

Abstract
BACKGROUND & AIMS
Non-alcoholic fatty liver disease affects up to 30% of the U.S. population, but the mechanisms underlying this condition are incompletely understood. We investigated how diet standardization and choline deficiency influence the composition of the microbial community in the human gastrointestinal (GI) tract and the development of fatty liver under conditions of choline deficiency.
METHODS
We performed a 2-month in-patient study of 15 female subjects who were placed on well-controlled diets in which choline levels were manipulated. We used 454-FLX pyrosequencing of 16S rRNA bacterial genes to characterize microbiota in stool samples collected over the course of the study.
RESULTS
The compositions of the GI microbial communities changed with choline levels of diets; each individual’s microbiome remained distinct for the duration of the experiment, even though all subjects were fed identical diets. Variations between subjects in levels of Gammaproteobacteria and Erysipelotrichi were directly associated with changes in liver fat in each subject during choline depletion. Levels of these bacteria, change in amount of liver fat, and a single nucleotide polymorphism that affects choline were combined into a model that accurately predicted the degree to which subjects developed fatty liver on a choline-deficient diet.
CONCLUSIONS
Host factors and GI bacteria each respond to dietary choline deficiency, although the gut microbiota remains distinct in each individual. We identified bacterial biomarkers of fatty liver that results from choline deficiency, adding to the accumulating evidence that GI microbes have a role in metabolic disorders.
Keywords: Phosphatidylcholine, Bacteria, Choline metabolism, Hepatic steatosis
Intestinal microbes utilize nutrients and produce metabolites that influence a wide range of human phenotypes, including susceptibility to conditions such as obesity,1-2 insulin resistance,3-4 metabolic syndrome,3-4 liver steatosis,3-4 Crohn’s disease5 and cancer.6-7 Recent characterizations of the human gut microbiota have repeatedly observed that microbial communities are distinct, even among closely related individuals.8 Nevertheless, the specific interactions between gut microbes, the environment and the human host that select for different microbial communities in different people are largely unknown.
Many studies have reported direct links between diet and the structure of the gut microbiome in mouse models. One recent example observed that microbiome structure rapidly shifts in response to a change from a low-fat, plant-based diet to a high-sugar, high-fat diet, modifying both the available metabolic pathways and actual gene expression.9 This study and others like it have demonstrated that symbiotic microbes are key for access to and use of nutrients and energy from dietary sources1, 9-12 and have identified relationships between specific gut bacteria and host metabolism.9, 12 These studies have demonstrated the relationship between dietary exposure and microbial response, but extending these results from mouse models to humans is complicated by differences in gut structure and microbiome composition.9
A principal challenge in determining associations between the human gut microbiome and health is the difficulty of standardizing the diet during sampling.12 Our study overcomes this limitation by taking advantage of ongoing research that explores the effects of choline depletion in human subjects under a rigorously standardized diet in a hospital setting. Choline is an essential nutrient and a major methyl donor that supports physiological processes from normal metabolism to neurological development in the fetus.13 Human beings obtain choline from two major sources: diet and endogenous production in the liver. The level of choline in the American diet varies substantially, with one study reporting a median intake of 284 mg/day compared with the recommended daily intake of 550 mg/day.14 Low-choline diets have been associated with health problems in humans and in mouse models, including non-alcoholic fatty liver disease, neural tube defects, hepatic cancer and an increased risk of breast cancer.15 Furthermore, common single nucleotide polymorphisms (SNPs) in several genes have been shown to affect choline production and metabolism.16-17 One notable example is the gene, PEMT, which is important in the endogenous de novo synthesis of phosphatidylcholine. A common haplotype associated with a defective estrogen response element in PEMT’s promoter region disrupts this critical process.16-17
Gut bacteria can hydrolyze choline to form dimethylamine and trimethylamine.18-20 Phosphatidylcholine has been identified as a component of bacterial cell walls in approximately 10% of bacteria.21 To obtain phosphatidylcholine, prokaryotes utilize two pathways, the PMT pathway for endogenous biosynthesis and the PCS pathway that utilizes exogenous choline to produce phosphatidylcholine.22 Different bacterial types may have none, one or both pathways, and pathway specificity is important to microbe-host interactions.23
Our recent metabolomic analysis of the effects of choline deficiency on human beings has identified metabolites, some of bacterial origin, that differentiated subjects who experienced organ dysfunction associated with choline deficiency from those who did not.24 Other studies have used mouse models to determine bacterial influences on host metabolism and biochemistry,3-4, 25-29 and some have suggested that gut bacteria affect the bioavailability of dietary choline to the host.3, 29-30 One recent study used vancomycin to chemically knock-out gut microbes in mice and observed a variety of metabolic changes, including increased choline in feces.31. Another study reported on the effects from metabolic syndrome in a mouse model. These were ameliorated when antibiotics were administered to remove the gut microbiome.4 Taken together, these studies suggest that the gut microbiome influences an organism’s need for choline, as well as the health outcomes associated with choline deficiency. Our research seeks to further characterize this relationship.
Materials and Methods
Study Subjects
Healthy female subjects (n=15), a subset of those enrolled in an NIH-funded study (DK055895) investigating choline metabolism, were recruited to participate in a gut metagenomic study and provided informed consent (approved by the Institutional Review Boards at the University of North Carolina at Chapel Hill [UNC] and at the University of North Carolina at Charlotte [UNCC]). Inclusion was contingent on a good state of health, a body mass index (BMI) of 18–34, and no history of hepatic, renal, or other chronic system disease determined by physical examination and standard clinical laboratory tests. Individuals eating unusual diets that would interfere with the study, using drugs or medications known to alter liver metabolism or using choline-containing dietary supplements during the previous 3 months were excluded. Subjects were genotyped for the PEMT promoter SNP rs12325817 (supplemental table 1).
