

Abstract
Aphakia (lack of lens) is a rare human congenital disorder with its genetic etiology largely unknown. Even in model organisms, very few mutations are known to result in such a drastic ocular defect. In this study, we have shown that homozygous deletion of Nf1, the Ras GTPase gene underlying human neurofibromatosis type 1 syndrome, causes lens dysgenesis in mouse. Although early lens specification proceeded normally in Nf1 mutants, lens induction was disrupted due to deficient cell proliferation. Further analysis showed that extracellular signal-regulated kinase (ERK) signaling was initially elevated in the invaginating lens placode, but by the lens vesicle stage, ERK phosphorylation was significantly reduced. Only after intraperitoneal treatment of U0126, an inhibitor of ERK phosphorylation, was lens development restored in Nf1 mutants. Hyperactive Ras-mitogen-activated protein kinase (MAPK) signaling is known to cause neuro-cardiofacial-cutaneous (NCFC) syndromes in humans. As a member of NCFC family genes, Nf1 represents the first example that attenuation of Ras-MAPK kinase signaling pathway is essential for normal lens development.
INTRODUCTION
Human genetic studies have shown that hyperactivation of RAS-mitogen-activated protein kinase (MAPK) signaling is detrimental to embryonic development. Gain-of-function mutations in SHP2 are identified in more than 50% of patients with Noonan syndrome, a relatively common birth defect with facial, cardiovascular and skeletal abnormalities. Interestingly, activating mutations in SOS1 accounts for another 10% of Noonan syndrome patients and that in KRAS accounts for 5% (1–4). Moreover, phenotypic similarities have classified Noonan syndrome into a superfamily named neuro-cardiofacial-cutaneous (NCFC) syndromes that include neurofibromatosis type 1 syndrome caused by NF1, Costello syndrome caused by HRAS and cardio-facio-cutaneous syndrome caused by KRAS, BRAF, MEK1 or MEK2 (5–8). These results have identified the aberrant activation of the Ras-MAPK cascade as a common theme within these human congenital diseases.
Previous studies have also demonstrated that the fibroblast growth factor (FGF)-activated Ras-MAPK pathway is necessary for lens development. Transgenic expression of a dominant negative Fgfr1 inhibited cell proliferation in the lens pit and conditional deletion of Fgfr1–3, following lens induction, resulted in a vacant lens vesicle lacking lens fiber cells (9,10). In line with these results, impaired enzymatic modification of heparan sulfate, a critical co-receptor of FGF receptors (FGFRs), led to a hypoplastic eye phenotype ranging from microphthalmia to anophthalmia (11). Frs2α is an FGFR adaptor protein that recruits the protein tyrosine phosphatase Shp2, the docking protein Grb2 and the nucleotide exchange factor Sos to activate downstream Ras-MAPK pathways. Both the mutation of the Shp2-binding sites on Frs2α and the conditional knockout of Shp2 disrupted the lens-specific extracellular signal-regulated kinase (ERK) phosphorylation, resulting in lens hypoplasia (12,13).
Although the above studies have established that lens formation requires FGF-Ras-MAPK signaling, it is still unclear whether lens development is susceptible to the Ras-MAPK hyperactivation. In this study, we report that increasing Ras-ERK signaling in the murine model of neurofibromatosis type 1 syndrome (NF1) indeed disrupts lens development. Nf1 is a GTPase-activating protein (GAP) that assists in the hydrolysis of Guanosine triphosphate (GTP)-bound Ras to Guanosine diphosphate-bound Ras, a reaction that suppresses Ras activity (6). Loss-of-function mutations in human NF1 result in the autosomal dominant familial cancer syndrome characterized by multiple discrete dermal neurofibromas in addition to optic and central nervous system gliomas. Ras-MAPK activity has been shown to be up-regulated in NF1-null cells (14,15). However, the highly conserved Drosophila Nf1 has been shown to modulate cyclic adenosine monophosphate-protein kinase A (cAMP-PKA) signal transduction, and the Nf1-null phenotype of insects can be reversed by activation of the cAMP-PKA pathway (16,17). A similar role of Nf1 in the cAMP-PKA pathway has recently been reported in mice, and mammalian cell culture studies indicate that Nf1 could also regulate cytoskeletal rearrangement in a GAP-independent manner, underscoring the complexity of Nf1 function in intracellular signaling (18–20). In analyzing the mouse Nf1 knockouts, we have observed a specific lens dysgenesis phenotype, which can be traced to abnormal lens induction. We showed that cell proliferation, but not differentiation, was adversely affected in the Nf1-null lens. Although ERK phosphorylation was initially up-regulated as the lens placode invaginates in the E10.5 Nf1 mutants, by E11.5, both ERK phosphorylation and lens vesicle development were disrupted. More importantly, intraperitoneal treatment of the E10.5 Nf1 embryos with U0126, a MAPK kinase inhibitor, restored lens development. Therefore, attenuation of the Ras-MAPK pathway by Nf1 is essential for embryonic lens development.
RESULTS
Defective lens development in the Nf1 mutants
We observed a highly penetrant ocular abnormality ranging from reduced lens size to anophthalmia in E11.5–12.5 Nf1 homozygous mutants, at least 2 days before they would die of cardiac defects (21). Among 32 Nf1 eye samples analyzed, six eyes (19%) were normal, seven eyes (22%) had more than 50% reduction in lens size and two samples (6%) had lost the entire eye structure (Fig. 1A–E; data not shown). The remaining 17 eyes (53%) lacked a lens but retained a retina, which had collapsed to occupy the space normally filled by the lens (Fig. 1F). This suggested that Nf1 lens dysgenesis occurred even in the presence of a developing retina. In the Nf1 lens that did form at E12.5, we observed expression of Prox1, a homeobox transcription factor crucial for lens fiber cell differentiation and elongation (Fig. 1H, arrowhead). Consistent with this, the lens differentiation marker αA crystallin was detected even in a small cluster of Nf1 lens cells that appeared to be attached to the overlying ectoderm (Fig. 1K, arrowhead). Thus, lenses differentiated normally once they were formed in the Nf1 mutants.
