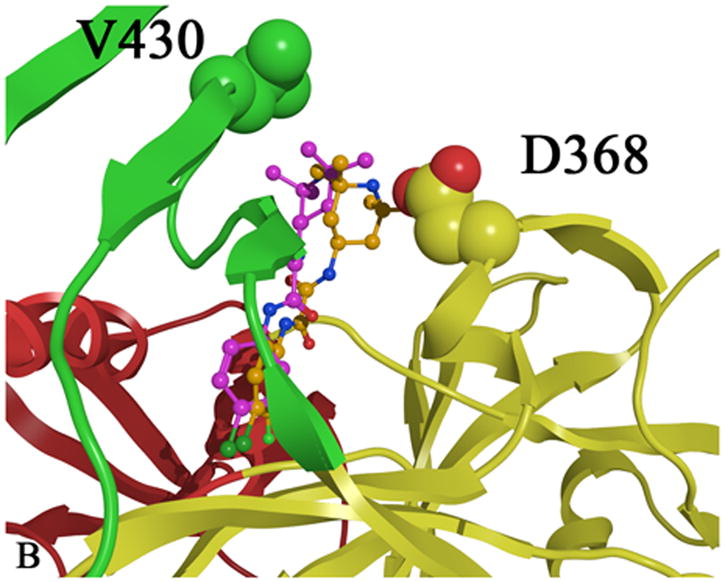
A) CD4 bound (cyan) to gp120 with the gp120 domains colored as follows: inner domain (red); outer domain (yellow) and bridging sheet (green). Carbon atoms are colored by domain color while non-carbon atoms are colored as follows: oxygen (red) and nitrogen (blue). B) Two plausible docked conformations of 1 (orange and purple) bound in the Phe 43 cavity of gp120 (1G9M). Docking indicates that the p-chloro-m-fluoro-benzenyl binds at the bottom of the Phe 43 cavity, while the tetramethyl-piperidine forms hydrophobic interactions in the vestibule of the cavity between D368 and V430.
The two compounds NBD-556 (41) and NBD-557,18 first discovered by Zhao et al. utilizing high-throughput screening, as well as analogues of these compounds prepared in our laboratory, 19 have been shown to compete with CD4 binding. The chemotype of the NBD series consists of three essential regions: Region I, a para-substituted phenyl ring; Region II, an oxalamide linker; and Region III, a tetramethyl-piperidine (Figure 2). Modification in region I with various para-substitutions indicates that size and electron withdrawing character are determinants for binding affinity.19,20 The Br and Cl para-substituted phenyl NBD compounds achieved the best binding affinity (cf. Kd =2.2 μM and 3.7 μM, respectively).24 Study of Region I meta-substitutions within the context of the p-Cl analogue, indicates that a fluorine substituent is preferred over Cl, OH, CH3 or CF3 substitutions, whereas ortho substitution in Region I abolished the ability of the compounds to bind gp120 (Supplemental Tables 1-6). Further synthesis and testing of other diverse Region I analogues also suggested the low probability of enhancing binding affinity in the base of the Phe 43 cavity. Thus far, studies of para and meta-substituted NBD compounds with comparable binding affinities demonstrate a wide capacity to act as a CD4 antagonist (i.e., to inhibit HIV-infection of CD4+ cells) and as a CD4 agonist (i.e., to promote CCR5 binding and enhance viral infection in the absence of CD4).19 Assaying compounds on CD4-deficient Cf2Th-CCR5 cells to measure the enhancement of viral infection is a biological indicator of the extent to which a compound mimics CD4. Importantly, NBD mimicry of CD4-gp120 binding is further demonstrated by isothermal titration calorimetry (ITC), revealing a large unfavorable entropy change, compensated by a larger favorable change in enthalpy; thus, NBD analogues induce conformational changes in the HIV-1 envelope glycoproteins with a similar thermodynamic profile to those induced by CD4. Although CD4 mimics enhance viral infection in CD4-deficient cells by inducing gp120 conformational change, HIV-1 entry and infection is inhibited in CD4-expressing cells.19,21 A recent study of this mechanism with NBD analogue 1, (Table 1, Kd =0.76 μM) demonstrates that HIV-1 infection is inhibited via induction of a short-lived activated state in addition to specific competition for CD4 binding.22
Figure 2.
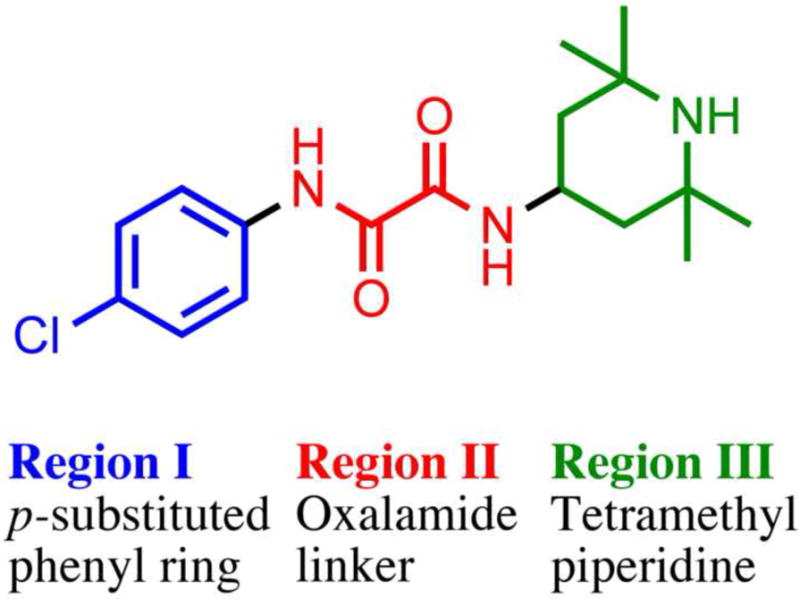
NBD-556 (41) chemotype defined with Region I, Region II and Region III.
Table 1.
