

Abstract
Sphingosine kinases catalyze the transfer of phosphate from ATP to sphingosine to generate sphingosine 1-phosphate, an important bioactive lipid molecule that mediates a diverse range of cell signaling processes. The conventional assay of sphingosine kinase enzymatic activity uses [γ-32P]ATP and sphingosine as substrates with the radiolabeled S1P product recovered by organic extraction, displayed by thin-layer chromatography and quantified by liquid scintillation counting. While this assay is sensitive and accurate, it is slow and labor intensive and thus precludes the simultaneous screening of more than a few inhibitor compounds. Herein we describe a 96 well assay for sphingosine kinases that is rapid and reproducible. Our method, which takes advantage of the limited solubility of S1P, detects radioactive S1P adhering to the plate by scintillation proximity counting. Our procedure obviates extraction into organic solvents, post-reaction transfers and chromatography. Further, our assay enables assessment of both inhibitors and substrates, and can detect endogenous sphingosine kinase activity in cell and tissue extracts. The sphingosine kinase kinetic parameter, Km, and the Ki values of inhibitors determined with our assay and the conventional assay were indistinguishable. These results document that our assay is well suited for the screening of chemical libraries of sphingosine kinase inhibitors.
Keywords: sphingosine kinase, Sphingosine 1-phosphate, high through-put assay
INTRODUCTION
Sphingosine kinases (SphK1, SphK2) are phosphoryltransferases that catalyze the formation of sphingosine 1-phosphate (S1P) from ATP and D-erythro sphingosine. S1P, originally considered as an intermediate in the sphingolipid degradation pathway, is now known to be a signaling molecule. S1P has been linked to numerous cellular process including survival, cytoskeleton arrangement, cell motility, tumor invasion, angiogenesis, vascular maturation and trafficking of immune cells [1,2]. S1P exerts its effects after secretion into the extracellular milieu as a ligand for G-protein coupled receptors named S1P1–5, and possibly other intracellular targets.
Recognition of S1P’s role in cell survival led to the concept of a ‘sphingolipid rheostat’ (or biostat) that describes cellular mechanisms to balance the levels of S1P and its pro-apoptotic precursor, sphingosine and its precursor, ceramide [3,4]. The catalytic activity of the SphKs is central to the regulation of this balance, which has led to the suggestion that SphKs could be useful drug targets, particularly in the setting of neoplastic diseases. Such ideas have prompted a search for sphingosine kinase inhibitors.
Efficient discovery and characterization of SphK inhibitors has been hampered by the lack of a rapid assay for SphK activity. The standard assay for S1P utilizes [γ-32P]ATP and sphingosine as substrates and requires the isolation of radiolabeled S1P by extraction into acidified organic solvents and separation by TLC. Quantification is then carried out by liquid scintillation measurements of the scraped S1P bands. This method is time consuming because it requires many steps and thus limits the number of inhibitors that can be tested simultaneously. For example, in our laboratories, one individual can generate a maximum of 50 data points in 36 hours. Thus this assay is wholly inadequate to assess the synthetic output of a SphK inhibitor SAR (structure activity relationship) effort. We were prompted by these limitations to develop a higher throughput method for SphK activity measurements.
Several other SphK assays that obviate some of the problems associated with the standard method have been described. For example, HPLC-based methods can rapidly separate S1P from substrate but the method requires extraction into organic solvents and derivatization and therefore is ill-suited for high-throughput assays [5–7]. Vessey et al. used tritiated sphingosine as a substrate and a differential extraction technique to separate [3H]S1P from the substrate [8]. This method avoids the chromatography step but requires organic extraction. Roberts et al. described an assay system wherein biotinylated-sphingosine is used as a substrate and the [32P]S1P product is captured on streptavidin-coated membranes [9]. This approach avoids both the organic extraction and chromatography steps, but requires post-reaction manipulations, the use of individual scintillation vials and the expense of the biotinylated substrate. A number of authors have reported fluorescence-based methods for the measurement of SphK activity. These assays are much less time-consuming in terms of separation of product but require the use of unnatural substrates and are sometimes subjected to interference by the inhibitors being tested that are sometimes fluorescent themselves. For example, Billich et al. reported a fluorescence-based assay using NBD-labeled sphingosine as a substrate that requires post reaction liquid transfer and also organic extraction and the use of an expensive substrate [10]. Lee et al. developed a SphK assay using fluorescein-sphingosine relying on a mobility shift assay with laser induced fluorescein detection [11]. Vindugiriene et al. reported a SphK assay method based on detecting the formation of ADP using a bioluminescence approach that is an indirect method to determine the SphK activity and also based on detection of a fluorescent product [12]. Togame et al. reported a high throughput assay for SphK1 based on the absorption of S1P onto chemically modified plates (Aqua-Bind®, Asahi Techno Glass). This method does not require organic extraction or TLC separation, but it is not a single plate assay and requires multiple pipetting steps. In addition, kinetic parameters for substrates (e.g. ATP) and inhibitors (e.g. DMS) reported are inconsistent with published values [13].