Figure 1.

Embryonic Nf1−/− lens phenotype. (A–F) E12.5 Nf1−/− embryos and histology eye sections. The Nf1-null ocular phenotype ranged in severity from no lens formation to a smaller lens. (G–J) Cell differentiation in the Nf1−/− lens. Despite the reduction in lens size, both Prox1 and αA crystallin were expressed in the mutant lenses, indicating that lens differentiation was not inhibited in the Nf1-null mutants. Scale bars: 1 mm (A–C); 50 μm (D–L).
Loss of Nf1 disrupts lens vesicle formation
The severity and specificity of Nf1 lens defects led us to investigate the earliest developmental stage when the lens phenotype became apparent. Pax6 and Sox2 are two transcription factors critical for lens induction and morphogenesis (22). In the wild-type and the Nf1-null embryos, both proteins were up-regulated normally in a presumptive lens placode at E9.5 (Fig. 2A and B, arrowheads). Similarly, expression of AP2α, another important regulator of lens development, was also clearly present in the wild-type and the mutant (Fig. 2C and D, arrowheads) (23). By E10.0, however, when the lens pit was already formed in the wild-type, there was only a slight indentation in the Nf1 mutant lens ectoderm (Fig. 2E–H, arrowheads). The Nf1 mutant eventually formed a reduced lens pit at E10.5, but by this time, a closed lens vesicle had detached from the surface ectoderm in the wild-type embryo (Fig. 2I–L, arrowhead). Interestingly, despite the lens morphogenesis delay, we did not detect any reduction in Pax6, Sox2 and AP2α expression. In addition, Six3, an early eye determination gene, and Pitx2, a marker for neural-crest-derived periocular mesenchyme, were also expressed normally in the Nf1 mutants (Fig. 2M–P, arrowheads). We next analyzed cell apoptosis and proliferation in the Nf1−/− embryos to investigate the underlying mechanism of the lens phenotype. In E9.5 and 10.5 embryos, no significant difference in the number of terminal dUTP nick end labeling (TUNEL)-positive signals was observed between the wild-type and mutants (Fig. 3A–D). Quantification of BrdU-positive cells also failed to reveal any changes within the E9.5 lens ectoderm (Fig. 3E, F and I). However, at E10.5, the proportion of BrdU-positive lens cells in the Nf1 mutants was significantly reduced when compared with wild-type controls (Fig. 3G–I; P < 0.01). These results suggest that Nf1 does not control lens determination and differentiation, but it is required for cell proliferation underlying lens vesicle formation.
Figure 2.

Defective lens induction in Nf1−/− mutants. (A–D) Pax6, Sox2 and AP2α, all essential lens determination factors, were appropriately expressed in the Nf1 presumptive lens ectoderm at E9.5 (arrowheads). (E–H) At E10.0, lens placode invagination was reduced in the Nf1 mutants. (I–P) At E10.5, although the lens vesicle was formed in wild-type controls, the Nf1 mutants were arrested at the lens pit stage. Nevertheless, expression of the lens determination genes Pax6, Sox2, AP2α, Six3 and periocular marker transcription factor Pitx2 were unaffected (arrowheads). le, lens ectoderm; ov, optic vesicle; lp, lens pit; lv, lens vesicle. Scale bars: 50 μm.
Figure 3.

Lens cell proliferation was reduced in Nf1−/− mutants. (A–D) Lack of apoptosis defects in Nf1-null lens. Normal TUNEL staining in the Nf1 mutant surface ectoderm at E9.5 (arrowheads) as well as in the invaginating surface ectoderm at E10.5 (insets). (E–H) Cell proliferation quantified as the ratio of BrdU-positive cells versus total DAPI-positive cells in the E9.5 surface ectoderm (arrowheads) and E10.5 lens vesicle (insets). (I) Significant reduction in lens cell proliferation in Nf1 mutants compared with wild-type at E10.5 (P < 0.05; n = 4). Scale bar: 50 μm.
Hyperactive ERK signaling in E9.5 Nf1 head surface ectoderm
Because Nf1 negatively regulates Ras activities, we hypothesized that the downstream MAPK and PI3K signaling pathways could be up-regulated during lens induction in Nf1-null embryos. We first performed western blot analysis on E12.5 whole-embryo extracts and found that the phosphorylation level of ERK was indeed elevated in the mutants, whereas the phospho-AKT level remained unchanged (Fig. 4A). We thus focused on the ERK signaling pathway in our efforts to study the mechanism of Nf1-null lens phenotype. In whole-mount immunohistochemistry at E9.5, we observed phospho-ERK staining in the wild-type forebrain and branchial arch, where Fgfs are known be expressed. In the exact same staining conditions, however, Nf1 mutants exhibited consistently higher ERK phosphorylation throughout the head ectoderm, including the eye region (Fig. 4B and C). Two members of the Pea3 group of Ets transcription factors, Pea3 and Erm, are known downstream targets of Ras-ERK signaling in lens development (13). Detected by whole-mount RNA in situ hybridization, both genes were indeed expressed in the similar pattern as phospho-ERK (Fig. 4D–G). As expected, although there was little expression of Pea3 and Erm in the wild-type presumptive lens ectoderm at E9.5, both genes were clearly up-regulated in the Nf1 mutant eye region. Taken together, loss of Nf1 resulted in up-regulation of the Ras-MAPK signaling pathway at the beginning of lens induction.
