

Abstract
New chemotherapeutics are urgently needed to combat malaria. We previously reported on a novel series of antimalarial, ethylenediamine-based inhibitors of protein farnesyltransferase (PFT). In the current study, we designed and synthesized a series of second generation inhibitors, wherein the core ethylenediamine scaffold was varied in order to examine both the homology model of Plasmodium falciparum PFT (PfPFT) and our predicted inhibitor binding mode. We identified several PfPFT inhibitors (PfPFTIs) that are selective for PfPFT versus the mammalian isoform of the enzyme (up to 136-fold selectivity), that inhibit the malarial enzyme with IC50 values down to 1 nM, and that block the growth of P. falciparum in infected whole cells (erythrocytes) with ED50 values down to 55 nM. The structure–activity data for these second generation, ethylenediamine-inspired PFT inhibitors were rationalized by consideration of the X-ray crystal structure of mammalian PFT and the homology model of the malarial enzyme.
Introduction
Malaria is an infectious disease, prevalent primarily in the tropics and subtropics. With as many as 300–500 million cases reported each year, malaria causes between 1 and 3 million deaths annually, approximately 90% of which occur in Africa.1,2 Unfortunately, malaria mortality is increasing, especially in the highest risk group, African children.3 There are a number of likely reasons for this increase, the most important of which is increased resistance of malaria parasites to existing drugs.4–6 There is now a general consensus that new antimalarials are urgently needed.7
Transmitted by mosquitoes of the genus Anopheles, four species of the protozoal parasite Plasmodium are known to cause malaria in humans, namely falciparum, vivax, malariae, and ovale. Of these, P. falciparuma is the most virulent, and malaria mortality is almost exclusively attributable to infection by this species.1,7 Chloroquine, which is believed to disrupt heme polymerization, is one of the cheapest and most widespread drugs for malaria chemotherapy. However, P. falciparum has developed considerable resistance to chloroquine and to other antimalarial drugs, such as mefloquine and sulfadoxime/pyrimethamine,6,7 and in those countries that are affected most seriously, existing alternative chemotherapeutics are virtually unaffordable. Of significant concern is the identification of multidrug resistant strains of P. falciparum.5 The development of drug resistance is not the only cause of the increasing spread of malaria. Other factors also contribute to this worsening scenario, such as the resistance of the Anopheles mosquito to the pesticide DDT, the migration of refugee populations, and an ever-warming climate.8 The associated increase in malaria mortality has accelerated research into new antimalarial drugs, to disrupt not only conventional targets, such as heme polymerization, but also more novel targets, such as the biochemical pathways of fatty acid biosynthesis and mevalonate-independent isoprenoid biosynthesis.5 We believe that exploitation of these alternative targets will fast become essential, owing to the existence of multidrug resistant strains of P. falciparum coupled with the observation that the parasite readily mutates to develop resistance to new drugs (designed for conventional targets).5 Since the economic reality of the effective treatment of malaria is beyond the means of Third World countries, where this disease is most prevalent, this raises the need for inexpensive chemotherapeutics. Subsequently, while it is acknowledged that the majority of the cost of a new therapeutic lies in its clinical trials, to minimize the cost at the drug development stage and to expedite access to new antimalarials, there has been considerable research into the possible antimalarial activity of drugs designed for other diseases in a so-called “piggy-back” approach.9–14
Mammalian protein farnesyltransferase (PFT) is a key target for the antagonism of oncogenic Ras activity that is found in around 30% of human cancers,15 and a number of protein farnesyltransferase inhibitors (PFTIs) have shown antitumor activity, having progressed to phase II/III in clinical trials.16 PFT, a member of the prenyltransferase family, is one of three closely related heterodimeric zinc metalloenzymes (the others being the protein geranylgeranyltransferases I and II, PGGT-I and PGGT-II, respectively) that are important post-translational modification enzymes, catalyzing protein prenylation and subsequent membrane association.17 PFT catalyzes the transfer of a C15 isoprenoid (farnesyl) unit from farnesylpyrophosphate (FPP) to the free thiol of a cysteine residue within a specific CaaX tetrapeptide sequence, located at the C-terminus of the substrate protein (e.g., RasGTPase), where a = an aliphatic amino acid and X (which contributes to substrate specificity) = M, S, A, or Q. Chakrabarti et al. have identified prenylated proteins and associated prenyltransferase activity in P. falciparum and confirmed the viability of P. falciparum protein farnesyltransferase (PfPFT) as a new antimalarial target.9,18 Upon administration of mammalian-designed anticancer PFTIs to P. falciparum-infected erythrocytes, a reduction in the cellular levels of farnesylated proteins was observed coupled with lysis of the parasites.13,18 More recently, Van Voorhis and co-workers have identified two P. falciparum mutants, each with single amino acid substitutions (Y837C19 and G612A20) in PfPFT that map to the predicted inhibitor binding site, which show resistance to tetrahydroquinoline (THQ)-based PfPFT inhibitors both in vitro and in whole cells, further supporting PfPFT as the target for antimalarial activity.19,20 We10,21–23 and others14 have successfully adopted this “piggy-back” approach with several series of anticancer PFTIs and observed antimalarial activity in every case.10,21–23 Notably, in animal studies we have cured rats infected with malaria via oral dosing of our PFT-targeted THQ-based inhibitors,24 while Schlitzer et al. have cured mice infected with malaria by intraperitoneal injection of their benzophenone-based PFTIs.14 The now-complete genome of P. falciparum indicates an apparent lack of PGGT-I,25 suggesting that no alternative protein prenylation can occur upon PfPFT inhibition, which may explain the observation that PFTIs have been found to be significantly more toxic to plasmodial cells than to mammalian cells.13 Indeed, if PFTIs are to be effective antimalarials, plasmodium selectivity may be required, since the antiproliferative nature of PFTIs may preclude or at least restrict their use in children and pregnant women, the main target groups in malaria therapy.
Using the sequence alignment of PfPFT on the template crystal structure of rat PFT complexed with the nonsubstrate tetrapeptide inhibitor CVFM and the cosubstrate FPP, we have developed a homology model of the active site of PfPFT.19,26 This model reveals a large (~20 × 20 × 20 Å3), open, and predominantly hydrophobic cavity, with FPP extending across the cavity base, itself forming part of the binding surface for the enzyme substrate. Further inspection of the active site homology model indicates that there are four subpockets. In the first, the Zn(II) ion is chelated by three residues (Cys661, Asp659, His838), with a water molecule hydrogen-bonded between the terminal phosphate of FPP and Asp659, defining the limit of the Zn binding domain. Second and third are two well-defined, predominantly hydrophobic subpockets (Lys149, Asn317, Ser150, Phe151, where Asn317 and Ser150 form a hydrophilic domain at the deepest point; and Trp456, Trp452, Tyr837). The fourth subpocket is a larger hydrophilic domain formed by Arg564 and three water molecules participating in a hydrogen-bonded network between Ser449 and Gln152. We have previously reported on a series of ethylenediamine-based inhibitors that were predicted to allow simultaneous access to the four subpockets within the PfPFT active site.26,27 Lead inhibitors displayed excellent activity in vitro (IC50 < 1 nM) and toxicity toward cultured parasites in whole cells (ED50 < 100 nM). Furthermore, these PFTIs represent the first antimalarials to exhibit selectivity for plasmodial over mammalian PFT (up to 145-fold selectivity). With such potent data and plasmodium PFT-isoform selectivity already achieved, the main aim of the present research was to investigate the validity of the PfPFT active site homology model, as well as the initial docking studies reported in our previous work,26,27 by introducing alternative scaffolds with different rigidities/flexibilities and with different nitrogen–nitrogen distances into our ethylenediamine-based inhibitors. In turn, we hoped that our findings would assist in future, potent PfPFT inhibitor design.