We herein describe a simple and rapid SphK assay protocol that uses the natural substrate and requires no organic extraction, post-reaction liquid transfers or chromatographic separation. It takes advantage of the insolubility of S1P in aqueous medium and detects adherent [33P]S1P by scintillation proximity counting using 96-well plates embedded with scintillant (FlashPlates®, Perkin Elmer). The only special equipment required is a liquid scintillation counter capable of accepting standard 96 well plates. We document the utility of our method by documenting that Ki values obtained for a number of SphK inhibitors with this method are in close agreement with similar values obtained with the standard TLC-based method.
EXPERIMENTAL
Materials
D-erythro-sphingosine was purchased from Avanti Polar Lipids. The SphK inhibitors DMS, SKI II and SKI V were purchased from Sigma; [γ-32P]ATP, [γ-33P]ATP and FlashPlates were purchased from PerkinElmer. Compounds 9ab [20], 23, 28 [21] and 5c [22] were synthesized according to the published routes. In each case, the structures were confirmed by proton NMR and high resolution mass spectrometry. S1P, FTY720-phosphate and similar long chain amino phosphate compounds are routinely dissolved in DMSO : 1 M HCl (95:5, v/v) to 20 mM and then diluted 1: 20, with rapid mixing, into aqueous buffer containing 3% fatty acid free BSA” to yield a final concentration of 1 mM with respect to phospholipid.
Expression of recombinant sphingosine kinases
The expression vectors, pCMV6-XL4-SphK1 and pCMV6-XL6-SphK2 were obtained from Origene (Rockville, MD). SphK1 and SphK2 genes were amplified using the following primers: 5′-GACGACGACAAGATTTCCGCTCAAGTTCTGGGATTT and 5′-GAGGAGAAGCCCGGTTCAATGGTGATGGTGATGATG for SphK1: 5′-GACGACGACAAGATTAATGGACACCTTGAAGCAGAG and 5′-GAGGAGAAGCCCGGTTCAATGGTGATGGTGATGATG for SphK2. The respective cDNA fragments were purified by agarose gel electrophoresis and ligated into the pBacgus-2cp vector according to manufacturer’s protocol (Novagen). The resultant plasmids were analyzed by DNA sequencing to confirm that the SphK1 and SphK2 cDNAs were in the correct translational reading frame. Recombinant SphK1 and SphK2 baculovirus were prepared according to manufacturer’s protocol. Briefly, each expression vector (pBacgus-2cp-SphK1 or SphK2) was mixed with baculoviral genomic DNA into Sf9 cells and used to transfect 6 × 107 Sf9 cells. At 3–5 days post-infection, the medium was retrieved and used to infect another batch of Sf9 cells. This process was repeated until expression of SphK1 and SphK2 was detected by Western blotting. Cells were then collected, resuspended in SphK buffer and homogenized using a Dounce homogenizer. The homogenate was clarified by centrifugation at 21,000×g in a microcentrifuge, aliquoted, and stored at −80°C. In some cases, SphK1 and SphK2 were purified using Ni2+-agarose affinity chromatography. Protein was quantified using the BCA method (Bio-Rad).