Figure 4.
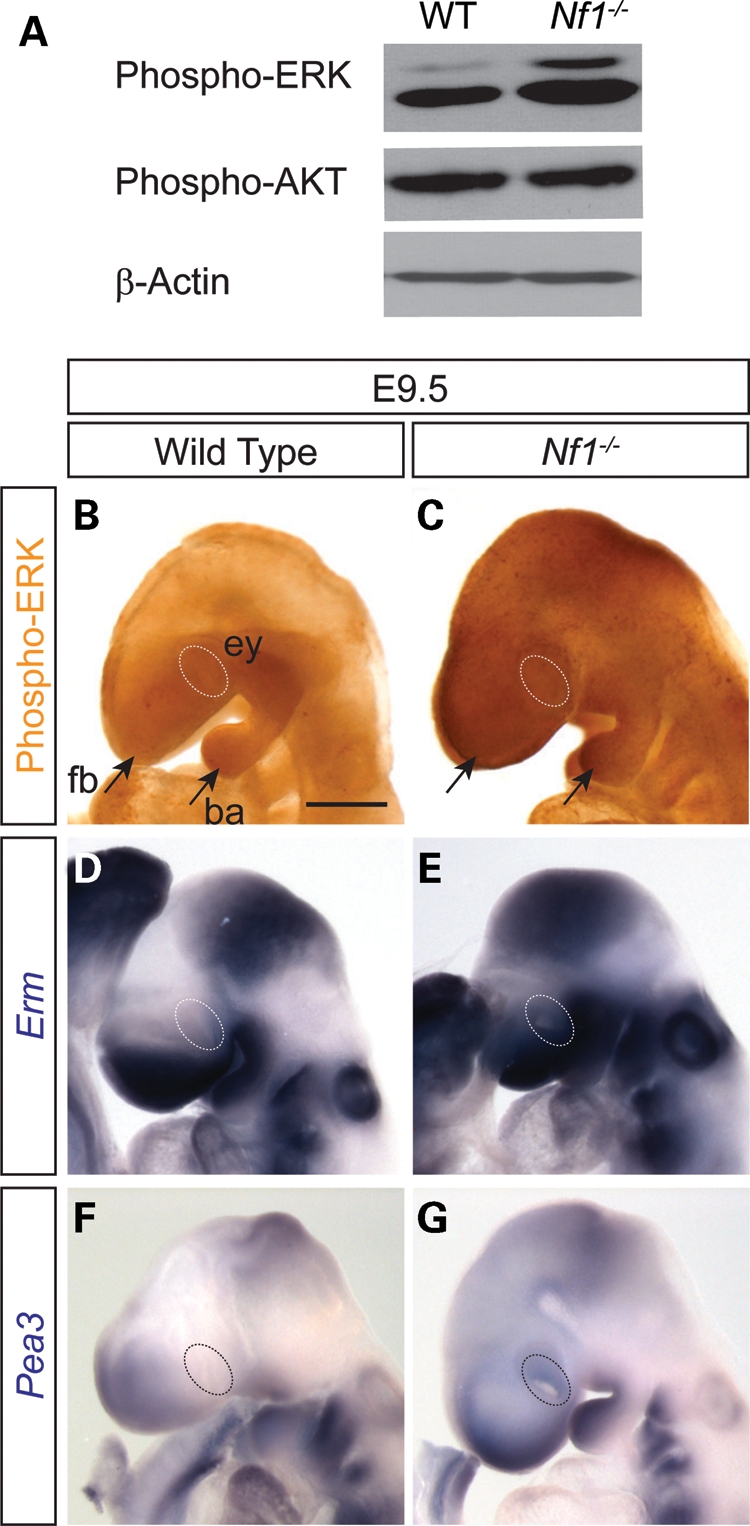
Nf1 mutation resulted in increasing ERK signaling in lens ectoderm. (A) Western blot analysis affirmed that phospho-ERK, but not phospho-Akt, was up-regulated in whole-embryo lysates of E12.5 Nf1 mutants. (B, C) E9.5 whole-mount immunohistochemistry showed that phospho-ERK was up-regulated throughout the Nf1 mutant head surface ectoderm compared with the wild-type (dotted ovals outline ocular region). (D–G) Ras-ERK signaling response genes, Erm and Pea3, were also induced in the Nf1-null lens ectoderm (dotted ovals). Scale bars: 50 μm (A, B, E, F); 1 mm (C, D, G, H).
Induction of Sprouty2 suppressor and attenuation of ERK signaling
We next examined ERK phosphorylation at E10.5, when the lens ectoderm invaginates to form the lens pit. By immunohistochemistry on sections, the Nf1 mutant lens pit again displayed much more intense phospho-ERK staining than the wild-type control (Fig. 5A and B, arrowheads). Interestingly, Sprouty2, an antagonist of the Ras-MAPK signaling pathway, was also up-regulated in the E10.5 Nf1 lens (Fig. 5C and D, insets). This is consistent with a previous report that inactivation of Nf1, although initially promoted Ras-ERK signaling, eventually induced a general negative feedback mechanism mediated by Sprouty proteins (24). At E11.5, although phospho-ERK staining remained strong in the wild-type lens vesicle, the small lens pit in the Nf1 mutants exhibited much weaker ERK phosphorylation (Fig. 5E and F, arrowheads). As a result, E11.5 Nf1 mutants also lost the expression of ERK signaling response gene Erm (Fig. 5G and H, insets). Therefore, compared with the consistent ERK signaling in wild-type lens, ERK signaling in Nf1 mutants was initially up-regulated in the lens ectoderm, but ultimately attenuated at the lens vesicle stage.