First, we illustrate the scaffold modifications that we chose to investigate and then comment on their abilities to dock in the PfPFT active site homology model using the computer modeling program GOLD.28 We describe the syntheses of these novel inhibitors and then present in vitro PfPFT inhibition data, as well as two sets of whole cell data of erythrocytes infected with either the 3D7 (chloroquine-sensitive) or the K1 (chloro-quine-resistant, pyrimethamine-resistant) strain of P. falciparum. To conclude the manuscript, we utilize our computational and experimental data to present quantitative structure–activity relationship (QSAR) models of PfPFT and discuss their implications.
Design
Previous research identified that our most potent ethylene-diamine-based inhibitors were functionalized as in 1 and where varying the R group proved critical to inhibitor potency. As reference compounds, we selected five derivatives of 1 with the following R groups: a, R = benzyl; b, R = 2-methylbenzyl; c, R = thiophen-3-ylmethyl; d, R = N-Boc-piperidin-4-ylmethyl; e = N-(2-pyrimidinyl)-piperidin-4-ylmethyl. The ethylenediamine-alternative scaffolds selected to test both our predicted inhibitor binding mode and the PfPFT active site homology model are illustrated in Figure 1. These fall into three categories with respect to their rigidities/flexibilities relative to the ethylenediamine scaffold (1). The first category possesses only 1,3-diaminopropane (2), which is acyclic and, with the additional methylene group, more flexible than ethylenediamine. The second category includes gem-dimethylethylenediamine (3), 2-aminoethanamide (4), and 3-aminopropanamide (5), which are acyclic and more rigid than ethylenediamine. The third category incorporates (±)-cis-1,2-diaminocyclopentane (6) and (±)-trans-1,2-diaminocyclopentane (7), (±)-cis-1,3-diaminocyclopentane (8) and (±)-trans-1,3-diaminocyclopentane (9), and (±)-cis-1,4-diaminocyclohexane (10) and (±)-trans-1,4-diaminocyclohexane (11), all of which are cyclic and more rigid than ethylenediamine and exhibit gradually increasing distances between the scaffold nitrogens. The corresponding rigid (±)-cis- and (±)-trans-1,2-diaminocyclobutanes and the (±)-cis- and (±)-trans-1,2-diaminocyclopropanes were deemed too unstable because of the “push–pull effect” and were not investigated.29
Figure 1.

The various scaffolds used in this study as alternatives to the ethylenediamine scaffold in 1.
Computational docking experiments were performed using the GOLD 3.1 software package.28 First, InsightII30 was used to draw structures (R = Bn), energy-minimize them and to prepare our homology model of the active site of PfPFT for use in GOLD. Novel ligands were assumed to maintain a similar binding mode to that hypothesized for 1a;26,27 the two imidazole rings and the cyanoaniline were loosely constrained to their associated binding pockets for each compound (for full details, see Experimental Methods). Visualization of GOLD low energy docked poses was performed with InsightII. In all figures presented (Figure 2 and Supporting Information Figures 1–8), the binding site of PFT has been surfaced, in which the enzyme cosubstrate farnesylpyrophosphate (FPP) forms part of that binding surface.
Figure 2.

Low energy docked conformations (GOLD28) of inhibitors loosely constrained to the predicted binding mode of 1a26,27 Docked inhibitors are colored by atom type and are overlaid by the predicted docking pose of 1a in yellow. Farnesylpyrophosphate (FPP) is shown in red. Hydrophobic surface residues are colored red, hydrophilic residues and structural waters are blue, and the Zn2+ ion is pink: (A) 1a docked alone; (B) (±)-6a overlaid with 1a; (C) (±)-9a overlaid with 1a.
The GOLD docking studies predicted that many of these alternative scaffolds should not be as well tolerated as the parent ethylenediamine scaffold in the active site of PfPFT (Figure 2 and Supporting Information Figures 1–8. As Supporting Information Figure 1 illustrates, the 1,3-diaminopropyl scaffold (inhibitor 2a) is accommodated well in the active site, although the extra methylene appears to require the inhibitor to buckle in order to allow all four N-substituents to reach their predicted subpockets. We anticipated that inhibitors derived from this scaffold may bind PfPFT well but not as potently as the parent ethylenediamine-based inhibitors. As expected, docking of the gem-dimethylethylenediamine-based inhibitor (Supporting Information Figure 2, inhibitor 3a) illustrates that all four subpockets can be reasonably accessed by the four N-derivatives in much the same way as the parent ethylenediamine scaffold, suggesting that these compounds may be potent inhibitors of PfPFT. While the amide-based scaffolds (Supporting Information Figure 3, inhibitor 4a) are also predicted to bind well, the constraints we imposed in the GOLD docking experiments have caused the increase in hydrophilicity of the core scaffold to be ignored, which may lead to an erroneous result due to the hydrophobic environment in which the inhibitor scaffold is predicted to bind. The rigid cis-1,2- (Figure 2B inhibitor 6a), trans-1,2- (Supporting Information Figure 4, inhibitor 7a), and cis-1,3-diaminocyclopentyl scaffolds (Supporting Information Figure 5, inhibitor 8a), and the cis-1,4-diaminocyclohexyl (Supporting Information Figure 7, inhibitor 10a) scaffold do not fit well in the active site because of their constrained structures that prevent simultaneous access by all four N-substituents into the four subpockets. These observations suggest that compounds with these scaffolds may be poor inhibitors of PfPFT. On the other hand, trans-1,3-diaminocyclopentyl-(Figure 2C, inhibitor 9a) and trans-1,4-diaminocyclohexyl-(Supporting Information Figure 6, inhibitor 11a) derivatives appear to be reasonably well accommodated, so we predicted these inhibitors may bind well to PfPFT. However, the increasing scaffold nitrogen–nitrogen distance may prove detrimental with bulkier R groups.