SphK Assay
Assays were carried out in 96-well FlashPlates® (PerkinElmer) as previously reported [14] with slight modifications. Briefly, each reaction consisted of 0.2 ml of SphK buffer [14] supplemented with D-erythro-sphingosine, 250 μM [γ-33P]ATP (0.7 μCi per well, specific activity 7.2 Ci/mmol) and cell lysate (2 μg of protein). After incubation at 37°C for indicated times, plates were washed twice with phosphate-buffered saline and bound radionuclide measured using a microplate scintillation counter (TopCount, Packard).
Determination of Kinetic Parameters
For the determination of the Km for sphingosine, the concentration of ATP was fixed at 250 μM and sphingosine was varied from 0–50 μM. The Km for ATP was obtained in the presence of 5–10 μM sphingosine and ATP amounts ranging from 0–600 μM. Background activity, determined by the absence of enzyme or sphingosine substrate, was negligible (<100 cpm/well). SphK kinetic constants were obtained by measuring the initial velocity of catalysis (less than 5% of the substrate was consumed) and fitting the data to the Michaelis-Menten equation by non linear regression using GraphPad Prism 5.01 (GraphPad Software). For Ki determinations, a concentration of inhibitor that produces about 50% inhibition at sphingosine concentrations near the Km for sphingosine (i.e. 5–10 μM) was chosen. For each inhibitor, Ki was calculated using the following equation:
where [I] is the concentration of inhibitor; and K′m and Km are the Michaelis-Menten constants in presence and absence of inhibitor, respectively.
RESULTS AND DISCUSSION
Our goal was to develop a higher throughput sphingosine kinase assay useful for the analysis of multiple SphK inhibitors. A fundamental problem in designing any kinase assay is detecting the product without interference from either substrate. When considering lipid kinases, an obvious solution is to separate the phosphorylated lipid from radiolabeled ATP on the basis of their differential partitioning between aqueous and organic solvents, but this is a laborious and time consuming step that we wished to avoid. The widely used conventional TLC-based assay is limited to obtaining Ki values of 1–2 compounds per day, whereas a 96 well plated-based assay would allow us to generate Ki values of 10–12 compounds per day. To develop a plate-based assay, we took advantage of the extreme insolubility of S1P in aqueous media as a basis for the separation of S1P from the highly water soluble radiolabeled ATP. Unlike sphingosine, S1P is nearly insoluble in aqueous media probably because of its vicinal amino/phosphate groups, indeed, S1P is only sparingly soluble in any single solvent at room temperature; we routinely dissolve compounds such as S1P in acidified DMSO (see experimental). We found that S1P generated in plastic wells effectively precipitates onto the well walls and can be separated from radiolabeled ATP simply by washing the wells with aqueous medium. Use of multiwell plastic plates that contain scintillant embedded in the walls enable measurement of trapped S1P by scintillation spectrometry.
To test this assay we first established the minimum number of washes that were required to completely remove ATP from the wells. Using two washes, we found that wells containing SphK exhibited wash-resistant radioactivity as compared to control wells without SphK (i.e. lysates from untransfected cells). We interpreted this result as the presence of insoluble S1P precipitate. We then compared the amounts of S1P detected by the traditional, TLC-based method and our method. We found, as shown in Fig. 1, that our method yields about half the number of cpm as the TLC-based assay. We traced this difference to a lower efficiency of radioactivity measured by proximity scintillation. We found that the addition of scintillation fluid to the wells of the FlashPlate increased the cpm about 1.6 fold, to a level similar to that obtained using a TLC-based assay. Because we wanted to develop a high-throughput assay for the evaluation of SphK inhibitors, we omitted the addition of scintillation fluid thus avoiding a time-consuming step. Moreover, the diminished sensitivity is not relevant for the evaluation of competitive SphK inhibitors, which is based on the Km shift in presence of inhibitor.
Figure 1.
