

Abstract
Background
J Wave Syndromes have emerged conceptually to encompass the pleiotropic expression of J point abnormalities including Brugada syndrome (BrS) and early repolarization syndrome (ERS). Recently, KCNJ8, which encodes the cardiac KATP Kir6.1 channel, has been implicated in ERS following the identification of a functionally uncharacterized missense mutation, S422L. Here, we sought to further explore KCNJ8 as a novel susceptibility gene for J wave syndromes.
Methods
Using PCR, DHPLC, and direct DNA sequencing, comprehensive open reading frame/splice site mutational analysis of KCNJ8 was performed in 101 unrelated patients with J wave syndromes including 87 with BrS and 14 with ERS. 600 healthy individuals were examined to assess allelic frequency for all variants detected. KCNJ8 mutation(s) were engineered by site directed mutagenesis and co-expressed heterologously with SUR2A in COS-1 cells. Ion currents were recorded using whole cell configuration of the patch-clamp technique.
Results
One BrS case and one ERS case hosted the identical missense mutation, S422L that was reported previously. KCNJ8-S422L involves a highly conserved residue and was absent in 1200 reference alleles. Both cases were negative for mutations in all known BrS- and ERS-susceptibility genes. The KATP current of Kir6.1-S422L mutation was increased significantly over the voltage range of 0 mV to 40 mV compared to Kir6.1-WT channels (p < 0.05, n=16-21).
Conclusions
These findings further implicate KCNJ8 as a novel J wave syndrome-susceptibility gene and a marked gain-of-function in the cardiac KATP Kir6.1 channel secondary to KCNJ8-S422L as a novel pathogenic mechanism for the phenotypic expression of both BrS and ERS.
Keywords: Idiopathic ventricular fibrillation, early ventricular repolarization, J wave syndromes, ion channels, sudden cardiac death, KATP channels, genetic diseases
INTRODUCTION
The early repolarization pattern in the 12-lead electrocardiogram (ECG) is characterized by an elevation of the QRS-ST junction, better known as “J point”, and for a long time was considered a benign abnormality common in young healthy men and athletes. However, data obtained in a large multicenter study reported a 30% increase in the prevalence of an early repolarization pattern, especially J point elevation in the inferolateral leads in cases with idiopathic ventricular fibrillation (IVF) compared with controls1. Recently, Antzelevitch and Yan have proposed heritable J Wave Syndromes as a new conceptual framework for electrocardiographic/arrhythmic phenotypes involving J-point/QRS-ST abnormalities2. According to this classification scheme, the heritable J wave syndromes include Brugada syndrome (BrS) and three different subtypes of Early Repolarization Syndrome (ERS) distinguished by the spatial localization of the early repolarization pattern.
Over the past decade, molecular sleuthing to elucidate the pathogenic substrates for these J wave syndromes as well as for idiopathic ventricular fibrillation (IVF) has yielded 8 genes associated with either BrS or IVF: SCN5A, GPD1L, CACNA1C, CACNB2B, SCN1B, KCNE3, SCN3B, and DPP6 3-8. Perturbations in SCN5A and SCN3B have been implicated in both BrS and IVF without any discernible Brugada ECG pattern evidencing the pleiotropic expression that is now recognized for several of the channelopathy-susceptibility genes.
Recently, a novel missense mutation, S422L, in the KCNJ8-encoded Kir6.1 alpha subunit of the ATP-sensitive potassium (KATP) channel was reported in a young female with VF secondary to ERS but KCNJ8-S422L’s functional properties were not investigated9. Notwithstanding, Kir6.1 is clearly expressed in cardiomyocytes10-12 though its functional role in ventricular repolarization remains controversial. In this study, we examined KCNJ8 as a candidate gene involved in the pathogenesis of J wave syndromes.
METHODS
Study Participants
We examined a cohort of 101 unrelated J wave syndrome cases including 87 with BrS and 14 with ERS that were referred to either the Windland Smith Rice Sudden Death Genomics Laboratory at Mayo Clinic, Rochester, MN or the Molecular Cardiology Laboratory, Fondazione IRCCS Policlinico San Matteo, Pavia, Italy for BrS/IVF genetic testing. Following receipt of written consent for this Mayo Foundation Institutional Review Board and Fondazione IRCCS Policlinico S. Matteo, Pavia, Italy Medical Ethical Committee-approved protocol, genomic DNA was extracted from peripheral blood lymphocytes using the Purgene DNA extraction kit (Gentra, Inc, Minneapolis, MN, USA).
Mutational Analysis
Comprehensive open reading frame/splice site mutational analysis of KCNJ8 was performed using polymerase chain reaction (PCR), denaturing high performance liquid chromatography (DHPLC), and direct DNA sequencing as described previously 13. Six hundred healthy individuals (1200 reference alleles), including 100 African-Americans and 200 Caucasians from the Human Genetic Cell Repository and 300 additional European Caucasian Controls, were examined to assess allelic frequency for all non-synonymous variants detected.