We hypothesized that if a structure with a diamino-based scaffold (whose nitrogens have been functionalized with four previously optimized groups) is loosely constrained to our predicted binding mode and subsequently docks well in the active site homology model, then that structure should bind well experimentally. In turn, this should be reflected by potent enzyme inhibition data. Conversely, structures that are predicted to bind less well should be poorer inhibitors. By testing this hypothesis over a range of different scaffolds that modulate the inhibitors’ abilities to simultaneously access all four predicted binding subpockets within the PfPFT active site, we should be able to garner enough information to develop a quantitative structure–activity relationship (QSAR) model and thereby validate both our predicted inhibitor binding mode and the homology model itself. It is our hope that this information will assist in future PfPFT inhibitor design.
Chemistry
1,3-Diaminopropane-Based Inhibitors (2a–e)
These inhibitors were prepared as in Scheme 1. N-Carbobenzyloxy-3-amino-1-propanol (12) was converted to its THP ether derivative (with dihydropyan (DHP) and catalytic pyridinium p-toluene-sulfonate (PPTS)), after which the Cbz protecting group was removed under standard hydrogenolytic conditions (H2 and 10% Pd/C) to furnish primary amine 13. Nucleophilic aromatic substitution of 13 with p-fluorobenzonitrile, followed by N-alkylation of secondary aniline 14 with 5-chloromethyl-1-methyl-1H-imidazole·HCl (15) under optimized conditions gave tertiary aniline 16 in a yield of 81% (or 97% based on recovered starting material (brsm)). Acid-catalyzed methanolysis of the THP protecting group furnished primary alcohol 17, which was coupled to the secondary sulfonamides 19a–e (prepared as described in Schemes 2 and 3) under Mitsunobu conditions, employing diisopropyl azodicarboxylate (DIAD)/triphenylphosphine (PPh3) as the redox system, to give PfPFT inhibitors 2a–e in excellent yields. Due to the success of the Mitsunobu reaction coupled with its convergent effect on the overall syntheses of these 1,3-diaminopropane-based PfPFTIs, we designed syntheses of the remaining PfPFTIs that also incorporated Mitsunobu as the final step.
Scheme 1a.

a (a) DHP, cat. PPTS, CH2Cl2, 0 °C → rt, 16 h, 85%; (b) H2, 10% Pd/C, MeOH, rt, 1 h, 82%; (c) p-fluorobenzonitrile, DIPEA, DMSO, 120 °C, 24 h, 87%; (d) (1) NaH, DMF, 0 °C, 30 min; (2) 15, 0 °C → rt, 3 h, 81% (or 97% brsm); (e) p-TsOH ·H2O, MeOH, rt, 1 h, 88%; (f) 19a–e, DIAD, PPh3, THF, rt, 1 h, 61–98%.
Scheme 2a.

a (a) RNH2, DIPEA, CH3CN, 0 °C → rt, 16 h, 81–93%.
Scheme 3a.

a (a) Boc2O, cat. DMAP, THF, rt, 16 h, 99%; (b) H2, 10% Pd/C, EtOH, rt, 16 h, 100%; (c) N-(2-pyrimidinyl)-4-hydroxymethylpiperidine, PPh3, DIAD, THF, rt, 16 h; (d) TFA/CH2Cl2, 1:1, rt, 3 h, 92% (two steps).
Sulfonamides for Mitsunobu Reactions (19a–e, 21)
The secondary sulfonamides 19a–d required for the Mitsunobu reactions (e.g., step f in Scheme 1) were prepared in simple one-step procedures from 1-methyl-1H-imidazole-4-sulfonyl chloride (18) and the respective primary amines in yields ranging from 81% to 93% (Scheme 2). Due to the unavailability of N-(2-pyrimidinyl)-4-aminomethylpiperidine and the need for the more acidic sulfonamide 21 in the synthesis of 3-aminoethanamide derivatives 5a and 5d as well as the trans-1,2-diaminocyclopentyl derivatives 7a–e, sulfonamide 19e was prepared by a different route (Scheme 3). Accordingly, treatment of 19a with Boc2O led to the fully derivatized sulfonamide 20 in quantitative yield. Subsequent hydrogenolysis of 20 furnished N-tert-butoxycarbonyl-1-methyl-1H-imidazole-4-sulfonamide (21), whose NH was expected to have a lower pKa than that of simple N-alkyl secondary sulfonamides, such as 19a, whose pKa values are just on the cusp (pKa ≈ 12) of acidic nucleophiles (NuH) allowed in the Mistunobu reaction. The enhanced acidity of 21 was subsequently found to resolve troublesome Mitsunobu reactions (see sections on the syntheses of 5a,d and 7a–e). Additionally, compound 21 was successfully coupled to N-(2-pyrimidinyl)-4-hydroxymethylpiperidine, giving 22, which, after Boc deprotection with TFA, yielded sulfonamide 19e in an excellent two-step yield of 92%.
gem-Dimethylethylenediamine-Based Inhibitors (3a–e)
The hindered gem-dimethylethylenediamine derivatives 3a–e were furnished as shown in Scheme 4. Nucleophilic aromatic substitution of 2-amino-2-methylpropan-1-ol (23) with p-fluorobenzonitrile in a sealed vessel at 180 °C for 48 h afforded 24 in 40% yield. Attempted chemoselective benzylation of the primary hydroxyl of 24 with NaH and BnBr led to a mixture of products. However, alternative protection of the alcohol with tert-butyldiphenylsilylchloride (TBDPSCl), employing imidazole (Im) as base and nucleophilic catalyst, gave the desired compound 25 in excellent yield. The zinc-binding imidazole group was next installed by treatment of secondary aniline 25 with NaH at 0 °C followed by addition of 5-chloromethyl-1-methyl-1H-imidazole·HCl (15) as before to furnish tertiary aniline 26. Quantitative deprotection of the TBDPS group with tetra-n-butylammonium fluoride (TBAF) was observed, and the resultant primary alcohol was condensed with the five sulfonamides 19a–e under Mitsunobu conditions at 45 °C to afford the final PfPFTIs 3a–e in 50–68% yields.
Scheme 4a.

a (a) p-fluorobenzonitrile, DIPEA, DMSO, 180 °C, 48 h, 40%; (b) TBDPSCl, Im, DMF, 45 °C, 18 h, 90%; (c) (1) NaH, DMF, 0 °C, 30 min; (2) 15, 0 °C → rt, 3 h, 53% (or 82% brsm); (d) TBAF, THF, 0 °C → rt, 2 h, 95%; (e) 19a–e, DIAD, PPh3, THF, 45 °C, 18 h, 50–68%.
2-Aminoethanamide-Based Inhibitors (4a, 4d) and 3-Aminopropanamide-Based Inhibitors (5a, 5d)
Due to earlier findings that incorporation of an amide bond into the inhibitor scaffold is not tolerated,31 we decided to make just two derivatives of each amide-containing scaffold, with a small (a, R = Bn) and a large (d, R = N-Boc-piperidin-4-ylmethyl) R group. The syntheses of these 2-aminoethanamide- (4a, 4d) and 3-aminopropanamide-based PfPFT inhibitors (5a, 5d) are presented in Scheme 5 and are described in full in Supporting Information.