Figure 5.

Induction of Sprouty2 expression and attenuation of ERK signaling during Nf1 lens induction. (A–D) As ERK phosphorylation was up-regulated in the E10.5 Nf1 mutant lens pit, Ras signaling inhibitor Sprouty2 was also induced. (E–H) At E11.5, Nf1 mutant lens exhibited significantly reduced phospho-ERK staining and Erm expression. Scale bars: 50 μm (A–D); 1 mm (E–H).
Inhibition of ERK activation restored lens development in Nf1−/− mice
Because we observed significant biological defects at E10.5, a time point when ERK activation was maximal in Nf1-deficient lens tissues, we hypothesized that hyperactivation of ERK at the onset of lens induction caused eventual lens developmental failure. To test this hypothesis, we targeted the ERK pathway with a pharmacological MEK inhibitor U0126 to attempt rescue of the Nf1 mutant lens phenotype. U0126 is a potent and specific inhibitor of MEK, the dual specificity kinase for ERK. To validate the efficacy of the MEK inhibitor within our model, we first injected U0126 (5 mg/kg body weight) or vehicle alone [5% dimethyl sulfoxide (DMSO) in 0.05 m phosphate-buffered saline (PBS)] intraperitoneally into pregnant females at E10.5 and dissected out the embryos 2 h later. As expected, vehicle treatment did not alter the hyperactive ERK signaling in Nf1 mutants compared with the wild-type (Fig. 6A and B, arrowheads). In drug-treated embryos, however, both the wild-type and the mutants exhibited significant reduction in ERK phosphorylation, although moderate phospho-ERK staining was still preserved in Nf1 lens (Fig. 6C and D, arrowheads). We next injected a single dose of U0126 or vehicle control into pregnant females at E9.5 and dissected out the embryos at E12.5. Interestingly, the U0126 treatment indeed led to an increase in the percentage of Nf1 mutants that developed lens, but the effect did not reach statistical significance when compared with the vehicle control (Fig. 6I). However, when the pregnant females were injected at E10.5 when ERK signaling appeared to peak in Nf1 lens primordial, lenses were eventually formed in all six U0126-treated Nf1−/− eyes at E12.5 (P = 0.0143) (Fig. 6E–I). Thus, pharmacological suppression of ERK signaling at the lens vesicle stage was sufficient to rescue lens induction defects in Nf1 mutants.
Figure 6.

Treatment with MEK inhibitor U0126 rescued the Nf1 lens phenotype. (A–D) Validation of MEK inhibitor U0126 at E10.5. E10.5 Nf1 pregnant females were injected intraperitoneally with either MEK inhibitor U0126 or vehicle (see Materials and Methods) and embryos dissected out after 2 h. Phospho-ERK staining was reduced in both U0126-treated wild-type and Nf1 mutant lenses at E10.5 compared with vehicle-treated embryos (arrowheads). (E–H) Nf1-null lens phenotype rescued after U0126 treatment. E10.5 pregnant Nf1−/+ females were injected intraperitoneally with MEK inhibitor U0126 and embryos dissected out E12.5. Hematoxylin and eosin staining shows lens formed in Nf1−/− mutants treated with U0126 treatment compared with vehicle-treated embryos. (H) Lens formation was scored for vehicle- versus drug-treated E9.5 and E10.5 embryos and statistical significance was calculated by Fisher's exact test. NS, not significant. Scale bars: 50 μm.
DISCUSSION
To date, no signaling molecule mutations have been identified in human aphakia, whereas in mouse, only a handful of gene knockouts, including Bmp4, Bmp7, Frs2α and Ndst1, are known to cause lens dysgenesis (11,12,25–27). In this study, we have investigated the Nf1 gene, whose human homolog underlies the common genetic disorder neurofibromatosis type 1. NF1 has been studied extensively for its role in human development and cancer, but likely because homozygous NF1 fetuses are expected to die in uteri, the full impact of NF1 in human eye development has not been explored. Using a mouse model of NF1, we have shown for the first time that Nf1 homozygous deletion results in aphakia or even anophthalmia, thus establishing Nf1 as a novel genetic factor in congenital lens defects. It is noteworthy that Nf1 resides in the same FGF-Ras-ERK signaling cascade as Frs2α and Ndst1, and systemic knockout of all three genes specifically disrupt lens induction. Taken together, these findings support the critical role of FGF-Ras-ERK signaling in early lens development.