Modification of Region II (Linker)
| Compd | Analogue | na | IC50b (μM) | Kdc (μM) | Activation of Viral Infectivityd |
|---|---|---|---|---|---|
| 1 | 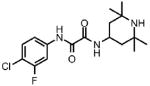 |
67 | 74.8 ± 5.4 | 0.76 | 1.0 |
| 2 |  |
1 | 33.0 | 2.2 | 0.09 |
| 3 | 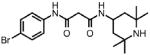 |
1 | >100.0 | N.D. | 0.0 |
| 4 | 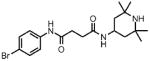 |
1 | >100.0 | N.D. | 0.0 |
| 5 |  |
1 | >100.0 | N.D. | 0.0 |
| 6 |  |
1 | >100.0 | N.D. | 0.0 |
| 7 |  |
1 | >100.0 | N.D. | 0.0 |
Each compound was assayed in triplicate and is reported as a mean for one experiment. For multiple experiments the means and standard deviations are reported. The number of times independent experiments were performed is indicated by n.
The compound concentrations that inhibited 50% of virus infection (IC50) was determined by infecting Cf2Th-CD4/CCR5 cells with 10,000 RT units of wild-type HIV-1YU2 virus expressing luciferase with increasing concentrations of the compound. Compounds labeled non-specific in the Supplementary Tables where found to have comparable IC50’s when assayed against viruses with the ampotrophic murine leukemia virus (A-MLV) envelop glycoproteins.
Kd’s were measured by isothermal titration calorimetry once, unless otherwise indicated in parentheses.
Activation of viral infectivity was determined by infecting Cf2Th-CCR5 cells with recombinant HIV-1YU2 in the presence of NBD analogs. The luciferase activity in the target cells incubated with each compound was divided by that in the cells incubated with JRC-II-191 to obtain the relative activation of infectivity. ND indicates not determined. NB indicates no detectable binding.
Changes to HIV gp120 residues (E370A, S375N) that flank the Phe 43 cavity confer resistance to NBD-556, supporting the model that NBD-556 binds within this cavity.23 Mutagenesis data and the SAR results of meta and para phenyl substitution of the NBD core, combined with molecular modeling,19,20 support the hypothesis that the region I aromatic ring binds in the base of the gp120 Phe 43 cavity. Studies of mutations in the vestibule of the Phe 43 cavity, however, had opposing effects: a change of aspartate to alanine at position 368 increased the binding affinity, enhancement of CD4− cell infection, and inhibition of CD4+ cell infection by 1; by contrast, decreases in affinity, viral enhancement in CD4− cells, and viral inhibition of CD4+ cells of 1 were observed when valine at position 430 was substituted with an alanine.19 This mutational data suggested that key protein-ligand interactions in the vestibule could be manipulated to optimize the inhibitory properties of the NBD compounds. A recent report exploring SAR of region III also suggests the importance of the piperdine moiety to CD4 mimicry and anti-HIV activity. 24
Inspired by the possibility of exploiting NBD interactions in the gp120 vestibule, in conjunction with the unmet need for CD4-gp120 inhibitors as therapeutic modalities to prevent HIV entry, we turned to NBD SAR results for further exploration. The goal was to identify new chemotypes of 1 that would maintain similar or enhanced gp120 binding affinity as measured by ITC, inhibit viral infection, and exhibit favorable pharmaceutical characteristics. We report here on Region I and II SAR and focus on protein-ligand interactions in Region III of 1, which presumably contacts the vestibule of the CD4-binding pocket (Figure 1). To facilitate Region III SAR development, two virtual screening methods structure-based docking (GOLD),25,26 and ligand-based, shape similarity matching (ROCS),27,28 were employed to identify analogues of the structurally complex tetramethyl-piperidine moiety of 1. Synthesis and biological evaluation of the computationally designed analogues and resulting biological profiles were employed to investigate the effects of the structural modifications on CD4-gp120 binding and inhibition of viral entry.
2. Results and Discussion
Prior to conducting virtual screening studies we synthesized and tested many Region I compounds (c.f. compounds in Madani et al.19 and Supplemental Tables 1-6) demonstrating that modification of the phenyl group in the base of the Phe 43 cavity was poorly tolerated. An optimal p-chloro, m-fluoro phenyl ring substitution pattern (1, Table 1) was necessary for enhanced binding in the base of the gp120 cavity19. We thus sought to explore variations in the oxalamide linker of Region II. Several compounds were prepared (2-7) and evaluated as inhibitors of viral entry. Inhibition was measured as an IC50 in CD4-expressing Cf2Th-CCR5 target cells in the presence of different concentrations of the NBD analogue. The capacity of the analogue to replace CD4 in viral infection was measured with a recombinant HIV-1 expressing firefly luciferase pseudotyped with different envelope glycoproteins, incubated with CD4-deficient Cf2Th-CCR5 cells, and then normalized to the enhancement seen for 1. Analogues that inhibited or enhanced viral infection in CD4-expressing, or CD4-deficient Cf2Th-CCR5 cells, respectively, were assessed by ITC. As summarized in Table 1, analogues with changes to the linker did not inhibit gp120-CD4 binding. We thus focused on the synthesis and testing of piperidine analogues of 1. Removal of gem-dimethyl groups (Table 2) indicated the importance of the dimethyl groups at positions 2 and 6 of the piperidine ring (cf. 9-13). However, addition of an isopropyl group to the piperidyl amine (11), or a carbon between the piperidine ring and oxalamide nitrogen (14-16), also led to retention of binding affinity. The carbocyclic analogues 17 and 18 failed to exhibit inhibition of gp120-CD4 binding. Furthermore, replacement of the piperidine amine 1 by an oxygen (19) or an N-oxide (20) also reduced binding.
Table 2.