Scheme 5a.

a (a) (1) 3-Methyl-3H-imidazole-4-carbaldehyde, AcOH, 4 Å molecular sieves, MeOH, rt, 1 h; (2) NaCNBH3, rt, 16 h, 63%; (b) benzyloxyacetyl chloride (or 3-benzyloxypropanoyl chloride), pyridine, CH2Cl2, rt, 2 h, 87–94%; (c) Zn(CN)2, 10 mol % Pd(PPh3)4, cat. Zn(OAc)2, cat. Zn dust, DMF, 120 °C, 2 h, 85–89%; (d) H2, 10% Pd/C, 0.5% conc HCl (v/v), EtOH, rt, 90 min, 84–94%; (e) 19a or 19d, PPh3, DIAD, THF, rt, 1 h, 52% for 4a, or 20% for 4d; or 21, PPh3, DIAD, THF, rt, 1 h, 56% for 34; (f) TFA/CH2Cl2, 1:1, rt, 3 h, 97%; (g) RBr, Cs2CO3, DMF, rt, 16–36 h, 50–96%.
(±)-cis-1,2-Diaminocyclopentane-Based Inhibitors ((±)-6a–e) and (±)-trans-1,2-Diaminocyclopentane-Based Inhibitors ((±)-7a–e)
The (±)-cis-1,2-diaminocyclopentyl-based inhibitors (±)-6a–e were furnished by following the synthetic steps described in Scheme 6. Racemic (±)-trans-2-benzyloxy-cyclopentylamine ((±)-36) was arylated with p-fluorobenzonitrile, using an excess of the aryl fluoride to compensate for reduced reactivity due to steric hindrance. N-Alkylation of the resultant secondary aniline (±)-37 with 15 as before furnished (±)-38 in a moderate yield of 56% (or 89% brsm), which was then smoothly debenzylated under optimized conditions (1 atm of H2, catalyst 10% Pd/C, 0.5% concentrated HCl in EtOH (v/v), 1 h) to furnish secondary alcohol (±)-39. The proposed final step in this synthesis, Mitsunobu reaction of (±)-39 with the secondary sulfonamides 19a–e, proved unsuccessful, leading to elimination rather than substitution, presumably due to the sterically encumbering tertiary amine and/or the low acidities of the sulfonamides (pKa ≈ 12) that places them at the uppermost limit of allowed nucleophiles for the Mitsunobu reaction. However, even with the more acidic sulfonamide 21 (Scheme 3), we again observed only elimination. Likewise, alternative, more powerful Mitsunobu redox systems such as N,N,N′,N′-tetramethylazodicarboxamide (TMAD)/tri-n-butylphos-phine (TBP) and cyanomethyl-tri-n-butylphosphorane (CMBP) proved fruitless. We next considered the steric hindrance in the desired transformation and found that Mitsunobu reaction of (±)-39 with diphenylphosphorylazide (DPPA), which involves the smaller and linear azide ion as the nucleophile, was more successful. These conditions led to a 3:2 inseparable mixture of the azide (substitution) product (±)-40a, wherein the usual inversion of stereochemistry had occurred, and the alkene (elimination) product (±)-40b. Staudinger reduction of azide (±)-40a in the mixture led to the anticipated increase in polarity, enabling facile separation from alkene (±)-40b and furnishing primary amine (±)-41 in a yield of 56% for the two steps. Subsequently, sulfonylation of (±)-41 afforded secondary sulfonamide (±)-42 in good yield, which was then alkylated with a series of bromides (or iodides) to give the (±)-cis-1,2-diaminocyclopentyl PfPFT inhibitors (±)-6a–e in poor to good yields. Importantly, for the two benzylic and thiophen-3-ylmethyl bromides, this reaction had to be performed under dilute (0.01 M) conditions to reduce the facile quaternization of the Zn(II)-binding imidazole τ-nitrogen (Nτ-alkylation), a side reaction that competed effectively with the desired sulfonamide alkylation because of considerable steric hindrance around the sulfonamide NH.
Scheme 6a.
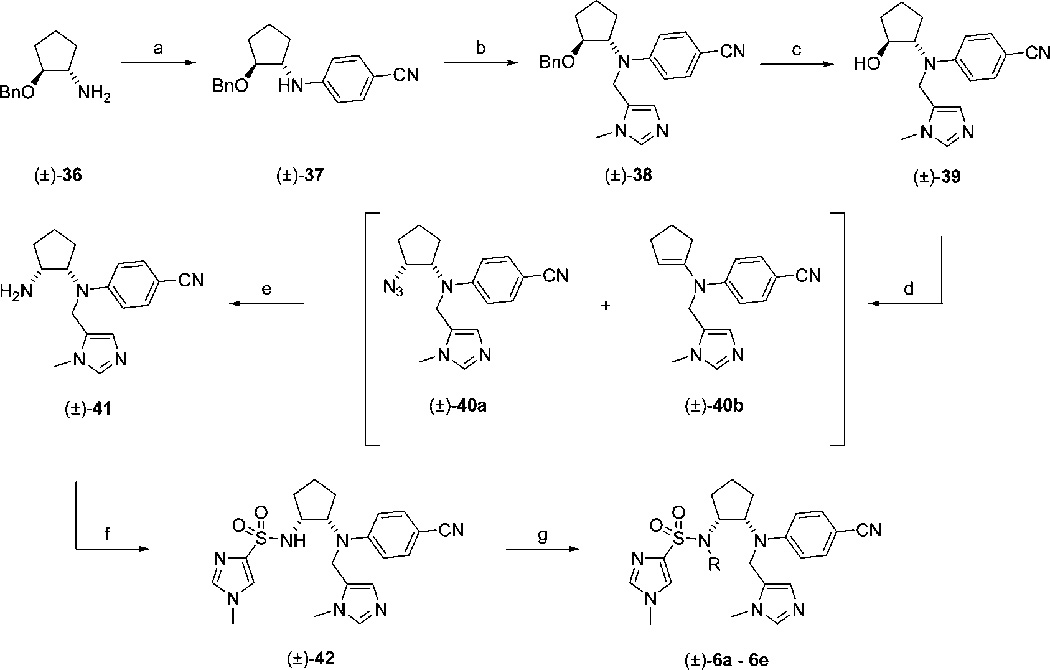
a (a) p-Fluorobenzonitrile, DIPEA, DMSO, 120 °C, 24 h, 87%; (b) (1) NaH, DMF, 0 °C, 30 min; (2) 15, 0 °C → rt, 3 h, 56% (or 89% brsm); (c) H2, 10% Pd/C, 0.5% conc HCl, EtOH, rt, 1 h, 87%; (d) PPh3, DIAD, DPPA, THF, 0 °C → rt, 16 h; (e) (1) PPh3, THF, rt, 1 h; (2) H2O, 65 °C, 7 h, 56% (combined yield for steps d and e); (f) 18, DIPEA, CH3CN, 0 °C → rt, 16 h, 75%; (g) RBr (or RI), Cs2CO3, DMF, rt, 16 h to 7 days, 22–81%.