Dietary manipulation
After admission to the Clinical and Translational Research Center (CTRC) at UNC Hospitals, subjects were continuously supervised to assure protocol compliance. Participants were fed study diets, prepared in-house to protocol specifications.32 Total food intake was adjusted to be isocaloric and to provide adequate intakes of macro- and micronutrients. (supplemental table 2)
During the baseline study phase (Figure 1), all participants were fed a conventional diet of normal foods containing 550 mg choline · 70 kg body weight · day [the current adequate intake (AI) 33], 50 mg betaine · 70 kg body weight · day. After 10 days of this initial diet, subjects entered the choline-depletion phase during which they were fed a low-choline diet containing <50 mg choline · 70 kg body weight · day and 6 mg betaine · 70 kg body weight · day, as confirmed by chemical analysis of a sample of duplicate food portions.34-35 Periodic measurements of urinary choline and betaine concentrations35 were made to confirm dietary compliance. Subjects consumed the depletion diet until they developed organ dysfunction associated with choline deficiency or for 42 days if they did not develop dysfunction. Subjects were deemed to have organ dysfunction if they had a more than 5-fold increase in serum creatine phosphokinase (CPK) activity 36; a more than 1.5-fold increase in aspartate aminotransferase (AST) or alanine aminotransferase (ALT); or an increase in liver fat content of >28% during the choline-depletion diet and if these elevated measures resolved when dietary choline was restored.
Figure 1. Experimental Design.

Participants were fed a controlled research diet that included adequate daily choline intake during the baseline period (red). During depletion, subjects were fed a diet very low in choline until they demonstrated signs of deficiency or for a maximum of 42 days (grey). The 10-day repletion diet was very rich in choline (green). Arrows indicate timing of stool samples.
During the depletion phase, if functional markers indicated choline deficiency associated organ dysfunction, subjects were transitioned to a high choline repletion diet, containing 850 mg choline · 70 kg body weight · day for 10 days. Subjects who did not manifest signs of organ dysfunction after 42 days of the low-choline diet were likewise advanced to the choline repletion diet for 10 days.
Clinical assessment
Blood and urine samples were taken and laboratory tests were performed on each subject at screening, on day 1, and at the end of each dietary phase, as well as every 3–4 days for the duration of the study to monitor the depletion and repletion status. These laboratory analyses (conducted at the McLendon Clinical Laboratory at UNC Hospitals; Clinical Laboratory Improvement Act and College of American Pathologists accredited) included measurements of AST, ALT, and CPK.
Fatty liver
Liver fat was measured by magnetic resonance imaging (MRI) at the beginning and end of the baseline diet, after 21 and 42 days of the low-choline diet, and at the end of the repletion period. Liver fat content was estimated by MRI with a Vision 41.5T clinicalMRsystem (Siemens, Malvern, PA) by using a modified “In and Out of Phase” procedure.37-38 This approach used the differences in transverse magnetization intensity after an ultrabrief time interval (FLASH; echo time _2.2 and 4.5 ms, flip angle_80 °, and repetition time_140 ms). Processing of successive FLASH MRI images with software from Siemens Medical Solutions (Malvern, PA) was used to estimate fat content. Quantification of organ fat content was based on measurements across 5 images per subject and standardized using similarly measured images of spleen fat content to create a liver fat to spleen fat ratio (LF:SF), based on the assumption that spleen fat remained constant and could be used to normalize liver fat results (supplemental table 3). A 28% increase in liver fat from B1 to D2 time points was the predetermined threshold for indicating organ dysfunction secondary to choline deficiency. Liver fat measurements were subject to being mistimed by a couple of days because of MRI unavailability.
Dietary Compliance
Our diets were well tolerated by subjects in this study. If subjects did not tolerate the diet, their participation in the study was terminated. No side effects were observed other than those associated with the removal of choline (hepatic dysfunction and muscle damage). Symptoms typically resolved within days when dietary choline was re-introduced. All subjects who completed our study were unaware of the diet treatment they received; none felt sick or abnormal. No study-related physical illness occurred in any subject, nor were there any serious adverse events.
Sample collection and preparation
Time-series stool samples were collected as indicated (Figure 1) based on the instructions provided to study personnel and study subjects. Samples were frozen at −80°C and shipped on dry ice to The University of North Carolina at Charlotte where they were stored at −80°C. Samples were thawed on ice and metagenomic DNA was extracted using QIAamp DNA Stool Mini-kit. Extracted DNA was quantified using a NanoDrop ND-1000 spectrophotometer and frozen at −20°C to await further preparation.
ARISA sample preparation and analysis
Automatic Ribosomal Intergenic Sequence Analysis39 (ARISA) was used to detect microbial signatures in our samples. Using extracted DNA, the intergenic region between the 16S rRNA and the 23S rRNA genes was PCR amplified for each of two technical replicates using universal bacterial primers. (Supplemental Methods) Samples were run on an Applied Biosystems 3130 genetic analyzer and resulting spectra were analyzed and data vectors from all samples were compared using hierarchical clustering with custom JAVA code.
Sequencing sample preparation and sequence data management
Extracted DNA was thawed and used to PCR amplify the V1-V2 hypervariable regions of the 16S rRNA gene. The PCR products for 454 tagged sequencing were prepared with primers and thermalcycling parameters described in Fierer et al40 (Supplemental Methods). PCR products were sent to the Environmental Genomics Core Facility at the University of South Carolina for 454-FLX pyrosequencing. The resulting ~213,000 sequences were subjected to quality control standards (see supplemental methods) and sequences that did not meet quality criteria were removed from the dataset (supplemental table 4).
