

Abstract
Duchenne muscular dystrophy, the most common form of childhood muscular dystrophy, is caused by X-linked inherited mutations in the dystrophin gene. Dystrophin deficiencies result in the loss of the dystrophin–glycoprotein complex at the plasma membrane, which leads to structural instability and muscle degeneration. Previously, we induced muscle-specific overexpression of Akt, a regulator of cellular metabolism and survival, in mdx mice at pre-necrotic (<3.5 weeks) ages and demonstrated upregulation of the utrophin–glycoprotein complex and protection against contractile-induced stress. Here, we found that delaying exogenous Akt treatment of mdx mice after the onset of peak pathology (>6 weeks) similarly increased the abundance of compensatory adhesion complexes at the extrasynaptic sarcolemma. Akt introduction after onset of pathology reverses the mdx histopathological measures, including decreases in blood serum albumin infiltration. Akt also improves muscle function in mdx mice as demonstrated through in vivo grip strength tests and in vitro contraction measurements of the extensor digitorum longus muscle. To further explore the significance of Akt in myofiber regeneration, we injured wild-type muscle with cardiotoxin and found that Akt induced a faster regenerative response relative to controls at equivalent time points. We demonstrate that Akt signaling pathways counteract mdx pathogenesis by enhancing endogenous compensatory mechanisms. These findings provide a rationale for investigating the therapeutic activation of the Akt pathway to counteract muscle wasting.
INTRODUCTION
Duchenne muscular dystrophy (DMD) is the most common and severe form of childhood muscular dystrophy, caused by X-linked inheritance of mutations in the dystrophin gene. Mdx mice, an animal model for DMD, are also deficient in dystrophin, a crucial structural protein that is essential for assembly of the dystrophin–glycoprotein complex (DGC) at the myofiber sarcolemma (1–7). In the absence of dystrophin, the mechanical connection between the intracellular actin cytoskeleton and the extracellular matrix (ECM) is lost, resulting in reduced sarcolemmal stability during muscle contractions (8,9). The increased susceptibility of the sarcolemma leads to microfissures, causing an influx of calcium, aberrant activation of calcium-dependent proteases and, eventually, onset of apoptosis or cellular necrosis (10,11). Mdx mice undergo periods of myofiber degeneration followed by recuperative regeneration of new myofibers beginning at 3 weeks of age (12). The severity of pathology peaks at 6 weeks of age and stabilizes thereafter, and mdx animals have a lifespan comparable with wild-type (WT) mice (13–17).
Despite ongoing cycles of degeneration and regeneration, mdx mice do not have the severe loss of ambulation characteristic of DMD patients. The milder phenotype in mdx mice may be due, in part, to the ability of utrophin (utrn) to compensate for the loss of dystrophin in murine skeletal muscle. Utrophin, an autosomal homolog of dystrophin, is normally restricted to the neuromuscular junction in WT skeletal muscle where it forms the utrophin-glycoprotein complex (UGC). However, in dystrophin deficiency, utrophin levels are increased in muscle and the UGC is more broadly localized around the sarcolemma, thereby partially compensating for the loss of dystrophin (18). Mdx mice overexpressing the utrophin transgene experience nearly complete rescue from dystrophic pathophysiology (19,20). Upregulation of integrins at the sarcolemma can also compensate for the loss of dystrophin. The significant role of integrin is highlighted by elegant experiments showing that integrin overexpression can significantly reduce disease pathology in dystrophin and utrophin double-null (mdx:utrn−/−) mice (21). Development of human therapies that target mechanisms to increase levels of utrophin and integrin is of special interest.
A potential mediator of these compensatory proteins is the Akt signaling pathway, which is a ubiquitous regulator of cell cycle and proliferation, cell growth and hypertrophy, metabolism and apoptosis (reviewed in 22). Also known as protein kinase B, the Akt family of serine/threonine kinases is activated downstream of cell surface receptor tyrosine kinases and the phosphoinositide 3-kinase [PI(3)K] pathway. Numerous studies have demonstrated that the Akt1 isoform induces muscle hypertrophy in vitro and in vivo through activation of the mammalian target of rapamycin (mTOR) pathway (23–26). Akt1 has also been shown to be crucial for the differentiation of myoblasts into fused myofibers (27). We and others have established that the same PI(3)K/Akt signaling pathways responsible for inducing skeletal muscle hypertrophy (24) are also activated in many forms of muscular dystrophy (28). Furthermore, we have recently demonstrated that direct manipulation of Akt in normal, WT mice induces skeletal muscle hypertrophy while decreasing adipose mass (29). Taken together, these previous reports support a role for activation of Akt in dystrophic muscle as a key mediator of the hypertrophic response both in the promotion of muscle hypertrophy and in blood vessel recruitment.
In dystrophic muscle, elevated Akt signaling has been associated with advanced dystrophy and peak stages of muscle hypertrophy (28). Increases in Akt activation may be a compensatory response that may serve as an early biomarker of pathology. Mdx mice induced to upregulate Akt by genetic ablation of myostatin or overexpression of insulin-like growth factor 1 (IGF-1) exhibit increases in muscle fiber area and force generation (30,31). The multi-faceted signaling roles of Akt1 make it an interesting candidate for promotion of myofiber growth and survival that may counteract the progressive muscle wasting seen in DMD.
Two important adhesion complexes are known to significantly ameliorate the effects of dystrophin deficiency in mdx skeletal muscle. The integrins are a ubiquitous family of integral membrane proteins that overlap with the UGC in compensatory overexpression in mdx (reviewed in 32). The α7β1D isoforms of integrin are normally restricted to the neuromuscular and myotendinous junctions in WT muscle but are thought to form part of the compensatory response in mdx muscles due to their increased expression in dystrophin deficiency (33–35). Like the DGC/UGC, α7β1D integrin stabilizes the sarcolemma by linking the ECM and cytoskeleton. In normal adult mice, utrophin expression is restricted to the neuromuscular and myotendinous junctions (7,36). Relative to WT muscle, mdx mice express slightly higher levels of utrophin around the sarcolemma, which is thought to partially compensate for the loss of dystrophin (18,36). Transgenic overexpression of α7 integrin in mdx:utrn−/− muscle significantly elevates extrasynaptic levels of integrin (21).
In our previous investigation, we showed that overexpression of skeletal muscle-specific Akt1 in young, pre-necrotic mdx mice increased expression of the UGC and integrin around the sarcolemma (37). In addition to upregulation of the UGC, we demonstrated that Akt increased mRNA and protein levels of α7 and β1D integrin as well as dysferlin, a calcium-regulated membrane repair protein (37). Improving levels of several adhesion complexes at the sarcolemma were sufficient to rescue sarcolemmal fragility as documented by reduced infiltration of Evan's Blue Dye (EBD) into mdx myofibers (37). However, central nucleation, which occurs in regenerating myofibers after dystrophic degeneration, was not reduced after Akt treatment. Reductions in pathological central nucleation in mdx may have been masked by spontaneous myofiber regeneration, which also occurred in WT muscle upon Akt induction (37). The conclusion from these data is that Akt expression is an important regulator of muscle regeneration in the absence of pathology and degeneration.
Based on our previous studies, we sought to determine whether Akt could influence expression of compensatory adhesion complexes in dystrophin-deficient mice after the onset of pathology. In determining the potential of utrophin-based therapy for DMD patients after onset of pathology, it is crucial to elucidate whether Akt can elevate utrophin levels at the sarcolemma during later stages of disease. In order to address this issue, we delayed our Akt treatment paradigm in dystrophin-deficient mdx mice to 6 weeks of age during the peak pathological state. We found that Akt improves membrane integrity as well as physiological functions of muscle in mdx mice.
RESULTS
Generation of mdx mice with skeletal muscle-specific inducible Akt expression
Mice were engineered to express the TRE-myrAkt1 and MCK-rtTA transgenes, both of which are required to induce conditional, skeletal muscle-specific expression of Akt1 (29,37). Mice with either the TRE-myrAkt1 or MCK-rtTA transgene are referred to as single transgenic (STG) and serve as controls since they lack the means for conditional activation of Akt. Male mice with both transgenes (DTG) were mated with WT and mdx females to yield male offspring of four genotypes: WT STG, WT DTG, mdx STG and mdx DTG. Doxycycline (DOX), a tetracycline derivative used to control expression of the reverse-tetracycline transactivator under control of a modified muscle creatine kinase (MCK) promoter, was administered in the drinking water from 6 to 9 weeks of age to induce Akt1 transgene expression in DTG mice. This time frame represents the peak necrotic stage of pathology in mdx mice (Fig. 1A).
Figure 1.

Exogenous Akt induction increases muscle mass without affecting body mass. (A) Schematic illustration of Akt treatment paradigm over the time course of mdx disease progression. STG and DTG mice of both WT and mdx genotypes were treated with DOX to overexpress exogenous Akt in DTG skeletal muscle. Mice were treated with DOX at 6 weeks of age, after the onset of mdx muscle pathology that begins at 3 weeks of age. At 9 weeks of age (3 weeks of DOX treatment), mice underwent functional tests and tissue collection. (B) Mdx DTG mice exhibit a significant increase in total body mass when compared with their STG counterparts, whereas WT DTG masses do not change significantly. There was a significant effect of WT/mdx (ANOVA, P < 0.003) and STG/DTG (ANOVA, P < 0.004) on body mass. Results of post hoc comparisons using Bonferroni-corrected α-level are as follows: *P < 0.001 when compared with WT STG; •P < 0.001 when compared with mdx STG. Data displayed in box plots are the median (line in box), mean (circle), 25th and 75th percentile (box) and minimum and maximum values (whiskers; n= 6–10 mice in WT groups, 16–17 mice in mdx groups). (C) Both WT DTG and mdx DTG mice display significant increases in quadriceps mass 3 weeks post-Akt activation (n= 4–9 mice in each group). There was no significant effect of WT/mdx; however, there was a significant effect of STG/DTG (ANOVA, P < 0.0001) and an interaction between mdx and DTG (ANOVA, P < 0.004). Results of post hoc comparisons using Bonferroni-corrected α-level are as follows: *P < 0.002 when compared with WT STG; #P < 0.007 when compared with WT DTG; •P < 0.0001 when compared with mdx STG.