In order to prepare the corresponding trans analogues, (±)-39 was reacted with p-nitrobenzoic acid under Mitsunobu conditions (Scheme 7), which again led to an approximate 3:2 mixture of ester (substitution product) and alkene (elimination product). No attempt at purification was made at this stage, and LiOH·H2O was added directly to the reaction mixture. After 3 h, the ester hydrolysis was complete. Purification of the reaction mixture gave the cis product (±)-43 in 39% yield for the two steps, whose inverted stereochemistry was evident from the comparison of 1H NMR spectra of (±)-39 and (±)-43. Again, the subsequent Mitsunobu reaction with secondary sulfonamide 19a was unfruitful. However, reaction with the alternative sulfonamide 21 was a success, likely due to a combination of the increased acidity of 21 relative to the other sulfonamides 19a–e and reduced steric hindrance in the substitution step of the Mitsunobu reaction. Silica gel flash column chromatography furnished (±)-44 in approximately 90% purity, and subsequent removal of the Boc group with TFA, followed by the usual purification, generated pure secondary sulfonamide (±)-45 in a yield of 63% for the two steps. Finally, alkylation of sulfonamide (±)-45 with the five bromides as before gave the final (±)-trans-1,2-diaminocyclopentyl PfPFT inhibitors (±)-7a–e in good yields.
Scheme 7a.

a (a) p-Nitrobenzoic acid, PPh3, DIAD, THF, rt, 16 h; (b) LiOH·H2O, THF/MeOH/H2O, 3:1:1, rt, 3 h, 39% (two steps); (c) 21, PPh3, DIAD, THF, 45 °C, 16 h; (d) TFA/CH2Cl2, 1:1, rt, 3 h, 63% (two steps); (e) RBr, Cs2CO3, DMF, rt, 16 h to 7 days, 75–85%.
(±)-cis-1,3-Diaminocyclopentane-Based Inhibitors ((±)-8a–e) and (±)-trans-1,3-Diaminocyclopentane-Based Inhibitors ((±)-9a–e)
The 1,3-diaminocyclopentyl inhibitors (±)-8a-–e and (±)-9a–e were prepared as shown in Scheme 8. After quantitative O-silylation of hepta-1,6-dien-4-ol (46) with TB-DPSCl, the cyclopentane scaffold was constructed by treatment with Grubbs’s catalyst to give 48. Subsequent oxidation with m-CPBA in CH2Cl2 gave cis epoxide 49 and trans epoxide 50 in an approximate 1:1 ratio, in a combined yield of 84%. Reductive opening of the epoxide ring of 49 with H2 and a range of catalysts, including 10% Pd/C, Pd(OH)2, or PtO2, were all attempted but were very slow even at pressures of up to 70 psi of H2. Alternative treatment with LiAlH4 was successful, furnishing racemic (±)-51 in 93% yield. On the other hand, while executing the reductive opening of the epoxide ring of diastereomeric compound 50, LiAlH4 also caused the surprising removal of the TBDPS protecting group to give (±)-trans-cyclopentane-1,3-diol ((±)-56) in 84% yield. Nucleophilic aromatic substitution test reactions of the primary amino analogue of (±)-51, specifically 1-tert-butyldiphenylsilyloxy-2-aminocyclopentane, with p-fluorobenzonitrile, led to removal of the silicon protecting group, presumably by the action of liberated fluoride ion, and subsequent O-arylation. Therefore, single re-silylation of (±)-56 was not attempted; monobenzylation of (±)-56 under phase transfer conditions was instead effected to give (±)-57 in excellent yield.
Scheme 8a.

a (a) TBDPSCl, Im, THF, 45 °C, 16 h, 99%; (b) Grubbs’s first generation catalyst, CH2Cl2, rt, 3 days, 63%; (c) m-CPBA, CH2Cl2, 0 °C → rt, 16 h, 43% (49), 41% (50); (d) LiAlH4, THF, 0 °C, 2 h, 93% ((±)-51) or 84% ((±)-56); (e) BnBr, NaH, DMF, 0 °C → rt, 16 h, 98%; (f) TBAF, THF, 0 °C → rt, 3 h, 97%; (g) PPh3, DIAD, DPPA, THF, rt, 16 h, 88% (from (±)-52) or 92% (from (±)-57); (h) (1) PPh3, THF, rt, 1 h; (2) H2O, 65 °C, 7 h, 94% ((±)-53) or 97% ((±)-58); (i) p-fluorobenzonitrile, DIPEA, DMSO, 120 °C, 2 d, 48 h, 99% (from (±)-53) or 94% (from (±)-58); (j) (1) NaH, DMF, 0 °C, 30 min; (2) 15, 0 °C → rt, 3 h, 52% ((±)-54) or 47% ((±)-59); (k) H2, 10% Pd/C, 0.5% conc HCl (v/v), EtOH, rt, 1 h, 83% ((±)-55) or 85% ((±)-60); (l) 19a–e, PPh3, DIAD, THF, rt, 16 h, 32–79%; (m) (1) NaH, THF, 0 °C, 1 h; (2) BnBr, TBAI, 0 °C → rt, 16 h, 95%.
In order to keep the two syntheses, which were already being conducted in tandem, as similar as possible, we executed a two-step protecting group exchange on O-TBDPS derivative (±)-51 to give O-benzyl compound (±)-52 (95%, two steps). Mitusnobu conditions effected conversion of the hydroxyl of (±)-52 to the inverted azide whose reduction to primary amine (±)-53 was accomplished with PPh3 and water (Staudinger reaction). Nucleophilic aromatic substitution of (±)-53, followed by N-alkylation as before gave tertiary aniline (±)-54, which was then subjected to optimized hydrogenolytic cleavage conditions to furnish secondary alcohol (±)-55 in a yield of 83%. Finally, coupling of (±)-55 to the series of secondary sulfonamides 19a–d with PPh3 and DIAD proceeded in poor to good yields to afford the (±)-trans-1,3-diaminocyclopentyl PfPFT inhibitors (±)-8a–d. Due to the limited solubility of sulfonamide 19e and the already low yields for this final Mitsunobu step, compound 8e had to be prepared in the same manner as that used to prepare the trans-1,2-diaminocyclopentane-based inhibitors. Specifically, (±)-55 was coupled to sulfonamide 21 under standard Mitsunobu conditions, after which the Boc group was removed by TFA, and then the resultant secondary sulfonamide was alkylated with N-(2-pyrimidinyl)-4-iodomethylpiperidine, furnishing (±)-8e. For the syntheses of the (±)-trans-1,3-diaminocyclopentyl PfPFT inhibitors (±)-9a–e, the synthetic transformations were identical from (±)-57, as depicted in Scheme 8.