Despite the similarities in their ocular phenotypes, Nf1, Frs2α and Ndst1 mutants have distinctively different underlying mechanisms. Although both Frs2α and Ndst1 are necessary for activation of FGF-Ras-ERK signaling, as a negative regulator of Ras, Nf1 is expected to suppress FGF-Ras-ERK signaling. Indeed, we have showed that Nf1 mutants initially up-regulate ERK phosphorylation and its downstream response genes in the presumptive lens ectoderm. Interestingly, ERK signaling is greatly diminished later at the lens vesicle stage and Nf1 lens development is eventually aborted. These observations may appear to conflict with previous transgenic overexpression studies which show that constitutively activated Ras promotes ERK signaling and lens hyperplasia (28,29). One likely reason for this discrepancy is the timing of Ras transgene expression, which was detected in lens only as early as E11.5. On the other hand, by disrupting one of the many Ras GAPs, Nf1 mutation enhances embryonic Ras-ERK signaling only moderately (see western blot analysis in Fig. 4A). In contrast, the transgenic approach could potentially elevate Ras levels significantly beyond the physiologic range. The magnitude of Ras expression is clearly important, because previous studies have shown that oncogenic Kras could either promote ERK signaling when overexpressed in tumor cells or suppress ERK signaling when expressed at physiologic level in mouse embryonic fibroblasts (30,31). Similarly in vivo, oncogenic mutation in the endogenous Kras locus could induce Ras signaling antagonists, such as Sprouty2 and Mkp3, in the lung and intestine, eventually leading to MAPK signaling attenuation (32,33). In fact, the knockdown of NF1 in human fibroblasts has also been shown to trigger a global Ras signaling suppression, which is mediated by the increasing expression of Ras-negative regulators, such as Ras GAPs and Sprouty proteins (24). These studies underscore the dynamic interplay between Ras and its negative feedback mechanisms, which could result in up-regulation or attenuation of downstream MAPK signaling in a Ras dosage-dependent manner.
Taken together, we propose that lens induction can only occur within a permissive zone of ERK signaling, which is precisely targeted by endogenous FGF signaling during normal lens development (Fig. 7). Although Nf1 mutation raises the overall ERK signaling only moderately as shown in E12.5 whole-embryo extracts, when coupled with the existing FGF signaling, it elevated ERK signaling beyond the permissive zone in the lens vesicle, triggering lens development failure. In this process, we speculate that the strong negative feedback response mediated by Sprouty and other Ras antagonists may be responsible for the eventual suppression of ERK signaling. In our pharmacological intervention scheme, U0125 injection at E9.5 was unable to rescue Nf1 lens development because it suppressed ERK signaling before it reached the lens induction zone. At E10.5, however, U0125 effectively corrects Nf1 ERK hyperactivation, allowing successful lens development. Our results thus demonstrate that Nf1-induced ERK signaling hyperactivation, in addition to ERK signaling deficiency, can disrupt lens induction. As NF1 belongs to the NCFC syndrome family genes that cause hyperactive Ras-MAPK signaling in human, mutations in the NCFC genes may also underlie human aphakia.
Figure 7.

A model for defective lens induction in Nf1 mutant. In wild-type embryos, FGF signaling promotes Ras-ERK signaling (blue line) to reach the lens induction zone (green area), which is necessary for the initiation of lens development. In Nf1 mutants, however, ERK signaling (red line) was elevated above the permissive boundary at E10.5, inducing Sprouty-mediated negative feedback response and eventual ERK signaling attenuation. This drastic swing could be avoided by U0126 treatment which suppressed excessive ERK signaling (yellow line), allowing normal lens development.
MATERIALS AND METHODS
Mice
The Nf1+/− mouse, previously described as Nf1n31/+, contains a targeted disruption of exon 31 to mimic human NF1 mutations (21). It was kindly provided by Drs Fengchun Yang and Wade Clapp (Herman B. Wells Center for Pediatric Medicine, School of Medicine, Indiana University) and maintained in C57BL/6 background. The presence of a vaginal plug was considered 0.5 days post-coitum, or E0.5. All experimental procedures involving mice were humanely performed in accordance with the Laboratory Animal Research Center at Indiana University (LARC).
Histology, immunohistochemistry and western blot
Hematoxylin and eosin histology on paraffin sections, fluorescent immunohistochemistry on cryosections and western blot were carried out as reported previously (11,13,34). The following antibodies were used: mouse monoclonal anti-Pax6 (1:10) (Developmental Studies Hybridoma Bank, Department of Biological Sciences, University of Iowa, Iowa City, IA, USA), rabbit polyclonal anti-αA crystalline (1:1000) (kindly provided by Samuel Zigler, National Institute of Health, Bethesda, MD, USA), rabbit polyclonal anti-AP2α (1:1000) (Santa Cruz Biotechnology, Santa Cruz, CA, USA), rabbit polyclonal anti-Sox2 (1:800) (Cat. #AB5603; Chemicon International, Billerica, MA, USA), rabbit polyclonal anti-Prox1 (1:500) (Cat. #PBR-238C; Covance, Berkeley, CA, USA), anti-β-actin (Cat. #A5441; Sigma, St Louis, MO, USA) (1:5000), rabbit polyclonal anti-phospho-ERK1/2 (Cat. #9101) (1:1000) and anti-phospho-Akt (Cat. #3787) (1:1000) (all from Cell Signaling Technology, Beverly, MA, USA). Immunohistochemistry of murine sections with anti-phospho-ERK1/2 was performed using the Cyanine 3 TSA™ Plus Fluorescence System according to the manufacturer's instructions (Cat. #NEL744; NEN Life Science Products, Boston, MA, USA). Secondary antibodies for all experiments, except phospho-ERK1/2 section-immunohistochemistry, were either Alexa Fluor 488 (1:250)- or Alexa Fluor 555 (1:500)-conjugated anti-mouse and anti-rabbit IgG (Jackson ImmunoResearch, West Grove, PA, USA).