Piperidine Analogues (For Tables 2-8 only substitutions R1, R2 and the Ring are varied)
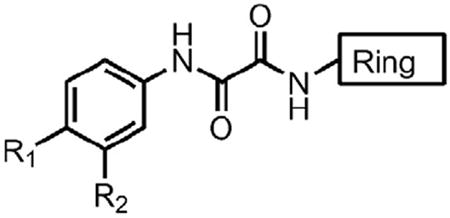 | |||||||
|---|---|---|---|---|---|---|---|
| Compd | R1 | R2 | Ring | n | IC50 (μM) | Kd (μM) | Activation of Viral Infectivity |
| 8 | Cl | F | 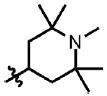 |
49 | 65.8 ± 7.1 | 0.3 (5) | 2.1 |
| 9 | Cl | F | 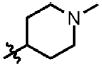 |
1 | >100 | 9.4 | 0.1 |
| 10 | Cl | F | 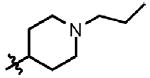 |
1 | >100 | 4.2 | 0.2 |
| 11 | Cl | F | 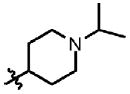 |
1 | >100 | 1.9 | 0.0 |
| 12 | Cl | F | 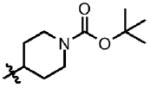 |
1 | >100 | N.D. | 0.0 |
| 13 | Cl | F | 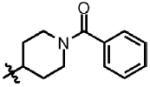 |
1 | >100 | N.D. | 0.0 |
| 14 | Cl | F | 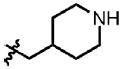 |
1 | >100 | 3.0 | 0.0 |
| 15 | Cl | F | 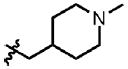 |
1 | >100 | 4.3 | 0.3 |
| 16 | Cl | F |  |
1 | >100 | 3.4 | 0.2 |
| 17 | Cl | F | 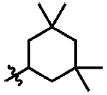 |
1 | >100.0 | N.B. | 0.1 |
| 18 | Cl | F | 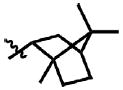 |
1 | >100.0 | N.B. | 0.0 |
| 19 | Cl | F | 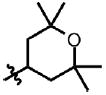 |
1 | >100.0 | 1 | 0.0 |
| 20 | Cl | F | 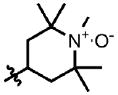 |
1 | >100.0 | 1.3 | 0.23 |
As Region I and II modifications did not improve inhibition, we chose a computational approach to identify new analogues of 1. As previously reported, mutations D368A and V430A in the vestibule of the cavity respectively enhance and decrease the effect of 1 in CD4-negative, CCR5-expressing cells.19 This result suggests that the opening of the gp120 cavity is a suitable target for manipulation of gp120-ligand interactions, thus we chose to focus on identifying additional Region III analogues. Two orthogonal virtual screening methods were used to identify suitable analogues of the tetramethyl-piperidine moiety, that, when conjugated to the core of 1, would enhance binding affinity and expand the SAR of Region III in the gp120 vestibule.
The first virtual screening method relied on the CD4-bound gp120 crystal structure (1G9M) for structure-based drug design.15 The similarity of the thermodynamic profile of CD4 and 1 binding to gp120 provided the rationale for using the CD4-bound gp120 crystal form. Docking of 1 indicated two likely binding poses to gp120 (see Figure 1). Based on the orientation of the oxalamide linker of 1, the tetramethyl-piperidine was predicted to bind either in proximity to D368 or V430. With ample room in the vestibule to accommodate larger groups, we searched for suitable amine building blocks for conjugation. We chose over 300 primary amine building blocks from selected commercial vendors for the docking study. These amines were conjugated in silico to the oxalamide core of 1, and docked with GOLD25,26 to gp120 (1G9M). Amine building blocks were considered for synthesis based on a combination of SlogP less than 4.5, possible commercial availability, and a Goldscore26 value equivalent to or better than that achieved with 1. Several compounds were identified in this manner, synthesized (see supporting material) and demonstrated to achieve a range of inhibition for CD4-gp120 binding (see Tables 3, 4, 5; inactive compound classes are tabulated in Supplemental Tables 2 and 3). While these compounds did not exhibit increased binding affinity as measured by ITC, several analogues displayed improved IC50 (25), decreased enhancement of viral infectivity in CD4− target cells (39), and/or established a new chemotype (30).
Table 3.
Analogs based on virtual screening via docking
| Compd | R1 | R2 | Ring | n | IC50 (μM) | Kd (μM) | Activation of viral infectivity |
|---|---|---|---|---|---|---|---|
| 21 | Cl | F | 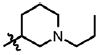 |
5 | 58 ± 18.6 | 3.3 | 0.5 ± 0.2 |
| 22 | Cl | F | 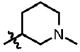 |
1 | >100.0 | 4.2 | 0.7 ± 0.3 |
| 23 | Cl | F | 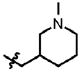 |
1 | >100.0 | 2.5 | 0.3 |
| 24 | Cl | F | 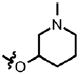 |
1 | >100.0 | ND | 0.0 |
| 25 | Cl | F | 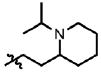 |
1 | 27 | 2.3 | 0.0 |
| 26 | Cl | F | 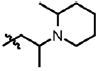 |
1 | >100 | ND | 0.0 |
| 27 | Cl | F | 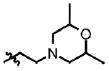 |
1 | >100.0 | ND | 0.0 |
| 28 | Cl | F | 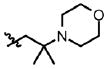 |
1 | >100.0 | ND | 0.0 |
| 29 | Cl | F |  |
1 | >100 | ND | 0.1 |
Table 4.
Analogs based on virtual screening via docking
| Compd | R1 | R2 | Ring | n | IC50 (μM) | Kd (μM) | Activation of viral infectivity |
|---|---|---|---|---|---|---|---|
| 30 | Cl | F |  |
5 | 64.5 ± 21.9 | 4.0 | 0.1 |
| 31 | Cl | F |  |
1 | >100 | ND | 0.0 |
| 32 | Cl | F |  |
1 | >100 | ND | 0.0 |
| 33 | Cl | F | 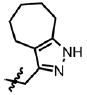 |
1 | >100 | ND | 0.1 |
| 34 | Cl | F | 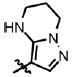 |
1 | >100 | ND | 0.0 |
| 35 | Cl | F | 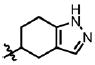 |
1 | >100 | NB | 0.0 |
| 36 | Cl | F | 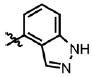 |
1 | >100 | ND | 0.0 |
| 37 | Cl | F |  |
1 | >100 | ND | 0.0 |
Table 5.
