

Abstract
The purpose of our study was to determine the relationship between mutant huntingtin (Htt) and mitochondrial dynamics in the progression of Huntington's disease (HD). We measured the mRNA levels of electron transport chain genes, and mitochondrial structural genes, Drp1 (dynamin-related protein 1), Fis1 (fission 1), Mfn1 (mitofusin 1), Mfn2 (mitofusin 2), Opa1 (optric atrophy 1), Tomm40 (translocase of outermembrane 40) and CypD (cyclophilin D) in grade III and grade IV HD patients and controls. The mutant Htt oligomers and the mitochondrial structural proteins were quantified in the striatum and frontal cortex of HD patients. Changes in expressions of the electron transport chain genes were found in HD patients and may represent a compensatory response to mitochondrial damage caused by mutant Htt. Increased expression of Drp1 and Fis1 and decreased expression of Mfn1, Mfn2, Opa1 and Tomm40 were found in HD patients relative to the controls. CypD was upregulated in HD patients, and this upregulation increased as HD progressed. Significantly increased immunoreactivity of 8-hydroxy-guanosine was found in the cortical specimens from stage III and IV HD patients relative to controls, suggesting increased oxidative DNA damage in HD patients. In contrast, significantly decreased immunoreactivities of cytochrome oxidase 1 and cytochrome b were found in HD patients relative to controls, indicating a loss of mitochondrial function in HD patients. Immunoblotting analysis revealed 15, 25 and 50 kDa mutant Htt oligomers in the brain specimens of HD patients. All oligomeric forms of mutant Htt were significantly increased in the cortical tissues of HD patients, and mutant Htt oligomers were found in the nucleus and in mitochondria. The increase in Drp1, Fis1 and CypD and the decrease in Mfn1 and Mfn2 may be responsible for abnormal mitochondrial dynamics that we found in the cortex of HD patients, and may contribute to neuronal damage in HD patients. The presence of mutant Htt oligomers in the nucleus of HD neurons and in mitochondria may disrupt neuronal functions. Based on these findings, we propose that mutant Htt in association with mitochondria imbalance and mitochondrial dynamics impairs axonal transport of mitochondria, decreases mitochondrial function and damages neurons in affected brain regions of HD patients.
INTRODUCTION
Huntington's disease (HD) is a neurodegenerative disease with an autosomal dominant inheritance that strikes humans in midlife. HD is characterized by involuntary movements, chorea, dystonia, changes in personality and cognitive decline (1–4). Key features found in postmortem brain tissues of HD patients include the loss of medium spiny neurons in the basal ganglia and pyramidal neurons in the cortex and hippocampus. Mutant huntingtin (Htt) aggregates have been found in affected regions of the brain in HD patients and in mouse models of HD (4,5). The extent of mutant Htt aggregates in selective neuronal loss is still not completely understood.
HD is caused by a genetic mutation, resulting in an expanded polyglutamine (or polyQ) that encodes repeats in exon 1 of the HD gene. In persons affected by HD, the number of polyQ repeats ranges from 36 to 120, whereas in unaffected persons, polyQ repeats range from 6 to 35 (3). The progression of HD has been found to correlate inversely with the number of polyQ repeats. Htt, a product of the HD gene, is a 350 kDa protein, ubiquitously expressed in the brain and peripheral tissues (1,2). In the HD brain, Htt is localized mainly in the cytoplasm; however, a small portion of mutant Htt localizes in subcellular organelles, including the plasma membrane, mitochondria, lysosomes and endoplasmic reticulum (6–13). The nature and mechanism of translocation of mutant Htt, particularly mutant Htt oligomers, to the subcellular organelles are not fully understood.
Mutant polyQ aggregates have been extensively reported in HD and other polyQ repeat-associated diseases (1). More recently, formations of oligomers, fibrils and protofibrils have been found in cell cultures and HD transgenic mice (14–18). Mutant oligomeric proteins are toxic, and these proteins have been found to enter subcellular organelles, such as mitochondria in neurons from patients with Alzheimer's disease (19). However, mutant Htt oligomers and their association with mitochondria have not been studied in HD patients.
Several cellular pathways have been proposed, to explain the causes of HD pathogenesis, including: transcriptional dysregulation, N-methyl-d-aspartate receptor activation, the interaction of expanded polyQ repeat proteins with other proteins in the central nervous system and abnormal mitochondrial bioenergetics and defective axonal trafficking. Among these, mitochondrial abnormalities and defective bioenergetics appear to be strongly involved in HD progression (4).
Increasing evidence supports the involvement of mitochondria in HD pathogenesis. Several studies using positive emission tomography scans of brains from HD patients and control subjects revealed a marked decrease in glucose utilization in the striatum of patients (20–25). Studies using magnetic resonance imaging of postmortem brains from HD patients revealed a progressive atrophy of the striatum, caudate nucleus, putamen, globus pallidus and thalamus compared with images of postmortem brain from age-matched control subjects (26–29). Recent biochemical studies of mitochondria in striatal neurons from postmortem brains taken from late-stage HD patients revealed reduced activity in several components of oxidative phosphorylation, including complexes II, III and IV of the electron transport chain (30–32). Recent studies of HD knockin striatal cells and lymphoblasts from HD patients revealed that expanded polyQ repeats are associated with low levels of mitochondrial adenosinetriphosphate (ATP) and decreased mitochondrial ADP uptake, suggesting that repeat expansions associated with mitochondrial functional defects (33). In addition, several studies of mitochondrial trafficking in cortical neurons from HD mice revealed mutant Htt aggregates impairing mitochondrial movement (13,34,35).
Recent studies using postmortem brains of HD patients (36), peripheral cells (fibroblasts, lymphoblasts and myoblasts) from HD patients (37,38) and cell models of HD (38,39) revealed structural abnormalities in mitochondria in neurons, suggesting that defective mitochondria may be responsible for neuronal damage in HD. Mitochondria are cytoplasmic organelles and provide essential energy (ATP) to all mammalian cells, including neurons. They are synthesized in the cell body and are transported down axons and dendrites to serve energy demands of the cell (40). If mitochondria in the cell body are damaged due to mutant Htt in striatal and cortical neurons, these defective mitochondria may be transported to synaptic terminals via natural mitochondrial trafficking, where they could produce low levels of ATP due to their degradation and could cause synaptic degeneration (41). The transport of healthy mitochondria to synaptic terminals may be critical and necessary for the delivery of adequate ATP. Very little is known about synaptic degeneration and how mutant Htt damages mitochondria and, in turn, the synapses to which the damaged mitochondria are trafficked.
Mitochondrial shape and structure are maintained by two opposing forces: fission and fusion (42). In a healthy neuron, fission and fusion mechanisms balance equally and imbalance between fission and fusion leads to abnormal mitochondrial dynamics. Mitochondria alter their shape and size to move, through mitochondrial trafficking, from the cell body to the axons, dendrites and synapses via anterograde motion, and back to the cell body via retrograde motion. Fission and fusion are controlled by evolutionarily conserved, large GTPases belonging to the family of dynamin. Fission is controlled by the dynamin-related protein 1 (Drp1) and the mitochondrial fission 1 (Fis1) protein. Most Drp1 protein is localized in the cytoplasm, but a small part punctuates the outer membrane, which promotes mitochondrial fragmentation (4). Fis1 is localized in the outer membrane of mitochondria (43). Drp1 and Fis1 are activated by an increase in free radicals in mitochondria, which is critical for mitochondrial fission. Mitochondrial fusion is controlled by three GTPase proteins: two outer-membrane-localized proteins Mfn1 and Mfn2 (mitofusin 1 and 2) and one inner-membrane-localized protein Opa1 (optric atrophy 1) (43). The C-terminal part of Mfn1 mediates oligomerization between Mfn molecules of adjacent mitochondria and facilitates mitochondrial fusion. Mitochondrial fusion protects cells from toxic effects that are known to associate with mutant Htt and mitochondrial DNA to allow functional complementation of mitochondrial DNA, proteins and metabolites (43). However, the precise link between mutant Htt and mitochondrial structural changes is not clear. Further, the role of mitochondrial dynamics in HD progression is still unclear. In the present study, we sought to determine abnormal mitochondrial dynamics/mitochondrial damage in relation to mutant Htt in HD patients.
The goals of the present study were: (i) to measure the mRNA and protein levels of Drp1, Fis1 (fission), Mfn1, Mfn2, Opa1 (fusion) and CypD (cyclophilin D) (matrix) in postmortem brains from control subjects (controls) and HD patients, (ii) to assess the RNA expression abundance of mitochondrial electron transport chain genes (complexes I, III, IV and V) in postmortem brains from HD patients, (iii) to localize mitochondrial proteins and mutant Htt aggregates and oligomers in brain sections from HD patients, (iv) to assess oxidative DNA damage in postmortem brains from HD patients and (v) to identify abnormal mitochondrial dynamics and mitochondrial damage in relation to mutant Htt in brain specimens from these patients.
RESULTS
mRNA expression of mitochondrial genes
To determine the role of mitochondrial structural genes in HD pathogenesis, using quantitative real-time RT–PCR with Sybr-Green chemistry, we measured mRNA fold changes for Drp1, Fis1, Mfn1, Mfn2, Opa1, Tomm40 (translocase of outermembrane 40) and CypD in the frontal cortex specimens from six patients with grade III and IV HD and three controls (Table 1). We found increased expression of Drp1 and Fis1, and CypD in brain specimens from HD patients and decreased expression of Mfn1, Mfn2 and Opa1, indicating the presence of abnormal mitochondrial dynamics.
Table 1.
mRNA fold changes of mitochondrial fission/fusion and matrix genes in frontal cortex brain specimens from grade III and grade IV HD patients
| HD brains—pathology stage | Drp1 | Fis1 | Mfn1 | Mfn2 | Opa1 | CypD | TOMM40 |
|---|---|---|---|---|---|---|---|
| HD3 patient 1 | 10.9 | 7.79 | −4 | −3.44 | −4.34 | 9.91 | 1.14 |
| HD3 patient 2 | 3.62 | 6.86 | −2.5 | −4.7 | −2.63 | 31.19 | 1.25 |
| HD3 patient 3 | 2.8 | 13.75 | −2.85 | −50 | −2.85 | 24.5 | −1.07 |
| Mean fold change in HD3 patients | 5.8 | 9.5 | −3.1 | −19.4 | −3.3 | 21.9 | 1.1 |
| HD4 patient 1 | 5.96 | 36.18 | −2 | −33.3 | −1.61 | 26.6 | 2.67 |
| HD4 patient 2 | 7.04 | 32.38 | −2.85 | −50 | −1.61 | 20.71 | 5.8 |
| HD4 patient 3 | 4.35 | 30.75 | −2.5 | −4.34 | −1.58 | 51.5 | 2.53 |
| Mean fold change in HD4 patients | 5.8 | 33.1 | −2.45 | −29.2 | −1.6 | 32.9 | 3.7 |
Fission genes
Overall, mitochondrial fission was upregulated in the brain specimens from HD patients, but there was some heterogeneity. As shown in Table 1, mRNA fold changes for Drp1 were greater in brain specimens at grade III and IV HD compared with brain specimens from controls; the mRNA expression ranged from 2.8- to 10.9-fold. Interestingly, Fis1 expression levels were very high, between 6.86- and 36.18-fold.