RNA in situ hybridization
RNA in situ hybridization experiments for whole-mount embryos and cryosections were performed as detailed in our previous studies (11,13,34). Antisense probes were generated from cDNA for Pax6, Erm and Pea3 (both generously provided by Bridget Hogan, Duke University Medical Center, Durham, NC, USA), Six3 (generously provided by Guillermo Oliver, St Jude Children's Research Hospital, Memphis, TN, USA), Sprouty2 (generously provided by Gail Martin, University of California, San Francisco, CA, USA) and Pitx2 (a generous gift from Valerie Dupé, Faculté de Médecine, Institut de Génétique et Développement, Université de Rennes 1, Rennes Cedex, France). At least three samples were analyzed for each genotype.
BrdU and TUNEL analyses
BrdU analysis was performed as described previously (11). The cell proliferation rate was calculated as the ratio of counted BrdU-positive cells versus 4′,6-diamidino-2-phenylindole (DAPI)-positive cells and analyzed by Student's t-test. TUNEL staining was performed with the In situ Cell-Death Detection Kit (Cat. #11-684-795; Roche, Indianapolis, IN, USA).
Whole-mount phospho-ERK immunohistochemistry
Whole-mount phospho-ERK immunohistochemistry was performed as described previously (35). Briefly, 9.5-day-old embryos were quickly dissected out into ice-cold PBS and fixed in 8% paraformaldehyde overnight at 4°C. Embryos were then washed in PBS containing 0.5% Igepal CA-630 (Cat. #18896; Sigma) and dehydrated in methanol, followed by a 1.5 h bleaching-blocking incubation in a mixture of methanol:30% hydrogen peroxide solution (5:1) at 4°C. Next, embryos were rehydrated into PBSST (5% natural goat serum and 0.1% Triton-X in PBS) for 1 h at 4°C, prior to overnight incubation in rabbit polyclonal anti-phospho-Erk1/2 primary antibody (1:350) (Cat. #9101; Cell Signaling Technology). Embryos were then washed at 4°C with PBSST, followed by overnight incubation in biotinylated secondary antibody and the Vectastain® ABC agent (Cat. #PK6100; Vector Laboratories, Burlingame, CA, USA). For signal detection, embryos were incubated in 0.3 mg/ml DAB (Sigma FAST™ 3,3′-diaminobenzidine) (Cat. #D4168-50SET; Sigma) at room temperature for 20 min. Embryos were photographed with a Leica DFC400 camera mounted on a Leica M165FC dissecting microscope.
Treatment of mouse embryos with MEK inhibitor
Pregnant females at E9.5 or E10.5 were injected intraperitoneally with MEK inhibitor U0126 (Cat. #9903; Cell Signaling Technology) at a dose of 5 mg/kg body weight (36), and embryos were collected for paraffin histology at E12.5. For U0126 preparation, 5.0 mg of U0126 MEK 1/2 inhibitor was dissolved in a total volume of 1.31 ml (0.655 ml of DMSO and 0.655 ml of 1 m PBS) as the stock solution, which was further diluted with 12 ml of water prior to injecting the mice. Injection of the vehicle alone (5% DMSO in 0.05 m PBS) was used as a control. Fisher's exact test was used to examine the significance of the contingency between drug- and vehicle-treated mutants.
FUNDING
The work was supported by the National Institutes of Health (EY017061).
ACKNOWLEDGEMENTS
The authors thank Drs Wade Clapp, Valerie Dupé, Bridget Hogan, Guillermo Oliver, Gail Martin, Fengchun Yang and Samuel Zigler for mice and reagents, Kristina Hertzler for editing and members of Zhang Lab for discussions.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Roberts A.E., Araki T., Swanson K.D., Montgomery K.T., Schiripo T.A., Joshi V.A., Li L., Yassin Y., Tamburino A.M., Neel B.G., et al. Germline gain-of-function mutations in SOS1 cause Noonan syndrome. Nat. Genet. 2007;39:70–74. doi: 10.1038/ng1926. doi:10.1038/ng1926. [DOI] [PubMed] [Google Scholar]
- 2.Schubbert S., Zenker M., Rowe S.L., Boll S., Klein C., Bollag G., van der Burgt I., Musante L., Kalscheuer V., Wehner L.E., et al. Germline KRAS mutations cause Noonan syndrome. Nat. Genet. 2006;38:331–336. doi: 10.1038/ng1748. doi:10.1038/ng1748. [DOI] [PubMed] [Google Scholar]
- 3.Tartaglia M., Mehler E.L., Goldberg R., Zampino G., Brunner H.G., Kremer H., van der Burgt I., Crosby A.H., Ion A., Jeffery S., et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat. Genet. 2001;29:465–468. doi: 10.1038/ng772. doi:10.1038/ng772. [DOI] [PubMed] [Google Scholar]
- 4.Tartaglia M., Pennacchio L.A., Zhao C., Yadav K.K., Fodale V., Sarkozy A., Pandit B., Oishi K., Martinelli S., Schackwitz W., et al. Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nat. Genet. 2007;39:75–79. doi: 10.1038/ng1939. doi:10.1038/ng1939. [DOI] [PubMed] [Google Scholar]