Cloning of Human KCNJ8 and Mutagenesis
Human heart cDNA was created using human heart total RNA14 and SuperScript First-Strand cDNA Synthesis System for RT-PCR (Invitrogen, Carlsbad, CA). The reaction was performed according to the manufacturer’s protocol. The KCNJ8 (Kir6.1) gene was amplified from the human heart cDNA by PCR using forward primer 5’-ATGTTGGCCAGAAAGAGTATCATC-3’ and reverse primer 5’-TCATGATTCCGATGTGTTTTGATT-3’. The human Kir6.1 PCR product was first TOPO cloned into pCR2.1 vector (Invitrogen) and then subcloned into mammalian expression vector pIRES2-EGFP (Clontech, Pal Alto, CA) by a single EcoRI site. Kir6.1-S422L was generated by using a Quick Change Site-Directed Mutagenesis kit (Stratagene) with the following primers: Kir6.1-S422L forward 5’-CCAGAAGGAAATCAAAACACATTGGAATCA-3’ and Kir6.1-S422L reverse 5’-TGATTCCAATGTGTTTTGATTTCCTTCTGG-3’. The cDNA sequence of Kir6.1-WT and Kir6.1-S422L in the constructs was verified by sequencing analysis.
Transfection and cell culture
COS-1 cells were co-transfected with the mammalian expression vector pIRES2-EGFP containing human Kir6.1-WT (1 mcg), or human Kir6.1-S422L (1 mcg) with 1 mcg mouse full-length SUR2A cDNA15 using FuGENE®6 Transfection Reagent (Roche Diagnostics; Indianapolis, IN) according to the manufacturer’s instructions. Transfected cells were cultured in 35- mm diameter cell-culture dish with Dulbecco’s modified Eagle’s medium, as previously described15.
Electrophysiology and Data Analysis
After 48-72 hours of transfection, the cells expressing green fluorescence protein were selected for recording the whole cell current at room temperature (22°C ~24°C). Axopath 200A amplifier and pClamp version 10.2 (Axon Instruments, Union City, California, USA) were used. Patch pipettes were drawn from borosilicate glass (World Precision Instruments Incorporated, Sarasota, Florida, USA) with a resistance of 2 to 3 MΩ when filled with recording solutions. The bath (extracellular) solution contained (in mM) 140 NaCl, 5 KCl, 1 MgCl2, 1 CaCl2, and 10 HEPES, (pH 7.4 set with NaOH). The pipette (intracellular) solution contained (in mM) 120 K-aspartate, 25 KCl, 1 MgCl2 10 EGTA, and 10 HEPES, (pH 7.2 set with KOH). The whole cell current was generated by clamp pulses from a holding potential of -40 mV to voltages ranging from -100 to 40 mV in 20-mV steps for 260 ms. The currents were filtered at 1 KHz and sampled at 5 KHz. Data were digitally stored for off-line analysis using pClamp10.2 software (Axon Instruments Inc.). After the cell membrane rupture, a extracellular 100 μM pinacidil (Parke Davis, Ann Arbor, Michigan, USA) was applied for obtaining the maximal inwardly rectifying potassium channel current (Ik, usually within 2 minutes). The whole cell current could be blocked partially by 20 μM glibenclamide (Sigma-Aldrich). The glibenclamide-sensitive current was interpreted as KATP current and was normalized by cell capacitance to obtain the whole cell current density.
Statistical Analysis
All data points are shown as the mean value and the bars represent the standard error of the mean (S.E.M.). Determinations of statistical significance were performed using a Student t test for comparisons of two groups. A p-value < 0.05 was considered statistically significant.
RESULTS
Among these 101 unrelated patients referred for genetic testing following diagnosis of a particular J wave syndrome, 87 had a referral diagnosis of BrS and 14 had a referral diagnosis of ERS. Overall, 93% were Caucasian, 81% were male, and the average age at diagnosis was 36±14 years. Thirty of the BrS cases (34%) patients exhibited a spontaneous type 1 Brugada ECG pattern while 2 ERS patients displayed J-point elevation that localized to the lateral leads (ERS1), 4 localizing to inferior leads (ERS2), 3 had more of a global pattern (ERS3), and 5/14 ERS patients had J-point elevation confined to the right precordial leads but did not meet ECG criteria for BrS (ERS4)2.
Mutational Analysis
A KCNJ8 1265 C>T nucleotide substitution, which yielded the previously reported missense mutation S422L (Serine [S] to Leucine [L] at position 422), was identified in 2 cases (Figure 1A). KCNJ8-S422L was absent in 1200 references alleles (p=0.02) and involves a highly conserved residue that localizes to the Kir6.1 channel’s intracellular carboxyl (C)-terminus (Figure 1B, 1C). Neither KCNJ8-S422L positive case had any identifiable open reading frame/splice site mutations in any of the known BrS-/IVF-susceptibility genes (data not shown).