The remaining 194,781 sequences were assigned to Operational Taxonomic Units (OTUs) at ≥ 97% sequence similarity using the RDP-II infernal aligner and complete linkage clustering from the RDP web-based pipeline.41 Sequences were assigned taxonomy using RDP classification software.42 A minimum 50% threshold confidence score 43 was used to include sequences in each taxonomic group. Sequence counts were analyzed using logged proportion abundance, standardized to the average sample size across all samples (Supplemental Methods).
Statistical Analysis
General descriptive statistics used all available samples, 74 samples across 15 subjects. For statistical analysis, two subjects with an incomplete sample set (29, 04) were left out of any statistical test where a missing sample was required for the analysis.
Sequence counts were standardized to the average sample size across all samples and the resulting sequence frequencies were logged (adding 1 to each sequence to avoid logging zeros). For the calculation and an example, see Supplemental Methods.
Hierarchical clustering of OTUs at 97% and of ARISA binned signal intensities were conducted using Ward’s method on standardized logged sequence proportions. Statistical analyses on RDP classifications were conducted at the most inclusive taxonomy for which results were identified (at the class level) to ensure that the highest possible numbers of sequences were used in comparisons (Supplemental Methods). All p-values were corrected for multiple comparisons using an adjusted p-value (Supplemental Methods).
In Figure 5C, we used Principal Components Analysis (PCA) to build a simple linear model which related gut metagenomic sequencing and LF:SF ratio changes. We conducted the PCA on the two taxa at the class level (Gammaproteobacteria and Erysipelotrichi) with the highest R2 values when regressed against LF:SF ratio change (supplemental Table 5). We used the first component of the PCA as a regressor against the % change in LF:SF ratio from B1 to D2 (Fig. 5C). In our final model (Figure 5D), we included the subject genotype for the PEMT SNP with B1 abundance levels for Gammaproteobacteria and Erysipelotrichi to conduct the PCA. The wild-type genotype value was set to 1 and, for either heterozygous or homozygous genotype, the value was set to 2. Because such models are subject to over-fitting, we performed permutation procedures that produced a permuted p-value to assess the validity of our results (Supplemental Methods).
Figure 5. Gammaproteobacteria and Erysipelotrichi abundance, particularly when combined with subject geneotype, predicts choline deficiency induced fatty liver.




Regressions of A. Gammaproteobacteria B1 abundance and B. Erysipelotrichi B1 abundance against the Liver fat/Spleen fat (LF:SF) % change from baseline (B1) to choline deficient (D2) diet. C. Regression of PCA1 from PCA of Gammaproteobacteria and Erysipelotrichi B1 abundance against the LF:SF % change from B1 to D2. D. Regression of PCA1 from PCA of PEMT genotype for rs12325817, Gammaproteobacteria and Erysipelotrichi B1 abundance (6B) against the LF:SF % change from B1 to D2.
Statistical analyses were conducted using the R statistical package (R version 2.7.2 (2008-08-25)), JMP® 8.0 software for Microsoft Windows (SAS Institute) and Microsoft® Office Excel 2003.
Results
Experimental Design
Healthy adult female subjects (n=15; supplemental table 1) were brought into the hospital and fed a proscribed experimental diet that included three dietary phases: 1) a standard research diet providing the current recommended level of choline, 2) a diet with very low choline and 3) a diet that included significant levels of choline to restore subjects’ choline levels (Figure 1). Stool samples were obtained at time points reflecting dietary changes (Figure 1), and the gut microbiome was characterized from these samples. Although we recognize that the composition of mucosal-adherent microbes could differ from stool sample results,44 invasive procedures required to sample that component were not advisable for this study. By sampling under controlled conditions as choline levels were manipulated, we observed the effects of dietary challenge on microbial community stability, identifying changes in bacterial taxa that were coincident with controlled dietary manipulations and disease states in human beings.
Distinctly individual gut microbial communities
Accumulating evidence has documented the diversity of gut microbial community composition between individuals, even those who are closely related.8 If dietary differences primarily defined gut microbial community structure, placing subjects on common diets would erase some of those differences. To test the prediction that a common diet leads to a homogenous microbial community, we conducted hierarchical clustering on Operational Taxonomic Units (OTUs), groups of sequences identified as having at least 97% sequence similarity. Our samples clustered perfectly by subject (P = 6.78E-65; Supplemental Methods) regardless of sampling time point (Figure 2A). A profile of each subject’s microbiome obtained from ARISA, a DNA fingerprinting technology that does not involve direct sequencing (supplemental figure 1), also found nearly perfect clusters by subject (Figure 2B). The highly similar clustering patterns observed from these two distinct methods ensures that our results are not an artifact caused by sequencing errors that may be associated with pyrosequencing methodology.45 These observations demonstrate that differences between individuals were maintained despite subjects being fed a common diet and that the established adult gut microbiome does not undergo wholesale, common change in response to short-term, common dietary perturbations.
Figure 2. Hierarchical clustering of gut microbiome samples.