Muscle expressing the TRE-myrAkt1/MCK-rtTA transgenes (DTG) has been shown to undergo muscle fiber hypertrophy upon transgene activation, leading to a 5% increase in lean muscle mass and a decrease in adipose mass without affecting total body weight (29). We found that Akt activation in our treatment paradigm did not affect body mass in WT mice (Fig. 1B). However, in mdx DTG mice, Akt transgene activation was associated with an 8% increase in total body mass when compared with mdx STG mice (Fig. 1B). Isolated quadriceps muscles, which contain predominantly type IIb fibers, were weighed to examine whether Akt transgene activation influenced muscle mass. Both WT DTG and mdx DTG quadriceps were notably heavier than their STG counterparts (77 and 31% increases in average mass, respectively), correlating with Akt-induced muscle hypertrophy (Fig. 1C). Additionally, mdx STG quadriceps were markedly (31%) larger than those of WT STG due to pseudohypertrophy in mdx muscle caused by tissue inflammation, fibrosis and adipose deposition (Fig. 1C). WT DTG and mdx DTG quadriceps masses were not significantly different from one another, reflecting a plateau in muscle growth due to the Akt transgene (Fig. 1C).
Exogenous Akt activation reduces histological measures of mdx pathophysiology
Dystrophic muscle is characterized by the presence of myofiber degeneration, immune cell infiltration and increased connective tissue deposition. Transverse quadriceps sections were stained with hematoxylin and eosin (H&E) to examine the effect of Akt transgene activation on mdx pathology (Fig. 2A and B). Mdx quadriceps displayed signs of muscle damage, although regions of pathology were significantly reduced in mdx DTG after Akt induction (Fig. 2A and B). Central nucleation, a marker for myofiber regeneration, was elevated (11%) in WT DTG muscles when compared with low (1%) central nucleation in WT STG (Fig. 2C). In contrast, the percentage of centrally nucleated fibers was slightly reduced in mdx DTG quadriceps when compared with mdx STG (51 and 60% of fibers, respectively), although this difference was not statistically significant (Fig. 2C). The effect of Akt on central nucleation in mdx DTG muscle was confounded by Akt-induced spontaneous regeneration in WT mice and regeneration induced as a consequence of degeneration in mdx mice. We found that Akt activation increased median cross-sectional areas by 2- and 2.5-fold in WT and mdx myofibers, respectively, when compared with their STG counterparts (Fig. 2D), which is consistent with previous reports of Akt-induced muscle hypertrophy (29,37). Approximately 30% of DTG myofibers were at least 8000 µm2 in cross-sectional area, whereas <1% of STG myofibers exhibited cross-sectional areas of 8000 µm2 or greater (Fig. 2D). Median fiber areas were as follows: 2292 µm2 (WT STG), 5365 µm2 (WT DTG), 1938 µm2 (mdx STG) and 4803 µm2 (mdx DTG).
Figure 2.

Post-degenerative Akt induction improves mdx histopathology. (A) Transverse quadriceps muscle cryosections were stained with H&E and examined by light microscopy for histological changes upon Akt activation. In the representative images, numerous necrotic regions are present in mdx STG sections as visualized by areas of intense purple staining (denoted with *) that are not present in normal, healthy muscle (WT STG). Akt activation in mdx muscle exhibits significant reduction of myofiber necrosis (mdx DTG). No changes in overall muscle histology were observed in WT muscle upon Akt induction (WT DTG). Bar, 1 mm. (B) Insets of transverse quadriceps from (A) show modest reductions in central nucleation, a marker for muscle regeneration, in mdx DTG when compared with mdx STG controls. Constitutive Akt activation in mdx and WT DTG mice increases fiber size variability and induces muscle fiber hypertrophy. Bar, 50 µm. (C) Centrally nucleated fibers (% of total fibers) were quantified as a measure of muscle regeneration. Constitutive Akt expression has differential effects on regeneration in WT and mdx muscle. There was a significant effect of WT/mdx (ANOVA, P < 0.0001), no significant effect of STG/DTG and an interaction between the two factors (ANOVA, P < 0.006). WT DTG mice exhibited significant increases in regenerated fibers compared with WT STG controls. Mdx mice had elevated central nucleation when compared with WT mice, but did not exhibit significant differences between transgenic groups (*P < 0.003 when compared with WT STG; #P < 0.005 when compared with WT DTG; Bonferroni-corrected α-level). Fibers counted per whole quadriceps ranged from 1200 to 6000 fibers (n= 4–9 individual quadriceps in each group). (D) Cross-sectional fiber areas were measured from quadriceps muscles to describe Akt-mediated hypertrophy. Both WT and mdx DTG mice exhibited a 2-fold increase in median myofiber area when compared with STG counterparts. Data are displayed as mean ± SEM (300 adjacent fibers traced per muscle, n= 4 in each group).
The loss of dystrophin and the DGC at the sarcolemma results in membrane disruption during muscle contraction. The EBD assay, which traces infiltration of blood serum albumin into compromised muscle fibers, is an indicator of myofiber damage and, indirectly, necrosis. Mdx STG muscle exhibited elevated levels of EBD-positive fibers when compared with the remaining genotypes (Fig. 3A and B). Mdx DTG mice had markedly lower levels of EBD-positive fibers and were comparable with WT controls (Fig. 3A and B). The results from our EBD analysis suggest that exogenous Akt introduction after the onset of necrotic stages can significantly reduce mdx pathology.
Figure 3.

Post-degenerative Akt induction restores sarcolemmal stability in mdx muscle. (A) EBD, which binds to blood serum albumin, infiltrates compromised sarcolemmas and was used to detect damaged myofibers. EBD-positive fibers in transverse quadriceps are shown in red fluorescence. Indirect immunofluorescence was used to visualize laminin in the ECM (green). When compared with mdx STG quadriceps, mdx DTG mice have fewer patches of degenerating fibers. Bar, 100μm. (B) EBD-positive myofibers were quantitated (% of total fibers) in transverse quadriceps sections. There was a significant effect of WT/mdx (ANOVA, P < 0.0001) and an interaction between the two WT/mdx and STG/DTG factors (ANOVA, P < 0.0001). Constitutive Akt activation significantly reduces EBD infiltration in mdx DTG mice to nearly WT levels when compared with mdx STG. In contrast, Akt significantly increases EBD infiltration in WT DTG mice when compared with WT STG controls (*P < 0.005 when compared with WT STG; #P < 0.002 when compared with WT DTG; •P < 0.0001 when compared with mdx STG; Bonferroni-corrected α-level; n= 6–9 individual quadriceps in WT groups, 12–14 quadriceps in mdx groups).
Akt activation increases levels of several compensatory adhesion complexes
In order to determine the level of Akt overexpression after 3 weeks of transgene activation, we immunoblotted total skeletal muscle lysates with Akt antibodies. Induction of the Akt transgene was restricted to DTG muscle, while STG controls with either the TRE-myrAkt1 or MCK-rtTA transgene lacked conditional Akt activation (Fig. 4), as previously documented for this model system (29,37). Total Akt levels were elevated 3-fold in DTG muscles relative to their STG controls. These data are consistent with our previous finding in which the 3-week treatment of WT and mdx mice began at the pre-necrotic stage (37). The levels of Akt expression in DTG muscle that we observed represent the maximal level of Akt induction, which occurs as early as 3 days post-DOX treatment (29). DTG muscle exhibited a >20-fold increase in P-Akt levels compared with identically treated STG controls (Fig. 4). Transgene expression occurs primarily in type IIb fast twitch/glycolytic fibers, probably due to the enhancement of the MCK promoter in these fibers (29,38).
Figure 4.