Fusion genes
As shown in Table 1, mRNA fold changes were downregulated for Mfn1, Mfn2 and Opa1 in brain specimens from all subjects with HD, compared with the mRNA fold changes in tissues from the control brain specimens. However, the genes from the patients with HD exhibited variability of downregulation: Mfn1 expression ranged from −2.5- to −4.0-fold in brain specimens from patients with grade III HD, and from −2- to −2.85-fold in brain specimens from patients with grade IV HD. Mfn2 expression ranged from −3.44- to −50.0-fold in patients with grade III HD and from −4.3 to −50.0 in patients with grade IV HD. Opa1 expression ranged from −2.63- to −4.34-fold in patients with grade III HD and from −1.58 to −1.61 in patients with grade IV HD. Tomm40 expression was upregulated in five out of six cases, ranging from 1.14- to 5.80-fold.
Matrix gene
In brain specimens from all six patients with HD, CypD was upregulated, from 9.91- to 51.5-fold changes, indicating that increased CypD expression may be associated with mitochondrial structural damage in HD neurons (Table 1).
Mitochondrial electron transport chain genes
To determine the effects of mutant Htt on mitochondrial-encoded genes, we used quantitative real-time RT–PCR analysis to calculate mRNA fold changes for the six genes that represent all complexes of oxidative phosphorylation (Table 2).
Table 2.
mRNA fold changes of mitochondrial-encoded electron transport chain genes in frontal cortex brain specimens from grade III and grade IV HD patients
| HD brains—pathology stage | Complex I, NADH-sub5 | Complex III, cytochrome b | Complex IV |
Complex V, ATP6 | ||
|---|---|---|---|---|---|---|
| COX1 | COX2 | COX3 | ||||
| HD3 patient 1 | 1.08 | 1.25 | 1.59 | 1.09 | 1.36 | 1.60 |
| HD3 patient 2 | 2.04 | 2.92 | 3.70 | 3.59 | 3.32 | 3.53 |
| HD3 patient 3 | 3.61 | 1.14 | 2.38 | 2.59 | 2.66 | 2.53 |
| Mean fold change | 2.24 | 1.77 | 2.55 | 2.42 | 2.44 | 2.55 |
| HD4 patient 1 | 1.96 | 1.90 | 3.37 | 2.26 | 2.28 | 3.48 |
| HD4 patient 2 | 1.39 | 1.10 | 2.12 | 1.73 | 1.83 | 2.91 |
| HD4 patient 3 | 1.22 | 1.05 | 1.95 | 1.56 | 1.85 | 2.52 |
| Mean fold change | 1.52 | 1.35 | 2.48 | 1.85 | 1.98 | 2.97 |
Complex I
The mean mRNA fold changes were higher (2.24 for grade III HD and 1.52 for grade IV HD patients) for NADH subunit 5 relative to control subjects (Table 2), suggesting that mRNA expressions were altered early in the disease progression. Further, patient–patient mitochondrial gene expression varied in both grade III and IV HD patients (Table 2). Mutant polyQ length and/or mitochondrial DNA alterations may be responsible for this heterogeneity.
Complex III
For HD3 patients, the mean mRNA fold change in cytochrome b was 1.77, and for HD4 patients, it was 1.35, but the cytochrome b levels were the lowest among all mitochondrial-encoded genes studied.
Complex IV
As shown in Table 3, mRNA levels were greater in all three subunits of cytochrome oxidase for both grade III and IV HD patients. The mean mRNA fold change was 2.55 for grade III HD patients and 2.48 for grade IV HD patients in subunit 1; 2.42 for grade III HD patients and 1.85 for grade IV HD patients in subunit 2; and 2.44 for grade III HD patients and 1.98 for grade IV HD patients in subunit 3 (Table 2). The higher mRNA levels may represent a compensatory response to decreased mitochondrial function.
Table 3.
Demographic details of postmortem brain specimens from HD patients and control subjects obtained from the Harvard Tissue Resource Center
| Case type | Age/sex | Postmortem interval (h) | Stage of HD brains |
|---|---|---|---|
| Control 1 | 58/F | 17.8 | 0 |
| Control 2 | 53/M | 20 | 0 |
| Control 3 | 54/M | 24 | 0 |
| HD3 patient 1 | 78/M | 18 | III |
| HD3 patient 2 | 54/F | 12.9 | III |
| HD3 patient 3 | 75/M | 14 | III |
| HD4 patient 1 | 53/M | 23 | IV |
| HD4 patient 2 | 48/M | 16 | IV |
| HD4 patient 3 | 45/F | 11 | IV |
Complex V
The mRNA levels of ATPase 6 were greater in all HD patients relative to control subjects. The mean mRNA fold change was 2.55 for grade III HD patients and 2.97 for grade IV HD patients, indicating that more mRNA is produced for ATPase 6 in grade IV HD patients (Table 2). Further, the mean mRNA levels of ATPase 6 in grade III and IV HD patients were highest among all mitochondrial-encoded genes studied.
Western blot analysis of frontal cortex tissues of mitochondrial proteins
To determine whether mitochondrial structural proteins are abnormally expressed as HD progresses, we used western blot and densitometry analyses to quantify Drp1, Fis1, Mfn1, Mfn2, Opa1, Tomm40 and CypD in the frontal cortical tissues from patients with grade III and IV HD.
Fission proteins
Our protein data agreed with our real-time RT–PCR findings. Drp1 levels were significantly increased in grade III HD patients (P< 0.04) and grade IV HD (P< 0.004) patients compared with the Drp1 levels in controls. Fis1 protein levels also were significantly increased in patients at grade III HD (P< 0.004) and grade IV HD (P< 0.01) relative to the Fis1 protein levels in controls (Fig. 1).
Figure 1.

Western blot analysis of mitochondrial fission, fusion and matrix proteins in the frontal cortex of HD patients and controls. Drp1 levels were significantly increased in grade III (P< 0.04) and grade IV (P< 0.004) HD patients compared with levels in control subjects, as were Fis1 protein levels (grade III, P< 0.004; grade IV, P< 0.01). In contrast, Mfn1 proteins were significantly decreased in grade III (P< 0.0002) and grade IV (P< 0.002) HD patients compared with control subjects, as were Mfn2 levels (grade III, P< 0.005; grade IV, P< 0.002). Tomm40 levels were significantly increased in HD patients at grade III (P< 0.02) and grade IV (P< 0.004), relative to controls. CypD was significantly increased in HD patients at grade III (P< 0.01) and grade IV (P< 0.001). Error bars indicate mean ± SEM.
Fusion proteins
In contrast to fission proteins, fusion proteins were lower in patients with HD relative to controls. Mfn1 protein levels were significantly lower in the brain samples from grade III HD (P< 0.0002) and grade IV HD (P< 0.002) patients. Mfn2 protein levels were also significantly lower in the brain samples from grade III HD (P< 0.005) and grade IV HD (P< 0.002) patients. Opa1 protein levels were lower, but did not reach statistical significance (Fig. 1). Tomm40 levels were significantly higher in grade III HD (P< 0.02) and grade IV HD patients (P< 0.004), relative to controls.
Matrix protein
The level of CypD was significantly higher in patients at grade III HD (P< 0.01) and grade IV HD (P< 0.001), compared with the level in controls (Fig. 1).
Our immunoblotting findings agreed with our real-time RT–PCR analysis. Overall, we found higher levels of fission proteins and lower levels of fusion proteins in all of brain tissues from HD patients compared with control subjects, indicating abnormal mitochondrial dynamics in the HD patients that we studied.
Western blot analysis of mitochondrial proteins in HD patients
To determine whether altered mitochondrial proteins are confined to HD-affected regions of the brain, we performed western blot analysis of mitochondrial proteins, using tissues from the striatum, frontal cortex and cerebellum of grade III HD patients. We found significantly more fission proteins in the striatum and frontal cortex relative to the cerebellum. Drp1 levels were significantly higher in the striatum (P< 0.003) and in the frontal cortex (P< 0.01) relative to the cerebellum. Fis1 protein levels also were significantly increased in the striatum (P< 0.01) and the frontal cortex (P< 0.01) relative to the cerebellum in the grade III HD patients (Fig. 2A).
Figure 2.

Western blot analysis of mitochondrial fission, fusion and matrix proteins in the striatum, frontal cortex and cerebellum in HD patients and controls. (A) Grade III HD patients. Drp1 levels were significantly increased in the striatum (P< 0.003) and frontal cortex (P< 0.01) compared with the cerebellum, as were Fis1 levels (striatum, P< 0.01; frontal cortex, P< 0.01). Mfn1 levels were significantly decreased in the frontal cortex (P< 0.04) compared with the cerebellum, similar to Mfn2 protein levels (striatum, P< 0.01; frontal cortex, P< 0.03). Tomm40 levels were significantly increased in the frontal cortex (P< 0.004) relative to cerebellum, and CypD was significantly increased in both the striatum (P< 0.04) and the frontal cortex (P< 0.001) compared with the cerebellum. The data are expressed as mean ± SEM. (B) Control subjects. The levels of fission proteins, Drp1 and Fis1, did not change significantly in the striatum and frontal cortex compared with the cerebellum. The levels of fusion proteins Mfn1, Mfn2 and Opa1 and matrix protein CypD also did not change in the frontal cortex compared with the cerebellum. P-values are given on the top of each bar.
Fusion proteins were lower in the striatum and the frontal cortex relative to the cerebellum in grade III HD patients, and Mfn1 protein levels were significantly lower in the frontal cortex relative to the cerebellum (P< 0.04). Mfn2 protein levels were lower in the striatum (P< 0.01) and the frontal cortex (P< 0.03) relative to the cerebellum. Tomm40 levels were higher in the striatum and the frontal cortex relative to the cerebellum in grade III HD patients, but the significance level was not reached for the striatum (Fig. 2A).
The matrix protein CypD was significantly higher in the striatum (P< 0.04) and the frontal cortex (P< 0.001) relative to the cerebellum in grade III HD patients (Fig. 2A).