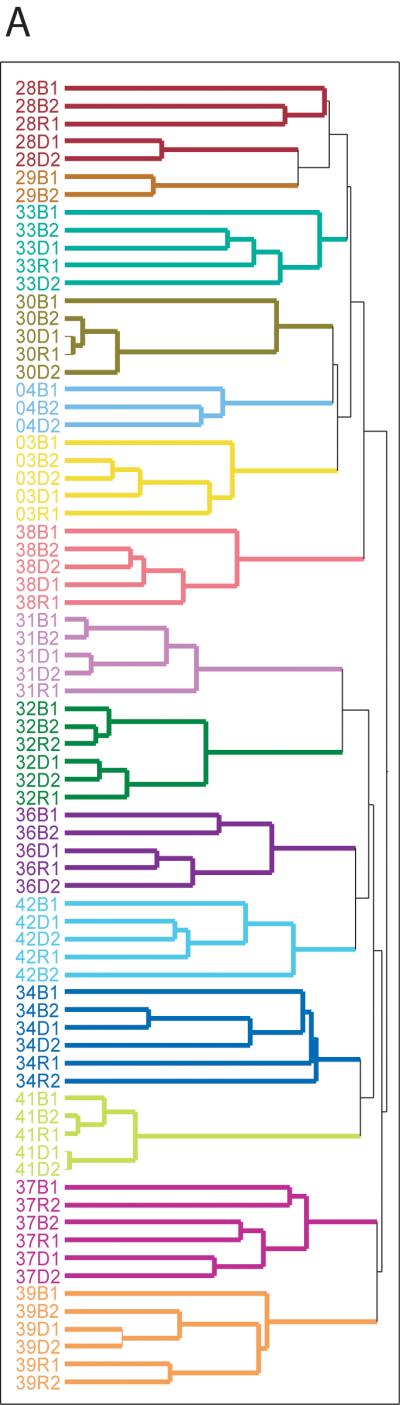
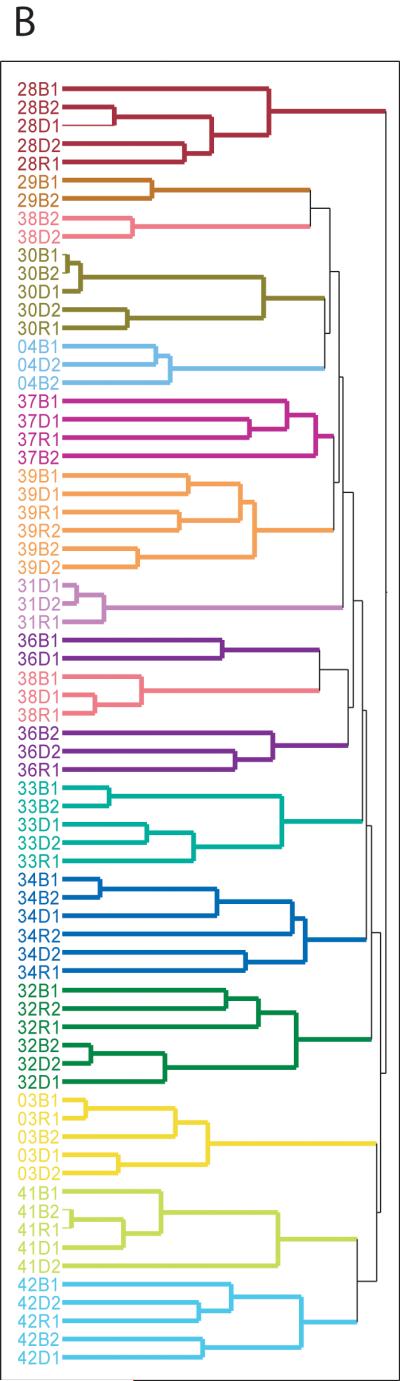
A. Hierarchical clustering based on OTUs at 97% sequence similarity. Samples are colored by subject. B. Hierarchical clustering based on ARISA profiles, a DNA fingerprinting technique.
Effects of diet on the human gut microbiome
Although our subjects’ microbiota remained distinct, we observed shifts in microbial community composition within subjects as their diets were altered. When bacterial proportions were plotted for each sampling time point in each subject, changes were visually apparent (Figure 3). Bacterial classes with the highest abundance, such as Bacteroidia and Clostridia, were ubiquitous, whereas less abundant classes, such as Fusobacteria and Bacilli, were present in only a few subjects.
Figure 3. Diagrams of bacterial sequence proportions by time point at the Class level.


The key shows ordering of time points in the ring: B1-baseline, B2-standard diet, D1-choline-deficient diet, D2-end of choline-deficient diet, R1-high dietary choline and R2-second high dietary choline sample (subjects 34, 32, 37, 39). Blue subject labels designate those who developed fatty liver (liver fat change ≥ 28%) on a choline deficient diet; those with red labels did not. Subjects 4 and 29 do not have all samples, and time points are labeled.
We measured phylogenetic differences between dietary time points using Unifrac46 to assess the degree to which a phylogenetic tree for each sample differed in branch lengths from a subject’s “community” tree, constructed from all sequences from that subject. In six subjects, the p-values from these comparisons revealed that the microbial community present when subjects consumed an ad libitum diet at baseline was more phylogenetically unique than would be expected by chance based on a threshold of P ≤ 0.05 (supplemental table 6), indicating that switching from an ad libitum diet to a balanced, standardized research diet substantially altered gut microbiome composition in a subset of our subjects.