Analogues based on virtual screening via docking
| Compd | R1 | R2 | Ring | n | IC50 (μM) | Kd (μM) | Activation of Viral Infectivity |
|---|---|---|---|---|---|---|---|
| 38 | Cl | F | 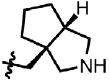 |
5 | 68.3 ± 9.6 | 3.1 | 0.0 |
| 39 | Cl | F | 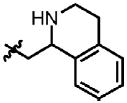 |
1 | 50.4 ± 14.2 | 1.2 | 0.6 |
| 40 | Cl | F |  |
1 | >100 | N.D. | 0.0 |
A second complimentary approach, shape-based virtual screening with the ROCS algorithm,27,28 was also employed to expand SAR. This strategy relies on a modeled ligand conformation to generate new analogues as opposed to selection based on docked protein-ligand interactions in GOLD. The docked conformation of NBD-556 (41), as reported by Madani et al.,19 was employed as a query in the ROCS searches of the Zinc database comprised of 2 million drug-like molecules.29,30 Based on SAR of Region I only compounds containing a p-Cl substituted aromatic group were considered to explore SAR of Region II and III. Among the 21 analogues purchased and tested (Supplemental Table 5), only 4231 inhibited CD4-gp120 binding. The combined Tanimoto and Color scores40 show that 42 was the second highest in the ordered set (Supplemental Table 5) and further reiterated the importance of the oxalamide in Region II in conferring inhibition. The virtual screen was repeated with an unpublished crystal structure of NBD-557 bound to gp120, however the 14 purchased compounds (Supplemental Table 6) did not inhibit viral infection. When the 1-ethyl pyrrolidine 43 was coupled to the core of 1, a binding affinity comparable to 41 was achieved without the accompanying enhancement of viral infectivity. Exploration via docking of similar pyrrolidine building blocks in silico followed by synthesis, produced 44-48, which possessed IC50 values comparable to 43 without enhancing viral infectivity (see Table 6).
Table 6.
Compounds identified using ROCS shape-based virtual screening
| Compd | R1 | R2 | Ring | n | IC50 (μM) | Kd (μM) | Activation of Viral Infectivity |
|---|---|---|---|---|---|---|---|
| 41 | Cl | H | 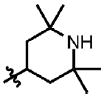 |
67 | 73.7 | 3.7 (5) | 1.0 |
| 42 | Cl | H | 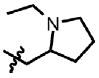 |
1 | 58.6 | N.D. | 0.0 |
| 43 | Cl | F | |
3 | 85.2 ± 14.8 | 2.1 | 0.0 |
| 44 | Cl | F | 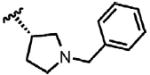 |
1 | > 100 | N.D. | 0.0 |
| 45 | Cl | F | 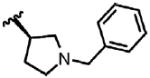 |
2 | 58.5 ± 35.8 | N.B. | 0.0 |
| 46 | Cl | F | 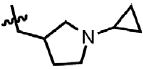 |
3 | 33.5 ± 9.1 | 1.7 | 0.3 |
| 47 | Cl | F | 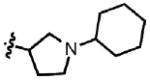 |
9 | 57.1 ± 14.2 | 2.4 | 0.48 ± 0.18 |
| 48 | Cl | F | 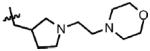 |
1 | > 100 | N.D. | 0.1 |
The ROCS shape-based virtual screening strategy was also repeated using only the tetramethyl-amino-piperidine from 1 to identify suitable amine building blocks available in the Zinc database.29,30 In this case both the docked model from 1 and a later crystal structure of tetramethyl-amino-piperidine moiety were employed as ROCS queries. Amine building blocks identified were conjugated in silico and docked with GOLD. Several active analogues 49-54 were obtained upon synthesis (Table 7). Follow-up ROCS queries based on the docked conformation of the spiro-piperidine of 49 led to compounds 50-52, which were prepared and assayed. Subsequent ROCS queries of the docked conformation for the pyrrolidine of 43 yielded compounds 53-54, which were also synthesized and tested.
Table 7.
Compounds identified using ROCS shape-based virtual screening
| Compd | R1 | R2 | Ring | n | IC50 (μM) | Kd (μM) | Activation of viral infectivity |
|---|---|---|---|---|---|---|---|
| 49 | Cl | F | 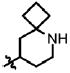 |
5 | 38.5 ± 10.1 | 0.6 | 2.6 ± 0.4 |
| 50 | Cl | F | 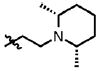 |
3 | 75.5 ± 23.3 | 1.6 | 0.2 ± 0.2 |
| 51 | Cl | F | 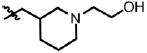 |
1 | 27.6 ± 7.2 | 3.2 | 0.0 |
| 52 | Cl | F | 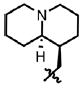 |
1 | >100 | ND | 0.0 |
| 53 | Cl | F | 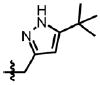 |
1 | >100 | ND | 0.1 |
| 54 | Cl | F |  |
1 | 76.6 ± 5.7 | 2.6 | 1.5 |
The primary mode of screening entails the functional evaluation of the IC50 values combined with the activation of viral infectivity of the analogues in CD4-expressing Cf2Th-CCR5 cells.19 Analogues that displayed improved IC50 or activation of viral infectivity compared to 1, 8 or 41 were considered for further measurement by ITC. Overall, the NBD analogues with novel piperidine moieties did not display appreciable improvement with respect to the already modest IC50 values. However, the activation of viral infectivity on Cf2Th-CCR5 cells, devoid of CD4 receptors, did provide a measure of the analogues’ capacity to mimic CD4 interactions with gp120, and to activate HIV infection.19 Surprisingly, several compounds with IC50 value and binding affinity similar to 41 were ineffective at enhancing viral infectivity (cf. 46, 47, 50, and 51). Madani et al. have previously reported that small variants in phenyl ring substitution impact viral enhancement and the observed entropy changes associated with gp120 structuring. This subset of Region III analogues demonstrates a similar phenomenon, a desirable property for gp120 focused entry inhibitors. The magnitude of activation of viral infectivity correlated with the binding affinity (Kd) for several of the analogues (cf. 49 and 54), but did not correlate with the IC50 values for inhibition of HIV-1 infection of CD4+ target cells. Discrepancies between measured IC50 and Kd reflect differences in binding to the gp120 trimer and monomer in viral and ITC binding assays, respectively. Importantly, one compound (49) did yield a sub-micromolar binding affinity, demonstrated an activation of viral infectivity similar to 1, and maintained inhibition as assessed by IC50. The structurally similarity of analogue 49 to 1 suggests that only one of the gem-dimethyl groups is required for efficient binding to gp120. We also note that the ITC measurements provided an assessment of the binding affinity independent of functional activity, in order to assess the suitability of the analogues for future synthetic modification.