- 5.Aoki Y., Niihori T., Kawame H., Kurosawa K., Ohashi H., Tanaka Y., Filocamo M., Kato K., Suzuki Y., Kure S., et al. Germline mutations in HRAS proto-oncogene cause Costello syndrome. Nat. Genet. 2005;37:1038–1040. doi: 10.1038/ng1641. doi:10.1038/ng1641. [DOI] [PubMed] [Google Scholar]
- 6.Cichowski K., Jacks T. NF1 tumor suppressor gene function: narrowing the GAP. Cell. 2001;104:593–604. doi: 10.1016/s0092-8674(01)00245-8. doi:10.1016/S0092-8674(01)00245-8. [DOI] [PubMed] [Google Scholar]
- 7.Niihori T., Aoki Y., Narumi Y., Neri G., Cave H., Verloes A., Okamoto N., Hennekam R.C., Gillessen-Kaesbach G., Wieczorek D., et al. Germline KRAS and BRAF mutations in cardio-facio-cutaneous syndrome. Nat. Genet. 2006;38:294–296. doi: 10.1038/ng1749. doi:10.1038/ng1749. [DOI] [PubMed] [Google Scholar]
- 8.Rodriguez-Viciana P., Tetsu O., Tidyman W.E., Estep A.L., Conger B.A., Cruz M.S., McCormick F., Rauen K.A. Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science. 2006;311:1287–1290. doi: 10.1126/science.1124642. doi:10.1126/science.1124642. [DOI] [PubMed] [Google Scholar]
- 9.Zhao H., Yang T., Madakashira B.P., Thiels C.A., Bechtle C.A., Garcia C.M., Zhang H., Yu K., Ornitz D.M., Beebe D.C., et al. Fibroblast growth factor receptor signaling is essential for lens fiber cell differentiation. Dev. Biol. 2008;318:276–288. doi: 10.1016/j.ydbio.2008.03.028. doi:10.1016/j.ydbio.2008.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Faber S.C., Dimanlig P., Makarenkova H.P., Shirke S., Ko K., Lang R.A. Fgf receptor signaling plays a role in lens induction. Development. 2001;128:4425–4438. doi: 10.1242/dev.128.22.4425. [DOI] [PubMed] [Google Scholar]
- 11.Pan Y., Woodbury A., Esko J.D., Grobe K., Zhang X. Heparan sulfate biosynthetic gene Ndst1 is required for FGF signaling in early lens development. Development. 2006;133:4933–4944. doi: 10.1242/dev.02679. doi:10.1242/dev.02679. [DOI] [PubMed] [Google Scholar]
- 12.Gotoh N., Ito M., Yamamoto S., Yoshino I., Song N., Wang Y., Lax I., Schlessinger J., Shibuya M., Lang R.A. Tyrosine phosphorylation sites on FRS2alpha responsible for Shp2 recruitment are critical for induction of lens and retina. Proc. Natl Acad. Sci. USA. 2004;101:17144–17149. doi: 10.1073/pnas.0407577101. doi:10.1073/pnas.0407577101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pan Y., Carbe C., Powers A., Feng G.-S., Zhang X. Sprouty2-modulated Kras signaling rescues Shp2 deficiency during lens and lacrimal gland development. Development. 2010;137:1085–1093. doi: 10.1242/dev.042820. doi:10.1242/dev.042820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Basu T.N., Gutmann D.H., Fletcher J.A., Glover T.W., Collins F.S., Downward J. Aberrant regulation of ras proteins in malignant tumour cells from type 1 neurofibromatosis patients. Nature. 1992;356:713–715. doi: 10.1038/356713a0. doi:10.1038/356713a0. [DOI] [PubMed] [Google Scholar]
- 15.DeClue J.E., Papageorge A.G., Fletcher J.A., Diehl S.R., Ratner N., Vass W.C., Lowy D.R. Abnormal regulation of mammalian p21ras contributes to malignant tumor growth in von Recklinghausen (type 1) neurofibromatosis. Cell. 1992;69:265–273. doi: 10.1016/0092-8674(92)90407-4. doi:10.1016/0092-8674(92)90407-4. [DOI] [PubMed] [Google Scholar]
- 16.Guo H.F., The I., Hannan F., Bernards A., Zhong Y. Requirement of Drosophila NF1 for activation of adenylyl cyclase by PACAP38-like neuropeptides. Science. 1997;276:795–798. doi: 10.1126/science.276.5313.795. doi:10.1126/science.276.5313.795. [DOI] [PubMed] [Google Scholar]
- 17.The I., Hannigan G.E., Cowley G.S., Reginald S., Zhong Y., Gusella J.F., Hariharan I.K., Bernards A. Rescue of a Drosophila NF1 mutant phenotype by protein kinase A. Science. 1997;276:791–794. doi: 10.1126/science.276.5313.791. doi:10.1126/science.276.5313.791. [DOI] [PubMed] [Google Scholar]
- 18.Brown J.A., Gianino S.M., Gutmann D.H. Defective cAMP generation underlies the sensitivity of CNS neurons to neurofibromatosis-1 heterozygosity. J. Neurosci. 2010;30:5579–5589. doi: 10.1523/JNEUROSCI.3994-09.2010. doi:10.1523/JNEUROSCI.3994-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buchanan M.E., Davis R.L. A distinct set of Drosophila brain neurons required for neurofibromatosis type 1-dependent learning and memory. J. Neurosci. 2010;30:10135–10143. doi: 10.1523/JNEUROSCI.0283-10.2010. doi:10.1523/JNEUROSCI.0283-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Corral T., Jimenez M., Hernandez-Munoz I., Perez de Castro I., Pellicer A. NF1 modulates the effects of Ras oncogenes: evidence of other NF1 function besides its GAP activity. J. Cell. Physiol. 2003;197:214–224. doi: 10.1002/jcp.10349. doi:10.1002/jcp.10349. [DOI] [PubMed] [Google Scholar]