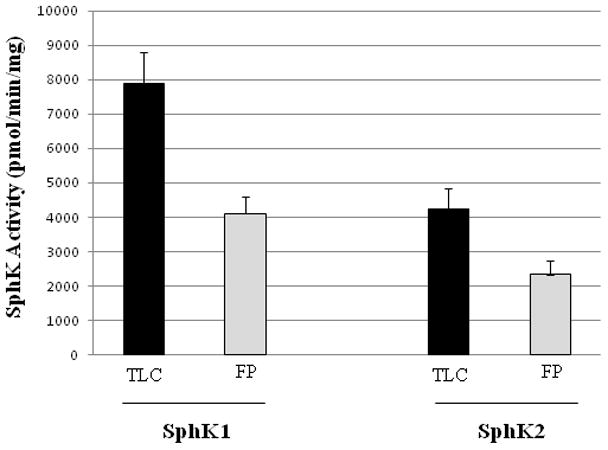
Comparison between the current method (“FlashPlate”) and the classical method (“TLC”) in terms of their sensitivities of S1P detection. The amounts of S1P produced in SphK1- and SphK2-catalyzed reactions were measured using both methods. Reactions were carried out for 20 min using 10 μM sphingosine and 250 μM [γ-33P]ATP as substrates. Values are means of triplicate measurements ± SD. Black and gray bars represent TLC- and FlashPlate-based assays, respectively.
Further evidence for the accuracy of our assay was obtained by measuring the kinetic parameters of SphK 1 and 2 for both sphingosine and ATP. As shown in Fig. 2, we obtained curves having the characteristic shape of enzyme-mediated reactions, i.e. rectangular hyperbolas. Moreover, no product was observed in the absence of sphingosine (< 100 cpm, not shown). Fitting the experimental points to the Michaelis-Menten equation by non-linear regression resulted in Km values that were in very close agreement with previously measured values. Specifically, the Km values with respect to sphingosine were 12 μM and 6 μM for SphK1 and SphK2, which is in good agreement with previously reported values of 5–17 μM for SphK1 [15,16] and 3–5 μM for SphK2 [17]. For ATP, our Km values were 125 μM and 79 μM for SphK1 and SphK2, which are also in the range of values published previously [16,17]. Additional evidence for an enzyme-mediated reaction, as shown in Fig. 3, is the linearity of the activity as a function of both time and amount of enzyme. These data also document that our method has a linear response up to about 30 nanomoles of S1P (Pane B, SphK 1, 4 μg point). We also evaluated the reproducibility of this method by calculating the coefficients of variation (σ/μ) for the Km and Vmax and found them to be 6.5 % and 3%, respectively (n= 5). Our assay yielded a high signal-to-background ratio, small variations and Z factor generally exceeding 0.67 (data not shown).
Figure 2.
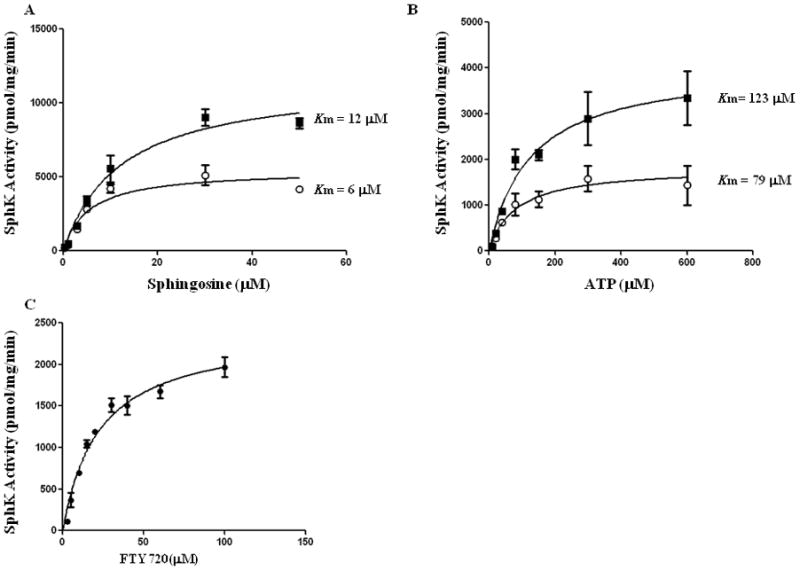
Kinetic analysis of SphK1 and SphK2 using the FlashPlate method. Activity as a function of sphingosine (panel A) or ATP (panel B) was measured with the FlashPlate method and the Michaelis-Menten constants (Km) were calculated for each substrate using non-linear regression. In both cases, purified SphK1 (filled squares) and SphK2 (open circles) were used. SphK2 activity was measured also as a function of selective SphK2 substrate, FTY720 (panel C). Experimental values are means ± SD of three independent measurements. Fitted values are shown as a continuous line. Calculated Km values are shown next to the corresponding plots.