Figure 1. Molecular elucidation of KCNJ8-S422L.
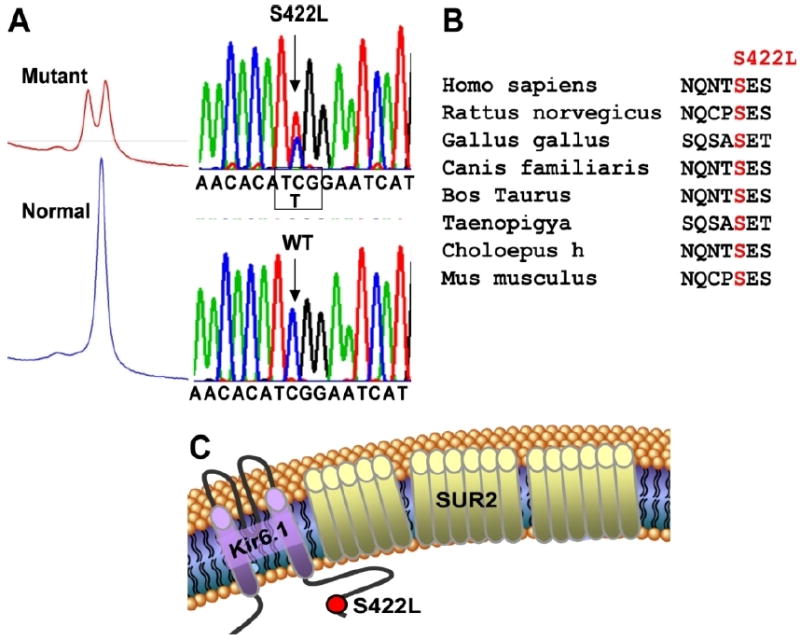
A. Top panel, the proband’s DHPLC profile and DNA sequence chromatogram showing a C>T substitution at position 1265 of KCNJ8 resulting in a Serine (S) to Leucine (L) substitution at position 422 (S422L). Bottom panel, the normal DHPLC profile and DNA sequence chromatogram.
B. Conservation across species for S422 in Kir6.1
C. Linear topology of the Kir6.1/SUR2 complex showing the predicted localization of the S422L missense mutation to the C-terminus. SUR2= Sulfonylurea receptor type 2.
KCNJ8-S422L Positive Cases
Case 1
A 21-year-old otherwise healthy male suddenly lost consciousness while exercising. He had no family history of sudden cardiac death, syncope or seizure disorder. A complete cardiac work-up was performed including electrocardiogram, echocardiogram, stress test and cardiac catheterization. His 12-lead ECG exhibited intraventricular conduction delay, normal QT intervals (QTc ranging from 404 – 438 ms), dynamic ST changes with T-wave inversion in the right precordial leads and intermittent J point elevation in limb lead III, aVF and the precordial leads V1-V4 (Figure 2). The T wave abnormalities were less prominent during exercise testing but failed to normalize (data not shown). In addition, the exercise test did not reproduce syncope or induce arrhythmia. All other tests were normal. An internal cardiac defibrillator was implanted as secondary prevention. Six years after his ICD implantation, he received an appropriate ICD shock for monomorphic VT during an easy bike ride after sleep deprivation from a trans-Pacific flight (Figure 2C).
Figure 2. Case 1 Electrocardiographic Phenotype.
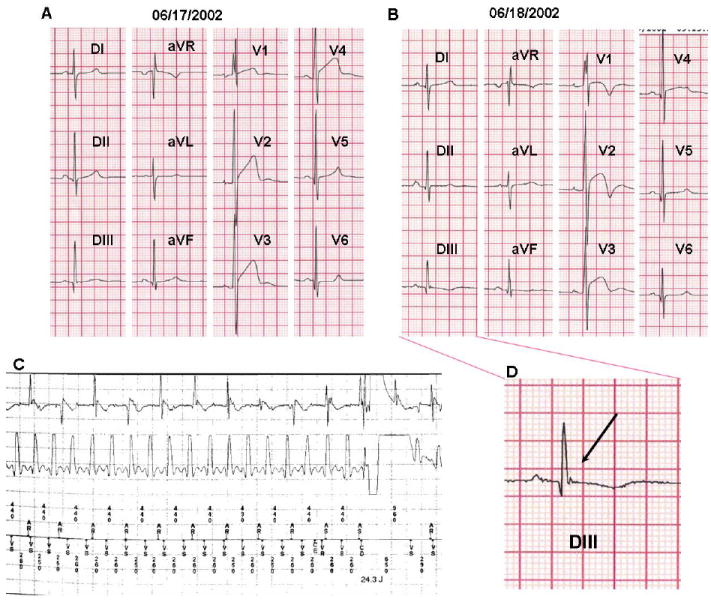
12-lead ECGs of case 1 showing intraventricular conduction delay (V1), important dynamic repolarization abnormalities in leads V1-V4 (A, B) and intermittent J waves in the inferior leads (A,B,D).