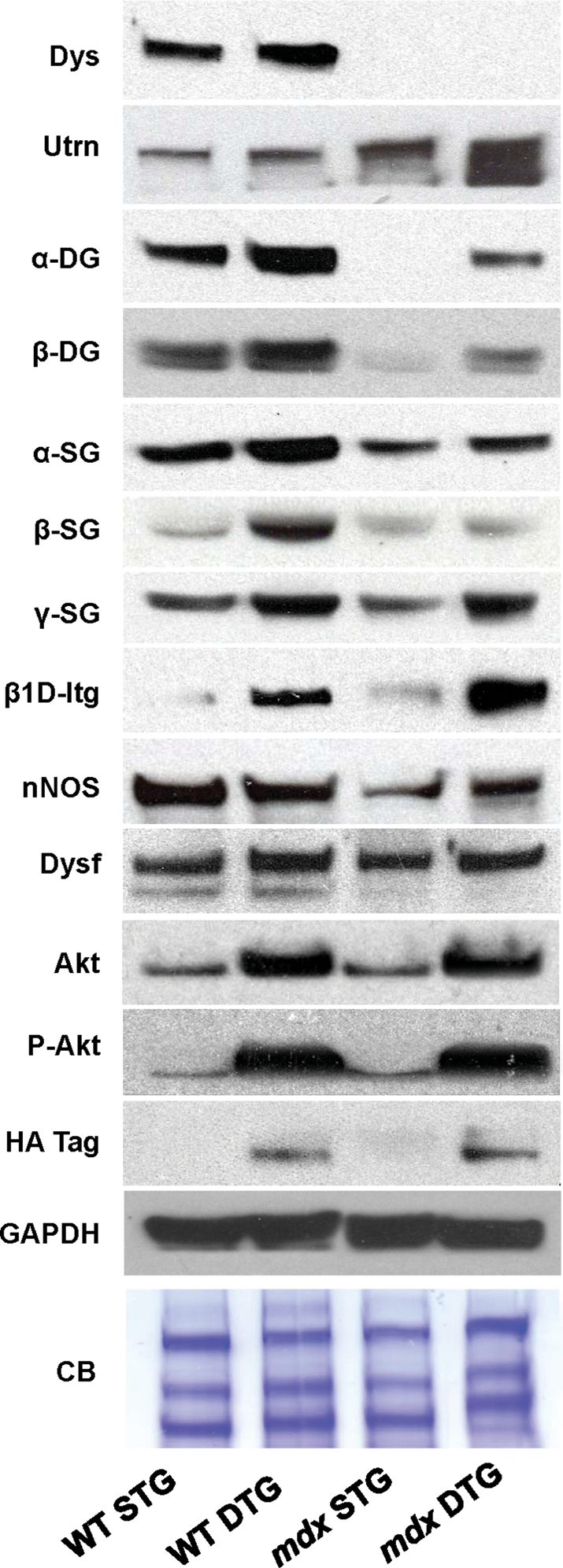
Akt increases abundance of adhesion complex components and membrane repair proteins. Total skeletal muscle preparations in 9-week-old mice were analyzed by immunoblotting with the indicated antibodies. Utrophin and its associated proteins in the utrophin-glycoprotein complex (UGC), α- and β-DG, as well as alpha-, beta- and gamma-sarcoglycan (α-SG, β-SG and γ-SG) are restored to nearly WT levels. Dystrophin (dys) expression is absent in mdx tissue. β1D integrin (β1D-Itg) is substantially increased in mdx DTG mice, above even WT DTG levels. Dysferlin (Dysf), a calcium-modulated membrane repair protein, is mildly increased in mdx DTG mice. Total neuronal nitric oxide synthase (nNOS, cytosolic and membrane bound) is moderately increased in mdx DTG mice but not in WT DTG mice, when compared with STG counterparts. Phosphorylated Akt (P-Akt) and the HA tag, which was engineered on the Akt transgene, are shown for the functional identification of exogenous Akt. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and Coomassie blue (CB) staining of the protein gel are shown as loading controls.
In addition to anti-apoptotic signaling, Akt activation can promote dystrophic myofiber survival by increasing the expression of several adhesion complexes, most notably integrin and the UGC (37,39). Within the UGC, utrophin replaces dystrophin and associates with the dystroglycans (α- and β-DG), which are in turn stabilized by the sarcoglycan (SG)–sarcospan subcomplex (including α-, β-, γ- and δ- SG; reviewed in 39). In fetal and developing muscle fibers, utrophin is expressed around the entire sarcolemma, whereas utrophin is restricted to the postsynaptic regions in adult skeletal muscle (40,41). Since increased expression of the UGC or integrin can rescue membrane fragility in mdx muscle, we examined whether these adhesion complexes were altered by Akt transgene activation in adult muscle as a feasible mechanism for sarcolemmal membrane improvements observed in the mdx DTG mice. In our analysis, we examined total protein levels by immunoblotting skeletal muscle lysates in addition to investigating membrane expression of each protein by indirect immunofluorescence.
In 9-week-old mice, Akt activation increased dystrophin levels in WT skeletal muscle to an average of 1.5-fold in relation to STG controls, as observed in immunoblots of total muscle lysates (Fig. 4). Utrophin levels were increased at an average of 3-fold in mdx DTG compared with its STG control (Fig. 4). β-DG, which associates with both utrophin and dystrophin, was not significantly elevated in WT DTG mice but was increased in mdx DTG mice at levels similar to WT STG mice (Fig. 4). Levels of α-DG, which serves as a receptor for ECM ligands, were significantly increased in WT DTG and mdx DTG mice relative to STG counterparts (Fig. 4). Akt induction also increased levels of SG proteins with respect to their STG controls in both mdx and WT STG muscles (Fig. 4). Importantly, we found that activation of Akt in older mice restores the UGC to a functional level of rescue at the sarcolemma, attributed to the elevated levels of utrophin–dystroglycan complex and, to a lesser degree, the SG–sarcospan subcomplex. We also found that β1D integrin expression dramatically increases in both WT and mdx DTG skeletal muscles relative to STG controls, as revealed by immunoblot analysis (Fig. 4). Furthermore, the UGC and β1D integrin are expressed homogenously and robustly at the sarcolemma in DTG muscles (Fig. 5), which promote sarcolemma stability. Due to the lack of commercially available high-titer antibody, we were unable to probe for the α7 integrin subunit.
Figure 5.

Akt restores protein complexes to the extrasynaptic sarcolemma. Selected integral and peripheral membrane proteins were visualized using indirect immunofluorescence on transverse quadriceps sections. Akt induction leads to increased presence of the DGC in WT DTG mice. In mdx DTG muscle, extrasynaptic UGC and integrin expression is evident. Integrin, which is not a component of the DGC/UGC, is also increased in DTG mice when compared with STG counterparts. nNOS is also modestly increased at the sarcolemma in WT and mdx DTG muscle, although staining is quite heterogeneous. Bar, 25μm.
In addition to adhesion complexes that promote sarcolemma stability, Akt1 activation leads to increased synthesis of accessory proteins that promote myofiber survival and function. Dysferlin, a calcium-dependent membrane repair protein, was only mildly increased in DTG muscle relative to STG controls (Fig. 4). We also observed significant elevations in neuronal nitric oxide synthase (nNOS) at the sarcolemma in WT DTG muscle (Fig. 5) that was not accompanied by a change in total nNOS protein levels (Fig. 4), suggesting that increasing levels of the DGC provided additional anchorage for nNOS at the surface membrane. However, the effects of Akt expression were much more modest in mdx DTG muscle where nNOS expression at the sarcolemma was heterogeneous (Figs 4 and 5).
Akt-mediated amelioration of dystrophic pathology improves muscle function
In order to determine whether restoration of the UGC to the sarcolemma affects function, we measured several physiological parameters in Akt-treated muscle. First, we recorded force capacity of forelimbs of treated 9-week-old mice using a standard grip strength assay. Under the conditions of our assays, average grip strength over a series of five measurements was unaffected by Akt expression in WT mice (Fig. 6A). However, mdx DTG mice had statistically significantly greater average grip strength performances when compared with mdx STG mice, although the degree of improvement in mdx DTG did not reach WT levels (Fig. 6A). We were unable to detect differences in average grip strength between WT STG and WT DTG groups.
Figure 6.

Akt expression improves force production and resistance to fatigue. (A) Nine-week-old male mice were subjected to forelimb grip strength tests to evaluate the effect of exogenous Akt on muscular strength. There was a significant effect of WT/mdx (ANOVA, P < 0.0001) and a significant effect of STG/DTG (ANOVA, P < 0.02). Overall, WT mice perform significantly better than mdx mice in average grip strength, measured in Newtons (N). WT STG and DTG mice genotypes perform similarly; however, mdx DTG performed better than mdx STG (values obtained from an average of five trials, *P < 0.0001 when compared with WT STG; #P < 0.0001 when compared with WT DTG; •P < 0.003 when compared with mdx STG; Bonferroni-corrected α-level; n= 6 WT DTG mice and 12–16 mice each in other groups). (B) Forelimb grip strength results from five individual trials are represented as a percentage value of the first trial. Three-way mixed ANOVA with repeated measures found a significant effect of WT/mdx (ANOVA, P < 0.0001), a significant interaction of WT/mdx and STG/DTG (ANOVA, P < 0.0001), a significant effect of trial number within subjects (ANOVA, P < 0.0001) and a significant interaction between trial number and WT/mdx (ANOVA, P < 0.0001). WT STG and DTG grip strength performance did not decline through sequential trials. Both mdx STG and DTG mice showed a significant decline in grip strength by the last trial (*P < 0.0001 within subjects when compared with trial 1, #P < 0.0001 when compared with WT groups; Bonferroni-corrected α-level). Mdx DTG mice displayed significantly elevated grip strength after five trials when compared with mdx STG mice (•P < 0.004; Bonferroni-corrected α-level; n= 4 WT DTG mice and 10–13 mice each in all other groups). Data are represented as mean ± SEM. (C) Immunoblot analysis of protein lysates from several muscle groups show that Akt transgene expression occurs in both slow and fast twitch muscles after 3 weeks of DOX treatment. Blots were probed with antibodies to the HA tag that was engineered onto the Akt transgene. Muscles shown are gastronemius, soleus, tibialis anterior and extensor digitorum longus (EDL). Note that all DTG muscle groups express robust levels of transgenic Akt, which is not present in muscle from STG mice. (D) Muscle strength was further characterized by in vitro stimulation of the EDL. Maximal force (g) generated in response to single stimuli and 300 ms tetanic stimulation at ascending frequencies was normalized to EDL mass (mg) as shown. Resting muscle length was set at the length at which single supramaximal stimuli produced maximal twitch tension. At frequencies of 40 Hz and higher, the mdx curves differ significantly from one another (*P < 0.02 or smaller, Student's t-test), and both differed significantly from either WT muscle. WT STG and DTG values were not statistically different after normalization for muscle weight (n= 5 for WT groups and 8–10 for mdx groups). (E) Effects of transgene expression on muscle contractile fatigue in EDL muscles from mdx mice. Curves plot the maximal tetanic strength in response to 100 Hz stimulation for 300 ms, every 2 s, for 5 min. There was a significant effect of the repeated stimulations on fatigue (ANOVA, P < 0.001). Expression of the Akt transgene significantly slows and reduces the extent of fatigue in mdx DTG in comparison with mdx STG muscles (ANOVA, P < 0.02; n= 6–7 muscles in each group).