Western blot analysis of mitochondrial proteins in control subjects
To determine whether mitochondrial structural protein changes are confined only to HD patients, we performed western blot analysis of mitochondrial proteins, using tissues from the striatum, frontal cortex and cerebellum of control subjects. As shown in Figure 2B, we did not find significantly altered proteins between the affected (cortex and striatum) and unaffected (cerebellum) regions. These results suggest that the presence of mutant Htt is needed in order to alter mitochondrial structural and subsequent abnormal mitochondrial dynamics in HD patients.
Western blot analysis of mutant Htt oligomers
To determine the extent of mutant Htt oligomers in HD brains, we performed western blot analysis of tissues from the frontal cortex of patients at grade III and IV HD, and controls, using the oligomer-specific antibody A11. We found several mutant Htt oligomers of different sizes, including 15, 25, 50 and 60 kDa sizes (Fig. 3A). Levels of oligomeric Htt were higher in the brains from HD patients, indicating that mutant oligomer formation and production may be dependent on disease progression.
Figure 3.

Western blot analysis of mutant Htt oligomeric proteins in grade III and IV HD patients and control subjects. (A) Western blot using A11 antibody. Three distinct bands of oligomers (15, 25 and 50 kDa) were found in each grade of HD, and all three oligomeric bands were significantly increased in grade III and grade IV HD patients compared with control subjects. (B) Dot blot analysis. Dot blot immunoreactivity was significantly increased in grade III (P< 0.04) and grade IV (P< 0.02) HD patients compared with control subjects. Error bars indicate mean ± SEM.
We found three distinct bands of oligomers (15, 25 and 50 kDa) in HD patients, in addition to a faint 60 kDa band (Fig. 3A). Significantly higher levels of 15 kDa bands were found in grade III (P< 0.001) and grade IV HD (P< 0.04) patients. We found significantly increased 25 kDa bands in our grade III (P< 0.01) and grade IV (P< 0.04) HD patients, and 50 kDa in our grade III (P< 0.001) and grade IV (P< 0.03) HD patients.
Using the oligomer-specific A11 antibody, we performed quantitative dot blot analysis of brain specimens from HD patients at stages III and IV. As shown in Figure 3B, significantly higher levels of oligomers were found in tissues from patients with grade III (P< 0.04) and grade IV (P< 0.02) HD relative to controls. Overall, these immunoblotting findings indicate that mutant Htt oligomeric levels in HD patients increase as the disease progresses and that this increase may contribute to abnormal protein interactions and neuronal damage in persons with HD.
Immunofluorescence analysis of HD patients
Fission proteins
To localize Drp1 and Fis1 in HD patients at different stages of disease progression, we performed immunofluorescence analyses of Drp1 and Fis1 in frontal cortex sections from HD patients at grade III and IV, and from controls. As shown in Figure 4, Drp1 immunoreactivity was higher in brain specimens from HD patients relative to controls. However, brain specimens from grade III HD patients showed the greatest immunoreactivity, indicating a progressive activation of Drp1 in HD pathogenesis. We also found increased immunoreactivity of Fis1 in brain specimens from grade III and IV HD patients compared with those from controls (Fig. 4). These findings concurred with our quantitative real-time RT–PCR findings and our immunoblotting data of Drp1 and Fis1. Our fission protein findings suggest the presence of abnormal mitochondrial dynamics in HD patients.
Figure 4.

Immunofluorescence analysis of mitochondrial proteins Drp1, Fis1, Mfn1, Mfn2, Opa1 and CypD. Immunoreactivities of Drp1 and Fis1 were increased in frontal cortex sections from grade III (B, Drp1; E, Fis1) and IV HD (C, Drp1; F, Fis1) patients relative to control subjects (A, Drp1; D, Fis1). Mfn1, Mfn2 and Opa1 expressions were decreased in frontal cortex sections from grade III (H, Mfn1; K, Mfn2) and grade IV HD patients (I, Mfn1; L, Mfn2) compared with control subjects (G, Mfn1; J, Mfn2). Immunoreactivity of Opa1 was decreased in grade III (N) and grade IV (O) patients relative to control subjects (M). CypD expression was increased in frontal cortex sections from grade III (Q) and grade IV HD (R) patients relative to control subjects (P). Arrows indicate increased immunoreactivity of proteins. Images were photographed at ×40.
Fusion proteins
To localize fusion proteins in brain specimens from HD patients at grade III and IV and from controls, we conducted immunofluorescence analysis of Mfn1, Mfn2 and Opa1. As shown in Figure 4, we found the lowest levels of Mfn1, Mfn2 and Opa1 expression in brain specimens from both grade III and IV HD patients, and the immunoreactivity of Mfn2 and Opa1 was the lowest in grade IV HD patients. These findings agree with our real-time RT–PCR and immunoblotting data, indicating the presence of abnormal mitochondrial dynamics in HD pathogenesis.
Matrix protein
We found greater immunoreactivity of CypD in brain sections from grade III HD and grade IV HD patients relative to controls (Fig. 4), indicating that mitochondrial structural damage is present in HD patients.
Immunofluorescence analysis of mitochondrial-encoded proteins
To identify the activity of mitochondria in grade III and IV HD patients and in controls, we performed immunofluorescence analysis of cytochrome oxidase 1 (COX1) and cytochrome b. Immunoreactivity of cytochrome b and COX1 were uniform throughout the neuron in brain specimens from controls. In contrast, immunoreactivity of cytochrome b and COX1 was not normal and was disrupted in brain specimens from grade III and IV HD patients. Using quantitative analysis, we found that cytochrome b levels were significantly lower in grade III (P< 0.02) and IV HD patients (P< 0.0001) compared with controls (Fig. 5). The COX1 levels were also significantly lower in grade III (P< 0.001) and IV HD patients (P< 0.0001) compared with controls (Fig. 6). The lower levels of cytochrome b and COX1 in patients at grade III HD and IV HD may due to the lower numbers of neurons found in HD patients; these lower levels may also suggest that mitochondrial function is lower in HD patients.
Figure 5.

(A) Mitochondrial-encoded proteins and cytochrome b immunoreactivity in frontal cortex sections from grade III (B) and grade IV HD (C) patients and controls (A). Uniform immunoreactivity of cytochrome b was found in the control subject (D). Interrupted immunoreactivity of cytochrome b (red arrows) was found in grade III (E) and grade IV (F) HD patients. Arrows indicate immunoreactivity. Images were photographed at ×40 (upper panel) and ×100 (lower panel). (B) Quantitative analysis of cytochrome b. Significantly decreased cytochrome b in grade III (P< 0.02) and grade IV (P< 0.0001) HD patients. Error bars indicate mean ± SEM.
Figure 6.

(A) Mitochondrial-encoded protein COX1 expression in frontal cortex sections from grade III (B) and grade IV (C) HD patients and controls (A). Uniform immunoreactivity of COX1 was found in brain sections from control subjects (D). Interrupted immunoreactivity of COX1 was found in brain sections from grade III (E) and grade IV (F) HD patients. Arrows indicate immunoreactivity. Images were photographed at ×40 (upper panel) and ×100 (bottom panel). (B) Quantitative analysis of COX1. Significantly decreased cytochrome b in grade III (P< 0.001) and grade IV (P< 0.0001) HD patients. Error bars indicate mean ± SEM.
Immunofluorescence analysis of mutant Htt in HD patients
To determine mutant Htt localization in neurons from patients at grade III and IV HD and from controls, we conducted immunofluorescence analysis of brain specimens, using the mutant Htt-specific antibodies 2166 and MW8 (5). As shown in Figure 7A, we found intraneuronal inclusions in the MW8 antibody-stained brain sections from HD patients but not in those from controls. However, in the 2166 monoclonal antibody-stained sections, we found immunoreactivity of mutant Htt in the cell body, axons and nerve terminals, and increased immunoreactivity in brain specimens from HD patients (Fig. 7B).
Figure 7.

(A) Mutant Htt immunoreactivity in frontal cortex sections from grade III (B) and grade IV (C) HD patients and control subjects (A), using MW8 antibody. Neuronal inclusions were found in the nucleus (red arrows) in brain sections from grade III (E) and grade IV (F) HD patients. (B) Mutant Htt immunoreactivity, using MAB 2166 antibody. Increased cytoplasmic immunoreactivity of mutant Htt in grade III (H) and grade IV (I) HD patients. Arrows indicate immunoreactivity, particularly perinuclear staining of mutant Htt in neurons from HD patients. Images were photographed at ×20 (upper panel), ×100 (middle panel) and ×40 (bottom panel).
Immunofluorescence analysis of mutant Htt oligomers in HD patients
To determine the presence of mutant Htt oligomers, using A11 antibody, we conducted immunofluorescence analysis of brain sections from patients with grade III and IV HD and controls. As shown in Figure 8, we found oligomeric mutant Htt immunoreactivity in the cytoplasm, but it was mostly perinuclear. We also found nuclear localization of oligomeric mutant Htt.
Figure 8.

Oligomer-specific mutant Htt immunoreactivity in brain sections from grade III (B and C) and grade IV (D) HD patients and control subjects (A), using A11 antibody. Mutant Htt oligomers were present in the perinuclear region (G–K) and also within nucleus (H) in HD patients. Arrows indicate mutant Htt oligomers. Images were photographed at ×40 (upper panel) and ×100 (lower panel).
Immunofluorescence analysis of oxidative DNA damage in HD patients
To determine the presence of oxidative DNA damage in HD patients, we conducted immunofluorescence analysis of brain sections from HD patients, using the 8-hydroxy-guanosine (8-OHG) antibody. As shown in Figure 9A, we found greater immunoreactivity in grade III and IV HD patients relative to controls, indicating the presence of oxidative DNA damage in HD pathogenesis. Further, we quantitatively analyzed the immunoreactivity of 8-OHG in brain sections from grade III and IV HD patients and controls. As shown in Figure 9B, oxidative DNA damage was significantly higher in patients at grade III (P< 0.002) and grade IV HD (P< 0.0001) compared with controls. These findings suggest that oxidative DNA damage may be due to increased association of mutant Htt with mitochondria in HD neurons.
Figure 9.

(A) Immunoreactivity of 8-hydroxyguanosine (oxidative DNA damage) in frontal cortex sections from grade III (B) and grade IV (C) HD patients and controls (A). Arrows indicate increased immunoreactivity; images were photographed at ×40. (B) Quantitative analysis of 8-OHG. Significantly increased 8-OHG immunoreactivity was found in grade III (P< 0.002) and grade IV (P< 0.0001) patients. Error bars indicate mean ± SEM. (C) Triple-labeling analysis of mutant Htt (2166 antibody, D) and 8-OHG (oxidative DNA damage, E) and nuclear staining DAPI (F) and merged section (G) cortical section from grade III HD patients. White arrows indicate mutant Htt immunoreactivity and green arrows indicate 8-OHG immunoreactivity. (D) Enlarged triple-labeling analysis of mutant Htt (H), 8-OHG (I), DAPI (J) and merged section (K). Mutant Htt is colocalized with 8-OHG in neurons affected by HD, indicating that neurons expressing mutant are oxidatively damaged. Images were photographed at ×100. White arrows indicate mutant Htt immunoreactivity and green arrows indicate 8-OHG immunoreactivity.