C. ICD electrogram of VT- therapy with both atrial electrogram (top) and ventricular electrogram (bottom). The VT episode is unsuccessfully shocked; the arrhythmia was self-terminated before the second ICD therapy (electrogram not shown).
D. Magnification of the “J” wave observed in lead DIII from the 12-lead ECG shown in panel B.
Case 2
A 30-year-old otherwise healthy male was referred for further evaluation following an ECG that was suspicious for BrS. He had no family history of sudden cardiac death, syncope or seizure disorder. His initial ECG exhibited minor right bundle-branch-block with modest ST segment elevation, coved-type in V1 and saddle-back in V2. Flecainide provocation elicited a diagnostic type 1 Brugada ECG pattern (Figure 3A). Interestingly, a coved-type ECG pattern was not only present in V1 and V2, but was also evident in aVR and aVL. It has been reported that 11% of the patients with BrS have spontaneous early repolarization pattern in the inferior-lateral leads and this group of patients seemed to have a more severe phenotype, this case had BrS pattern in lateral but not in inferior leads16. The patient was asymptomatic for syncopal events; however episodes of palpitations, suggestive for supraventricular arrhythmias were described. An electrophysiological study was performed; an AV nodal reentrant tachycardia was induced and successfully ablated. Atrial fibrillation was easily inducible while ventricular arrhythmias were not inducible.
Figure 3. Case 2 Electrocardiographic Phenotype.
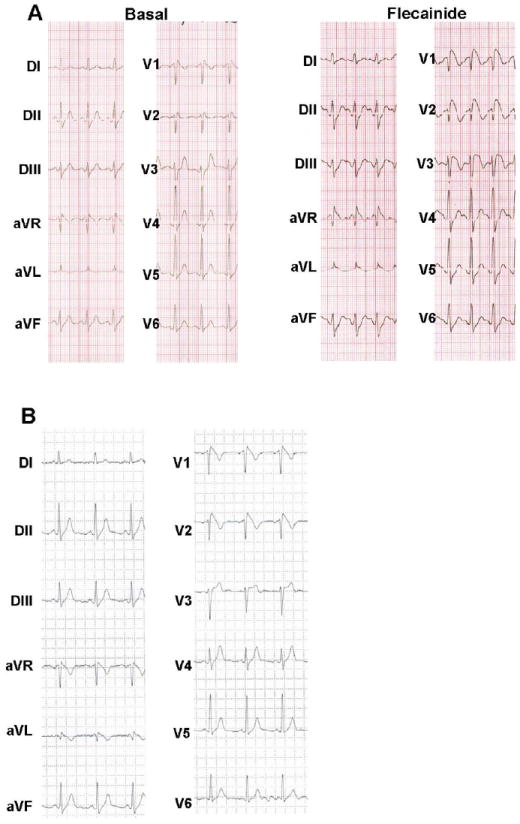
A. Flecainide challenge unmasked the typical coved type Brugada Syndrome ECG pattern in case 2.
B. Spontaneous type 1 Brugada ECG pattern recorded during 24 hour Holter monitoring in case 2.
The patient remained asymptomatic during four years and suffered again episodes of palpitations, different from the ones previously observed. His ECG exhibited abnormal ventricular repolarization and during 24-hour Holter monitoring, a spontaneously positive type 1 BrS ECG pattern appeared with coved-type ST segment elevation in V1, V2, aVR and aVL (Figure 3B). Both exercise stress testing and cardiac MRI were normal.
Heterologous Expression and Functional Characterization of KCNJ8-S422L
COS-1 cells transiently expressing the Kir6.1-WT or Kir6.1-S422L mutant and co-expressed with SUR2A were voltage clamped after 48-72 hours of incubation. Kir6.1-S422L mutant channels displayed an increase in glibenclamide-sensitive KATP current compared to Kir6.1-WT (Figure 4A, 4B). The IKATP densities were increased by ~60%-67% for the Kir6.1-S422L mutant compared to Kir6.1-WT over the voltage range of 0 mV to 40 mV (n=16-21, p<0.05, Figure 4C, 4D).
Figure 4. Marked gain-of-function for Kir6.1-S422L channels.