In addition to examining the decline in average grip strength, we evaluated the grip performance relative to the first peak grip performance of each animal across five serial grip strength trials. Normalizing each trial to the first peak grip strength measurement permitted an examination of fatigue independent of the magnitude of force production. WT mice displayed consistent grip strength performances throughout all five trials, regardless of the transgenic group (Fig. 6B), supporting the conclusion that they were resistant to fatigue during the course of the experiment. Mdx mice exhibited peak grip strength values during the first trial that were comparable with WT levels, but both STG and DTG fatigued quickly as evidenced by dramatic force deficits in each subsequent trial (Fig. 6B). Importantly, mdx DTG mice were resistant to the dramatic loss of relative force when compared with mdx STG mice (Fig. 6B).
We further tested the extent of muscle strength and fatigability through electrophysiological stimulation of isolated extensor digitorum longus (EDL) muscles. Previous studies have shown that exogenous Akt expression is the highest in fast twitch/glycolytic muscle groups with predominantly type II fibers (29). We found that mice treated with DOX for 3 weeks displayed robust expression of exogenous Akt in EDL as shown by immunoblots of muscle protein lysates using antibodies to the hemagglutinin (HA) tag engineered onto the transgene (Fig. 6C). Immunoblots of lysates harvested from the gastrocnemius, soleus and tibialis anterior muscles are shown for comparison (Fig. 6C). Analysis of absolute force amplitudes suggests that Akt induction improves strength in both WT and mdx muscles (data not shown). However, after normalization to muscle mass, specific force produced by EDL muscles from WT STG and DTG mice exhibited similar performances in force amplitude (Fig. 6D). Akt expression in mdx EDL produced higher specific force amplitudes compared with STG counterparts at peak tension (100 Hz) as well as all other stimulation frequencies from 40 to 150 Hz (Fig. 6D). Mdx mice displayed reduced force production relative to WT mice, and improvements observed in mdx DTG mice were not sufficient to reach WT STG levels (Fig. 6D). Mdx EDL muscles were also examined for fatigue as revealed by the percent decline in amplitude relative to the first amplitude throughout serial stimulations (Fig. 6E). Modest improvements in resistance to fatigue were evident in mdx DTG relative to STG controls.
Protective effect of Akt following cardiotoxin injury
In order to determine whether Akt affects cell survival and myofiber regeneration after acute muscle damage, we used the cardiotoxin (CTX) model of muscle injury in WT STG and DTG mice. CTX, a cytotoxic venom obtained from Naja nigricollis, causes pore formation at the sarcolemma, followed by membrane depolarization and eventual degeneration due to Ca2+-dependent apoptosis (42). Adult WT mice were pre-treated with DOX for 2 weeks prior to CTX injury to induce Akt expression and continually treated until tissue harvesting (Fig. 7A). Histological surveys of muscle sections stained with H&E revealed that WT STG muscles underwent degeneration at 2 days following CTX injury, whereas WT DTG muscles already showed regions of small, centrally nucleated fibers and mononuclear cell infiltration at the same time point (Fig. 7B). Analysis of contralateral phosphate-buffered saline (PBS)-injected quadriceps revealed no substantial histological damage caused by injection (data not shown). Centrally nucleated fibers and mononuclear cells appeared in WT STG tissue by 4 days following injury and persisted for 7 days in both WT STG and DTG muscles (Fig. 7B).
Figure 7.

Akt activation reduces severity of CTX-induced injury. (A) Schematic timeline depicting the course of DOX treatment and CTX injury in WT STG and DTG mice. Following 2 weeks of Akt activation, quadriceps were injected with CTX and PBS contralaterally. Quadriceps were harvested for histological analysis prior to injury and 1, 2, 4 and 7 days post-CTX injury as indicated. (B) Transverse quadriceps sections were stained with H&E to detect comprehensive damage due to CTX. DTG mice show reduced histopathology at 2, 4 and 7 days following injury when compared with their STG counterparts. Central nucleation appears at 2 days post-injury in DTG mice, whereas STG mice develop central nucleation at day 4. Bar, 50 µm. (C) Quadriceps were surveyed for sarcolemmal disruption and indirectly for necrosis using the EBD tracer assay. Beginning at 1 day post-injury, WT STG and DTG muscles exhibit extensive damage as indicated by the presence of widespread EBD-positive fibers (red fluorescence). By day 7 post-injury, EBD infiltration persists in STG mice, while it is dramatically reduced in DTG mice. Laminin (green) is stained to delineate the ECM. Bar, 100 µm. (D) Newly regenerating myofibers were detected with indirect labeling of dMHC (green). DTG mice show an accelerated regenerative response to injury, as demonstrated through the appearance of dMHC-positive fibers at 2 days following injury. dMHC-positive fibers appear in STG mice at 4 days post-injury. Laminin is depicted with red fluorescence. Bar, 100 µm.
We further examined the rate of myofiber regeneration after injury-induced degeneration by tracking the appearance of EBD- and developmental myosin heavy chain- (dMHC) positive fibers, the latter being a marker for newly regenerating fibers. At 1 and 2 days following injury, WT STG injured quadriceps exhibited infiltration of EBD into nearly all myofibers, while regenerating dMHC-positive cells were not found (Fig. 7C and D). WT DTG quadriceps displayed the same level of injury at 1 day post-injury as evidenced by nearly widespread EBD infiltration. However, by 2 days post-injury, we observed a reduction in EBD-positive fibers and widespread dMHC staining in WT DTG quadriceps (Fig. 7C and D). At 4 days, WT STG mice presented a histological profile comparable with that of WT DTG at 2 days following injection, including reduced EBD infiltration and appearance of dMHC fibers. At subsequent time points, EBD infiltration was sparse, signifying the end of degenerative phases due to CTX injury, and regenerative dMHC cells persisted at lower levels in both genotypes until 7 days post-injection (Fig. 7C and D). Taken together, we conclude that myogenic Akt upregulation not only confers protection against membrane damage but also improves myofiber regeneration.
DISCUSSION
In our current study, we demonstrate that overexpression of constitutively active Akt1 can restore sarcolemmal integrity, promote muscle regeneration and improve functional parameters in muscle at post-necrotic stages of disease. Because the pathogenesis of DMD progresses with age, it is critical to determine the stages of disease at which therapeutic intervention is effective. The muscle-specific, inducible transgene system allowed us to control the timing of exogenous Akt treatment. We delayed onset of Akt expression to a time period after myofiber development and maturation. By postponing the introduction of Akt, we were able to model a therapeutic scenario comparable with later courses of disease progression that would not be possible with a conventional transgenic system. We find that Akt targets multiple pathways that protect against degeneration and promote regeneration in dystrophin-deficient muscle.
Blaauw et al. (43) previously reported that they were unable to identify improvements in EBD infiltration and central nucleation using tamoxifen-induced myr Akt-ER transgenic mice. However, they found reduced force deficits in Akt-mdx gastrocnemius muscle following eccentric contractions in skinned and intact fibers (43,44). Interestingly, Blaauw et al. (43) reported that rapamycin treatment, which blocks phosphorylation of Akt target p70S6K kinase, did not alter the protective effects of Akt on eccentric contraction-induced force drop in mdx muscle. There are many differences in our two Akt overexpression model systems. First, the tamoxifen-induced model system allows expression in fast and slow twitch fibers. Our system is limited to fast twitch fibers and the levels of P-Akt transgene activation vary in different muscle groups. Secondly, we document improvements in skeletal muscle membrane damage that were not observed in the tamoxifen-induced system. Thirdly, we perform an extensive analysis on the effects of the Akt transgene on the expression of many adhesion complexes in muscle. There is great value to examination of both model systems in that the recombinant protein and/or the chemical induction methods may have unintended effects on muscle.
In our Akt mdx mouse model, we have previously shown that upregulation of Akt signaling amplifies activation of p70S6K, a downstream effector of the mTOR pathway of protein synthesis, and increases synthesis of two compensatory protein adhesion complexes, the UGC and the integrins (37). Both complexes bind laminin in the ECM to confer structural stability and promote dystrophic myofiber survival. Previous studies have shown that type IIb muscle fibers are most susceptible to dystrophic pathology (45) and that treatment with the β2-adrenoceptor agonist formoterol can prevent contraction-induced injury in mdx fast twitch muscles (46). Thus, it is particularly advantageous that our transgene expression is most active in type IIb fibers where we have shown that it improves resistance of these fibers to sarcolemmal damage. It is unclear whether the protective effects of Akt in dystrophin deficiency observed in the tamoxifen-inducible (43,44) and DOX-inducible (37) models involve similar mechanisms, but further investigation of both systems will lead to insightful knowledge on the role of Akt in muscle repair and regeneration.