Previously we demonstrated that NBD compounds are sensitive to gp120 mutations in and near the Phe 43 cavity.19 As reported, both D368A and S375A mutations, in isolation or in combination, increased binding affinity of compound 1 to gp120 variant envelopes. Similar assessment of 49 and 50 (Table 8) suggests that, in the vestibule of the Phe 43 cavity, D368A has a negative effect on 49 and 50 binding, while the S375A mutation in the base of the cavity increases the ability of these analogues to inhibit CD4-gp120 binding. In the absence of a crystal structure of the gp120-ligand complex, the experimental data suggests a difference in binding within the gp120 vestibule for analogues 49 and 50 compared to 1. Furthermore, predicted binding modes via docking of 49 and 50 to gp120 (1G9M) suggests that subtle differences in binding mode can affect the inhibitory capabilities of these analogues (Figure 3).
Table 8.
Mutational sensitivity of NBD analogs
| Compd | R1 | R2 | Ring | n | Wild-type IC50μM | D368A IC50μM | S375A IC50μM |
|---|---|---|---|---|---|---|---|
| 1 | Cl | F | |
67 | 74.8 ± 5.4 | 29.1 ± 9.9 | 9.6 ± 0.5 |
| 49 | Cl | F | |
5 | 38.5 ± 10.2 | 86.9 | 9.3 ± 1.4 |
| 50 | Cl | F | |
3 | 75.5 ± 23.8 | 83.2 | 34.5 ± 5.5 |
Figure 3.
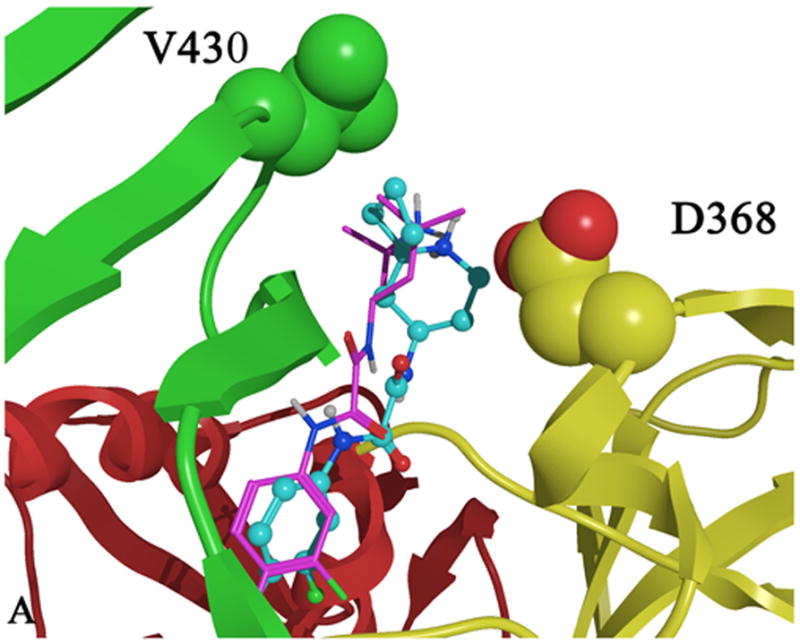
Docked conformations of A) 1 (purple), 49 (blue) and of B) 1 (purple) 50 (orange) bound in the Phe 43 cavity of gp120 (1G9M). Predicted binding modes and mutagenesis data suggest that 49 and 50 may bind differently in the Phe 43 cavity vestibule. Residues D368 and V430 are shown as space filling models with carbon atoms colored by domain color while non-carbon atoms are colored as follows: oxygen (red) and nitrogen (blue). The domains are colored as follows: inner domain (red); outer domain (yellow) and bridging sheet (green).
3. Conclusions
The discovery that NBD analogues inhibit gp120-CD4 binding,19 mimic CD4-induced conformational changes in gp120,21 and inhibit HIV infection by inducing a short-lived activated state of the virus22 argued for further SAR explorations of the NBD chemotype. Ortho substitution on the phenyl ring of 41 and changes to the oxalamide core of 1 led to analogues that failed to inhibit CD4-gp120 binding (Table 1 and Supplemental Tables 1-6). Further exploration and testing of shape-based analogues of 1 did not yield additional leads (Supplemental Tables 5 and 6) necessitating expanding SAR of the Region III. The steric bulk of the piperidine gem-dimethyl groups precludes facile modification, thus inspiring a computational approach to identify suitable structural moieties, that when incorporated onto the core of 1, would maintain similar affinity for gp120 as determined by ITC and, ideally, continue to inhibit viral entry in a single-round viral infectivity assay. Two orthogonal screening approaches using the three-dimensional properties of 1 and gp120 were pursued concurrently to increase the likelihood of discovering novel NBD analogues that bind the highly flexible gp120 envelope protein. Furthermore, ROCS 3-D approaches have been previously reported to be more effective then 2-D searches in producing diverse scaffolds.28 The ROCS shape-based screening leverages both the shape and chemotype of a bioactive compound (1) yielding analogues containing diverse moieties. In contrast, GOLD docking to a CD4-bound gp120 crystal structure permitted exploration of compounds that might employ novel protein-ligand interactions in the cavity vestibule. Virtual screening via docking with GOLD or ROCS shape-based matching, in conjunction with both the purchase of commercial building blocks and synthesis, produced several analogues (cf. 21, 25, 30, 38, 39, 43, 47, 49, 50, and 51) that have comparable binding activity profiles to 1 and 41. GOLD docking to a CD4-bound gp120 crystal structure permitted exploration of compounds that might employ novel protein-ligand interactions in the cavity vestibule. For the number of analogues screened and tested, the docking strategy led to a larger set of inactive analogues. Moreover, the ROCS shape-based searches exploiting similarity in size, shape and chemotype to 1, identified fewer analogues for synthesis and testing. Pleasingly, one promising analogue was identified from each method: 39 by docking and 49 by shape based-similarity. From these data, we conclude that a basic amine comprises a major requirement for effective binding in the vestibule of Phe 43 cavity. Furthermore, the activation of viral infectivity is very sensitive to modification of the vestibule-binding moiety (cf. 25, 39, 43, 50). Although a potent (<100 nM) analogue has not as yet been identified, the SAR from the analogues reported herein demonstrates that gp120 binding affinity and viral infectivity are dramatically affected by specific protein-ligand interactions in the vestibule of the gp120 cavity. Continued use of this computational approach to identify new Region III heterocyclic chemotypes, along with synthetic optimization, thus holds promise of identifying novel compounds with better binding profiles than 1.