To quantify any common impact of dietary changes on frequencies of individual taxa, we compared differences in bacteria abundance between study time points that corresponded to changes in diet (Figure 1): the ad libitum diet (B1) vs. the controlled research diet (B2), the ad libitum diet (B1) vs. the choline deficient diet (D2), the ad libitum diet (B1) vs. the high choline repletion diet (R1) and the choline deficient diet (D2) vs. the high choline repletion diet (R1). Using paired t-tests, we found no common change in abundance of any taxon from one time point to another after correcting for multiple comparisons (supplemental table 7). These results indicate that, while subjects experienced changes to their microbial community on exposure to a new diet (Figure 3), there is little compelling evidence for a common pattern of change across subjects for individual types of bacteria. Nevertheless, test results before multiple comparison correction for Gammaproteobacteria (P = .006) and Betaproteobacteria (P = .008) from D2 to R1 indicate a potential impact either from restoring dietary choline (supplemental table 7) or possibly from removing the soy shake from the repletion diet (Supplemental Methods). Based on these results, we examined the differences in Gammaproteobacteria abundance between time points D2 and R in greater detail.
We plotted the distributions of Gammaproteobacteria for all time points (Figure 4). At D2, where subjects had been on an extended diet containing very low daily choline levels (50 mg), subjects exhibited a variety of Gammaproteobacteria abundance levels. When high dietary choline levels were restored to subjects’ diets at the R1 time point, abundance levels in all but two subjects dropped to zero. This suggests that Gammaproteobacteria may be inhibited by very high levels of dietary choline. Although evidence for such a direct effect of choline on Gammaproteobacteria is not unprecedented in the literature,47 further evidence will be necessary to confirm this observation at a threshold of statistical significance that survives correction for multiple tests.
Figure 4. Distribution of Gammaproteobacteria abundance by time point by subject.

Plot of logged standardized sequence frequencies (log10) for Gammaproteobacteria colored by subject.
Relationships among choline deficiency, fatty liver, gut microbes and host genotype
We next examined whether each taxon’s abundance levels at the B1 (baseline) time point, an “experiment-free” condition prior to dietary changes, could predict how subjects would respond to dietary choline insufficiency at D2 (end of the depletion phase). Abundance levels of two taxa, Gammaproteobacteria (Figure 5A) and Erysipelotrichi (Figure 5B), were correlated to the percentage change in the LF:SF ratio from B1 to D2. Gammaproteobacteria showed the strongest correlation, and the negative association with liver fat changes survived correction for multiple comparisons at a 5% false discovery rate (R2 = .5679, P = .00118, adjusted p-value .011) (supplemental table 5). These results suggest that a subject’s baseline levels (ad libitum diet) of this taxon predict the degree of subject susceptibility to fatty liver when dietary choline is deficient.
Although both Gammaproteobacteria and Erysipelotrichi abundance levels offered some predictive power for fatty liver development in our subjects, we wished to explore whether multivariate models could better explain subjects’ susceptibility. Principal Components Analysis (PCA) provides a simple method to combine Gammaproteobacteria and Erysipelotrichi B1 abundance levels as explanatory variables. A regression of the first principal component generated from this analysis against the percentage change in the LF:SF ratio from B1 to D2 improved predictive value for fatty liver susceptibility over either taxa alone (Figure 5C). To correct for the possibility of model over-fitting, we completed one million permutations of this analysis (Supplemental Methods), and the resulting permutation derived p-values supported the observed correlation.
To further refine our model, we borrowed insight from previous studies that demonstrated that subject genotype for the PEMT SNP influences susceptibility to choline deficiency induced fatty liver.16-17 Results from Welch’s t test (Figure 6A) confirmed that this SNP genotype would be an effective predictor of liver fat changes in our subjects (P = .0028). We combined the PEMT SNP genotype values with B1 abundance information from Gammaproteobacteria and Erysipelotrichi (Supplemental Methods) using PCA (Figure 6B). The first principal component from this analysis proved to be highly correlated with the change in LF:SF ratio (Figure 5D) for all our subjects (R2 = 0.8388, P = 2.4e-06). A permutation based p-value, designed to correct for over-fitting remained highly significant (P = 7.0e-04), demonstrating that these three factors together (Figure 5D), are a powerful predictor of the physiological effects of choline deficiency. We obtained similar results using multivariate regression (supplemental table 8).
Figure 6. The PEMT SNP affects risk of developing choline deficiency induced fatty liver.


A. Variability plot of PEMT genotype for SNP rs12325817 and the % change in Liver fat/Spleen fat ratio (LF:SF). WT, wildtype; HET, heterozygous; HO, homozygous. B. PCA of PEMT genotype for rs12325817, Gammaproteobacteria and Erysipelotrichi B1 abundance. Open circles indicate subjects who did not develop fatty liver; closed squares are subjects who did.
Discussion
Our study demonstrates the importance of longitudinal experimental design and rigorous dietary control to identify changes in the gut microbiome that have potentially significant ramifications for human nutrition and health. We have verified previous findings 8, 48 that gut microbial communities are distinctly individual and have demonstrated that there is little generalized convergence between subjects on a common diet over a two-month time period (Figure 2). Although gut microbiota remained characteristically individual, dietary changes did produce some effects, altering overall gut microbial composition when subjects shifted from their normal diets and changing the abundance of specific microbes when the essential nutrient, choline, was perturbed (Figure 3). The lack of statistically significant general microbial community convergence between subjects suggests that immigration of bacteria from food or the environment was not a major contributor to microbiome composition in our subjects.