Double-labeling analysis of mutant Htt and 8-OHG in HD brains
To determine whether mutant Htt localizes with the immunoreactivity of 8-OHG, we conducted double-labeling analysis of mutant Htt and 8-OHG in brain specimens from the frontal cortex of grade III and IV HD patients and from controls. As shown in Figure 9C, immunoreactivity of mutant Htt was colocalized with 8-OHG immunoreactivity, indicating that neurons overexpressing mutant Htt are oxidatively damaged. Our findings also suggest that mutant Htt is involved in oxidative damage during disease progression.
Double-labeling analysis of mutant Htt and CypD in HD brains
To determine whether mutant Htt localizes in the mitochondrial matrix of neurons from the frontal cortex of HD patients at grade III and IV and of controls, we conducted double-labeling analysis, using the 2166 antibody and CypD. As shown in Figure 10, immunoreactivity of mutant Htt was colocalized with CypD immunoreactivity, indicating the presence of mutant Htt in the mitochondrial matrix of HD patients but not of controls.
Figure 10.

Triple-labeling analysis of mutant Htt (2166 antibody, A and E), CypD (B and F) and nuclear staining DAPI (C and G) and merged section (D and H) in cortical sections from grade III HD patients. Arrows indicate mutant Htt immunoreactivity colocalized with CypD. Images were photographed at ×40 (upper panel) and ×100 (bottom panel).
Double-labeling analysis of mutant Htt oligomers and COX1 in HD brains
To determine whether mutant Htt oligomers localize in the mitochondria of frontal cortex brain specimens from HD patients, we conducted double-labeling analysis, using A11 and COX1. As shown in Figure 11A, immunoreactivity of mutant Htt oligomers was colocalized with COX1 immunoreactivity, indicating the presence of mutant Htt oligomers in mitochondria, in HD patients but not in controls.
Figure 11.

(A) Triple-labeling immunofluorescence analysis of COX1 (A and E), mutant Htt oligomers (B and F), DAPI (C and G) in HD patients. Images (D) and (H) show merged section. Arrows indicate mutant Htt oligomeric immunoreactivity colocalized with COX1 immunoreactivity. Images were photographed at ×40 (upper panel) and ×100 (bottom panel). (B) Double-labeling analysis of mutant Htt oligomers (A) and nuclear marker histone3 (B) and merged section (C) in grade III HD patients. White arrows indicate histone3 mutant Htt oligomeric immunoreactivity, and red arrows indicate mutant Htt oligomers in the nucleus. Images were photographed at ×40 (upper panel) and ×100 (bottom panel).
Double-labeling analysis of mutant Htt oligomers and histone3 in HD brains
To determine whether mutant Htt oligomers localize in the nucleus, we conducted double-labeling analysis of frontal cortex specimens from grade III and IV HD patients, using A11 and the histone3 antibody, which is specific for the nuclear matrix. As shown in Figure 11B, immunoreactivity of mutant Htt oligomers was colocalized with histone3 immunoreactivity, indicating the presence of mutant Htt oligomers in the nucleus.
DISCUSSION
We investigated mitochondrial structural proteins, oxidative DNA damage and mutant Htt oligomers in postmortem brain specimens from grade III and IV HD patients and controls. We found high levels of fission genes, low levels of fusion genes and high levels of CypD selectively in striatum and cortex specimens from HD patients used in this study. We found significantly greater oxidative DNA damage in HD patients than in controls. We also found mutant Htt oligomers in the cortex of HD patients, and that the levels of oligomers increased as the disease progressed. These findings allow us to speculate that increased mutant Htt oligomers may be involved in mitochondrial fragmentation, abnormal mitochondrial dynamics and oxidative DNA damage in neurons affected by HD.
We investigated mitochondrial structural proteins, oxidative DNA damage and mutant Htt oligomers in postmortem brain specimens from grade III and IV HD patients and controls. We found high levels of fission genes, low levels of fusion genes and high levels of CypD selectively in striatum and cortex specimens from HD patients. We found significantly greater oxidative DNA damage in HD patients than in controls. We also found mutant Htt oligomers in the cortex of HD patients, and that the levels of oligomers increased as the disease progressed. These findings lead to the conclusion that increased mutant Htt oligomers may be involved in mitochondrial fragmentation, abnormal mitochondrial dynamics and oxidative DNA damage in neurons affected by HD.
Abnormal mitochondrial dynamics
Overall, decreased mRNA levels in the fusion genes were found in patients at grade III and IV HD. Among these proteins, Mfn2 was the most downregulated, particularly in grade IV HD patients, indicating that mitochondrial fusion decreases in late-stage HD pathogenesis. In contrast, increased mRNA levels in the fission genes were found in HD patients at both grades III and IV. Interestingly, mean increased mRNA levels of Fis1 were higher in grade IV HD patients (33.1-fold) than in grade III HD patients (9.5-fold), indicating the involvement of mitochondrial fragmentation and abnormal mitochondrial dynamics in late-stage HD pathogenesis. Mean Drp1 mRNA levels were similar in grade III (5.8-fold) and IV (5.8-fold) HD patients, suggesting that Drp1 upregulation is involved throughout HD pathogenesis. The mRNA levels of Tomm40 were higher in grade IV than in grade III HD patients, indicating that Tomm40 is involved in late-stage HD pathogenesis.
Interestingly, mRNA levels of CypD were higher in brain specimens from HD patients at both grades of HD compared with controls, suggesting that higher mRNA expression of CypD may be an early change in HD progression and may be critical for mitochondrial structural damage in late-stage HD. Overall, the increased mRNA levels of fission genes, the elevated CypD and the lower mRNA levels of fusion genes suggest abnormal mitochondrial dynamics in late-stage HD patients.
Abnormal mitochondrial proteins and impaired mitochondrial dynamics
Immunostaining analysis of Drp1 and Fis1 in brain specimens from grade III and IV HD patients and controls revealed greater immunoreactivity of Drp1 and Fis1, suggesting that high levels of fission proteins may be responsible for mitochondrial fragmentation and mitochondrial swelling that have been reported in HD neurons (39). Our findings about the greater immunoreactivity of Drp1 and Fis1 are consistent with the recent studies that compared Drp1 in HD patients (36) with other HeLa cells that express expanded polyQ repeats (39) and that found Drp1 increased in HD patients. Wang et al. (39) found less mitochondrial movement and less fusion in cells expressing expanded (78 or 138) polyQ repeats relative to cells expressing 28 polyQ repeats.
Overall, findings from our study, together with those from other studies, suggest that fission proteins are selectively elevated in neurons affected by HD. Further, abnormal mitochondrial dynamics have been reported in other neurodegenerative diseases, including Alzheimer's (44–46) and Parkinson's (47–50), indicating that mutant proteins' association with mitochondria (51) may cause mitochondrial structural and functional damage in neurodegenerative diseases.
Further, several studies suggest that mitochondrial abnormalities are present in peripheral cells from HD patients, indicating mitochondrial damage is associated with HD. Squitieri et al. (37) found increased defective mitochondria in peripheral cells (including fibroblasts and lymphoblasts) from HD patients relative to peripheral cells from control subjects, indicating that mitochondrial morphological changes are associated with polyQ repeats. Further, Costa et al. (38) found increased mitochondrial fragmentation and broken cristae in HD lymphoblasts. They also found that damaged mitochondrial morphology increased as the number of polyQ repeats and the dosage of the mutant HD gene increased. Furthermore, they characterized mitochondrial striatal progenitor cells from HD knockin mice that carried 110 polyQ repeats, finding a 15% increase in mitochondrial fragmentation, in HD knockin mice relative to the progenitor cells from control mice. Findings from their study further support the involvement of polyQ-dependent mitochondrial structural changes in HD.
Our findings of lower levels of fusion proteins, particularly Mfn2, in brain specimens from patients with late-stage HD concurred with an earlier study that reported decreased fusion proteins in HeLa cells that overexpressed expanded polyQ repeats (39). In addition, our findings that Mfn2 and Opa1 have lower immunoreactivity in grade III and IV HD patients relative to controls further confirm the notion that mitochondrial dynamics are impaired in HD patients. Increased levels of fission proteins and decreased levels of fusion proteins may lead to changes in mitochondrial structure and function. The increase in fission proteins and the decrease in fusion proteins collectively produce cytotoxicity that is induced by Htt proteins containing expanded polyQ repeats. The polyQ repeats are probably mediated by the impaired mitochondrial dynamics, and the increased polyQ repeats probably result in increased mitochondrial fragmentation and selective neuronal damage.
In a comparative western blot analysis of Drp1, Fis1, Mfn1, Mfn2 and Opa1, we found higher Drp1 and Fis1 levels and lower Mfn1 and Mfn2 levels in HD-affected brain regions (striatum and cortex) but not in the unaffected region (cerebellum). CypD protein levels were also selectively higher in the affected brain regions of HD patients. It is interesting to note that we found mutant Htt aggregates and mutant Htt oligomers only in the HD-affected brain regions, where we also found abnormal mitochondrial dynamics. Further, protein levels of Drp1, Fis1, Mfn1, Mfn2, Opa1 and CypD are unchanged in the striatum and cortex (affected in HD) relative to the cerebellum (unaffected in HD) of control subjects, which further supports that altered mitochondrial proteins in the affected regions of HD brains are due to mutant Htt. In other words, abnormal mitochondrial dynamics were absent in brain specimens from our control subjects.
Overall, we found increased levels of fission and matrix proteins and decreased levels of fusion proteins selectively in the affected regions (cortex and striatum) of HD brains, where increased numbers of mutant Htt aggregates and oligomers were also found (18), leading us to conclude that mutant Htt oligomers are critically involved in damaging mitochondria both structurally and functionally in HD progression and pathogenesis.
Mutant Htt oligomers and abnormal mitochondrial dynamics
A11 antibody has been reported to recognize oligomeric mutant proteins in neurodegenerative diseases, including Alzheimer's (52) and Parkinson's (53). However, the use of oligomeric antibody has been limited to Alzheimer's and Parkinson's diseases. In the present study, we used oligomer-specific antibody and studied mutant Htt oligomers in HD brains. Mutant Htt oligomers were found selectively in the frontal cortex and striatum of grade III and IV HD patients and the presence of these oligomers shows an increased trend with disease progression. Interestingly, abnormal mitochondrial dynamics was found in the brain regions that showed mutant Htt oligomers, indicating that mutant Htt oligomers may be involved in abnormal mitochondrial dynamics.