4. Methods
4.1. Small Molecule Modeling
Molecules were constructed in MOE (MOE Molecular Operating Environment Chemical Computing Group, version 2005.06 (Montreal, Canada) (http//www.chemcomp.com), ionized using MOE’s WashMDB function, and hydrogens were added.32 The small molecule conformation was minimized to a gradient of 0.01 in the MMFF94x33,34 force field using a distance-dependent dielectric constant of 1.
4.2. Protein Modeling
Protein modeling
Using the X-ray crystal structure of the CD4-bound HIV-1 gp120 core15 (PDB code 1G9M), hydrogen atoms were added and tautomeric states and orientations of Asn, Gln and His residues were determined with Molprobity.35-36 Hydrogens were added to crystallographic waters using MOE.32 The OPSLAA37 force field in MOE was used and all hydrogens were minimized to an rms gradient of 0.01, holding the remaining heavy atoms fixed. A stepwise minimization followed for all atoms, using a quadratic force constant (100) to tether the atoms to their starting geometries; for each subsequent minimization, the force constant was reduced by a half until 0.25. This was followed by a final cycle of unrestrained minimization. In later stages of virtual screening the coordinates of the unpublished crystal structure of the compound NBD-55718 bound to gp120 and 48d were used in modeling studies (unpublished results, Young Do Kwon and P.D. Kwong).
4.3. Docking Calculations
Initial docking calculations of compound 1 proceeded with Glide, subsequent virtual screenings were conducted with GOLD.
Glide (4.018) Water, isopropanol, fucose and N-acetyl D-glucosamine molecules were removed from the coordinates of the minimized protein (1G9M) as described above. The protein was then passed through the protein preparation utility in Glide38,39 using the OPSLAA37 force field and a water solvation model with extended cutoffs. All heavy atoms were constrained with a parabolic potential of 100 kJ/Å. One-hundred iterations of Polak-Ribiere conjugate gradient (PRCG) minimization were applied. The binding site was defined based on the positions of CD4 Phe 43 and the isopropanol molecule from the 1G9M crystal structure. The Glide grids were computed with a box center at 28.10, -12.35, 81.57 and an inner and outer box range of 14 Å and 36 Å, respectively. Docking calculations were performed in standard sampling mode with maxkeep 5,000 and maxref 1,000.
GOLD (version 3.2).26 The binding site was defined by using the docked conformation of NBD-556 produced with Glide. Docking calculations were performed with crystallographic water molecules in the cavity. Analysis of initial docking calculations with all six water molecules (HOH 6, HOH 77, HOH 134, HOH 313, HOH 327 and HOH 343 as numbered in the crystal structure, 1G9M15) toggled on and off during docking and the estimated free energy of binding in GOLD indicated that three crystallographic waters behaved as integral parts of the protein (HOH 6, HOH 327 and HOH 343) and three were likely to be displaced (HOH 77, HOH 134 and HOH 313). Water HOH 6 forms a bridging hydrogen bond between the backbone carbonyls of Gly 473 and Trp 427, HOH 343 forms a bridging hydrogen bond between Val 425 and Asn 377, and HOH 327 hydrogen bonds with Ser 375 in the base of the cavity. Subsequent docking calculations were performed with HOH 343 left on, while waters HOH 6 and HOH 327 were turned on with hydrogen atoms spun. One hundred genetic algorithm (GA) docking runs were performed with the following parameters:initial_virtual_pt_match_max=3.5, diverse_solutions=1,divsol_cluster_size=1, and divsol_rmsd=1.5. All other parameters were set as defaults.
4.4. ROCS Virtual Screening
Flipper from Open Eye was used to expand compounds with unspecified chirality prior to generation of conformers. Using Omega (version 2.2.1)40 from Open Eye with default parameters, a maximum of 50 low energy conformers for all compounds in the Zinc Database (version 7)30 were generated and stored in sd files of approximately 10,000 molecules. ROCS27,28,40 searches were run using 3D coordinates from the docked binding mode of the teramethy-piperidine portion of JRC-II-191. The Implicit Mills Dean41 force field was used to match chemotypes as well as shape. A maximum of 2000 hits were saved for each query and were ranked by a combination of Tanimoto and the scaled Color Score (ComboScore). Primary amines were selected from the set of hits, conjugated in silico and were docked with GOLD28 and scored with a mass-corrected Goldscore. Compounds that reiterated the binding mode of the p-Cl-m-F-phenyl oxalamide moiety of JRC-II-191 were considered for purchase and synthesis.
4.5. Synthesis
The preparation of identified analogues selected via the above screening efforts was conducted as follows: the commercially available 4-chloro-3-fluoroaniline was treated with triethylamine followed by ethyl oxalylchloride to give the requisite ethyl ester. The ester was then coupled to several commercially available amines in EtOH under microwave conditions (150 °C, 1 h). In some instances hydrolysis of the ester followed by EDC-mediated coupling was required. Detailed procedures and characterization data are provided within the Supplemental Material.