(±)-cis-1,4-Diaminocyclohexyane-Based Inhibitors ((±)-10a, (±)-10d) and (±)-trans-1,4-Diaminocyclohexane-Based Inhibitors ((±)-11a, (±)-11d)
Scheme 9 illustrates the synthetic steps pursued in order to furnish the (±)-cis-1,4- ((±)-10a, (±)-10d) and the (±)-trans-1,4-diaminocyclohexyl ((±)-11a, (±)-11d) PfPFT inhibitors, for which only the R = Bn and R = N-Boc-piperidin-4-ylmethyl derivatives were prepared. (±)-cis-1,4-Diaminocyclohexane ((±)-61) was mono-arylated with p-fluorobenzonitrile in 77% yield to give (±)-62, which was then sulfonylated with 1-methyl-1H-imidazole-4-sulfonyl choride (18) to furnish 63. Chemoselective alkylation of the sulfonamide NH with benzyl bromide and N-Boc-4-bromomethylpiperidine was accomplished without incident, affording (±)-64a and (±)-64d, respectively. Finally, the secondary anilines were alkylated with 15 to yield the target PfPFT inhibitors (±)-10a and (±)-10d in moderate yields, where the mass balance was recovered starting material. Starting from (±)-trans-1-tert-butoxycarbo-nylamino-4-aminocyclohexane ((±)-65), the trans isomers (±)-11a and (±)-11d were furnished in a similar fashion.
Scheme 9a.

a (a) p-fluorobenzonitrile, DIPEA, DMSO, 120 °C, 48 h, 77% (for (±)-62) or 89% (from (±)-65); (b) 18, DIPEA, CH3CN, rt, 16 h, 76% (for (±)-63) or 86% (for (±)-66); (c) RBr, Cs2CO3, DMF, rt, 16 h to 3 days, 82–99%; (d) (1) NaH, DMF, 0 °C, 30 min; (2) 15, 0 °C → rt, 12 h, 61–69% (92–95% brsm); (e) TFA/CH2Cl2, 1:1, rt, 30 min, 100%.
Results and Discussion
Ethylenediamine-Based Inhibitors (1a–e)
For comparison with the 10 series of inhibitors in this study, we present a table that includes previously published data on the corresponding ethylenediamine-based inhibitors.26,27 Table 1 shows percentage enzyme inhibition of PfPFT by compounds 1a–e at 50 and 5 nM inhibitor concentration. Also shown are ED50 data, which are the required inhibitor concentration to inhibit 50% of the growth of parasites (two strains of P. falciparum: 3D7 and K1) in whole cells (erythrocytes), as determined through the incorporation of tritium-labeled hypoxanthine (see Experimental Methods for details). As indicated in Table 1, all inhibitors exhibited potent inhibition of PfPFT in vitro (≥74% inhibition at 5 nM) and proved highly effective antimalarials in whole cells (several ED50 values <100 nM).
Table 1.
Enzyme Inhibition and Whole Cell Data for Ethylenediamine-Based Inhibitors 1a–ea
 | |||||
|---|---|---|---|---|---|
| Compound | Inhibition of PfFTase (%) | ED50 (nM)a | |||
| Number | R | 50 nM | 5 nM | 3D7 | K1 |
| 1a |  |
98 | 86 | 349 | 375 |
| 1b |  |
99 | 95 | 93 | 150 |
| 1c |  |
100 | 91 | 150 | 240 |
| 1d | 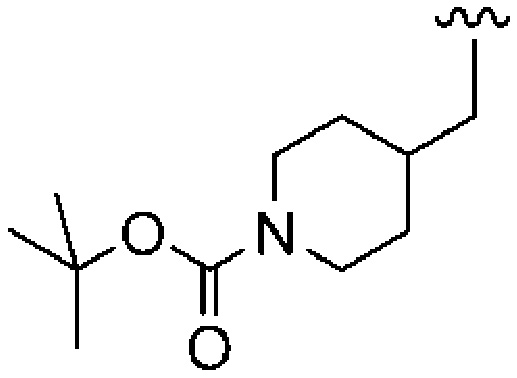 |
96 | 81 | 88 | 54 |
| 1e |  |
96 | 74 | 130 | 85 |
ED50 = effective dose of inhibitor required to decrease P. falciparum growth (in infected erythrocytes) by 50%.
1,3-Diaminopropane-Based Inhibitors (2a–e)
By comparison of the percentage enzyme inhibition data for the 1,3-diaminopropane-based inhibitors (Table 2) with the corresponding data for the ethylenediamine scaffold derivatives (Table 1), compounds 2a, 2b, and 2c were all much poorer inhibitors of PfPFT. However, with the larger N-Boc-piperidin-4-ylmethyl (2d) and N-(2-pyrimidinyl)-piperidin-4-ylmethyl derivatives (2e), inhibition of the enzyme was almost the same as that for the corresponding ethylenediamine derivatives (e.g., 2d, 73% inhibition at 5 nM, vs 1d, 81% inhibition at 5 nM), suggesting that the reduction in activity caused by the additional methylene in the scaffold may have been offset by incorporating a large R group. Indeed, compound 2d was an especially potent inhibitor of PfPFT, with an IC50 of 1 nM (Table 7). ED50 data reflect the variation in PfPFT inhibitor potency, supporting inhibition of PfPFT as the target for antimalarial activity, with 2d proving the most effective antimalarial of this series in whole cells (ED50 = 330 nM (3D7), 190 nM (K1)). A low energy docked conformation of 2a overlaid with that of 1a (Supporting Information Figure 1) suggested that the ethylenediamine-based derivative should be a much better fit in the homology model of the PfPFT active site and that 2a with its extra methylene unit in the scaffold should be too long to bind as well as 1a, requiring a degree of buckling of the scaffold to enable simultaneous access of the four N-appendages into the four subpockets. This prediction appears to have been confirmed experimentally, particularly in the case of the smaller R group derivatives.
Table 2.
Enzyme Inhibition and Whole Cell Data for 1,3-Diaminopropane-Based Inhibitors 2a–ea
 | |||||
|---|---|---|---|---|---|
| Compound | Inhibition of PfFTase (%) | ED50 (nM) | |||
| Number | R | 50 nM | 5 nM | 3D7 | K1 |
| 2a |  |
71 | 16 | 3100 | 2650 |
| 2b |  |
76 | 24 | 2900 | 2900 |
| 2c |  |
73 | 18 | 2800 | 2200 |
| 2d |  |
94 | 73 | 330 | 190 |
| 2e |  |
93 | 62 | 1300 | ND |
ND = not determined because of limited stocks of PfPFT enzyme or Plasmodium-infected whole cell cultures.
Table 7.
Comparative P. falciparum and Rat PFT Inhibition Data for a Series of Inhibitors Where R = N-Boc-piperidin-4-ylmethyl
 | ||||
|---|---|---|---|---|
| Compound | IC50 (nM)a | Selectivityb | ||
| Number | X | Plasmodium | rat | |
| 1d |  |
1.2 | 140 | 117 |
| 2d |  |
1 | ND | - |
| 3d |  |
1.1 | 150 | 136 |
| 6d |  |
350 | >1000 | >2.9 |
| 7d |  |
10 | >1000 | >100 |
| 9d |  |
3.1 | ND | - |
Inhibitor concentration required to decrease P. falciparum or rat PFT activity by 50%. ND = not determined.