Our results are presented in the context of non-alcoholic fatty liver disease estimated prevalence as high as 30% in the U.S. population49-50 and observed interrelationships between choline, high fat diets, fatty liver, insulin resistance, diabetes and other metabolic syndrome conditions. Previous literature suggests that changes associated with menopause in women, particularly when combined with relevant SNPs, may disrupt endogenous choline production, increase a subject’s need for dietary choline and predispose post-menopausal women to developing fatty liver.16-17, 51-52 If this model is correct, the subjects who deplete most on a choline deficient diet are those who are least efficient at endogenous choline production and are, therefore, the most dependent on obtaining needed choline from their normal diets. In Figure 5C, 3 subjects (33, 29 and 04) did not show the same strong correlation as the other subjects when the combination of Gammaproteobacteria and Erysipelotrichi abundance was plotted against LF:SF ratio changes. All 3 of these subjects, however, are WT for a SNP in the PEMT gene, which suggests that they are in the group of women better able to endogenously synthesize phosphatidylcholine.52 Combining the SNP status of PEMT with the abundance of the two taxa produces a correlation with essentially no outliers (Figure 5D). These data support a model in which subjects with the ability to endogenously produce phosphatidycholine are less dependent on the composition of the microbial community. While such a model will need further validation, the observation that Gammaproteobacteria abundance went to zero in all but two of our patients when dietary choline levels were high (Figure 4) lends further support to the assertion that members of this taxa are involved in choline-sensitive pathways that have implications for host health.
Animal models have also suggested a relationship between choline deficiency induced fatty liver and the gut microbiome. Dumas et al. described a microbiota-mediated mechanism underlying the development of fatty liver that mimicked choline deficiency in mice fed high fat diets and was also associated with insulin resistance.29 This mechanism was explained by microbial flora that disrupt choline bioavailability to the host by converting choline to methylamines, although no specific taxa were named. Increases in Proteobacteria, the phylum that includes Gammaproteobacteria, were also observed in mice that were fed high fat diets and that exhibited increased obesity.12 A study of metabolic endotoxemia, high-fat diets and obesity identified lipopolysaccharide (LPS), a phospholipid in the outer membrane of most gram-negative bacteria53, as a possible culprit in the chronic inflammation that accompanies metabolic dysfunction, insulin resistance and diabetes.28 Recent work by Kudo et al. implicated gut-derived bacterial endotoxin in up-regulation of TNF-alpha, apoptosis of primary hepatocytes and development of liver injury in a murine model of non-alcoholic steatohepatitis.54 Taken together, these studies provide support for the assertion that nutrient imbalance may trigger a bloom of inflammation-producing bacteria and concurrent metabolic dysfunction. The Gammaproteobacteria genera identified in our study, including Klebsiella spp., Enterobacter spp. and Eschericha spp., are known gram-negative bacteria with LPS-containing membranes.53 Their combined association with fatty liver development suggests that these mechanisms may be at work in our subjects, as well.
Even though we cannot yet assign cause and effect, our results suggest that host genotype and specific members of the microbial community are important predictors of susceptibility to choline deficiency induced fatty liver disease (Figure 5D). Defining the interrelationships between these bacteria, host genotype and choline metabolism could begin to establish the biological mechanisms through which the gut microbiome influences human health. Such work could ultimately yield important insights into the causes and risks associated with fatty liver disease and related, increasingly prevalent conditions, such as obesity, insulin resistance, diabetes and cardiovascular disease.
Supplementary Material
Acknowledgments
Grant support: NIH (DK055895 and DK056350) and Clinical and Translational Research Center (RR00046)
SZ and LF supervised the choline-depletion study and were responsible for all data and samples collected from human subjects.
TH performed DNA extraction, 16S-rRNA PCR and ARISA on stool samples.
RR analyzed the ARISA data.
MS and AF performed all other data analysis and drafted the manuscript.
All authors helped to edit the manuscript.
Abbreviations
- AI
recommended adequate intake value of a nutrient
- ARISA
Automated Ribosomal Intergenic Sequence Analysis
- 16S rRNA gene
ribosomal gene highly conserved between bacterial species
- BLAST
Basic Local Alignment Search Tool
- BMI
Body Mass Index
- CPK
creatine phosphokinase
- DRI
Dietary Reference Intake
- FDR
False Discovery Rate
- LF:SF
liver fat to spleen fat ratio
- LPS
lipopolysaccharide
- MRI
magnetic resonance imaging
- OTU
operational taxonomic unit
- PCA
Principal Components Analysis
- PCR
polymerase chain reaction
- RDP
Ribosomal Database Project
- SNP
single nucleotide polymorphism
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
All authors have no financial, professional or personal conflicts to disclose.