Figure 3.

Time and amount of enzyme dependence of S1P production assessed with the FlashPlate method. Values are means of triplicate measurements ± SD for SphK1 (filled squares) and SphK2 (open circles). (A) Time dependence: assays were carried out in the presence of 10 μM of sphingosine, 250 μM of ATP and SphK1 or SphK2 (2 μg of cell lysate protein) for the indicated times. (B) Amount of enzyme dependence: reactions were carried out as described above for 30 min in the presence of varying amounts of cell lysates as indicated in the figure.
To confirm the identity of the radioactive product generated by our method, we eluted the FlashPlate wells with warm methanol (37°C, 10 min) and analyzed the extracted material by TLC and radioautography. We found that under these conditions only one band was detected by autoradiography and this material migrated with an Rf identical to that of authentic S1P. Ninety percent of the bound radioactive material was eluted from the well, and 75% of this was recovered from the TLC plate as a single band. Similarly, 69% of the bound FTY720-P was recovered from the FlashPlate. We attribute the S1P/FTY720-P unaccounted for to unavoidable losses due to the insolubility of these molecules.
Our analyses suggest that the new method is suitable for the rapid screening of SphK inhibitors given its simplicity and its ability to generate satisfactory values for the kinetic parameters of SphKs. We tested this using published SphK inhibitors. As documented in Table 1, the Ki values obtained with our method are in agreement with those obtained in parallel using the TLC-base method. These results demonstrate the reliability of our method to screen SphK inhibitors.
Table 1.
Comparison between the current method (“FlashPlate”) and the conventional method (“TLC”) to assess the potency of SphK inhibitors. In both cases, potency was evaluated as the Ki of each inhibitor that was calculated from the observed Michaelis-Menten constants (Km) in the absence or presence of a fixed concentration of inhibitor ranging from 0.4 to 50 μM. The Ki value (in μM) is provided for each inhibitor for both SphK1 and SphK2. As Ki reflects the dissociation constant of the inhibitors for the enzyme, lower values indicate higher potency. Numbers in parenthesis refer to the literature citation for the inhibitors.
We tested, and ultimately discarded, several versions of our assay. For example we pre-coated the wells with cationic polylysine to increase S1P adherence, but this maneuver proved unnecessary. We considered also using FlashPlates pre-coated with streptavidin and substituting biotinylated sphingosine for sphingosine in the assay. This idea was judged impractical because of the higher cost of both biotinylated sphingosine and the streptavidin-coated plates. We tested our assay in standard 96-well plates (Perkin Elmer #6005280) using scintillation fluid. With this strategy, the Km values were similar to that obtained with FlashPlates (data not shown) but Vmax values were about 50% lower. Thus regular 96-well plates could be a less expensive alternative than FlashPates, albeit at the cost of reduced sensitivity and the additional pipeting step. We wondered whether the wash steps could be eliminated because [γ-33P]ATP (and any 33PO4 present) would not be in close proximity to the scintillant. However, we found that counting the plate without washing resulted in high background, presumably because emissions from phosphorus-33 were sufficiently energetic to activate the scintillant from a distance. We also found that increasing the number of washes resulted in S1P loss and established that two washes was the optimal number to remove ATP while retaining S1P. We note that the more commonly used and less expensive ATP radionuclide, [γ-32P]ATP, cannot be used in this assay. The reason being that the high energy of the phosphorus-32 emission produces unacceptable levels of radioactivity “spillover” among wells.