Ratio of rat to P. falciparum PFT IC50 values.
gem-Dimethylethylenediamine-Based Inhibitors (3a–e)
Discounting thiophene 3c, the gem-dimethylethylenediamine scaffold derivatives (Table 3) were as potent, if not more so, as the corresponding ethylenediamine compounds (e.g., 3a, 88% inhibition, vs la, 86% inhibition at 5 nM; and 3e, 93% inhibition, vs le, 74% inhibition at 5 nM). Especially potent whole cell activity, such as ED50 = 160 nM (3D7) and 55 nM (Kl) for 3a, parallels potent enzyme inhibition data in most cases, again supporting PfPFT as the relevant target for antimalarial activity. Indeed, 3a is one of our most potent, ethylenediamine-inspired antimalarials to date, and the improved whole cell activity relative to la (ED50 = 349 nM (3D7) and 375 nM (Kl)) may be a consequence of the increased hy-drophobicity of the scaffold, facilitating cellular entry. GOLD docking studies (Supporting Information Figure 2) suggested that these gem-dimethylethylenediamine-based inhibitors should be tolerated in the PfPFT active site as well as the parent ethylenediamines, and this has been supported experimentally. The reason for the increased potency of the gem-dimethylethylenediamine-based 3e in vitro may be due to additional hydrophobic contacts between the extra methyls of the inhibitor and the scaffold-binding region of the active site or to improved contacts of the p-cyanoaniline as a direct consequence of the Thorpe–Ingold effect32 or to a combination of both effects.
Table 3.
Enzyme Inhibition and Whole Cell Data for gem-Dimethyl ethyl enediamine-Based Inhibitors 3a–ea
 | |||||
|---|---|---|---|---|---|
| Compound | Inhibition of PfFTase (%) | ED50 (nM) | |||
| Number | R | 50 nM | 5 nM | 3D7 | K1 |
| 3a |  |
100 | 88 | 160 | 55 |
| 3b |  |
98 | 91 | 210 | 75 |
| 3c |  |
70 | ND | 2450 | ND |
| 3d |  |
87 | ND | 400 | 250 |
| 3e |  |
100 | 93 | 180 | 75 |
ND = not determined.
2-Aminoethanamide- and 3-Aminopropanamide-Based Inhibitors (4a,d and 5a,d)
All the amide derivatives (4a,d and 5a,d) (Figure 3) showed 0% enzyme inhibition at 50 nM and were not studied further. Even though GOLD docking studies suggested that these derivatives would be reasonably well accommodated in the PfPFT active site (Supporting Information Figure 3), we invoked several constraints that meant the increase in hydrophilicity of the scaffold would be ignored. The scaffold-binding region of the active site is hydrophobic, so it would be expected that a compound with a polar component in the scaffold, such as an amide bond, would be poorly tolerated. In addition, the rigidity incurred upon inclusion of the planar amide bond, which we hoped to offset by the addition of an extra methylene in the scaffold (4 → 5), and the amide-induced withdrawal of electrons from the p-cyanophenyl group (a moiety that is known to contribute particular potency to our inhibitors27) are two possible further reasons as to why these amide derivatives showed no inhibition of PfPFT at 50 nM.
Figure 3.

2-Aminoethanamide- (n = 1) and 3-aminopropanamide- (n = 2) based inhibitors.
(±)-cis- and (±)-trans-l,2-Diaminocyclopentane-Based Inhibitors ((±)-6a–e and (±)-7a–e)
As Table 4 shows, the (±)-cis-l,2-diaminocyclopentyl-based derivatives (±)-6a–e performed very poorly indeed, with little or no inhibition of PfPFT at 50 nM inhibitor concentration. Similarly, ED50 values were disappointing; in most cases they were at least an order of magnitude worse than their ethylenediamine-based parent compounds. These results were expected, since docking studies of (±)-6a in the homology model of the enzyme active site indicated that the constraint imposed by the cis 1,2-cyclic scaffold meant that only one of the two functionalized scaffold amines could project its appendages into its two predicted binding subpockets (Figure 2B).
Table 4.
Enzyme Inhibition and Whole Cell Data for (±)-cis-1,2-Diaminocyclopentane-Based Inhibitors (±)-6a–e and for (±)-trans-1,2-Diaminocyclopentane-Based Inhibitors (±)-7a–e
 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | Inhibition of PfFTase (%) | ED50 (nM) | Compound | Inhibition of PfFTase (%) | ED50 (nM) | ||||||
| Number | R | 50 nM | 5 nM | 3D7 | K1 | Number | R | 50 nM | 5 nM | 3D7 | K1 |
| 6a |  |
0 | 0 | >5000 | 2400 | 7a |  |
42 | 20 | 3100 | 2350 |
| 6b |  |
17 | 16 | 2800 | 2800 | 7b |  |
35 | 12 | 3000 | 3200 |
| 6c |  |
9 | 0 | >5000 | 4200 | 7c |  |
57 | 11 | 2700 | 2700 |
| 6d |  |
2 | 3 | 3000 | 1250 | 7d |  |
82 | 41 | 2650 | 600 |
| 6e |  |
0 | 10 | 3000 | 2500 | 7e |  |
28 | 12 | 2600 | 750 |
On the other hand, although the trans diastereoisomers (±)-7a–e (Table 4) did not perform better than the corresponding ethylenediamine-based inhibitors (Table 1), they were more active than their cis counterparts (±)-6a–e (Table 4) at both 50 and 5 nM inhibitor concentration; the most potent trans compound, (±)-7d, displayed 41% enzyme inhibition at 5 nM (cf. 81% for 1d and 3% for 6d). This improvement in activity in the enzyme assay was also reflected in superior whole cell activity, with (±)-7d exhibiting an ED50 of 600 nM for Kl strain (cf ED50 = 1250 nM for (±)-6d (Kl)). These trends in the experimental results (cis-l,2-diaminocyclopentane vs trans-1,2– diaminocyclopentane vs ethylenediamine) appear to be mirrored in the GOLD docking studies. While the trans-l,2-diaminocyclopentyl derivatives were predicted (Supporting Information Figure 4) to be unable to simultaneously access all four subpockets as effectively as the corresponding ethylenediamine-based inhibitors, the trans derivatives ((±)-7a–e) appeared to exhibit better complementarity to the active site than did their more sterically encumbered cis counterparts ((±)-6a–6e).