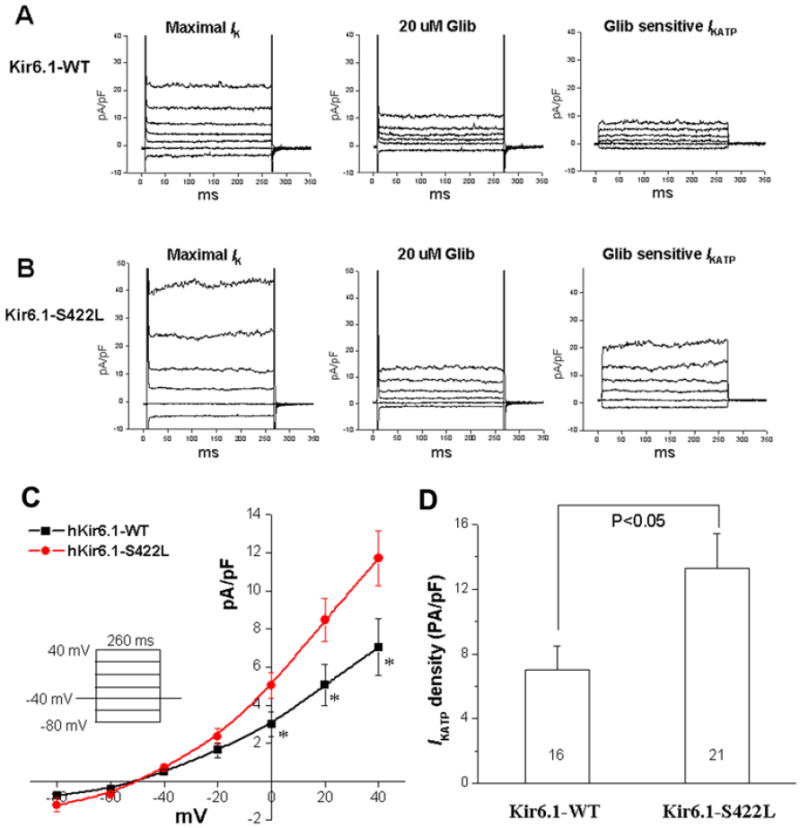
Comparing the glibenclamide (Glib)-sensitive KATP current of Kir6.1-S422L with Kir6.1-WT. (A) Examples of whole cell current traces of Kir6.1-WT recorded after 1-2 minutes perfusion of 100 μM pinacidil (maximal Ik, left), in the presence of 20 μM Glib (middle), and the Glib-sensitive IKATP obtained by subtraction (right). (B) Examples of whole cell current traces of Kir6.1-S422L recorded after 1-2 minutes perfusion of 100 μM pinacidil (maximal Ik, left), in the presence of 20 μM Glib (middle), and the Glib-sensitive IKATP obtained by subtraction (right). (C) Summary data of the current voltage plot for the Glib-sensitive IKATP at the voltages tested (insert, n=16-21 cells. * p<0.05). (D) Bar graphs showing the mean IKATP densities from Kir6.1-WT and Kir6.1-S422L channels elicited by a test pulse from holding potential of -40 mV to 40 mV for 260 ms. IKATP density of Kir6.1-S422L mutant channels was increased significantly compared with Kir6.1-WT channels.
DISCUSSION
KATP channels in heart, more questions than answers
KATP channels belong to the inwardly rectifying K+ channels family (Kir’s) and is gated directly by intracellular ATP and ADP, coupling the metabolic state of the cell with its electrical activity and hence contractility17,18. Structurally, KATP channels are heteromeric octamers, composed of 4 inwardly rectifying subunits, either KCNJ8-encoded Kir6.1 or KCNJ11-encoded Kir6.2 and 4 sulphonylurea receptors (SURs), a member of the ATP-binding cassette (ABC) transporter family. In contrast to the other Kir subfamilies, Kir6.1 and Kir6.2 require the SUR subunits19 to form functional K+ channels at the cell surface20.
It has been suggested that the main KATP channel in cardiomyocytes is composed by Kir6.2/SUR2A and the primary role of KATP channels in heart is cardioprotection under metabolic stress, such ischemia, anoxia or metabolic inhibition. This concept was derived from knockout mice and recombinant channel pharmacology18,21-24. However, several observations have revealed some mismatches in the understanding of the molecular components and functional roles of KATP channels in cardiac physiology.
More than 50 human mutations in the Kir6.2 alpha subunit have been reported and the pancreatic phenotype observed is neonatal diabetes (gain-of-function mutations) or congenital hyperinsulinism (loss-of-function mutations). Despite the presence of Kir6.2 in the heart, neither cardiac abnormalities or arrhythmia phenotypes have ever been described in these cases25,26. Despite the large amount of evidence confirming the role of KATP channels in cardioprotection during metabolic stress24, the SUR1-/- and SUR2-/- knockout mice were found to be more tolerant of global ischemia–reperfusion than control mice, further challenging our understanding of KATP channels22,27,28.
Several studies have shown that Kir6.1 is also highly expressed in cardiomyocytes10,11,29-31, but the role of this alpha subunit in cardiac action potential is not well understood. Kir6.1-null mice exhibit ST segment elevation followed by atrioventricular block and early sudden cardiac death11. In addition, Kir6.1 null mice demonstrated hypercontractile coronary arteries and have provided an animal model for Prinzmetal angina11. So far, this phenotype has not been translated to humans though as KCNJ8 has not been established as a Prinzmetal angina-susceptibility gene32.