We asked whether our assay was sensitive to the total amount of protein present in the wells. This is an important consideration if tissue homogenates are to be tested with the method. We performed the assay in presence of different amounts of BSA (1, 3, 10, 30, 100, 300, and 1,000 μg). We found no interference up to 100 μg (data not shown). In presence of >300 μg BSA, there was 60% reduction on S1P binding to the wells rendering our method unusable at high protein concentrations. We also tested the effect of Triton X-100, a detergent commonly used to discriminate between SphK1 and SphK2 activities because it selectively inhibits SphK2 while activating SphK1. Unfortunately we found that 0.05% Triton X-100, the concentration used to inhibit SphK2, completely abolished the binding of S1P to the wells making our method not suitable for SphK1/SphK2 discrimination by this strategy. However, SphK2 can be assessed with a selective substrate such as FTY720 (Fig. 2c). The Km value determined for FTY720 was 22μM, which is consistent with the published value [23].
We further examined the sensitivity of the high-throughput assay with cell/tissue extracts to measure the endogenous sphingosine kinase activity using sphingosine or FTY720 as a substrate to discriminate between SphK 1 and 2. The Km values determined for sphingosine using U937 cell and mouse kidney extracts were 6 and 7 μM respectively (Fig. 4), which were consistent with published values [15, 16]. Similarly, the Km values for FTY720 using U937 cell and mouse kidney extracts were 19 and 16 μM, respectively (Fig. 4b).
Figure 4.
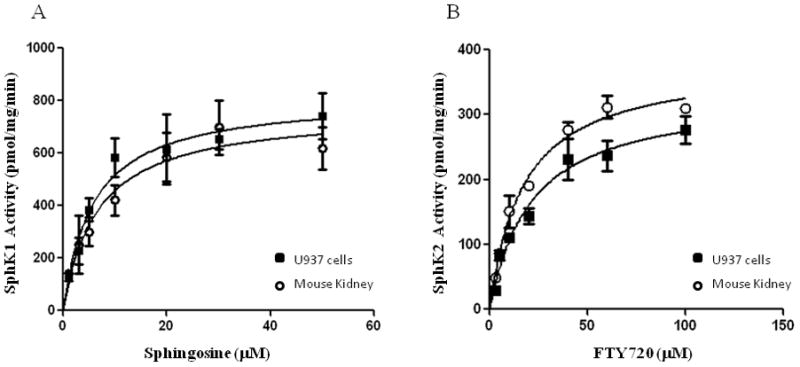
Determintation of sphingosine kinase activity of cell/tissue extracts using the FlashPlate method. Total sphingosine kinase activity of U937 cells (filled square) and mouse kidney (open circle) lysates was measured as a function of sphingosine. Michaelis-Menten constants (Km) was calculated using non-linear regression. Experimental values are averages of three independent experiments.
We considered also whether our assay protocol, which is based on the insolubility of S1P and S1P analogs, can be generalized to other lipid kinases such as ceramide kinase or the diacylglycerol kinases. We have not tested this idea, but we predict that it would be necessary to coat the wells (e.g. using poly-lysine, poly-arginine, or an alkyl amine) to enhance the selective capture of the phosphorylated lipid product. Finally, another strategy for S1P would be the use of sphingosine analog containing a high quantum yield fluorophore as a substrate, although that strategy would negate the ability to test alternate amino alcohol substrates (e.g. FTY720) and would be less sensitive than using radioactive ATP.
CONCLUSION
It has been a challenge to measure sphingosine kinase activity in a simple and rapid way. Our assay overcomes the fundamental problem of the TLC-based assay, which is limited to testing only a few inhibitors or substrates at a time. An ideal, but as yet un-realized, assay would use a 384 well plate and would require neither washes nor radionuclides. Our method approaches that ideal, but uses a 96 well format, requires a wash step and uses a radionuclide, although the phosphorus-33 is less problematic than the higher energy emitting phosphorus-32 employed in the standard TLC-based assay. Further, our assay is constrained by its upper limit of protein and the unavailability of Triton X-100 to discriminate between SphK1 and SphK2 activites. Nevertheless, our method is a substantial improvement over published assays in that it enables the rapid acquisition of Km values and thus the determination of Ki values for multiple SphK inhibitors. Furthermore the capacity of detecting endogenous SphK activity from crude cell/tissue extracts illustrates its usefulness in ex vivo analysis of signaling pathways involving sphingosine kinases.