Our double-labeling analysis using A11 oligomer-specific antibody and mitochondrial-encoded proteins, COX1 and cytochrome b, revealed colocalization of mutant Htt oligomers with COX1 and cytochrome b in cortical brain sections from HD patients, suggesting that mutant Htt oligomers are associated with mitochondria, and may be responsible for abnormal mitochondrial structural changes (38), impaired mitochondrial dynamics and oxidative damage found in HD brains (36 and present-study observations).
Our findings also indicate that mutant Htt aggregates and oligomers are mainly involved in HD progression. In addition, lower energy metabolism (the loss of body weight and brain weight) has been described a key event in HD progression and pathogenesis (54–59). Magnetic resonance studies suggest a progressive atrophy of the striatum (27,60) and reduced cortical lobes in patients with HD (61,62) relative to controls. Positron emission tomography studies revealed that decreased glucose utilization in the brain, particularly in the striatum, correlates with HD patients’ performance on cognitive tasks, including immediate recall, memory, verbal associate learning and executive functions, suggesting that cerebral glucose metabolism is defective in HD patients (20–22,25). However, the mechanistic link between lower levels of brain energy/glucose metabolism and selective neuronal damage has been unclear until now. Findings from our study, together with those from Kim et al. (36), provide compelling evidence that abnormal mitochondrial dynamics are selectively caused by mutant Htt oligomers in the striatum and cortex of HD patients. Further research, however, is needed to understand the mechanisms of mutant Htt oligomer fibril formation and the transport of these fibrils to subcellular organelles, such as mitochondria and nuclei. Further research is also needed to elucidate how mutant Htt oligomers selectively interfere with axonal mitochondrial-dependent transport in striatal and cortical neurons of HD patients.
Defective mitochondrial energetics and mitochondrial activity
To understand the role of mitochondrial abnormalities in HD pathogenesis, it is critical to determine the connection, if any, between altered mitochondrial gene expressions and HD pathogenesis in terms of mitochondrial-encoded polypeptides in the electron transport chain since these polypeptides play a role in oxidative phosphorylation. Increased expression of mitochondrial-encoded genes in complexes I, III, IV and V was found in patients with HD relative to controls, suggesting that this upregulation may be compensation for the loss of mitochondrial function caused by mutant Htt or may be somehow related to this loss of mitochondrial function. Either of these alternatives is supported by the decrease in the immunoreactivity of cytochrome b and COX1 that we found in neurons of the grade III and IV HD patients relative to controls. These decreases may be due to neuronal loss in grade III (at 20–50%) and grade IV (up to 80%) HD patients (63). Further, the increase in mRNA expression of electron transport chain genes (including COX1 and cytochrome b) in the surviving neurons may also be a compensatory response to mutant Htt aggregates.
Oxidative DNA damage in HD patients
We found significantly greater oxidative DNA damage brain specimens from grade III and IV HD patients. We also found oxidative DNA damage progressively increased in brain specimens from HD patients as HD progressed. Increased oxidative DNA damage and decreased cytochrome b and COX1 in HD patients clearly suggest that mitochondria are impaired in HD neurons and may be responsible for abnormal mitochondrial dynamics and impaired mitochondrial transport.
Mutant Htt immunoreactivity that colocalized with 8-OHG immunoreactivity indicates that neurons overexpressing mutant Htt are oxidatively damaged. These findings suggest that mutant Htt is involved in oxidative damage and mitochondrial dysfunction in HD, which is consistent with other research on impaired mitochondrial function, in which impaired mitochondrial bioenergetics was found in postmortem brains from HD patients (30,31,64,65).
Biochemical studies of HD knockin mice and HD cell lines suggest that calcium-induced mitochondrial permeability is a key factor in HD pathogenesis (8). Although some of these studies have attempted to associate increased polyQ repeat length with mitochondrial abnormalities and neuronal damage, there appears to be little evidence to support that view. Indeed, findings from our study provide evidence that mutant Htt oligomers are involved in mitochondrial abnormalities in HD because mutant Htt oligomers are associated with mitochondria, disrupt mitochondrial function and inhibit ATP production and damage neurons affected by HD. We propose that the missing link between mitochondrial dysfunction and HD progression/pathogenesis may be ‘mutant Htt oligomers in association with mitochondria’.
MATERIALS AND METHODS
Nine postmortem frozen and fixed brain specimens from the frontal cortex, striatum and cerebellum of HD patients and age-matched healthy subjects (control) were obtained from the Harvard Tissue Resource Center. Table 3 presents the characteristics of the brain specimens used in this study. Three specimens were from patients with grade III HD [graded according to Vonsattel et al. (63)], three specimens from patients with grade IV HD and three from controls.
Real-time RT–PCR quantification of mRNA expression of mitochondrial electron transport chain genes, mitochondrial fission, mitochondrial fusion and matrix genes
Using the reagent TRIzol (Invitrogen, Temicula, CA, USA), we isolated total RNA from cortical tissues of HD patients and controls. We used Primer Express Software (Applied Biosystems, Foster City, CA, USA) to design the oligonucleotide primers for the housekeeping genes β-actin and GAPDH, the mitochondrial electron transport chain genes, the mitochondrial structural genes, the fission genes (Drp1 and Fis1), the mitochondrial fusion genes (Mfn1, Mfn2, Opa1, Tomm40) and the mitochondrial matrix protein CypD. The primer sequences and amplicon sizes are listed in Supplementary Material, Table S1. Using SYBR-Green chemistry-based quantitative real-time RT–PCR, we measured mRNA expression of our study genes, following Manczak et al. (46). Briefly, 2 μg of DNAse-treated total RNA was used as starting material, to which we added 1 μl of oligo (dT), 1 μl of 10 mm dNTPs, 4 μl of 5× first-strand buffer, 2 μl of 0.1 m DTT and 1 μl of RNAseOUT. The reagents RNA, dT and dNTPs were mixed first, heated at 65°C for 5 min and chilled on ice until the remaining components were added. The samples were incubated at 42°C for 2 min and then 1 μl of Superscript II (40 U/μl) was added. The samples were then incubated at 42°C for 50 min, at which time the reaction was inactivated by heating the samples for 15 min at 70°C.
Using cDNA from HD patients and controls, we performed quantitative real-time PCR amplification reactions in a 25 μl volume of total reaction mixture, in an ABI Prism 7900 sequence detection system (Applied Biosystems). The reaction mixture consisted of 1× PCR-buffer-containing SYBR-Green; 3 mm MgCl2; 100 nm of each primer; 200 nm of dATP, dGTP and dCTP each; 400 nm of dUTP; 0.01 U/μl of AmpErase UNG and 0.05 U/μl of AmpliTaq gold. A cDNA template of 50 ng was added to each reaction mixture.
CT-values of β-actin and GAPDH were tested to determine the unregulated endogenous reference gene in HD patients and controls. The CT-value, described in Manczak et al. (46) and Gutala and Reddy (66), is an important quantitative parameter in real-time PCR analysis. All RT–PCR reactions were carried out in triplicate, and with no template control. The mRNA transcript level was normalized against β-actin and GAPDH at each dilution. The standard curve was the normalized mRNA transcript level, plotted against the log value of the input cDNA concentration at each dilution. Briefly, the comparative CT method involved averaging triplicate samples, which were taken as the CT values for β-actin, GAPDH and mitochondrial genes. β-Actin normalization was used in the present study because β-actin and CT values were similar for mitochondrial structural genes, and also allowed us to avoid the values of GAPDH. The ΔCT-value was obtained by subtracting the average β-actin CT-value from the average CT-value for the mitochondrial structural genes. The ΔCT of the controls was used as the calibrator. The fold change was calculated according to the formula 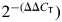 , where ΔΔCT is the difference between ΔCT and the ΔCT calibrator value.
, where ΔΔCT is the difference between ΔCT and the ΔCT calibrator value.
Western blot analysis of mitochondrial proteins
To determine whether mitochondrial protein levels are altered in HD patients relative to controls, we performed immunoblotting analyses of protein lysates from cortical tissues of HD patients and controls. To determine whether alterations in mitochondrial proteins are confined to affected brain regions in HD patients, we performed immunoblotting analysis of the striatum, frontal cortex and cerebellum specimens from grade III HD patients. Fifty micrograms of protein lysates were resolved on 10% PAGE gel (Invitrogen). The resolved proteins were transferred to nylon membranes (Millipore, San Diego, CA, USA) and then were incubated for 1 h at room temperature with a blocking buffer (5% dry milk dissolved in the TBST buffer). The nylon membranes were incubated overnight with the primary antibodies Drp1 (1:200 dilution, rabbit polyclonal; Santa Cruz Biotechnology, CA, USA), Fis1 (1:200, rabbit polyclonal; Protein Tech Group, Inc., Chicago, IL, USA), Mfn1 (1:300, rabbit polyclonal; Santa Cruz Biotechnology, Inc.), Mfn2 (1:300 rabbit polyclonal; Abcam, Cambridge, MA, USA), Tomm40 (1:300, rabbit polyclonal; Santa Cruz Biotechnology), Opa1 (1:500 mouse monoclonal; BD Biosciences, San Jose, CA, USA), CypD (1:1000; EMD4 Biosciences, San Diego, CA, USA) and β-actin (1:500 mouse monoclonal, Millipore). Details of antibody dilutions are given in Supplementary Material, Table S2.
The membranes were washed with the TBST buffer three times at 10 min intervals and then incubated for 2 h with appropriate secondary antibodies, followed by three additional washes at 10 min intervals. Proteins were detected with chemiluminescent reagents (Pierce Biotechnology, Rockford, IL, USA), and the bands from immunoblots were quantified on a Kodak scanner (ID Image Analysis Software, Kodak Digital Science, Kennesaw, GA, USA). Briefly, image analysis was used to analyze gel images captured with a Kodak Digital Science CD camera. The lanes were marked to define the positions and specific regions of the bands. An ID fine-band command was used to locate and to scan the bands in each lane and to record the readings. We analyzed the intensity of bands using ImageJ analysis and assessed statistical significance using Student's t-test with a significance level of P< 0.05. Western blot band intensities were expressed as mean ± standard error (mean ± SEM).