Sequence submissions: The original data file containing all pyrosequences has been submitted to the Short Read Archive at NCBI under accession number SRA012606.2 and is held pending publication. (http://www.ncbi.nlm.nih.gov/Traces/sra_sub/sub.cgi?&m=submissions&s=defaults)
References
- 1.Turnbaugh PJ, Ley RE, Mahowald MA, et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–31. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 2.Backhed F, Manchester JK, Semenkovich CF, et al. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc Natl Acad Sci U S A. 2007;104:979–84. doi: 10.1073/pnas.0605374104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Toye AA, Dumas ME, Blancher C, et al. Subtle metabolic and liver gene transcriptional changes underlie diet-induced fatty liver susceptibility in insulin-resistant mice. Diabetologia. 2007;50:1867–79. doi: 10.1007/s00125-007-0738-5. [DOI] [PubMed] [Google Scholar]
- 4.Vijay-Kumar M, Aitken JD, Carvalho FA, et al. Metabolic Syndrome and Altered Gut Microbiota in Mice Lacking Toll-Like Receptor 5. Science. 2010 doi: 10.1126/science.1179721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jansson J, Willing B, Lucio M, et al. Metabolomics reveals metabolic biomarkers of Crohn’s disease. PLoS One. 2009;4:e6386. doi: 10.1371/journal.pone.0006386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fox JG, Feng Y, Theve EJ, et al. Gut microbes define liver cancer risk in mice exposed to chemical and viral transgenic hepatocarcinogens. Gut. 2010;59:88–97. doi: 10.1136/gut.2009.183749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davis CD, Milner JA. Gastrointestinal microflora, food components and colon cancer prevention. J Nutr Biochem. 2009;20:743–52. doi: 10.1016/j.jnutbio.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turnbaugh PJ, Hamady M, Yatsunenko T, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–4. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Turnbaugh PJ, Ridaura VK, Faith JJ, et al. The Effect of Diet on the Human Gut Microbiome: A Metagenomic Analysis in Humanized Gnotobiotic Mice. Science Translational Medicine. 2009;1:6ra14–6ra14. doi: 10.1126/scitranslmed.3000322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turnbaugh PJ, Gordon JI. The core gut microbiome, energy balance and obesity. J Physiol. 2009;587:4153–8. doi: 10.1113/jphysiol.2009.174136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ley RE. Obesity and the human microbiome. Curr Opin Gastroenterol. 2010;26:5–11. doi: 10.1097/MOG.0b013e328333d751. [DOI] [PubMed] [Google Scholar]
- 12.Hildebrandt MA, Hoffmann C, Sherrill-Mix SA, et al. High-fat diet determines the composition of the murine gut microbiome independently of obesity. Gastroenterology. 2009;137:1716–24. e1–2. doi: 10.1053/j.gastro.2009.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zeisel SH, da Costa KA. Choline: an essential nutrient for public health. Nutr Rev. 2009;67:615–23. doi: 10.1111/j.1753-4887.2009.00246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bidulescu A, Chambless LE, Siega-Riz AM, et al. Repeatability and measurement error in the assessment of choline and betaine dietary intake: the Atherosclerosis Risk in Communities (ARIC) study. Nutr J. 2009;8:14. doi: 10.1186/1475-2891-8-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu X, Gammon MD, Zeisel SH, et al. High intakes of choline and betaine reduce breast cancer mortality in a population-based study. FASEB J. 2009;23:4022–8. doi: 10.1096/fj.09-136507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zeisel SH. Genetic polymorphisms in methyl-group metabolism and epigenetics: lessons from humans and mouse models. Brain Res. 2008;1237:5–11. doi: 10.1016/j.brainres.2008.08.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Song J, da Costa KA, Fischer LM, et al. Polymorphism of the PEMT gene and susceptibility to nonalcoholic fatty liver disease (NAFLD) FASEB J. 2005;19:1266–71. doi: 10.1096/fj.04-3580com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zeisel SH, da Costa KA, Fox JG. Endogenous formation of dimethylamine. Biochem. J. 1985;232:403–408. doi: 10.1042/bj2320403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zeisel SH, daCosta KA, LaMont JT. Mono-, di- and trimethylamine in human gastric fluid: potential substrates for nitrosodimethylamine formation. Carcinogenesis. 1988;9:179–81. doi: 10.1093/carcin/9.1.179. [DOI] [PubMed] [Google Scholar]
- 20.Zeisel SH, daCosta KA, Youssef M, et al. Conversion of dietary choline to trimethylamine and dimethylamine in rats: dose-response relationship. J. Nutr. 1989;119:800–4. doi: 10.1093/jn/119.5.800. [DOI] [PubMed] [Google Scholar]
- 21.Sohlenkamp C, Lopez-Lara IM, Geiger O. Biosynthesis of phosphatidylcholine in bacteria. Prog Lipid Res. 2003;42:115–62. doi: 10.1016/s0163-7827(02)00050-4. [DOI] [PubMed] [Google Scholar]
- 22.Martinez-Morales F, Schobert M, Lopez-Lara IM, et al. Pathways for phosphatidylcholine biosynthesis in bacteria. Microbiology. 2003;149:3461–71. doi: 10.1099/mic.0.26522-0. [DOI] [PubMed] [Google Scholar]
- 23.Aktas M, Wessel M, Hacker S, et al. Phosphatidylcholine biosynthesis and its significance in bacteria interacting with eukaryotic cells. Eur J Cell Biol. 2010 doi: 10.1016/j.ejcb.2010.06.013. [DOI] [PubMed] [Google Scholar]
- 24.Sha W, da Costa KA, Fischer LM, et al. Metabolomic profiling can predict which humans will develop liver dysfunction when deprived of dietary choline. FASEB J. 2010 doi: 10.1096/fj.09-154054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Backhed F, Crawford PA. Coordinated regulation of the metabolome and lipidome at the host-microbial interface. Biochim Biophys Acta. 2009 doi: 10.1016/j.bbalip.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wikoff WR, Anfora AT, Liu J, et al. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc Natl Acad Sci U S A. 2009;106:3698–703. doi: 10.1073/pnas.0812874106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Claus SP, Tsang TM, Wang Y, et al. Systemic multicompartmental effects of the gut microbiome on mouse metabolic phenotypes. Mol Syst Biol. 2008;4:219. doi: 10.1038/msb.2008.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cani PD, Bibiloni R, Knauf C, et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57:1470–81. doi: 10.2337/db07-1403. [DOI] [PubMed] [Google Scholar]