Acknowledgments
The authors gratefully acknowledge Dr. Robert Stoffel (Abbott Laboratories, Worcester, MA) for his suggestion to take advantage of the physiochemical properties of S1P in developing our assay. The authors thank Dr. Jose Tomsig, University of Virginia Department of Pharmacology, for his comments on the manuscript. This research was supported in part by research grants from the NIH (R01 GM067958) and the Ivy Foundation for Biomedical Research.
Abbreviations used
- SphK
sphingosine kinase
- S1P
sphingosine 1-phosphate
- ATP
adenosine triphosphate
- TLC
thin layer chromatography
- HPLC
high performance liquid chromatography
- DMS
N,N′-dimethylsphingosine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hla T, Lee MJ, Ancellin N, Paik JH, Kluk MJ. Lysophospholipids-receptor revelations. Science. 2001;294:1875–1878. doi: 10.1126/science.1065323. [DOI] [PubMed] [Google Scholar]
- 2.Spiegel S. Sphingosine 1-phosphate: a prototype of a new class of second messengers. J Leukoc Biol. 1999;65:341–344. doi: 10.1002/jlb.65.3.341. [DOI] [PubMed] [Google Scholar]
- 3.Cuvillier O, Pirianov G, Kleuser B, Vanek PG, Coso OA, Gutkind S, Spiegel S. Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature. 1996;381:800–803. doi: 10.1038/381800a0. [DOI] [PubMed] [Google Scholar]
- 4.Mandala SM, Thornton R, Tu Z, Kurtz MB, Nickels J, Broach J, Menzeleev R, Spiegel S. Sphingoid base 1-phosphate phosphatase: a key regulator of sphingolipid metabolism and stress response. Proc Natl Acad Sci U S A. 1998;95:150–155. doi: 10.1073/pnas.95.1.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caligan TB, Peters K, Ou J, Wang E, Saba J, Merrill AH., Jr A high-performance liquid chromatographic method to measure sphingosine 1-phosphate and related compounds from sphingosine kinase assays and other biological samples. Anal Biochem. 2000;281:36–44. doi: 10.1006/abio.2000.4555. [DOI] [PubMed] [Google Scholar]
- 6.Ruwisch L, Schäfer-Korting M, Kleuser B. An improved high-performance liquid chromatographic method for the determination of sphingosine-1-phosphate in complex biological materials. Naunyn Schmiedebergs Arch Pharmacol. 2001;363:358–363. doi: 10.1007/s002100000365. [DOI] [PubMed] [Google Scholar]
- 7.Min JK, Yoo HS, Lee EY, Lee WJ, Lee YM. Simultaneous quantitative analysis of sphingoid base 1-phosphates in biological samples by o-phthalaldehyde precolumn derivatization after dephosphorylation with alkaline phosphatase. Anal Biochem. 2002;303:167–175. doi: 10.1006/abio.2002.5579. [DOI] [PubMed] [Google Scholar]
- 8.Vessey DA, Kelley M, Karliner JS. A rapid radioassay for sphingosine kinase. Anal Biochem. 2005;337:136–142. doi: 10.1016/j.ab.2004.10.032. [DOI] [PubMed] [Google Scholar]
- 9.Roberts JL, Moretti PA, Darrow AL, Derian CK, Vadas MA, Pitson SM. An assay for sphingosine kinase activity using biotinylated sphingosine and streptavidin-coated membranes. Anal Biochem. 2004;331:122–129. doi: 10.1016/j.ab.2004.03.030. [DOI] [PubMed] [Google Scholar]
- 10.Billich A, Ettmayer P. Fluorescence-based assay of sphingosine kinases. Anal Biochem. 2004;326:114–119. doi: 10.1016/j.ab.2003.11.018. [DOI] [PubMed] [Google Scholar]