Western blot analysis of mutant Htt oligomers
To quantify mutant Htt oligomers in grade III and IV HD patients and in controls, we performed immunoblotting analyses of protein lysates from cortical tissues. To detect mutant Htt oligomers, protein lysates were prepared with the following buffers: 250 mm sucrose, 50 mm HEPES, 25 mm MgCl2, 0.5 mm dithiothereitol and protease inhibitors, 0.5 g/ml pepstatin, 0.5 g/ml leupeptin and 1 mm PMSF (phenylmethylsulfonyl fluoride). Fifty micrograms of protein lysates were resolved on a 4–12% gradient gel (Invitrogen). The resolved proteins were transferred to nitrocellulose membranes (Novax, Inc.) and then incubated for 1 h at room temperature with a blocking buffer (5% dry milk dissolved in the TBST buffer). The nitrocellulose membranes were incubated overnight with the A11 antibody (a mutant Htt oligomer-specific anti-rabbit polyclonal, 1:1000; Invitrogen) that recognizes mutant Htt oligomers in protein lysates from grade III and IV patients. Mutant Htt oligomers were detected with chemiluminescent reagents (Pierce Biotechnology). We quantified the bands from immunoblots using densitometry and ImageJ analysis and assessed statistical significance using Student's t-test with a significance level of P< 0.05.
Dot blot analysis
Ten micrograms of protein was spotted onto a nitrocellulose membrane that was then air-dried for 1 h. The membrane was soaked in 5% milk in TBST for 1 h to block non-specific sites and was then incubated with the A11 primary antibody (anti-rabbit polyclonal, 1:1000; Invitrogen) in the blocking solution for 1 h at room temperature. It was washed three times with TBST and then incubated with an anti-rabbit secondary antibody that was conjugated with HRP (horseradish peroxidase) of 1:10 000 dilution for 30 min at room temperature. The membrane was again washed three times with TBST and incubated with the ECL (enhanced chemiluminescence) reagent for 1 min before it was exposed to X-ray film. We quantified oligomer-specific dots from dot blots using ImageJ analysis and assessed statistical significance using Student's t-test.
Immunohistochemistry/immunofluorescence analysis
Using immunofluorescence techniques to localize mitochondrial structural proteins, mutant Htt monomers and oligomers and 8-OHG, we studied Drp1, Fis1, Mfn2, Opa1, CypD, COX1, cytochrome b, mutant Htt (using the MW8 antibody and monoclonal antibody 2166), mutant Htt oligomers (the A11 antibody), histone3 and 8-OHG proteins in frontal cortex specimens (Broadman area 9) from grade III HD patients (n= 3), grade IV HD patients (n = 3) and control subjects (n= 3). Brain specimens were embedded in paraffin, and sections were cut at 15 μm. We then deparaffinized the sections by washing them with xylene for 10 min and then for 5 min each, in a serial dilution of alcohol: 95, 70 and 50%. The sections were washed for 10 min with double-distilled H2O and three more times for 5 min each with phosphate-buffered saline (PBS) at pH 7.4.
To reduce the autofluorescence of brain specimens, we treated the deparaffinized sections with sodium borohydrate twice each for 30 min in a freshly prepared 0.1% sodium borohydrate solution dissolved in PBS at pH 8.0. We then washed the sections three times for 5 min each, with PBS at pH 7.4. To block the endogenous peroxidase, sections were treated for 15 min with 3% H2O2 and then with 0.5% Triton dissolved in PBS at pH 7.4. The sections were blocked for 1 h with a solution [0.5% Triton in PBS+10% goat serum+1% bovine serum albumin (BSA)]. They were incubated overnight at room temperature with the primary antibodies (see Supplementary Material, Table S3, for dilutions). On the day after primary antibody incubation, the sections were washed three times with a washing buffer (0.5% Triton in PBS) and then incubated with a secondary biotinylated anti-mouse antibody (1:200 dilution) (Vector Laboratories, Burlingame, CA, USA) or a biotinylated anti-rabbit antibody (1:200) for 1 h at room temperature. Sections were washed three times for 10 min each with PBS. The sections were incubated with the ABC solution (1:500 dilution, Vector Laboratories) for 30 min and then washed three times for 10 min each with PBS at pH 7.4. They were treated with tyramide-conjugated fluorescent dye Alexa 488 (green) or 594 (red) (Molecular Probes) for 10 min at room temperature and counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (1:1000; KPL, Gaithersburg, MD, USA) (blue) for nuclear labeling. They were cover-slipped with prolong gold per mount and photographed using a fluorescence microscope.
To assess oxidative DNA damage in HD patients, we quantified the immunoreactivity of 8-OHG in brain sections from controls, grade III and grade IV HD patients. Ten to 15 images were taken at 20 times the original magnification from each case, and positive immunoreactivity with fluorescence signal intensity of neurons was measured using ImageJ analysis.
For the analysis of mitochondrial-encoded proteins, cytochrome b and COX1, 10–15 images were taken at 20 times the original magnification from each three cases of control subjects, grade III and IV HD patients. Then images were analyzed by quantifying positive immunoreactivity with fluorescence signal intensity per neuron using ImageJ. We assessed statistical significance of 8-OHG, cytochrome b and COX1, using Student's t-test with a significance level of P< 0.05. The fluorescent signal intensities are expressed as mean ± SEM. We quantified signal intensity of positive immunoreactivities of cytochrome b and COX1 in neurons from grade III and grade IV HD patients compared with positive immunoreactivities of cytochrome b and COX1 in neurons from control subjects.
Double-labeling immunofluorescence analysis
We performed double-labeling analyses to determine whether neurons showing increased mutant Htt are more vulnerable to (i) oxidative damage, (ii) mutant Htt localized with mitochondria, (iii) mutant Htt oligomers localized in the nucleus or (iv) mutant Htt oligomers localized with mitochondria. These analyses involved immunostaining of the mutant Htt monoclonal antibody MAB2166 (mouse monoclonal) and an anti-goat 8-OHG antibody (goat polyclonal); mutant Htt MAB2166 and CypD (mouse monoclonal); oligomer-specific antibody A11 and nucleus marker, histone3 (rabbit polyclonal); and A11 and COX1.
As describe dearlier, brain sections from HD patients and controls were deparaffinized and also treated with sodium borohydrate to reduce autofluorescence.For the first labeling, the sections were incubated overnight at room temperature with primary antibodies (see Supplementary Material, Table S3, for dilutions), goat anti-8-OHG (1:400), histone3 (1:400), CypD (1:100) and COX1 (1:100). On the day after primary antibody incubation, the sections were washed with a washing buffer (0.05% Tween-20 in PBS) and then incubated with appropriate secondary antibodies (Supplementary Material, Table S3) for 1 h at room temperature. The sections were incubated with the ABC solution for 30 min, were washed three times for 10 min each with PBS at pH 7.4 and treated with tyramide-conjugated fluorescent dye Alexa 488 (green) for COX1, and Alexa 594 (red) for 8-OHG, histone3 and CypD for 10 min each at room temperature.
For the second labeling, the sections were blocked for 1 h with a blocking solution containing 0.5% Triton in PBS + 10% donkey serum + 1% BSA. Then they were incubated overnight at room temperature with primary antibodies (Supplementary Material, Table S3), the mutant Htt antibody (1:50) and the oligomer-specific antibody (1:50, A11). The sections were incubated with appropriate secondary antibodies and then treated with tyramide-conjugated fluorescent dye Alexa 488 or Alexa 594 for 10 min at room temperature. Finally, brain sections were stained with DAPI, cover-slipped with prolong gold per mount and photographed with a fluorescence microscope.
CONCLUSIONS
Mitochondrial dysfunction and oxidative stress have been described in HD, but the precise role of mitochondrial abnormalities and abnormal mitochondrial dynamics was unclear. In the present study, we investigated mitochondrial dynamics and oxidative DNA damage, using postmortem brains from patients at stages III and IV. We quantified the mRNA levels of the electron transport chain genes and the amount of mRNA and protein levels in the mitochondrial structural genes Drp1, Fis1, Mfn1, Mfn2, Opa1, Tomm40 and CypD in brain specimens. We quantified mutant Htt oligomers and studied the localization of Htt oligomers in the striatum and frontal cortex of HD patients. We studied mitochondrial structural proteins in the cortex, striatum and cerebellum of control brains to determine whether altered mitochondrial proteins in HD patients are associated with disease.
An increase in electron transport chain genes was found in HD patients, indicating that this increase may be a compensatory response to mitochondrial damage caused by mutant Htt. The increased expression of Drp1 and Fis1 and the decreased expression of Mfn1, Mfn2, Opa1 and Tomm40 were found in HD patients compared with controls. CypD was upregulated in HD patients, and this upregulation increased as HD progressed. Significantly increased immunoreactivity of 8-OHG was found in cortical specimens from stage III and IV HD patients relative to controls, suggesting that oxidative DNA damage increases in HD patients. In contrast, COX1 activity and cytochrome b were found to be significantly decreased in brain specimens from HD patients relative to controls, indicating the loss of mitochondrial function in HD patients. Immunoblotting analysis revealed 15, 25 and 50 kDa mutant Htt oligomers in brains of HD patients but not in brains of controls. All oligomeric forms were significantly higher in cortex tissues of HD patients. The increase in Drp1, Fis1 and CypD and the decrease in Mfn1 and Mfn2 may be responsible for abnormal mitochondrial dynamics that we found in the striatum and cortex of HD patients, and they may contribute to neuronal damage in HD patients. Formation of mutant Htt oligomers and their presence in the nucleus and in mitochondria may disrupt neuronal functions. Based on these findings, we propose that mutant Htt, associated with mitochondria, disrupts the balance of mitochondrial dynamics, impairs axonal transport of mitochondria, decreases mitochondrial function and damages neurons in affected brain regions in patients with HD.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
FUNDING
This research was supported by NIH grants AG028072, AG026051, RR00163 and S10RR024585, Alzheimer Association grant IIRG-09-92429, Vertex Pharmaceuticals and Medivation, Inc.