- 29.Dumas ME, Barton RH, Toye A, et al. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc Natl Acad Sci U S A. 2006;103:12511–6. doi: 10.1073/pnas.0601056103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zeisel SH, daCosta KA, Youssef M, et al. Conversion of dietary choline to trimethylamine and dimethylamine in rats: dose-response relationship. J Nutr. 1989;119:800–4. doi: 10.1093/jn/119.5.800. [DOI] [PubMed] [Google Scholar]
- 31.Yap IK, Li JV, Saric J, et al. Metabonomic and microbiological analysis of the dynamic effect of vancomycin-induced gut microbiota modification in the mouse. J Proteome Res. 2008;7:3718–28. doi: 10.1021/pr700864x. [DOI] [PubMed] [Google Scholar]
- 32.Busby MG, Fischer L, da Costa KA, et al. Choline- and betaine-defined diets for use in clinical research and for the management of trimethylaminuria. J Am Diet Assoc. 2004;104:1836–45. doi: 10.1016/j.jada.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 33.Institute of Medicine NAPU, editor. Dietary intakes for folate, thiamin, riboflavin, niacin, vitamin B12, panthothenic acid, biotin, and choline. National Academy Press; Washington, DC: 1998. pp. 390–422. [PubMed] [Google Scholar]
- 34.Zeisel SH, Mar MH, Howe JC, et al. Concentrations of choline-containing compounds and betaine in common foods. J Nutr. 2003;133:1302–7. doi: 10.1093/jn/133.5.1302. [DOI] [PubMed] [Google Scholar]
- 35.Koc H, Mar MH, Ranasinghe A, et al. Quantitation of choline and its metabolites in tissues and foods by liquid chromatography/electrospray ionization-isotope dilution mass spectrometry. Anal Chem. 2002;74:4734–40. doi: 10.1021/ac025624x. [DOI] [PubMed] [Google Scholar]
- 36.da Costa KA, Badea M, Fischer LM, et al. Elevated serum creatine phosphokinase in choline-deficient humans: mechanistic studies in C2C12 mouse myoblasts. Am J Clin Nutr. 2004;80:163–70. doi: 10.1093/ajcn/80.1.163. [DOI] [PubMed] [Google Scholar]
- 37.da Costa KA, Gaffney CE, Fischer LM, et al. Choline deficiency in mice and humans is associated with increased plasma homocysteine concentration after a methionine load. Am J Clin Nutr. 2005;81:440–4. doi: 10.1093/ajcn.81.2.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fishbein MH, Gardner KG, Potter CJ, et al. Introduction of fast MR imaging in the assessment of hepatic steatosis. Magn Reson Imaging. 1997;15:287–93. doi: 10.1016/s0730-725x(96)00224-x. [DOI] [PubMed] [Google Scholar]
- 39.Fisher MM, Triplett EW. Automated approach for ribosomal intergenic spacer analysis of microbial diversity and its application to freshwater bacterial communities. Appl Environ Microbiol. 1999;65:4630–6. doi: 10.1128/aem.65.10.4630-4636.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fierer N, Hamady M, Lauber CL, et al. The influence of sex, handedness, and washing on the diversity of hand surface bacteria. Proc Natl Acad Sci U S A. 2008;105:17994–9. doi: 10.1073/pnas.0807920105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cole JR, Chai B, Farris RJ, et al. The ribosomal database project (RDP-II): introducing myRDP space and quality controlled public data. Nucl. Acids Res. 2007;35:D169–D172. doi: 10.1093/nar/gkl889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Q, Garrity GM, Tiedje JM, et al. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–7. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu Z, DeSantis TZ, Andersen GL, et al. Accurate taxonomy assignments from 16S rRNA sequences produced by highly parallel pyrosequencers. Nucleic Acids Res. 2008;36:e120. doi: 10.1093/nar/gkn491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eckburg PB, Bik EM, Bernstein CN, et al. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–8. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kunin V, Engelbrektson A, Ochman H, et al. Wrinkles in the rare biosphere: pyrosequencing errors can lead to artificial inflation of diversity estimates. Environ Microbiol. 2010;12:118–23. doi: 10.1111/j.1462-2920.2009.02051.x. [DOI] [PubMed] [Google Scholar]
- 46.Lozupone C, Knight R. UniFrac: a New Phylogenetic Method for Comparing Microbial Communities. Appl. Environ. Microbiol. 2005;71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Selan L, Palma S, Scoarughi GL, et al. Phosphorylcholine impairs susceptibility to biofilm formation of hydrogel contact lenses. Am J Ophthalmol. 2009;147:134–9. doi: 10.1016/j.ajo.2008.07.032. [DOI] [PubMed] [Google Scholar]
- 48.Costello EK, Lauber CL, Hamady M, et al. Bacterial community variation in human body habitats across space and time. Science. 2009;326:1694–7. doi: 10.1126/science.1177486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Browning JD, Szczepaniak LS, Dobbins R, et al. Prevalence of hepatic steatosis in an urban population in the United States: Impact of ethnicity. Hepatology. 2004;40:1387–1395. doi: 10.1002/hep.20466. [DOI] [PubMed] [Google Scholar]
- 50.Wieckowska A, Feldstein AE. Diagnosis of nonalcoholic fatty liver disease: invasive versus noninvasive. Semin Liver Dis. 2008;28:386–95. doi: 10.1055/s-0028-1091983. [DOI] [PubMed] [Google Scholar]
- 51.Zeisel SH. Gene response elements, genetic polymorphisms and epigenetics influence the human dietary requirement for choline. IUBMB Life. 2007;59:380–7. doi: 10.1080/15216540701468954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.da Costa KA, Kozyreva OG, Song J, et al. Common genetic polymorphisms affect the human requirement for the nutrient choline. FASEB J. 2006;20:1336–44. doi: 10.1096/fj.06-5734com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Raetz CR, Whitfield C. Lipopolysaccharide endotoxins. Annu Rev Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kudo H, Takahara T, Yata Y, et al. Lipopolysaccharide triggered TNF-alpha-induced hepatocyte apoptosis in a murine non-alcoholic steatohepatitis model. J Hepatol. 2009;51:168–75. doi: 10.1016/j.jhep.2009.02.032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.