Recently, Morrissey and colleagues have shown that Kir6.1 is expressed in a sarcomeric striated pattern in epicardial ventricular myocytes while in midmyocardium and endocardium, Kir6.1 localizes to the coronary vasculature12. These observations are consistent with those observed nearly 20 years ago when epicardial dominant activation of KATP channels during ischemia was noted33,34. This heterogenous transmural distribution of Kir6.1 could explain the normal action potentials recorded previously in the Kir6.1 null mice11.
Functional investigations of human genetic variants have provided key insights into the physiological importance of essential proteins and represent an opportunity to dissect the mechanistic basis of human arrhythmogenic disease35. This KCNJ8-S422L mutation, now found in 3 unrelated cases with an electrocardiographic/arrhythmogenic phenotypic falling within the spectrum of J-wave syndromes, provides an excellent opportunity to dissect the possible physiological and pathological role(s) of Kir6.1 channels in cardiac ventricular repolarization.
Kir6.1 gain of function and the spectrum of J wave syndromes
Here, 2 unrelated cases, each expressing a different J wave syndrome, host precisely the same missense mutation, KCNJ8-S422L. Notably, KCNJ8-S422L was reported for the first time by Haissaguerre et al. albeit without any functional data in a high risk J wave syndrome case11. KCNJ8-S422L is highly conserved across species and has not been observed in nearly 2000 reference alleles, considering previous published data.
Heterologous expression of KCNJ8-S422L in COS1 cells evidenced a marked gain-of-function in the KATP current associated with Kir6.1-S422L channels when co-expressed in the setting of the predominant SUR isoform in cardiac ventricular tissue, SUR2A36. This gain-of-function coupled with the preferential epicardial distribution of Kir6.1 could explain the J-wave syndrome phenotype observed in the three unrelated patients known to host this specific missense mutation. We speculate that the KCNJ8-S422L gain of function mutation further accentuates epicardial KATP channel activity thereby shortening the action potential duration in the epicardium. This pathogenic substrate is reflected in the electrocardiogram by early ventricular repolarization in inferior and right precordial leads on the ECG. This mutation-accentuated transmural heterogeneity of KATP channels is a pathogenic mechanism similar to the one proposed in BrS-ST segment abnormalities, generated by the resultant accentuation in the transmural heterogeneity of ITO that emerges in the setting of sodium channel loss of function37. Akin to the pleotropic expression associated with SCN5A-E1784K missense mutation which is one of the most common LQT3-associated mutations and one of the most common BrS1-associated mutations38,39, we see pleiotropy in KCNJ8-S422L with phenotypic manifestation of BrS and ERS.
CONCLUSION
These findings further implicate not only KCNJ8 as a novel J wave syndrome-susceptibility gene but also a marked gain-of-function in the cardiac KATP Kir6.1 channel secondary to KCNJ8-S422L as a novel pathogenic mechanism for the phenotypic expression of both BrS and ERS. The precise role(s) of KATP currents to ventricular repolarization at rest and during stress in both health and disease and the exact architecture of the KATP channels in the heart require further scrutiny.
Acknowledgments
This work was supported by the Mayo Clinic Windland Smith Rice Comprehensive Sudden Cardiac Death Program (MJA), the University of Wisconsin Cellular and Molecular Arrhythmia Research Program (JCM), and the National Institutes of Health HD42569 (MJA), HL71092 (JCM), and 1PO1HL094291 (JCM and MJA).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rosso R, Kogan E, Belhassen B, et al. J-point elevation in survivors of primary ventricular fibrillation and matched control subjects: incidence and clinical significance. J Am Coll Cardiol. 2008;52:1231–8. doi: 10.1016/j.jacc.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 2.Antzelevitch C, Yan GX. J wave syndromes. Heart Rhythm. 2010;7:549–58. doi: 10.1016/j.hrthm.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen Q, Kirsch GE, Zhang D, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998;392:293–6. doi: 10.1038/32675. [DOI] [PubMed] [Google Scholar]