We propose that the abundance of adhesion complexes such as the UGC and integrins in adult mice strengthens the mechanical stability of the sarcolemma during contraction. However, the presence of these membrane proteins may augment signaling pathways that additionally contribute to myofiber growth and survival. The components of the DGC/UGC and the peripherally associated syntrophins and α-dystrobrevin contain phosphorylation sites and are known players in transmembrane signal transduction (reviewed in 47). For instance, β-dystroglycan, along with the integrins and caveolin-3, serves as a scaffold for adaptor proteins to bind and activate the Ras/MAPK pathway of cell growth, differentiation and survival (48–50). The SGs possess consensus sites for tyrosine phosphorylation, and SG-null animals have been shown to display aberrant signaling responses, implicating their importance in non-mechanical roles (51–53). Integrins have been shown to directly contribute to signaling processes that lead to myofiber hypertrophy and regeneration (54) as well as prevention of apoptosis (55) in mdx:utrn−/− animals. The histological and functional improvements that we observed in our mice may be linked to a myriad of complex signaling cascades that amplify the downstream benefits of Akt activation in dystrophin deficiency. Akt expression improved sarcolemma integrity in mdx DTG muscle. However, the effect of Akt on muscle regeneration is difficult to assess given the ongoing mdx pathology that was present at the time of Akt treatment. Evidence that Akt improves muscle regeneration is provided by CTX experiments in which pre-treatment of muscle with Akt improves muscle repair after acute injury.
Localization of nNOS to the sarcolemma through association with dystrophin can prevent exercise-induced fatigue through automodulation of blood flow (56–58). nNOS has also been shown to reduce membrane damage through anti-inflammatory actions in NOS transgenic/mdx mice (59), which may contribute to our observations of improved membrane stability and reduced mononuclear infiltration. Recent collaborative work from Davies and co-authors (60) reveals differences in the ability of dystrophin and utrophin to serve as anchorage sites for nNOS. Our work is consistent with these studies since we observed dramatic elevations in nNOS at the sarcolemma of WT DTG muscle probably due to increased levels of dystrophin upon Akt expression. Mdx DTG mice only exhibited modest changes in sarcolemmal nNOS, which lacked the homogeneous, uniform pattern of staining characteristic of normal muscle.
Additionally, Akt activation improves cell survival as well as regenerative responses to degeneration. Although Akt has been shown to promote MyoD-mediated muscle differentiation (61), exogenous Akt activation in satellite cells cannot be directly attributed to improved regenerative response because the MCK promoter is not expressed in undifferentiated satellite cells and myoblasts (reviewed in 62). Myogenic Akt activation has been previously shown to stimulate production of myokines that normalize metabolic parameters and induce revascularization after ischemic injury (29,63). It is feasible that myokine production downstream of Akt indirectly promotes satellite cell differentiation through paracrine signaling from mature myofibers (64) and autocrine amplification of IGF-1 receptor stimulation from differentiating myoblasts (61). In DMD muscle, fibroblasts deposit connective tissue after degeneration and secrete IGF-binding proteins that inhibit satellite cell regeneration (65).
Identifying key modulators of muscle regeneration and repair may reveal novel targets that, when properly regulated, prevent wasting while promoting growth and regeneration. Use of such targets is appealing since they have the potential to target a broad array of muscular dystrophies. Some therapeutic approaches, such as exon skipping and nonsense codon-suppressing drugs, can be limited by the need to target-specific genetic mutations. Many studies, including the current report, demonstrate that enhancing Akt activation promotes muscle hypertrophy (23–32,37). Furthermore, induction of Akt signaling in dystrophin-deficient muscle is particularly beneficial as it improves utrophin and integrin levels to restore membrane integrity caused by the loss of the sarcolemma–ECM connection. It is reasonable to speculate that Akt signaling is activated in most ‘rescue’ mechanisms in which muscle repair and growth are enhanced. By regulating endogenous pathways, Akt-targeted therapies have the potential to treat not only dystrophin-deficient muscular dystrophies, but also other inherited muscular dystrophies involving genetic mutations in the DGC and integrins. One major limitation to dramatic overexpression of Akt may be unregulated cell growth, as evidenced by the observation that many advanced tumors express high levels of activated Akt, and significant research efforts are aimed at identifying inhibitors of the cell cycle, growth and survival to prevent tumor progression (66). Whether long-term overexpression of Akt in skeletal muscle would promote similar effects is currently unknown and should be carefully considered before development of therapies that counteract the muscle wasting.
MATERIALS AND METHODS
Animal models
Female WT (C57BL/6J) and mdx mice (C57BL/10ScSnJ) were purchased from Jackson Laboratories (Bar Harbor, ME, USA). Male Akt DTG mice possessed the 1256 [3Emut] MCK-rtTA transgene expressing the reverse-tetracycline transactivator controlled by a mutated skeletal MCK promoter and the TRE-myrAkt1 transgene harboring the constitutively active form of the mouse Akt1 transgene controlled by a tetracycline-responsive promoter (29). Female WT and mdx mice were bred with founder Akt DTG males to produce the four genotypes used for comparison: (i) WT with a single transgene (WT STG), (ii) WT with both transgenes (WT DTG), (iii) mdx with a single transgene (mdx STG) and (iv) mdx with both transgenes (mdx DTG). Only male progeny were used in experiments.
For mdx-Akt transgenic comparisons, mice were treated starting at 6 weeks of age with 0.5 mg/ml DOX administered in drinking water. Following 3 weeks of treatment, mice were euthanized via inhalation of isoflurane anesthetic and tissues (blood, quadriceps, EDL, total skeletal muscle) were harvested. For CTX experiments, mice were treated with DOX at 12 weeks of age and sacrificed between 14 and 16 weeks of age (see below). Experimental procedures and animal maintenance were conducted with the approval of the Institutional Animal Care and Use Committee (IACUC) at UCLA.
Genotyping of Akt transgenic and mdx mice
Genomic DNA was isolated from mouse tail clippings using the DNeasy Blood and Tissue Kit (QIAGEN Inc., Valencia, CA, USA; #69506). Polymerase chain reaction (PCR) genotyping of WT and mdx dystrophin alleles was performed through the modified amplification-resistant mutation system (ARMS) assay (67,68). The MCK-rtTA and TRE-myrAkt1 transgenes were identified using PCR through previously described methods (37).
Histology
Quadriceps were dissected, weighed, mounted in optimal cutting temperature (OCT) tissue freezing medium (10.2% polyvinyl alcohol/4.3% polyethylene glycol), frozen in liquid nitrogen-cooled isopentane and stored at −80°C. For histological analyses, quadriceps were sectioned transversely at 8 µm in a CM 3050S cryostat (Leica Microsystems, Bannockburn, IL, USA) and mounted on Superfrost Plus positively charged slides (VWR International, West Chester, PA, USA; #48311-703). H&E staining was used for visualization of fibrosis, central nucleation and cross-sectional fiber area as previously described (37). Centrally nucleated fibers and cross-sectional fiber area were measured from digitized images captured with the AxioPlan 2 fluorescent microscope and AxioVision 4.8 software (Carl Zeiss Inc., Thornwood, NY, USA). Central nucleation was quantified as a percentage of centrally nucleated fibers over the total number of fibers in an entire transverse quadriceps section. Cross-sectional areas of fibers were sampled from 300 adjacent fibers in each quadriceps and calculated using the outline spline function.
EBD tracer assay
To measure sarcolemmal permeability, 50 µl per 10 mg of body weight of sterile EBD (10 mg/ml in PBS) was intraperitoneally injected into mice 18 h before tissue harvesting. For visualization of EBD-infiltrated myofibers in the quadriceps, cryosections were handled in the dark. Sections were fixed in ice-cold acetone prior to blocking with 3% bovine serum albumin and incubation with an anti-laminin primary antibody (Sigma, St Louis, MO, USA; #L 9393), which was detected with Alexa Fluor 488 (Invitrogen Corporation, Carlsbad, CA, USA; A11008, 1:200 dilution) labeled anti-rabbit antibody. Sections were mounted in VectaShield (Vector Laboratories, Burlingame, CA, USA; H-1000) and imaged using the AxioPlan 2 fluorescent microscope and AxioVision 4.8 software. Whole quadriceps mosaics were photographed, and sarcolemmal integrity was quantified through the percentage of EBD-positive fibers as a percentage of total fibers counted in the section.
Immunoblot analysis
Total skeletal muscles were collected, snap frozen in liquid nitrogen and stored at −80°C prior to use. Prior to protein lysate preparation, frozen skeletal muscles were crushed in liquid nitrogen with a mortar and pestle. Ice-cold RIPA lysis buffer (Thermo Scientific, Rockford, IL, USA; #89901) was modified by adding phosphatase inhibitors [1 mm sodium orthovanadate, 100nm okadaic acid and 5nm microcystin LR) and protease inhibitors (0.6 µg/ml pepstatin A, 0.5 µg/ml aprotinin, 0.5 µg/ml leupeptin, 0.75 mm benzamidine and 0.1 mm phenylmethylsulfonyl fluoride PMSF)]. Ten milliliters of modified RIPA buffer were added per gram of pulverized muscle, which was homogenized using a tissue miser at the lowest speed (Fisher Scientific, Pittsburgh, PA, USA). Homogenates were rotated for 1 h at 4°C, and centrifuged at 15 000g at 4°C for 15 min, after which the clarified supernatants were removed for use. Protein concentrations were measured using the DC Protein Assay (Bio-Rad, Hercules, CA, USA; #500-0111). Equal concentrations of protein (60 µg) were resolved through 4–20% gradient SDS–PAGE (Pierce, Rockford, IL, USA) and transferred to nitrocellulose membranes (Millipore, Billerica, MA, USA) for immunoblotting experiments.