4.6. Cell-based Infectivity Assays
4.6.1. General considerations
Compounds were dissolved in dimethyl sulfoxide (DMSO), and stored at 10 mM concentrations at -20°C. The compounds were diluted in Dulbecco Modified Eagle Medium (DMEM, Invitrogen) to create 1 mM solutions before use. Soluble CD4 (sCD4) was purchased from ImmunoDiagnostics (Woburn, MA). Human 293T embryonic kidney and canine Cf2Th thymocytes (ATCC) were grown at 37°C and 5% CO2 in DMEM (Invitrogen) containing 10% fetal bovine serum (Sigma) and 100 μg/mL of penicillin-streptomycin (Meditech, Inc.). Cf2Th cells stably expressing human CD4 and either CCR5 or CXCR442,43 were grown in medium supplemented with 0.4 mg/mL of G418 (Invitrogen) and 0.20 mg/mL of hygromycin B (Roche Diagnostics). Using the Effectene transfection reagent (Qiagen), 293T human embryonic kidney cells were cotransfected with plasmids expressing the pCMVΔP1ΔenvpA HIV-1 Gag-Pol packaging construct, the wild-type or mutant HIV-1YU2 envelope glycoproteins or the envelope glycoproteins of the control amphotropic murine leukemia virus (A-MLV), and the firefly luciferase-expressing vector at a DNA ratio of 1:1:3 μg. For the production of viruses pseudotyped with the A-MLV glycoprotein, a rev-expressing plasmid was added. The single-round, replication-defective viruses in the supernatants were harvested 24-30 hours after transfection, filtered (0.45 μm), aliquoted, and frozen at -80°C until further use. The reverse transcriptase (RT) activities of all viruses were measured as described previously.44
4.6.2. Assay of virus infectivity and drug sensitivity
Cf2Th/CD4-CCR5 or Cf2Th/CD4-CXCR4 target cells were seeded at a density of 6 × 103 cells/well in 96-well luminometer-compatible tissue culture plates (Perkin Elmer) 24 h before infection. On the day of infection, (1 to 100 μM) was added to recombinant viruses (10,000 reverse transcriptase units) in a final volume of 50 μL and incubated at 37°C for 30 minutes. The medium was removed from the target cells, which were then incubated with the virus-drug mixture for 2-4 hours at 37°C. At the end of this time point, complete medium was added to a final volume of 150 μL and incubated for 48 hours at 37°C. The medium was removed from each well, and the cells were lysed with 30 μL of passive lysis buffer (Promega) by three freeze-thaw cycles. An EG&G Berthold Microplate Luminometer LB 96V was used to measure luciferase activity in each well after the addition of 100 μL of luciferin buffer (15 mM MgSO4, 15 mM KPO4 [pH 7.8], 1 mM ATP, 1 mM dithiothreitol) and 50 μL of 1 mM D-luciferin potassium salt (BD Pharmingen).
4.7. Isothermal Titration Calorimetry
Isothermal titration calorimetric experiments were performed using a high-precision VP-ITC titration calorimetric system from MicroCal LLC. (Northampton, MA). The calorimetric cell (~1.4 mL), containing gp120 at a concentration of about 2 μM dissolved in PBS, pH 7.4 (Roche Diagnostics GmbH), with 2 % DMSO, was titrated with the different compounds dissolved in the same buffer at concentrations of 80 – 130 μM. The compound solution was added in aliquots of 10 μL at pre-set intervals. All solutions were degassed to avoid any formation of bubbles in the calorimeter during stirring. All experiments were performed at 25 °C. The heat evolved upon injection of compound was obtained from the integral of the calorimetric signal. The heat associated with the binding reaction was obtained by subtracting the heat of dilution from the heat of reaction. The individual binding heats were plotted against the molar ratio, and the values for the enthalpy change (ΔH) and association constant, Ka (Kd = 1/Ka), were obtained by nonlinear regression of the data.
Supplementary Material
Acknowledgments
We thank Irwin Chaiken and Wayne Hendrickson and all the members of the PO1 Consortium Structure-Based Antagonism of HIV-1 Envelope Function in Cell Entry. Funding was provided by NIH GM 56550 to JL, EF, ABS, and JS. A. Sugawara thanks the Japan Society for the Promotion of Science for research fellowship support. JL thanks the Pittsburgh Supercomputing Center for an allocation for computing resources #MCB090108.
Footnotes
Tables of inactive compounds, detailed synthetic procedures, and spectral data for new compounds is contained within the Supplementary Data, available on-line.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Barre-Sinoussi F, Chermann JC, Rey F, Nugeyre MT, Chamaret S, Gruest J, Dauguet C, Axler-Blin C, Vezinet-Brun F, Rouzioux C, Rozenbaum W, Montagnier L. Science. 1983;220:868. doi: 10.1126/science.6189183. [DOI] [PubMed] [Google Scholar]
- 2.Gallo RC, Salahuddin SZ, Popovic M, Shearer GM, Kaplan M, Haynes BF, Palker TJ, Redfield R, Oleske J, Safai B, et al. Science. 1984;224:500. doi: 10.1126/science.6200936. [DOI] [PubMed] [Google Scholar]
- 3.Kowalski M, Potz J, Basiripour L, Dorfman T, Goh WC, Terwilliger E, Dayton A, Rosen C, Haseltine W, Sodroski J. Science. 1987;237:1351. doi: 10.1126/science.3629244. [DOI] [PubMed] [Google Scholar]
- 4.Dalgleish AG, Beverley PC, Clapham PR, Crawford DH, Greaves MF, Weiss RA. Nature. 1984;312:763. doi: 10.1038/312763a0. [DOI] [PubMed] [Google Scholar]
- 5.Sattentau QJ, Moore JP. J Exp Med. 1991;174:407. doi: 10.1084/jem.174.2.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sattentau QJ, Moore JP, Vignaux F, Traincard F, Poignard P. J Virol. 1993;67:7383. doi: 10.1128/jvi.67.12.7383-7393.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dragic T, Litwin V, Allaway GP, Martin SR, Huang Y, Nagashima KA, Cayanan C, Maddon PJ, Koup RA, Moore JP, Paxton WA. Nature. 1996;381:667. doi: 10.1038/381667a0. [DOI] [PubMed] [Google Scholar]