- 21.Jacks T., Shih T.S., Schmitt E.M., Bronson R.T., Bernards A., Weinberg R.A. Tumour predisposition in mice heterozygous for a targeted mutation in Nf1. Nat. Genet. 1994;7:353–361. doi: 10.1038/ng0794-353. doi:10.1038/ng0794-353. [DOI] [PubMed] [Google Scholar]
- 22.Smith A.N., Miller L.A., Radice G., Ashery-Padan R., Lang R.A. Stage-dependent modes of Pax6–Sox2 epistasis regulate lens development and eye morphogenesis. Development. 2009;136:2977–2985. doi: 10.1242/dev.037341. doi:10.1242/dev.037341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.West-Mays J.A., Zhang J., Nottoli T., Hagopian-Donaldson S., Libby D., Strissel K.J., Williams T. AP-2alpha transcription factor is required for early morphogenesis of the lens vesicle. Dev. Biol. 1999;206:46–62. doi: 10.1006/dbio.1998.9132. doi:10.1006/dbio.1998.9132. [DOI] [PubMed] [Google Scholar]
- 24.Courtois-Cox S., Genther Williams S.M., Reczek E.E., Johnson B.W., McGillicuddy L.T., Johannessen C.M., Hollstein P.E., MacCollin M., Cichowski K. A negative feedback signaling network underlies oncogene-induced senescence. Cancer Cell. 2006;10:459–472. doi: 10.1016/j.ccr.2006.10.003. doi:10.1016/j.ccr.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Furuta Y., Hogan B.L. BMP4 is essential for lens induction in the mouse embryo. Genes Dev. 1998;12:3764–3775. doi: 10.1101/gad.12.23.3764. doi:10.1101/gad.12.23.3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Graw J. The genetic and molecular basis of congenital eye defects. Nat. Rev. Genet. 2003;4:876–888. doi: 10.1038/nrg1202. doi:10.1038/nrg1202. [DOI] [PubMed] [Google Scholar]
- 27.Wawersik S., Purcell P., Rauchman M., Dudley A.T., Robertson E.J., Maas R. BMP7 acts in murine lens placode development. Dev. Biol. 1999;207:176–188. doi: 10.1006/dbio.1998.9153. doi:10.1006/dbio.1998.9153. [DOI] [PubMed] [Google Scholar]
- 28.Burgess D., Zhang Y., Siefker E., Vaca R., Kuracha M.R., Reneker L., Overbeek P.A., Govindarajan V. Activated Ras alters lens and corneal development through induction of distinct downstream targets. BMC Dev. Biol. 2010;10:13. doi: 10.1186/1471-213X-10-13. doi:10.1186/1471-213X-10-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reneker L.W., Xie L., Xu L., Govindarajan V., Overbeek P.A. Activated Ras induces lens epithelial cell hyperplasia but not premature differentiation. Int. J. Dev. Biol. 2004;48:879–888. doi: 10.1387/ijdb.041889lr. doi:10.1387/ijdb.041889lr. [DOI] [PubMed] [Google Scholar]
- 30.Guerra C., Mijimolle N., Dhawahir A., Dubus P., Barradas M., Serrano M., Campuzano V., Barbacid M. Tumor induction by an endogenous K-ras oncogene is highly dependent on cellular context. Cancer Cell. 2003;4:111–120. doi: 10.1016/s1535-6108(03)00191-0. doi:10.1016/S1535-6108(03)00191-0. [DOI] [PubMed] [Google Scholar]
- 31.Tuveson D.A., Shaw A.T., Willis N.A., Silver D.P., Jackson E.L., Chang S., Mercer K.L., Grochow R., Hock H., Crowley D., et al. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell. 2004;5:375–387. doi: 10.1016/s1535-6108(04)00085-6. doi:10.1016/S1535-6108(04)00085-6. [DOI] [PubMed] [Google Scholar]
- 32.Haigis K.M., Kendall K.R., Wang Y., Cheung A., Haigis M.C., Glickman J.N., Niwa-Kawakita M., Sweet-Cordero A., Sebolt-Leopold J., Shannon K.M., et al. Differential effects of oncogenic K-Ras and N-Ras on proliferation, differentiation and tumor progression in the colon. Nat. Genet. 2008;40:600–608. doi: 10.1038/ngXXXX. doi:10.1038/ng.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shaw A.T., Meissner A., Dowdle J.A., Crowley D., Magendantz M., Ouyang C., Parisi T., Rajagopal J., Blank L.J., Bronson R.T., et al. Sprouty-2 regulates oncogenic K-ras in lung development and tumorigenesis. Genes Dev. 2007;21:694–707. doi: 10.1101/gad.1526207. doi:10.1101/gad.1526207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pan Y., Carbe C., Powers A., Zhang E.E., Esko J.D., Grobe K., Feng G.-S., Zhang X. Bud specific N-sulfation of heparan sulfate regulates Shp2-dependent FGF signaling during lacrimal gland induction. Development. 2008;135:301–310. doi: 10.1242/dev.014829. doi:10.1242/dev.014829. [DOI] [PubMed] [Google Scholar]
- 35.Corson L.B., Yamanaka Y., Lai K.-M.V., Rossant J. Spatial and temporal patterns of ERK signaling during mouse embryogenesis. Development. 2003;130:4527–4537. doi: 10.1242/dev.00669. doi:10.1242/dev.00669. [DOI] [PubMed] [Google Scholar]
- 36.Shukla V., Coumoul X., Wang R.H., Kim H.S., Deng C.X. RNA interference and inhibition of MEK-ERK signaling prevent abnormal skeletal phenotypes in a mouse model of craniosynostosis. Nat. Genet. 2007;39:1145–1150. doi: 10.1038/ng2096. doi:10.1038/ng2096. [DOI] [PubMed] [Google Scholar]