Membranes were probed with antibodies to the following proteins, listed with their working dilutions: dystrophin [Developmental Studies Hybridoma Bank (DSHB), Iowa City, IA, USA; MANDYS, 1:10], utrophin (DSHB; MANCHO3, 1:5), α-DG (Millipore; IIH6, 1:700), β-DG (DSHB; MANDAG2, 1:20), α-SG (Vector Laboratories; VP-A105, 1:100), γ-SG (Vector Laboratories; VP-G803, 1:200), β1D integrin (Chemicon International, Temecula, CA, USA; MAB1900, 1:200), nNOS (Invitrogen; #61-7000, 1:400), dysferlin (Abcam Inc., Cambridge, MA, USA; ab55988, 1:600), Akt (Cell Signaling Technologies, Beverly, MA, USA; #9272, 1:500), phosphorylated Akt (Ser 473, Cell Signaling Technologies; #9271, 1:500), HA tag (Sigma; H 3663, 1:5000) and GAPDH (Chemicon International; MAB374, 1:30,000). All primary antibodies were detected using horseradish peroxidase-conjugated secondary antibodies directed against mouse (GE Healthcare, Piscataway, NJ, USA; NA931V, 1:3,000), rabbit (GE Healthcare; NA934V, 1:3,000) or goat (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA; SC-2033, 1:3000) IgG. Immunoblots were developed using SuperSignal West Pico Chemiluminescent Substrate (Pierce; #34080). Relative changes in protein levels were quantified by densitometry analysis of immunoblot bands using the Alpha Imager 2200 and Alpha Imager v5.5 software (Alpha Innotech, Santa Clara, CA, USA).
Immunofluorescence
Indirect immunolabeling experiments using mouse monoclonal antibodies were conducted in combination with the Vector M.O.M Immunodetection Kit (Vector Laboratories; BMK-2202) according to the manufacturer's protocol. For α-DG staining, sections were fixed in pre-chilled 50% ethanol plus 50% acetic acid for 1min prior to washing and blocking steps. Transverse cryosections were incubated overnight with mouse antibodies to detect the following proteins, listed with their working dilutions: dystrophin (1:2), utrophin (1:5), α-DG (Millipore; VI A4-1, 1:40), β-DG (Vector Laboratories; BP-B205, 1:25), α-SG (1:30), β-SG (1:30), γ-SG (1:30), sarcospan [Rabbit 3 (69), 1:5], β1D integrin (1:25), nNOS (1:100) and dMHC (Novocastra, Newcastle upon Tyne, UK; NCL-MHCd, 1:40). Primary antibodies were detected via incubation with biotinylated secondary antibody against mouse (Vector Laboratories; BA-9200, 1:250) or rabbit (Vector Laboratories; BA-1000, 1:250) followed by fluorescein Avidin D (Vector Laboratories; A-2001, 1:250). Sections were mounted in VectaShield and imaged using the AxioPlan 2 fluorescent microscope and AxioVision 4.8 software. Images were taken under identical conditions so that appropriate comparisons could be made between the four different genotypes.
Forelimb grip strength measurements
One day prior to sacrifice, DOX-treated 9-week-old mice were subjected to forelimb grip strength tests using a horizontally positioned grip strength meter (Columbus Instruments, Columbus, OH, USA; DFIS2 Chatillon CE). Mice were lowered by the tail towards the metal pull bar on the apparatus. Upon grasping the bar with their forelimbs, mice were then pulled backwards in the horizontal plane. The procedure was repeated consecutively five times and the peak tension (N) of the five pulls was recorded as the grip strength value. Each animal was subjected to a total of five serial trials of five pulls each with 30 s of rest in between trials.
In vitro contraction force measurements
Mice were euthanized with isoflurane and the EDL muscles were quickly dissected out while being superfused with chilled mammalian Ringer's solution. One tendon was fixed to the bottom of a recording chamber, while the other tendon was attached to a Dynagage DG-600D capacitive tension transducer (Whittaker, North Hollywood, CA, USA). The chamber was perfused with room temperature (∼22°C), oxygenated mammalian Ringer containing: 119 mm NaCl, 5 mm KCl, 1 mm MgSO4, 5 mm NaHCO3, 1.25 mm CaCl2, 1 mm KH2PO4, 10 mm HEPES, 10 mm dextrose and 70 µl/100 ml insulin–transferrin–selenium A (Gibco; # 51300-044), with 30 µM d-tubocurarine chloride to block neuromuscular transmission. Direct muscle stimulation was achieved by passing current pulses between Pt plate electrodes placed on either side of the muscle. The muscle was held at the resting length at which twitch tension was maximal. Stimuli of 15–30 V were produced by a Grass S4 stimulator, driven by a computer-triggered Nihon Kohden stimulator that set the pulse duration (0.5 ms) and repetition frequency (300 ms trains of pulses at 1, 10, 20, 40, 60, 80, 100 and 150 Hz), repeated at 30 s intervals of 1–20 Hz, 2 min intervals at 30–60 Hz and 5 min intervals at >80 Hz. The output of the tension transducer was digitized at a 10 kHz sampling rate by a Digidata 1200A A/D converter and stored and analyzed with Clampfit 9.2 software (Axon Instruments). After a full recovery, fatigue was tested by 150 repetitions of tetanic stimulation at 100 Hz for 300 ms every 2s, totaling for 300 s. The entire recording sequence normally was completed in ∼90 min. The right EDL from each mouse was held in the same recording chamber and tested in the same way after completion of the measurements on the first, left EDL muscle. Whereas both left and right muscles usually gave similar results, data from the side corresponding to the greater maximum tension were used.
CTX analysis
For CTX experiments, 12-week-old male WT Akt transgenic mice were treated with 0.5 mg/ml DOX in drinking water. After 2 weeks of treatment, mice were anesthetized with isoflurane and were shaved on the hindlimbs for visualization of the quadriceps. CTX from N. nigricollis (CTX, 10 µm in PBS; EMD Chemicals, Gibbstown, NJ, USA; 217504) at a volume of 200 µl was injected deep into the quadriceps muscle, and 200 µl of PBS was injected into the contralateral quadriceps as a control. Mice were continually treated with DOX throughout injury recovery, and quadriceps were harvested at 1, 2, 4 and 7 days following injection, as well as day 0 prior to an injection.
For histological analysis, the entire quadriceps was surveyed at five levels from proximal to distal regions. For each level, 8μm transverse cryosections were collected in replicates of 50, followed by a collection of 1 mm of quadriceps tissue not used for histological analysis. Representative images from H&E staining, EBD fluorescence and dMHC indirect immunofluorescence reveal areas of the quadriceps with the maximum histological damage. To achieve dual label imaging of dMHC and laminin, tissue sections were extensively photobleached to eliminate EBD fluorescence prior to staining with laminin and dMHC antibodies.
Statistical analyses
For quadriceps mass, each data point was presented as the average mass of both left and right quadriceps of each animal. For central nucleation and EBD quantification, values from individual quadriceps were treated independently, because of variance in pathology observed within mdx animals. Statistical significance for all studies (with the exception of muscle strength time trials) was determined through two-way analysis of variance (ANOVA) on ranks. Two-way ANOVA was conducted in a 2 × 2 factorial design, with dystrophin genotype (WT and mdx) as the first factor and Akt transgene genotype (STG and DTG) as the second factor. Serial grip strength trials were compared using three-way mixed ANOVA with repeated measures to identify decline in strength. The two factors examined were dystrophin genotype and transgene genotype, with the third factor being the five trials within subjects. Fatigue measurements in mdx STG and DTG EDL muscles were analyzed using mixed model ANOVA with the factors of time as a continuous variable and genotype as a categorical variable. Post hoc pairwise comparisons were made using Student's t-test with Bonferroni correction using a familywise α-level of P< 0.05.
FUNDING
This work was supported by grants from the National Institutes of Health (HL81587 to K.W., AR48179-02 to R.H.C., P30AR057230-01 to R.H.C., P30AR057230-01 to M.J.S.); the Muscular Dystrophy Association USA (MDA3704 to R.H.C.) and a UCLA Graduate Division University Fellowship (to M.H.K.).