- 8.Feng Y, Broder CC, Kennedy PE, Berger EA. Science. 1996;272:872. [Google Scholar]
- 9.Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton RE, Hill CM, Davis CB, Peiper SC, Schall TJ, Littman DR, Landau NR. Nature. 1996;381:661. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- 10.Brasseur R, Cornet B, Burny A, Vandenbranden M, Ruysschaert JM. AIDS Res Hum Retroviruses. 1988;4:83. doi: 10.1089/aid.1988.4.83. [DOI] [PubMed] [Google Scholar]
- 11.Wyatt R, Sodroski J. Science. 1998;280:1884. doi: 10.1126/science.280.5371.1884. [DOI] [PubMed] [Google Scholar]
- 12.Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. Nature. 1998;393:648. doi: 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diskin R, Marcovecchio PM, Bjorkman PJ. Nat Struct Mol Biol. 2010;17:608. doi: 10.1038/nsmb.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moebius U, Clayton LK, Abraham S, Harrison SC, Reinherz EL. J Exp Med. 1992;176:507. doi: 10.1084/jem.176.2.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kwong PD, Wyatt R, Majeed S, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. Structure Fold Des. 2000;8:8. doi: 10.1016/s0969-2126(00)00547-5. [DOI] [PubMed] [Google Scholar]
- 16.Myszka DG, Sweet RW, Hensley P, Brigham-Burke M, Kwong PD, Hendrickson WA, Wyatt R, Sodroski J, Doyle ML. Proc Natl Acad Sci U S A. 2000;97:9026. doi: 10.1073/pnas.97.16.9026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leavitt SA, Schon A, Klein JC, Manjappara U, Chaiken IM, Freire E. Curr Protein Pept Sci. 2004;5:1. doi: 10.2174/1389203043486955. [DOI] [PubMed] [Google Scholar]
- 18.Zhao Q, Ma L, Jiang S, Lu H, Liu S, He Y, Strick N, Neamati N, Debnath AK. Virology. 2005;339:213. doi: 10.1016/j.virol.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 19.Madani N, Schon A, Princiotto AM, Lalonde JM, Courter JR, Soeta T, Ng D, Wang L, Brower ET, Xiang SH, Kwon YD, Huang CC, Wyatt R, Kwong PD, Freire E, Smith AB, 3rd, Sodroski J. Structure. 2008;16:1689. doi: 10.1016/j.str.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamada Y, Ochiai C, Yoshimura K, Tanaka T, Ohashi N, Narumi T, Nomura W, Harada S, Matsushita S, Tamamura H. Bioorg & Med Chem Lett. 2009;20:354. doi: 10.1016/j.bmcl.2009.10.098. [DOI] [PubMed] [Google Scholar]
- 21.Schon A, Madani N, Klein JC, Hubicki A, Ng D, Yang X, Smith AB, 3rd, Sodroski J, Freire E. Biochemistry. 2006;45:10973. doi: 10.1021/bi061193r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haim H, Si Z, Madani N, Wang L, Courter JR, Princiotto A, Kassa A, DeGrace M, McGee-Estrada K, Mefford M, Gabuzda D, Smith AB, 3rd, Sodroski J. PLoS Pathog. 2009;5:e1000360. doi: 10.1371/journal.ppat.1000360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yoshimura K, Harada S, Shibata J, Hatada M, Yamada Y, Ochiai C, Tamamura H, Matsushita S. J Virol. 2010;84:7558. doi: 10.1128/JVI.00227-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Narumi T, Ochiai C, Yoshimura K, Harada S, Tanaka T, Nomura W, Arai H, Ozaki T, Ohashi N, Matsushita S, Tamamura H. Bioorg & Med Chem Lett. 2010;20:5853–5858. doi: 10.1016/j.bmcl.2010.07.106. [DOI] [PubMed] [Google Scholar]
- 25.Jones G, Willett P, Glen RC, Leach AR, Taylor R. J Mol Biol. 1997;267:727. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 26.Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. Proteins. 2003;52:609. doi: 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
- 27.Grant JA, Gallardo MA, Pickup B. J Comput Chem. 1996;17:1653. [Google Scholar]
- 28.Rush TS, 3rd, Grant JA, Mosyak L, Nicholls A. J Med Chem. 2005;48:1489. doi: 10.1021/jm040163o. [DOI] [PubMed] [Google Scholar]
- 29.Irwin JJ, Shoichet BK. J Chem Inf Model. 2005;45:177. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zinc. 2006 http://zinc.docking.org/index.shtml.
- 31.Chembridge; San Diego, CA: http://www.chembridge.com/ [Google Scholar]
- 32.MOE. Molecular Operating Environment, Chemical Computing Group; Montreal Canada: 2008. http//www.chemcomp.com/ [Google Scholar]
- 33.Halgren TA. J Comput Chem. 1999;20:720. doi: 10.1002/(SICI)1096-987X(199905)20:7<720::AID-JCC7>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 34.Halgren TA. J Comput Chem. 1999;20:740. doi: 10.1002/(SICI)1096-987X(199905)20:7<720::AID-JCC7>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 35.Lovell SC, Davis IW, Arendall WB, III, de Bakker PIW, Word JM, Prisant MG, Richardson JS, Richardson DC. Proteins Struct Funct and Genet. 2003;50:437. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 36.Word J, Lovell S, Richardson J, Richardson D. J Mol Biol. 1999;285:1735. doi: 10.1006/jmbi.1998.2401. [DOI] [PubMed] [Google Scholar]
- 37.Jorgensen WL, Maxwell DS, Tirado-Rives J. J Am Chem Soc. 1996;117:11225. [Google Scholar]
- 38.Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. J Med Chem. 2004;47:1750. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 39.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. J Med Chem. 2004;47:1739. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 40.ROCS. OpenEye Scientific Software, Inc 3600 Cerrillos Road, Suite 1107, Santa Fe, NM 87507. 2008 www.eyesopen.com.
- 41.Mills JEJ, Dean PM. J Comput-Aided Mo l Des. 1996;10:607. doi: 10.1007/BF00134183. [DOI] [PubMed] [Google Scholar]
- 42.Mirzabekov T, Bannert N, Farzan M, Hofmann W, Kolchinsky P, Wu L, Wyatt R, Sodroski J. J Biol Chem. 1999;274:28745. doi: 10.1074/jbc.274.40.28745. [DOI] [PubMed] [Google Scholar]
- 43.Babcock GJ, Mirzabekov T, Wojtowicz W, Sodroski J. J Biol Chem. 2001;276:38433. doi: 10.1074/jbc.M106229200. [DOI] [PubMed] [Google Scholar]
- 44.Rho HM, Poiesz B, Ruscetti FW, Gallo RC. Virology. 1981;112:355. doi: 10.1016/0042-6822(81)90642-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.