ACKNOWLEDGEMENTS
We thank Dr Anda Cornea for her assistance with confocal microcopy.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Orr H.T., Zoghbi H.Y. Trinucleotide repeat disorders. Ann. Rev. Neurosci. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. doi:10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- 2.Bates G.P. History of genetic disease: the molecular genetics of Huntington disease—a history. Nat. Rev. Genet. 2005;6:766–773. doi: 10.1038/nrg1686. doi:10.1038/nrg1686. [DOI] [PubMed] [Google Scholar]
- 3.Reddy P.H., Williams M., Tagle D.A. Recent advances in understanding the pathogenesis of Huntington's disease. Trends Neurosci. 1999;22:248–255. doi: 10.1016/s0166-2236(99)01415-0. doi:10.1016/S0166-2236(99)01415-0. [DOI] [PubMed] [Google Scholar]
- 4.Reddy P.H., Mao P., Manczak M. Mitochondrial structural and functional dynamics in Huntington's disease. Brain Res. Rev. 2009;61:33–48. doi: 10.1016/j.brainresrev.2009.04.001. doi:10.1016/j.brainresrev.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ko J., Ou S., Patterson P.H. New anti-huntingtin monoclonal antibodies: implications for huntingtin conformation and its binding proteins. Brain Res. Bull. 2001;56:319–329. doi: 10.1016/s0361-9230(01)00599-8. doi:10.1016/S0361-9230(01)00599-8. [DOI] [PubMed] [Google Scholar]
- 6.Kegel K.B., Meloni A.R., Yi Y., Kim Y.J., Doyle E., Cuiffo B.G., Sapp E., Wang Y., Qin Z.H., Chen J.D., et al. Huntingtin is present in the nucleus, interacts with the transcriptional corepressor C-terminal binding protein, and represses transcription. J. Biol. Chem. 2002;277:7466–7476. doi: 10.1074/jbc.M103946200. doi:10.1074/jbc.M103946200. [DOI] [PubMed] [Google Scholar]
- 7.Kegel K.B., Sapp E., Yoder J., Cuiffo B., Sobin L., Kim Y.J., Qin Z.H., Hayden M.R., Aronin N., Scott D.L., et al. Huntingtin associates with acidic phospholipids at the plasma membrane. J. Biol. Chem. 2005;280:36464–36473. doi: 10.1074/jbc.M503672200. doi:10.1074/jbc.M503672200. [DOI] [PubMed] [Google Scholar]
- 8.Panov A.V., Gutekunst C.A., Leavitt B.R., Hayden M.R., Burke J.R., Strittmatter W.J., Greenamyre J.T. Early mitochondrial calcium defects in Huntington's disease are a direct effect of polyglutamines. Nat. Neurosci. 2002;5:731–736. doi: 10.1038/nn884. [DOI] [PubMed] [Google Scholar]
- 9.Choo Y.S., Johnson G.V., MacDonald M., Detloff P.J., Lesort M. Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum. Mol. Genet. 2004;13:1407–1420. doi: 10.1093/hmg/ddh162. doi:10.1093/hmg/ddh162. [DOI] [PubMed] [Google Scholar]
- 10.Truant R., Atwal R., Burtnik A. Hypothesis: Huntingtin may function in membrane association and vesicular trafficking. Biochem. Cell Biol. 2006;84:912–917. doi: 10.1139/o06-181. doi:10.1139/O06-181. [DOI] [PubMed] [Google Scholar]
- 11.Strehlow A.N., Li J.Z., Myers R.M. Wild-type huntingtin participates in protein trafficking between the Golgi and the extracellular space. Hum. Mol. Genet. 2007;16:391–409. doi: 10.1093/hmg/ddl467. doi:10.1093/hmg/ddl467. [DOI] [PubMed] [Google Scholar]
- 12.Atwal R.S., Xia J., Pinchev D., Taylor J., Epand R.M., Truant R. Huntingtin has a membrane association signal that can modulate huntingtin aggregation, nuclear entry and toxicity. Hum. Mol. Genet. 2007;16:2600–2615. doi: 10.1093/hmg/ddm217. doi:10.1093/hmg/ddm217. [DOI] [PubMed] [Google Scholar]
- 13.Orr A.L., Li S., Wang C.E., Li H., Wang J., Rong J., Xu X., Mastroberardino P.G., Greenamyre J.T., Li X.J. N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J. Neurosci. 2008;28:2783–2792. doi: 10.1523/JNEUROSCI.0106-08.2008. doi:10.1523/JNEUROSCI.0106-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wacker J.L., Zareie M.H., Fong H., Sarikaya M., Muchowski P.J. Hsp70 and Hsp40 attenuate formation of spherical and annular polyglutamine oligomers by partitioning monomer. Nat. Struct. Mol. Biol. 2004;11:1215–1222. doi: 10.1038/nsmb860. doi:10.1038/nsmb860. [DOI] [PubMed] [Google Scholar]
- 15.Poirier M.A., Li H., Macosko J., Cai S., Amzel M., Ross C.A. Huntingtin spheroids and protofibrils as precursors in polyglutamine fibrilization. J. Biol. Chem. 2002;277:41032–41037. doi: 10.1074/jbc.M205809200. doi:10.1074/jbc.M205809200. [DOI] [PubMed] [Google Scholar]
- 16.Thakur A.K., Jayaraman M., Mishra R., Thakur M., Chellgren V.M., Byeon I.J., Anjum D.H., Kodali R., Creamer T.P., Conway J.F., Gronenborn A.M., Wetzel R. Polyglutamine disruption of the huntingtin exon 1 N terminus triggers a complex aggregation mechanism. Nat. Struct. Mol. Biol. 2009;16:380–389. doi: 10.1038/nsmb.1570. doi:10.1038/nsmb.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Legleiter J., Mitchell E., Lotz G.P., Sapp E., Ng C., DiFiglia M., Thompson L.M., Muchowski P.J. Mutant huntingtin fragments form oligomers in a polyglutamine length-dependent manner in vitro and in vivo. J. Biol. Chem. 2010;285:14777–14790. doi: 10.1074/jbc.M109.093708. doi:10.1074/jbc.M109.093708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sathasivam K., Lane A., Legleiter J., Warley A., Woodman B., Finkbeiner S., Paganetti P., Muchowski P.J., Wilson S., Bates G.P. Identical oligomeric and fibrillary structures captured from the brains of R6/2 and knock-in mouse models of Huntington's disease. Hum. Mol. Genet. 2010;19:65–78. doi: 10.1093/hmg/ddp467. doi:10.1093/hmg/ddp467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manczak M., Anekonda T.S., Henson E., Park B.S., Quinn J., Reddy P.H. Mitochondria are a direct site of A beta accumulation in Alzheimer's disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 2006;15:1437–1449. doi: 10.1093/hmg/ddl066. doi:10.1093/hmg/ddl066. [DOI] [PubMed] [Google Scholar]
- 20.Kuhl D.E., Phelps M.E., Markham C.H., Metter E.J., Riege W.H., Winter J. Cerebral metabolism and atrophy in Huntington's disease determined by 18FDG and computed tomographic scan. Ann. Neurol. 1982;12:425–434. doi: 10.1002/ana.410120504. doi:10.1002/ana.410120504. [DOI] [PubMed] [Google Scholar]
- 21.Young A.B., Penney J.B., Starosta-Rubinstein S., Markel D.S., Berent S., Giordani B., Ehrenkaufer R., Jewett D., Hichwa R. PET scan investigations of Huntington's disease: cerebral metabolic correlates of neurological features and functional decline. Ann. Neurol. 1986;20:296–303. doi: 10.1002/ana.410200305. doi:10.1002/ana.410200305. [DOI] [PubMed] [Google Scholar]
- 22.Hayden M.R., Martin W.R., Stoessl A.J., Clark C., Hollenberg S., Adam M.J., Ammann W., Harrop R., Rogers J., Ruth T., et al. Positron emission tomography in the early diagnosis of Huntington's disease. Neurology. 1986;36:888–894. doi: 10.1212/wnl.36.7.888. [DOI] [PubMed] [Google Scholar]
- 23.Kuwert T., Lange H.W., Langen K.J., Herzog H., Aulich A., Feinendegen L.E. Cortical and subcortical glucose consumption measured by PET in patients with Huntington's disease. Brain. 1990;113:1405–1423. doi: 10.1093/brain/113.5.1405. doi:10.1093/brain/113.5.1405. [DOI] [PubMed] [Google Scholar]
- 24.Berent S., Giordani B., Lehtinen S., Markel D., Penney J.B., Buchtel H.A., Starosta-Rubinstein S., Hichwa R., Young A.B. Positron emission tomographic scan investigations of Huntington's disease: cerebral metabolic correlates of cognitive function. Ann. Neurol. 1998;23:541–546. doi: 10.1002/ana.410230603. doi:10.1002/ana.410230603. [DOI] [PubMed] [Google Scholar]
- 25.Powers W.J., Videen T.O., Markham J., McGee-Minnich L., Antenor-Dorsey J.V., Hershey T., Perlmutter J.S. Selective defect of in vivo glycolysis in early Huntington's disease striatum. Proc. Natl Acad. Sci. USA. 2007;104:2945–2949. doi: 10.1073/pnas.0609833104. doi:10.1073/pnas.0609833104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bamford K.A., Caine E.D., Kido D.K., Plassche W.M., Shoulson I. Clinical-pathologic correlation in Huntington's disease: a neuropsychological and computed tomography study. Neurology. 1989;39:796–801. doi: 10.1212/wnl.39.6.796. [DOI] [PubMed] [Google Scholar]
- 27.Aylward E.H., Brandt J., Codori A.M., Mangus R.S., Barta P.E., Harris G.J. Reduced basal ganglia volume associated with the gene for Huntington's disease in asymptomatic at-risk persons. Neurology. 1994;44:823–828. doi: 10.1212/wnl.44.5.823. [DOI] [PubMed] [Google Scholar]
- 28.Aylward E.H., Li Q., Stine O., Ranen N., Sherr M., Barta P.E., Bylsma F.W., Pearlson G.D., Ross C.A. Longitudinal change in basal ganglia volume in patients with Huntington's disease. Neurology. 1997;48:394–399. doi: 10.1212/wnl.48.2.394. [DOI] [PubMed] [Google Scholar]
- 29.Jernigan T.L., Salmon D.P., Butters N., Hesselink J.R. Cerebral structure on MRI. Part II: Specific changes in Alzheimer's and Huntington's diseases. Biol. Psychiatry. 1991;29:68–81. doi: 10.1016/0006-3223(91)90211-4. doi:10.1016/0006-3223(91)90211-4. [DOI] [PubMed] [Google Scholar]
- 30.Browne S.E., Bowling A.C., MacGarvey U., Baik M.J., Berger S.C., Muqit M.M., Bird E.D., Beal M.F. Oxidative damage and metabolic dysfunction in Huntington's disease: selective vulnerability of the basal ganglia. Ann. Neurol. 1997;41:646–653. doi: 10.1002/ana.410410514. doi:10.1002/ana.410410514. [DOI] [PubMed] [Google Scholar]
- 31.Tabrizin S.J., Cleeter M.W., Xuereb J., Taanman J.W., Cooper J.M., Schapira A.H. Biochemical abnormalities and excitotoxicity in Huntington's disease brain. Ann. Neurol. 1999;45:25–32. doi: 10.1002/1531-8249(199901)45:1<25::aid-art6>3.0.co;2-e. doi:10.1002/1531-8249(199901)45:1<25::AID-ART6>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 32.Fukui H., Moraes C.T. Extended polyglutamine repeats trigger a feedback loop involving the mitochondrial complex III, the proteasome and huntingtin aggregates. Hum. Mol. Genet. 2007;16:783–797. doi: 10.1093/hmg/ddm023. doi:10.1093/hmg/ddm023. [DOI] [PubMed] [Google Scholar]