ACKNOWLEDGEMENTS
The authors wish to thank Jamie L. Marshall and Dr Angela K. Peter for insightful discussion, Kyle Kern for statistical analysis, Amber Ocampo for expert technical assistance and Dr Stephanie White for critical reading of the manuscript.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Hoffman E.P., Brown R.H., Jr., Kunkel L.M. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 2.Campbell K.P., Kahl S.D. Association of dystrophin and an integral membrane glycoprotein. Nature. 1989;338:259–262. doi: 10.1038/338259a0. [DOI] [PubMed] [Google Scholar]
- 3.Ervasti J.M., Ohlendieck K., Kahl S.D., Gaver M.G., Campbell K.P. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature. 1990;345:315–319. doi: 10.1038/345315a0. [DOI] [PubMed] [Google Scholar]
- 4.Yoshida M., Ozawa E. Glycoprotein complex anchoring dystrophin to sarcolemma. J. Biochem. 1990;108:748–752. doi: 10.1093/oxfordjournals.jbchem.a123276. [DOI] [PubMed] [Google Scholar]
- 5.Ervasti J.M., Campbell K.P. Membrane organization of the dystrophin-glycoprotein complex. Cell. 1991;66:1121–1131. doi: 10.1016/0092-8674(91)90035-w. [DOI] [PubMed] [Google Scholar]
- 6.Ervasti J.M., Kahl S.D., Campbell K.P. Purification of dystrophin from skeletal muscle. J. Biol. Chem. 1991;266:9161–9165. [PubMed] [Google Scholar]
- 7.Ohlendieck K., Campbell K.P. Dystrophin-associated proteins are greatly reduced in skeletal muscle from mdx mice. J. Cell Biol. 1991;115:1685–1694. doi: 10.1083/jcb.115.6.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ervasti J.M., Campbell K.P. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J. Cell Biol. 1993;122:809–823. doi: 10.1083/jcb.122.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petrof B.J., Shrager J.B., Stedman H.H., Kelly A.M., Sweeney H.L. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl Acad. Sci. USA. 1993;90:3710–3714. doi: 10.1073/pnas.90.8.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turner P.R., Westwood T., Regen C.M., Steinhardt R.A. Increased protein degradation results from elevated free calcium levels found in muscle from mdx mice. Nature. 1988;335:735–738. doi: 10.1038/335735a0. [DOI] [PubMed] [Google Scholar]
- 11.Deconinck N., Dan B. Pathophysiology of duchenne muscular dystrophy: current hypotheses. Pediatr. Neurol. 2007;36:1–7. doi: 10.1016/j.pediatrneurol.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 12.McArdle A., Helliwell T.R., Beckett G.J., Catapano M., Davis A., Jackson M.J. Effect of propylthiouracil-induced hypothyroidism on the onset of skeletal muscle necrosis in dystrophin-deficient mdx mice. Clin. Sci. (Lond.) 1998;95:83–89. [PubMed] [Google Scholar]
- 13.Bulfield G., Siller W.G., Wight P.A., Moore K.J. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl Acad. Sci. USA. 1984;81:1189–1192. doi: 10.1073/pnas.81.4.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carnwath J.W., Shotton D.M. Muscular dystrophy in the mdx mouse: histopathology of the soleus and extensor digitorum longus muscles. J. Neurol. Sci. 1987;80:39–54. doi: 10.1016/0022-510x(87)90219-x. [DOI] [PubMed] [Google Scholar]
- 15.Torres L.F., Duchen L.W. The mutant mdx: inherited myopathy in the mouse. Morphological studies of nerves, muscles and end-plates. Brain. 1987;110:269–299. doi: 10.1093/brain/110.2.269. [DOI] [PubMed] [Google Scholar]
- 16.Coulton G.R., Morgan J.E., Partridge T.A., Sloper J.C. The mdx mouse skeletal muscle myopathy: I. A histological, morphometric and biochemical investigation. Neuropathol. Appl. Neurobiol. 1988;14:53–70. doi: 10.1111/j.1365-2990.1988.tb00866.x. [DOI] [PubMed] [Google Scholar]
- 17.DiMario J.X., Uzman A., Strohman R.C. Fiber regeneration is not persistent in dystrophic (MDX) mouse skeletal muscle. Dev. Biol. 1991;148:314–321. doi: 10.1016/0012-1606(91)90340-9. [DOI] [PubMed] [Google Scholar]
- 18.Matsumura K., Ervasti J.M., Ohlendieck K., Kahl S.D., Campbell K.P. Association of dystrophin-related protein with dystrophin-associated proteins in mdx mouse muscle. Nature. 1992;360:588–591. doi: 10.1038/360588a0. [DOI] [PubMed] [Google Scholar]
- 19.Tinsley J.M., Potter A.C., Phelps S.R., Fisher R., Trickett J.I., Davies K.E. Amelioration of the dystrophic phenotype of mdx mice using a truncated utrophin transgene. Nature. 1996;384:349–353. doi: 10.1038/384349a0. [DOI] [PubMed] [Google Scholar]
- 20.Tinsley J., Deconinck N., Fisher R., Kahn D., Phelps S., Gillis J.M., Davies K. Expression of full-length utrophin prevents muscular dystrophy in mdx mice. Nat. Med. 1998;4:1441–1444. doi: 10.1038/4033. [DOI] [PubMed] [Google Scholar]
- 21.Burkin D.J., Wallace G.Q., Nicol K.J., Kaufman D.J., Kaufman S.J. Enhanced expression of the alpha 7 beta 1 integrin reduces muscular dystrophy and restores viability in dystrophic mice. J. Cell Biol. 2001;152:1207–1218. doi: 10.1083/jcb.152.6.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Datta S.R., Brunet A., Greenberg M.E. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 23.Bodine S.C., Stitt T.N., Gonzalez M., Kline W.O., Stover G.L., Bauerlein R., Zlotchenko E., Scrimgeour A., Lawrence J.C., Glass D.J., Yancopoulos G.D. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat. Cell Biol. 2001;3:1014–1019. doi: 10.1038/ncb1101-1014. [DOI] [PubMed] [Google Scholar]
- 24.Rommel C., Bodine S.C., Clarke B.A., Rossman R., Nunez L., Stitt T.N., Yancopoulos G.D., Glass D.J. Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nat. Cell Biol. 2001;3:1009–1013. doi: 10.1038/ncb1101-1009. [DOI] [PubMed] [Google Scholar]
- 25.Pallafacchina G., Calabria E., Serrano A.L., Kalhovde J.M., Schiaffino S. A protein kinase B-dependent and rapamycin-sensitive pathway controls skeletal muscle growth but not fiber type specification. Proc. Natl Acad. Sci. USA. 2002;99:9213–9218. doi: 10.1073/pnas.142166599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takahashi A., Kureishi Y., Yang J., Luo Z., Guo K., Mukhopadhyay D., Ivashchenko Y., Branellec D., Walsh K. Myogenic Akt signaling regulates blood vessel recruitment during myofiber growth. Mol. Cell Biol. 2002;22:4803–4814. doi: 10.1128/MCB.22.13.4803-4814.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rotwein P., Wilson E.M. Distinct actions of Akt1 and Akt2 in skeletal muscle differentiation. J. Cell Physiol. 2009;219:503–511. doi: 10.1002/jcp.21692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peter A.K., Crosbie R.H. Hypertrophic response of Duchenne and limb-girdle muscular dystrophies is associated with activation of Akt pathway. Exp. Cell Res. 2006;312:2580–2591. doi: 10.1016/j.yexcr.2006.04.024. [DOI] [PubMed] [Google Scholar]
- 29.Izumiya Y., Hopkins T., Morris C., Sato K., Zeng L., Viereck J., Hamilton J.A., Ouchi N., LeBrasseur N.K., Walsh K. Fast/glycolytic muscle fiber growth reduces fat mass and improves metabolic parameters in obese mice. Cell Metab. 2008;7:159–172. doi: 10.1016/j.cmet.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wagner K.R., McPherron A.C., Winik N., Lee S.J. Loss of myostatin attenuates severity of muscular dystrophy in mdx mice. Ann. Neurol. 2002;52:832–836. doi: 10.1002/ana.10385. [DOI] [PubMed] [Google Scholar]
- 31.Barton E.R., Morris L., Musaro A., Rosenthal N., Sweeney H.L. Muscle-specific expression of insulin-like growth factor I counters muscle decline in mdx mice. J. Cell Biol. 2002;157:137–148. doi: 10.1083/jcb.200108071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burkin D.J., Kaufman S.J. The alpha7beta1 integrin in muscle development and disease. Cell Tissue Res. 1999;296:183–190. doi: 10.1007/s004410051279. [DOI] [PubMed] [Google Scholar]
- 33.Hodges B.L., Hayashi Y.K., Nonaka I., Wang W., Arahata K., Kaufman S.J. Altered expression of the alpha7beta1 integrin in human and murine muscular dystrophies. J. Cell Sci. 1997;110:2873–2881. doi: 10.1242/jcs.110.22.2873. [DOI] [PubMed] [Google Scholar]
- 34.Vachon P.H., Xu H., Liu L., Loechel F., Hayashi Y., Arahata K., Reed J.C., Wewer U.M., Engvall E. Integrins (alpha7beta1) in muscle function and survival. Disrupted expression in merosin-deficient congenital muscular dystrophy. J. Clin. Invest. 1997;100:1870–1881. doi: 10.1172/JCI119716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cohn R.D., Mayer U., Saher G., Herrmann R., van der Flier A., Sonnenberg A., Sorokin L., Voit T. Secondary reduction of alpha7B integrin in laminin alpha2 deficient congenital muscular dystrophy supports an additional transmembrane link in skeletal muscle. J. Neurol. Sci. 1999;163:140–152. doi: 10.1016/s0022-510x(99)00012-x. [DOI] [PubMed] [Google Scholar]
- 36.Khurana T.S., Watkins S.C., Chafey P., Chelly J., Tome F.M., Fardeau M., Kaplan J.C., Kunkel L.M. Immunolocalization and developmental expression of dystrophin related protein in skeletal muscle. Neuromuscul. Disord. 1991;1:185–194. doi: 10.1016/0960-8966(91)90023-l. [DOI] [PubMed] [Google Scholar]