- 4.Van Norstrand DW, Valdivia CR, Tester DJ, et al. Molecular and functional characterization of novel glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) mutations in sudden infant death syndrome. Circulation. 2007;116:2253–9. doi: 10.1161/CIRCULATIONAHA.107.704627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Delpon E, Cordeiro JM, Nunez L, et al. Functional effects of KCNE3 mutation and its role in the development of Brugada syndrome. Circ Arrhythm Electrophysiol. 2008;1:209–18. doi: 10.1161/CIRCEP.107.748103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hu D, Barajas-Martinez H, Burashnikov E, et al. A mutation in the beta 3 subunit of the cardiac sodium channel associated with Brugada ECG phenotype. Circ Cardiovasc Genet. 2009;2:270–8. doi: 10.1161/CIRCGENETICS.108.829192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cordeiro JM, Marieb M, Pfeiffer R, Calloe K, Burashnikov E, Antzelevitch C. Accelerated inactivation of the L-type calcium current due to a mutation in CACNB2b underlies Brugada syndrome. J Mol Cell Cardiol. 2009;46:695–703. doi: 10.1016/j.yjmcc.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Antzelevitch C. Genetic basis of Brugada syndrome. Heart Rhythm. 2007;4:756–7. doi: 10.1016/j.hrthm.2007.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haissaguerre M, Chatel S, Sacher F, et al. Ventricular fibrillation with prominent early repolarization associated with a rare variant of KCNJ8/KATP channel. J Cardiovasc Electrophysiol. 2009;20:93–8. doi: 10.1111/j.1540-8167.2008.01326.x. [DOI] [PubMed] [Google Scholar]
- 10.Wu SN, Wu AZ, Sung RJ. Identification of two types of ATP-sensitive K+ channels in rat ventricular myocytes. Life Sci. 2007;80:378–87. doi: 10.1016/j.lfs.2006.09.042. [DOI] [PubMed] [Google Scholar]
- 11.Miki T, Suzuki M, Shibasaki T, et al. Mouse model of Prinzmetal angina by disruption of the inward rectifier Kir6.1. Nat Med. 2002;8:466–72. doi: 10.1038/nm0502-466. [DOI] [PubMed] [Google Scholar]
- 12.Morrissey A, Rosner E, Lanning J, et al. Immunolocalization of KATP channel subunits in mouse and rat cardiac myocytes and the coronary vasculature. BMC Physiol. 2005;5:1. doi: 10.1186/1472-6793-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tester DJ, Will ML, Ackerman MJ. Mutation detection in congenital long QT syndrome: cardiac channel gene screen using PCR, dHPLC, and direct DNA sequencing. Methods Mol Med. 2006;128:181–207. doi: 10.1385/1-59745-159-2:181. [DOI] [PubMed] [Google Scholar]
- 14.Foell JD, Balijepalli RC, Delisle BP, et al. Molecular heterogeneity of calcium channel beta-subunits in canine and human heart: evidence for differential subcellular localization. Physiol Genomics. 2004;17:183–200. doi: 10.1152/physiolgenomics.00207.2003. [DOI] [PubMed] [Google Scholar]
- 15.Chutkow WA, Makielski JC, Nelson DJ, Burant CF, Fan Z. Alternative splicing of sur2 Exon 17 regulates nucleotide sensitivity of the ATP-sensitive potassium channel. J Biol Chem. 1999;274:13656–65. doi: 10.1074/jbc.274.19.13656. [DOI] [PubMed] [Google Scholar]
- 16.Sarkozy A, Chierchia GB, Paparella G, et al. Inferior and lateral electrocardiographic repolarization abnormalities in Brugada syndrome. Circ Arrhythm Electrophysiol. 2009;2:154–61. doi: 10.1161/CIRCEP.108.795153. [DOI] [PubMed] [Google Scholar]
- 17.Nichols CG. KATP channels as molecular sensors of cellular metabolism. Nature. 2006;440:470–6. doi: 10.1038/nature04711. [DOI] [PubMed] [Google Scholar]
- 18.Seino S, Miki T. Gene targeting approach to clarification of ion channel function: studies of Kir6.x null mice. J Physiol. 2004;554:295–300. doi: 10.1113/jphysiol.2003.047175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ashcroft FM, Gribble FM. Correlating structure and function in ATP-sensitive K+ channels. Trends Neurosci. 1998;21:288–94. doi: 10.1016/s0166-2236(98)01225-9. [DOI] [PubMed] [Google Scholar]
- 20.Pu JL, Ye B, Kroboth SL, McNally EM, Makielski JC, Shi NQ. Cardiac sulfonylurea receptor short form-based channels confer a glibenclamide-insensitive KATP activity. J Mol Cell Cardiol. 2008;44:188–200. doi: 10.1016/j.yjmcc.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Babenko AP, Aguilar-Bryan L, Bryan J. A view of sur/KIR6.X, KATP channels. Annu Rev Physiol. 1998;60:667–87. doi: 10.1146/annurev.physiol.60.1.667. [DOI] [PubMed] [Google Scholar]