- 33.Seong I.S., Ivanova E., Lee J.M., Choo Y.S., Fossale E., Anderson M., Gusella J.F., Laramie J.M., Myers R.H., Lesort M., MacDonald M.E. HD CAG repeat implicates a dominant property of huntingtin in mitochondrial energy metabolism. Hum. Mol. Genet. 2005;14:2871–2880. doi: 10.1093/hmg/ddi319. doi:10.1093/hmg/ddi319. [DOI] [PubMed] [Google Scholar]
- 34.Trushina E., Dyer R.B., Badger J.D., II, Ure D., Eide L., Tran D.D., Vrieze B.T., Legendre-Guillemin V., McPherson P.S., Mandavilli B.S., et al. Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol. Cell Biol. 2004;24:8195–8209. doi: 10.1128/MCB.24.18.8195-8209.2004. doi:10.1128/MCB.24.18.8195-8209.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chang D.T., Rintoul G.L., Pandipati S., Reynolds I.J. Mutant huntingtin aggregates impair mitochondrial movement and trafficking in cortical neurons. Neurobiol. Dis. 2006;22:388–400. doi: 10.1016/j.nbd.2005.12.007. doi:10.1016/j.nbd.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 36.Kim J., Moody J.P., Edgerly C.K., Bordiuk O.L., Cormier K., Smith K., Beal M.F., Ferrante R.J. Mitochondrial loss, dysfunction and altered dynamics in Huntington's disease. Hum. Mol. Genet. 2010;15:3919–3935. doi: 10.1093/hmg/ddq306. doi:10.1093/hmg/ddq306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Squitieri F., Falleni A., Cannella M., Orobello S., Fulceri F., Lenzi P., Fornai F. Abnormal morphology of peripheral cell tissues from patients with Huntington disease. J. Neural. Transm. 2010;117:77–83. doi: 10.1007/s00702-009-0328-4. doi:10.1007/s00702-009-0328-4. [DOI] [PubMed] [Google Scholar]
- 38.Costa V., Giacomello M., Hudec R., Lopreiato R., Ermak G., Lim D., Malorni W., Davies K.J., Carafoli E., Scorrano L. Mitochondrial fission and cristae disruption increase the response of cell models of Huntington's disease to apoptotic stimuli. EMBO Mol. Med. 2010;2:487–489. doi: 10.1002/emmm.201000102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang H., Lim P.J., Karbowski M., Monteiro M.J. Effects of overexpression of huntingtin proteins on mitochondrial integrity. Hum. Mol. Genet. 2009;18:737–752. doi: 10.1093/hmg/ddn404. doi:10.1093/hmg/ddn404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reddy P.H., Beal M.F. Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer's disease. Trends Mol. Med. 2008;14:45–53. doi: 10.1016/j.molmed.2007.12.002. doi:10.1016/j.molmed.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reddy P.H., Reddy T.P., Manczak M., Calkins M.J., Shirendeb U., Mao P. Dynamin-relate protein 1 and mitochondrial fragmentation in neurodegenerative diseases. Brain Res. Rev. 2010 doi: 10.1016/j.brainresrev.2010.11.004. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reddy P.H. Amyloid beta, mitochondrial structural and functional dynamics in Alzheimer's disease. Exp. Neurol. 2009;218:286–292. doi: 10.1016/j.expneurol.2009.03.042. doi:10.1016/j.expneurol.2009.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reddy P.H. Mitochondrial dysfunction in aging and Alzheimer's disease: strategies to protect neurons. Antioxid. Redox Signal. 2007;9:1647–1658. doi: 10.1089/ars.2007.1754. doi:10.1089/ars.2007.1754. [DOI] [PubMed] [Google Scholar]
- 44.Wang X., Su B., Lee H.G., Li X., Perry G., Smith M.A., Zhu X. Impaired balance of mitochondrial fission and fusion in Alzheimer's disease. J. Neurosci. 2009;29:9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009. doi:10.1523/JNEUROSCI.1357-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang X., Su B., Siedlak S.L., Moreira P.I., Fujioka H., Wang Y., Casadesus G., Zhu X. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc. Natl Acad. Sci. USA. 2008;105:19318–19323. doi: 10.1073/pnas.0804871105. doi:10.1073/pnas.0804871105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Manczak M., Mao P., Calkins M.J., Cornea A., Reddy A.P., Murphy M.P., Szeto H.H., Park B., Reddy P.H. Mitochondria-targeted antioxidants protect against amyloid-beta toxicity in Alzheimer's disease neurons. J. Alzheimers Dis. 2010;20(Suppl. 2):S609–S631. doi: 10.3233/JAD-2010-100564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Poole A.C., Thomas R.E., Andrews L.A., McBride H.M., Whitworth A.J., Pallanck L.J. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc. Natl Acad. Sci. USA. 2008;105:1638–1643. doi: 10.1073/pnas.0709336105. doi:10.1073/pnas.0709336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deng H., Dodson M.W., Huang H., Guo M. The Parkinson's disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc. Natl Acad. Sci. USA. 2008;105:14503–14508. doi: 10.1073/pnas.0803998105. doi:10.1073/pnas.0803998105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dagda R.K., Cherra S.J., III, Kulich S.M., Tandon A., Park D., Chu C.T. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J. Biol. Chem. 2009;284:13843–13855. doi: 10.1074/jbc.M808515200. doi:10.1074/jbc.M808515200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lutz A.K., Exner N., Fett M.E., Schlehe J.S., Kloos K., Lämmermann K., Brunner B., Kurz-Drexler A., Vogel F., Reichert A.S., et al. Loss of parkin or PINK1 function increases Drp1-dependent mitochondrial fragmentation. J. Biol. Chem. 2010;284:22938–22951. doi: 10.1074/jbc.M109.035774. doi:10.1074/jbc.M109.035774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Du H., Guo L., Fang F., Chen D., Sosunov A.A., McKhann G.M., Yan Y., Wang C., Zhang H., Molkentin J.D., et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nat. Med. 2008;14:1097–1105. doi: 10.1038/nm.1868. doi:10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kayed R., Head E., Thompson J.L., McIntire T.M., Milton S.C., Cotman C.W., Glabe C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;56:486–489. doi: 10.1126/science.1079469. doi:10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 53.Kostka M., Högen T., Danzer K.M., Levin J., Habeck M., Wirth A., Wagner R., Glabe C.G., Finger S., Heinzelmann U., et al. Single particle characterization of iron-induced pore-forming alpha-synuclein oligomers. J. Biol. Chem. 2008;283:10992–10003. doi: 10.1074/jbc.M709634200. doi:10.1074/jbc.M709634200. [DOI] [PubMed] [Google Scholar]
- 54.Kirkwood S.C., Su J.L., Conneally P., Foroud T. Progression of symptoms in the early and middle stages of Huntington disease. Arch. Neurol. 2001;58:273–278. doi: 10.1001/archneur.58.2.273. doi:10.1001/archneur.58.2.273. [DOI] [PubMed] [Google Scholar]
- 55.Mahant N., McCusker E.A., Byth K., Graham S. Huntington Study Group. Huntington's disease: clinical correlates of disability and progression. Neurology. 2003;61:1085–1092. doi: 10.1212/01.wnl.0000086373.32347.16. [DOI] [PubMed] [Google Scholar]
- 56.Hamilton J.M., Wolfson T., Peavy G.M., Jacobson M.W., Corey-Bloom J. Huntington Study Group. Rate and correlates of weight change in Huntington's disease. J. Neurol. Neurosurg. Psychiatry. 2004;75:209–212. doi: 10.1136/jnnp.2003.017822. doi:10.1136/jnnp.2003.017822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Phan J., Hickey M.A., Zhang P., Chesselet M.F., Reue K. Adipose tissue dysfunction tracks disease progression in two Huntington's disease mouse models. Hum. Mol. Genet. 2009;18:1006–1016. doi: 10.1093/hmg/ddn428. doi:10.1093/hmg/ddn428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aziz N.A., van der Burg J.M., Landwehrmeyer G.B., Brundin P., Stijnen T. EHDI Study GroupRoos, R.A. Weight loss in Huntington disease increases with higher CAG repeat number. Neurology. 2008;71:1506–1513. doi: 10.1212/01.wnl.0000334276.09729.0e. doi:10.1212/01.wnl.0000334276.09729.0e. [DOI] [PubMed] [Google Scholar]
- 59.Montoya A., Price B.H., Menear M., Lepage M. Brain imaging and cognitive dysfunctions in Huntington's disease. J. Psychiatry Neurosci. 2006;31:21–29. [PMC free article] [PubMed] [Google Scholar]
- 60.Bamford K.A., Caine E.D., Kido D.K., Plassche W.M., Shoulson I. Clinical–pathologic correlation in Huntington's disease: a neuropsychological and computed tomography study. Neurology. 1989;39:796–801. doi: 10.1212/wnl.39.6.796. [DOI] [PubMed] [Google Scholar]
- 61.Aylward E.H., Anderson N.B., Bylsma F.W., Wagster M.V., Barta P.E., Sherr M., Feeney J., Davis A., Rosenblatt A., Pearlson G.D., Ross C.A. Frontal lobe volume in patients with Huntington's disease. Neurology. 1998;50:252–258. doi: 10.1212/wnl.50.1.252. [DOI] [PubMed] [Google Scholar]
- 62.Dierks T., Linden D.E., Hertel A., Günther T., Lanfermann H., Niesen A., Frölich L., Zanella F.E., Hör G., Goebel R., Maurer K. Multimodal imaging of residual function and compensatory resource allocation in cortical atrophy: a case study of parietal lobe function in a patient with Huntington's disease. Psychiatry Res. 1999;90:67–75. [PubMed] [Google Scholar]
- 63.Vonsattel J.P., Myers R.H., Stevens T.J., Ferrante R.J., Bird E.D., Richardson E.P., Jr. Neuropathological classification of Huntington's disease. J. Neuropathol. Exp. Neurol. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. doi:10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- 64.Browne S.E., Ferrante R.J., Beal M.F. Oxidative stress in Huntington's disease. Brain Pathol. 1999;9:147–163. doi: 10.1111/j.1750-3639.1999.tb00216.x. doi:10.1111/j.1750-3639.1999.tb00216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Turner C., Schapira A.H. Mitochondrial matters of the brain: the role in Huntington's disease. J. Bioenerg. Biomembr. 2010;42:193–198. doi: 10.1007/s10863-010-9290-y. doi:10.1007/s10863-010-9290-y. [DOI] [PubMed] [Google Scholar]
- 66.Gutala R.V., Reddy P.H. The use of real-time PCR analysis in a gene expression study of Alzheimer's disease post-mortem brains. J. Neurosci. Methods, 2004;132:101–107. doi: 10.1016/j.jneumeth.2003.09.005. doi:10.1016/j.jneumeth.2003.09.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.