- 11.Lee KJ, Mwongela SM, Kottegoda S, Borland L, Nelson AR, Sims CE, Allbritton NL. Determination of sphingosine kinase activity for cellular signaling studies. Anal Chem. 2008;80:1620–1627. doi: 10.1021/ac702305q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vidugiriene J, Zegzouti H, Goueli SA. Evaluating the utility of a bioluminescent ADP-detecting assay for lipid kinases. Assay Drug Dev Technol. 2009;7:585–597. doi: 10.1089/adt.2009.0223. [DOI] [PubMed] [Google Scholar]
- 13.Togame H, Dodo R, Inaoka T, Reinemer P. Development of a simple and robust assay to screen for inhibitors of sphingosine kinases. Assay Drug Dev Technol. 2007;5:215–223. doi: 10.1089/adt.2006.049. [DOI] [PubMed] [Google Scholar]
- 14.Kharel Y, Lee S, Snyder AH, Sheasley-O’neill SL, Morris MA, Setiady Y, Zhu R, Zigler MA, Burcin TL, Ley K, Tung KS, Engelhard VH, Macdonald TL, Pearson-White S, Lynch KR. Sphingosine kinase 2 is required for modulation of lymphocyte traffic by FTY720. J Biol Chem. 2005;280:36865–36872. doi: 10.1074/jbc.M506293200. [DOI] [PubMed] [Google Scholar]
- 15.Olivera A, Kohama T, Tu Z, Milstein S, Spiegel S. Purification and characterization of rat kidney sphingosine kinase. J Biol Chem. 1998;273:12576–12583. doi: 10.1074/jbc.273.20.12576. [DOI] [PubMed] [Google Scholar]
- 16.Pitson SM, D’andrea RJ, Vandeleur L, Moretti PAB, Xia P, Gamble JR, Vadas MA, Wattenberg BW. Human sphingosine kinase : purification, molecular cloning and characterization of the native and recombinant enzymes. Biochem J. 2000;350:429–441. [PMC free article] [PubMed] [Google Scholar]
- 17.Liu H, Sugiura M, Nava VE, Edsall LC, Kono K, Poulton S, Milstien S, Kohama T, Spiegel S. Molecular cloning and functional characterization of a novel mammalian sphingosine kinase type 2 isoform. J Biol Chem. 2000;275:19513–19520. doi: 10.1074/jbc.M002759200. [DOI] [PubMed] [Google Scholar]
- 18.Edsall LC, Van Brocklyn JR, Cuvillier O, Kleuser B, Spiegel S. N. N-Dimethylsphingosine is a potent competitive inhibitor of sphingosine kinase but not of protein kinase C: modulation of cellular levels of sphingosine 1-phosphate and ceramide. Biochemistry. 1998;37:12892–12898. doi: 10.1021/bi980744d. [DOI] [PubMed] [Google Scholar]
- 19.French KJ, Schrecengost RS, Lee BD, Zhuang Y, Smith SN, Eberly JL, Yun JK, Smith CD. Discovery and evaluation of inhibitors of human sphingosine kinase. Cancer Res. 2003;63:5962–5969. [PubMed] [Google Scholar]
- 20.Xiang Y, Asmussen G, Booker M, Hirth B, Kane JL, Jr, Liao J, Noson KD, Yee C. Discovery of novel sphingosine kinase 1 inhibitors. Bioorg Med Chem Let. 2009;19:6119–6121. doi: 10.1016/j.bmcl.2009.09.022. [DOI] [PubMed] [Google Scholar]
- 21.Mathews TP, Kennedy AJ, Kharel Y, Kennedy PC, Nicoara O, Sunkara M, Morris AJ, Wamhoff BR, Lynch KR, Macdonald TL. Discovery, biological evaluation, and structure-activity relationship of amidine based sphingosine kinase inhibitors. J Med Chem. 2010;53:2766–2778. doi: 10.1021/jm901860h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wong L, Tang SS, Lam Y, Melendez AJ. Synthesis and evaluation of sphingosine analogues as inhibitors of sphingosine kinases. J Med Chem. 2009;52:3618–3626. doi: 10.1021/jm900121d. [DOI] [PubMed] [Google Scholar]
- 23.Billich A, Bornancin F, Devay P, Mechtcheriakova D, Urtz N, Baumruker T. Phosphorylation of the immunomodulatory drug FTY720 by sphingosine kinases. J Biol Chem. 2003;278:47408–47415. doi: 10.1074/jbc.M307687200. [DOI] [PubMed] [Google Scholar]