- 22.Zhang H, Flagg TP, Nichols CG. Cardiac sarcolemmal K(ATP) channels: Latest twists in a questing tale! J Mol Cell Cardiol. 2010;48(1):71–5. doi: 10.1016/j.yjmcc.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tong X, Porter LM, Liu G, et al. Consequences of cardiac myocyte-specific ablation of KATP channels in transgenic mice expressing dominant negative Kir6 subunits. Am J Physiol Heart Circ Physiol. 2006;291:H543–51. doi: 10.1152/ajpheart.00051.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zingman LV, Alekseev AE, Hodgson-Zingman DM, Terzic A. ATP-sensitive potassium channels: metabolic sensing and cardioprotection. J Appl Physiol. 2007;103:1888–93. doi: 10.1152/japplphysiol.00747.2007. [DOI] [PubMed] [Google Scholar]
- 25.Bryan J, Munoz A, Zhang X, Dufer M, Drews G, Krippeit-Drews P, Aguilar-Bryan L. ABCC8 and ABCC9: ABC transporters that regulate K+ channels. Pflugers Arch. 2007;453:703–18. doi: 10.1007/s00424-006-0116-z. [DOI] [PubMed] [Google Scholar]
- 26.Flechtner I, de Lonlay P, Polak M. Diabetes and hypoglycaemia in young children and mutations in the Kir6.2 subunit of the potassium channel: therapeutic consequences. Diabetes Metab. 2006;32:569–80. doi: 10.1016/S1262-3636(07)70311-7. [DOI] [PubMed] [Google Scholar]
- 27.Stoller D, Kakkar R, Smelley M, et al. Mice lacking sulfonylurea receptor 2 (SUR2) ATP-sensitive potassium channels are resistant to acute cardiovascular stress. J Mol Cell Cardiol. 2007;43:445–54. doi: 10.1016/j.yjmcc.2007.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elrod JW, Harrell M, Flagg TP, et al. Role of sulfonylurea receptor type 1 subunits of ATP-sensitive potassium channels in myocardial ischemia/reperfusion injury. Circulation. 2008;117:1405–13. doi: 10.1161/CIRCULATIONAHA.107.745539. [DOI] [PubMed] [Google Scholar]
- 29.Morrissey A, Parachuru L, Leung M, et al. Expression of ATP-sensitive K+ channel subunits during perinatal maturation in the mouse heart. Pediatr Res. 2005;58:185–92. doi: 10.1203/01.PDR.0000169967.83576.CB. [DOI] [PubMed] [Google Scholar]
- 30.Kuniyasu A, Kaneko K, Kawahara K, Nakayama H. Molecular assembly and subcellular distribution of ATP-sensitive potassium channel proteins in rat hearts. FEBS Lett. 2003;552:259–63. doi: 10.1016/s0014-5793(03)00936-0. [DOI] [PubMed] [Google Scholar]
- 31.Erginel-Unaltuna N, Yang WP, Blanar MA. Genomic organization and expression of KCNJ8/Kir6.1, a gene encoding a subunit of an ATP-sensitive potassium channel. Gene. 1998;211:71–8. doi: 10.1016/s0378-1119(98)00086-9. [DOI] [PubMed] [Google Scholar]
- 32.Emanuele E, Falcone C, Carabela M, et al. Absence of Kir6.1/KCNJ8 mutations in Italian patients with abnormal coronary vasomotion. Int J Mol Med. 2003;12:509–12. [PubMed] [Google Scholar]
- 33.Miyoshi S, Miyazaki T, Moritani K, Ogawa S. Different responses of epicardium and endocardium to KATP channel modulators during regional ischemia. Am J Physiol. 1996;271:H140–7. doi: 10.1152/ajpheart.1996.271.1.H140. [DOI] [PubMed] [Google Scholar]
- 34.Kubota I, Yamaki M, Shibata T, Ikeno E, Hosoya Y, Tomoike H. Role of ATP-sensitive K+ channel on ECG ST segment elevation during a bout of myocardial ischemia. A study on epicardial mapping in dogs. Circulation. 1993;88:1845–51. doi: 10.1161/01.cir.88.4.1845. [DOI] [PubMed] [Google Scholar]
- 35.Amin AS, Tan HL, Wilde AA. Cardiac ion channels in health and disease. Heart Rhythm. 2010;7:117–26. doi: 10.1016/j.hrthm.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 36.Davis-Taber R, Choi W, Feng J, et al. Molecular characterization of human SUR2-containing K(ATP) channels. Gene. 2000;256:261–70. doi: 10.1016/s0378-1119(00)00338-3. [DOI] [PubMed] [Google Scholar]
- 37.Morita H, Zipes DP, Wu J. Brugada syndrome: insights of ST elevation, arrhythmogenicity, and risk stratification from experimental observations. Heart Rhythm. 2009;6:S34–43. doi: 10.1016/j.hrthm.2009.07.018. [DOI] [PubMed] [Google Scholar]
- 38.Kapplinger JD, Tester DJ, Salisbury BA, et al. Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION long QT syndrome genetic test. Heart Rhythm. 2009;6:1297–303. doi: 10.1016/j.hrthm.2009.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kapplinger JD, Tester DJ, Alders M, et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm. 2010;7:33–46. doi: 10.1016/j.hrthm.2009.09.069. [DOI] [PMC free article] [PubMed] [Google Scholar]