- 37.Peter A.K., Ko C.Y., Kim M.H., Hsu N., Ouchi N., Rhie S., Izumiya Y., Zeng L., Walsh K., Crosbie R.H. Myogenic Akt signaling upregulates the utrophin-glycoprotein complex and promotes sarcolemma stability in muscular dystrophy. Hum. Mol. Genet. 2009;18:318–327. doi: 10.1093/hmg/ddn358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grill M.A., Bales M.A., Fought A.N., Rosburg K.C., Munger S.J., Antin P.B. Tetracycline-inducible system for regulation of skeletal muscle-specific gene expression in transgenic mice. Transgenic Res. 2003;12:33–43. doi: 10.1023/a:1022119005836. [DOI] [PubMed] [Google Scholar]
- 39.Blake D.J., Tinsley J.M., Davies K.E. Utrophin: a structural and functional comparison to dystrophin. Brain Pathol. 1996;6:37–47. doi: 10.1111/j.1750-3639.1996.tb00781.x. [DOI] [PubMed] [Google Scholar]
- 40.Clerk A., Morris G.E., Dubowitz V., Davies K.E., Sewry C.A. Dystrophin-related protein, utrophin, in normal and dystrophic human fetal skeletal muscle. Histochem. J. 1993;25:554–561. [PubMed] [Google Scholar]
- 41.Lin S., Burgunder J.M. Utrophin may be a precursor of dystrophin during skeletal muscle development. Brain Res. Dev. Brain Res. 2000;119:289–295. doi: 10.1016/s0165-3806(99)00165-0. [DOI] [PubMed] [Google Scholar]
- 42.Harris J.B. Myotoxic phospholipases A2 and the regeneration of skeletal muscles. Toxicon. 2003;42:933–945. doi: 10.1016/j.toxicon.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 43.Blaauw B., Mammucari C., Toniolo L., Agatea L., Abraham R., Sandri M., Reggiani C., Schiaffino S. Akt activation prevents the force drop induced by eccentric contractions in dystrophin-deficient skeletal muscle. Hum. Mol. Genet. 2008;17:3686–3696. doi: 10.1093/hmg/ddn264. [DOI] [PubMed] [Google Scholar]
- 44.Blaauw B., Canato M., Agatea L., Toniolo L., Mammucari C., Masiero E., Abraham R., Sandri M., Schiaffino S., Reggiani C. Inducible activation of Akt increases skeletal muscle mass and force without satellite cell activation. FASEB J. 2009;23:3896–3905. doi: 10.1096/fj.09-131870. [DOI] [PubMed] [Google Scholar]
- 45.Webster C., Silberstein L., Hays A.P., Blau H.M. Fast muscle fibers are preferentially affected in Duchenne muscular dystrophy. Cell. 1988;52:503–513. doi: 10.1016/0092-8674(88)90463-1. [DOI] [PubMed] [Google Scholar]
- 46.Gehrig S.M., Koopman R., Naim T., Tjoakarfa C., Lynch G.S. Making fast-twitch dystrophic muscles bigger protects them from contraction injury and attenuates the dystrophic pathology. Am. J. Pathol. 2010;176:29–33. doi: 10.2353/ajpath.2010.090760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rando T.A. The dystrophin-glycoprotein complex, cellular signaling, and the regulation of cell survival in the muscular dystrophies. Muscle Nerve. 2001;24:1575–1594. doi: 10.1002/mus.1192. [DOI] [PubMed] [Google Scholar]
- 48.Cary L.A., Guan J.L. Focal adhesion kinase in integrin-mediated signaling. Front. Biosci. 1999;4:D102–D113. doi: 10.2741/cary. [DOI] [PubMed] [Google Scholar]
- 49.Song K.S., Scherer P.E., Tang Z., Okamoto T., Li S., Chafel M., Chu C., Kohtz D.S., Lisanti M.P. Expression of caveolin-3 in skeletal, cardiac, and smooth muscle cells. Caveolin-3 is a component of the sarcolemma and co-fractionates with dystrophin and dystrophin-associated glycoproteins. J. Biol. Chem. 1996;271:15160–15165. doi: 10.1074/jbc.271.25.15160. [DOI] [PubMed] [Google Scholar]
- 50.Yang B., Jung D., Motto D., Meyer J., Koretzky G., Campbell K.P. SH3 domain-mediated interaction of dystroglycan and Grb2. J. Biol. Chem. 1995;270:11711–11714. doi: 10.1074/jbc.270.20.11711. [DOI] [PubMed] [Google Scholar]
- 51.Hack A.A., Cordier L., Shoturma D.I., Lam M.Y., Sweeney H.L., McNally E.M. Muscle degeneration without mechanical injury in sarcoglycan deficiency. Proc. Natl Acad. Sci. USA. 1999;96:10723–10728. doi: 10.1073/pnas.96.19.10723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hack A.A., Ly C.T., Jiang F., Clendenin C.J., Sigrist K.S., Wollmann R.L., McNally E.M. Gamma-sarcoglycan deficiency leads to muscle membrane defects and apoptosis independent of dystrophin. J. Cell Biol. 1998;142:1279–1287. doi: 10.1083/jcb.142.5.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Barton E.R. Impact of sarcoglycan complex on mechanical signal transduction in murine skeletal muscle. Am. J. Physiol. Cell Physiol. 2006;290:C411–C419. doi: 10.1152/ajpcell.00192.2005. [DOI] [PubMed] [Google Scholar]
- 54.Burkin D.J., Wallace G.Q., Milner D.J., Chaney E.J., Mulligan J.A., Kaufman S.J. Transgenic expression of {alpha}7{beta}1 integrin maintains muscle integrity, increases regenerative capacity, promotes hypertrophy, and reduces cardiomyopathy in dystrophic mice. Am. J. Pathol. 2005;166:253–263. doi: 10.1016/s0002-9440(10)62249-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu J., Burkin D.J., Kaufman S.J. Increasing alpha 7 beta 1-integrin promotes muscle cell proliferation, adhesion, and resistance to apoptosis without changing gene expression. Am. J. Physiol. Cell Physiol. 2008;294:C627–C640. doi: 10.1152/ajpcell.00329.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sander M., Chavoshan B., Harris S.A., Iannaccone S.T., Stull J.T., Thomas G.D., Victor R.G. Functional muscle ischemia in neuronal nitric oxide synthase-deficient skeletal muscle of children with Duchenne muscular dystrophy. Proc. Natl Acad. Sci. USA. 2000;97:13818–13823. doi: 10.1073/pnas.250379497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thomas G.D., Shaul P.W., Yuhanna I.S., Froehner S.C., Adams M.E. Vasomodulation by skeletal muscle-derived nitric oxide requires alpha-syntrophin-mediated sarcolemmal localization of neuronal nitric oxide synthase. Circ. Res. 2003;92:554–560. doi: 10.1161/01.RES.0000061570.83105.52. [DOI] [PubMed] [Google Scholar]
- 58.Lai Y., Thomas G.D., Yue Y., Yang H.T., Li D., Long C., Judge L., Bostick B., Chamberlain J.S., Terjung R.L., Duan D. Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J. Clin. Invest. 2009;119:624–635. doi: 10.1172/JCI36612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wehling M., Spencer M.J., Tidball J.G. A nitric oxide synthase transgene ameliorates muscular dystrophy in mdx mice. J. Cell Biol. 2001;155:123–131. doi: 10.1083/jcb.200105110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li D., Bareja A., Judge L., Yue Y., Lai Y., Fairclough R., Davies K.E., Chamberlain J.S., Duan D. Sarcolemmal nNOS anchoring reveals a qualitative difference between dystrophin and utrophin. J. Cell Sci. 2010;123:2008–2013. doi: 10.1242/jcs.064808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wilson E.M., Hsieh M.M., Rotwein P. Autocrine growth factor signaling by insulin-like growth factor-II mediates MyoD-stimulated myocyte maturation. J. Biol. Chem. 2003;278:41109–41113. doi: 10.1074/jbc.C300299200. [DOI] [PubMed] [Google Scholar]
- 62.Wamhoff B.R., Sinha S., Owens G.K. Conditional mouse models to study developmental and pathophysiological gene function in muscle. Handb. Exp. Pharmacol. 2007;178:441–468. doi: 10.1007/978-3-540-35109-2_18. [DOI] [PubMed] [Google Scholar]
- 63.Ouchi N., Oshima Y., Ohashi K., Higuchi A., Ikegami C., Izumiya Y., Walsh K. Follistatin-like 1, a secreted muscle protein, promotes endothelial cell function and revascularization in ischemic tissue through a nitric-oxide synthase-dependent mechanism. J. Biol. Chem. 2008;283:32802–32811. doi: 10.1074/jbc.M803440200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Haugen F., Norheim F., Lian H., Wensaas A.J., Dueland S., Berg O., Funderud A., Skalhegg B.S., Raastad T., Drevon C.A. IL-7 is expressed and secreted by human skeletal muscle cells. Am. J. Physiol. Cell Physiol. 2010;298:C807–C816. doi: 10.1152/ajpcell.00094.2009. [DOI] [PubMed] [Google Scholar]
- 65.Melone M.A., Peluso G., Galderisi U., Petillo O., Cotrufo R. Increased expression of IGF-binding protein-5 in Duchenne muscular dystrophy (DMD) fibroblasts correlates with the fibroblast-induced downregulation of DMD myoblast growth: an in vitro analysis. J. Cell Physiol. 2000;185:143–153. doi: 10.1002/1097-4652(200010)185:1<143::AID-JCP14>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 66.Liu P., Cheng H., Roberts T.M., Zhao J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 2009;8:627–644. doi: 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Amalfitano A., Chamberlain J.S. The mdx-amplification-resistant mutation system assay, a simple and rapid polymerase chain reaction-based detection of the mdx allele. Muscle Nerve. 1996;19:1549–1553. doi: 10.1002/(SICI)1097-4598(199612)19:12<1549::AID-MUS4>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 68.Rooney J.E., Welser J.V., Dechert M.A., Flintoff-Dye N.L., Kaufman S.J., Burkin D.J. Severe muscular dystrophy in mice that lack dystrophin and alpha7 integrin. J. Cell Sci. 2006;119:2185–2195. doi: 10.1242/jcs.02952. [DOI] [PubMed] [Google Scholar]
- 69.Miller G., Peter A.K., Espinoza E., Heighway J., Crosbie R.H. Over-expression of Microspan, a novel component of the sarcoplasmic reticulum, causes severe muscle pathology with triad abnormalities. J. Muscle Res. Cell Motil. 2006;27:545–558. doi: 10.1007/s10974-006-9069-2. [DOI] [PubMed] [Google Scholar]