(±)-cis- and (±)-trans-l,3-Diaminocyclopentane-Based Inhibitors ((±)-8a–e and (±)-9a–e)
As Table 5 shows, the (±)-cis-l,3-diaminocyclopentyl-based derivatives (±)-8a–e were not very active PfPFT inhibitors relative to the ethylenediamine parent compounds 1a–e (Table 1). Nonetheless, these inhibitors were more potent than the corresponding cis-l,2-diaminocyclopentyl derivatives (±)-6a–e (Table 4) and approximately as potent as the corresponding trans-l,2-diaminocyclopentyl-based inhibitors (±)-7a–e (Table 4). For example, compound (±)-8a exhibited 40% inhibition at 50 nM inhibitor concentration, compared with 0% for (±)-6a and 42% for (±)-7a. N-Boc-piperidin-4-ylmethyl compound (±)-8d was approximately as active as (±)-7d in terms of inhibition of PfPFT ((±)-8d, 33% inhibition at 5 nM, vs (±)-7d, 41% inhibition at 5 nM) but even more potent in whole cells with ED50 values of 300 nM (3D7) and 125 nM (Kl) for (±)-8d compared with ED50 values of 2650 nM (3D7) and 600 nM (Kl) for (±)-7d. GOLD docking studies correctly predicted that the cis-l,3-diaminocyclopentyl scaffold would not facilitate binding as well as the ethylenediamine scaffold (Supporting Information Figure 5) and also that simultaneous access to the four subpockets would be more reasonable than with the isomeric but more sterically congested cis-l,2-diaminocyclopentyls (compare Figure 2B and Supporting Information Figure 5).
Table 5.
Enzyme Inhibition and Whole Cell Data for (±)-cis-l,3-Diaminocyclopentane-Based Inhibitors (±)-8a–e and for (±)-trans-l,3-Diaminocyclopentane-Based Inhibitors (±)-9a–ea
 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | Inhibition of PfFTase (%) | ED50 (nM) | Compound | Inhibition of PfFTase (%) | ED50 (nM) | ||||||
| Number | R | 50 nM | 5 nM | 3D7 | K1 | Number | R | 50 nM | 5 nM | 3D7 | K1 |
| 8a |  |
40 | 4 | 700 | 800 | 9a |  |
93 | 65 | 340 | 145 |
| 8b |  |
24 | ND | 2400 | 2600 | 9b |  |
33 | ND | 2400 | 2200 |
| 8c |  |
15 | ND | 2400 | 2500 | 9c |  |
56 | ND | 1500 | 1200 |
| 8d |  |
83 | 33 | 300 | 125 | 9d |  |
97 | 86 | 310 | 80 |
| 8e |  |
6 | ND | 600 | 720 | 9e |  |
38 | ND | 2600 | 1500 |
ND = not determined.
As was the case with the 1,2-cyclopentyl derivatives, the trans-l,3-diaminocyclopentyl-derived inhibitors (±)-9a–e were more active (Table 5) than their analogous cis-l,3-diaminocyclopentyl diastereomers (±)-8a–e. Especially noteworthy are compounds (±)-9a and (±)-9d, which were approximately as active as the corresponding ethylenediamine inhibitors: a, 93% inhibition at 50 nM for (±)-9a vs 98% for la; d, 86% inhibition at 5 nM for (±)-9d vs 81% for 1d. These potent enzyme inhibition data are reflected in potent whole cell data, such as ED50 = 80 nM (Kl) for (±)-9d, making (±)-9d one of our most effective ethylenediamine-inspired antimalarials to date. These experimental findings confirm the docking studies, which suggested that the trans-l,3-diaminocyclopentyl scaffold would allow access by all four N-substituents to each of the predicted subpockets in a similar manner to the ethylenediamine scaffold and were, therefore, predicted to be good inhibitors of PfPFT (Figure 2C).
(±)-cis and (±)-trans-l,4-Diaminocyclohexane-Based Inhibitors ((±)-10a, (±)-10d, (±)-11a, and (±)-11d)
For the cyclohexyl scaffold, we made only two derivatives of each diastereoisomer, incorporating either the small benzyl group or the larger N-Boc-piperidin-4-ylmethyl group; the percentage enzyme inhibition data and whole cell ED50 data are shown in Table 6. The cis-l,4-diaminocyclohexyl derivative (±)-10a exhibited limited inhibition of PfPFT (23% inhibition at 50 nM), while the trans isomer (±)-11a was more active (73% inhibition at 50 nM). Both isomers were less potent than the parent ethylenediamine-based inhibitor la, which was predicted by GOLD docking studies. In the case of (±)-10a, the cis configuration of the scaffold appeared to render it difficult for both the scaffold nitrogens to deliver their appendages into the proposed binding pockets (Supporting Information Figure 6). Conversely, in the case of (±)-11a, the trans configuration was predicted to facilitate simultaneous access to all four subpockets, although the greater interscaffold nitrogen–nitrogen distance appeared to be less optimal than for the parent inhibitor la (Supporting Information Figure 7), possibly accounting for the slightly worse percentage inhibition data. N-Boc-piperdin-4-ylmethyl derivatives (±)-10d and (±)-11d demonstrated no inhibitory activity against PfPFT, which is likely a consequence of the large scaffold coupled with the bulky R group rendering these inhibitors too big to access the active site.
Table 6.
Enzyme Inhibition and Whole Cell Data for (±)-cis-l,4-Diaminocyclohexane-Based Inhibitors (±)-10a and (±)-10d and (±)-trans-l,4-Diaminocyclohexane-Based Inhibitors (±)-11a and (±)-11d
 | ||||
|---|---|---|---|---|
| Compound | Inhibition of PfFTase at 50 nM (%) |
ED50 (nM) K1 |
||
| Number | X | R | ||
| 10a |  |
 |
23 | 2750 |
| 10d |  |
0 | 650 | |
| 11a |  |
 |
73 | 1800 |
| 11d |  |
0 | 1200 | |
Selectivity
Previously reported PfPFT inhibitors have demonstrated poor selectivity for parasitic PFT over mammalian PFT or are highly selective for the mammalian enzyme.9,10,18 Although PFT inhibitors have demonstrated limited toxicities to mammalian cells at concentrations required to effect a therapeutic response,10 the antiproliferative nature of PFT inhibitors may restrict their use by children and pregnant women, two of the main target groups in malaria therapy. Hence, the selective inhibition of parasitic PFT may prove mandatory in order to realize safe and effective antimalarial PFT inhibitors. We previously reported on the selectivity of our ethylenediamine-based inhibitors for PfPFT over rat PFT; IC50 values revealed several inhibitors with greater than 100-fold selectivity for the parasitic PFT.27 The amino acid sequences of rat and human PFT are 95% identical with complete sequence and structural conservation around the active site.33 While the aim of this research was not to design ever more Plasmodium-selective PFT inhibitors, it is worth noting that modification to the inhibitor scaffold has had no detrimental effect on PFT selectivity. Specifically, for a series of the N-Boc-piperidin-4-ylmethyl derivatives (selected as representative examples of inhibitors bearing the alternative scaffolds) greater than 100-fold selectivity for parasitic over mammalian PFT was observed (Table 7). In particular, compound 3d represents one of our most potent (PfPFT IC50 = 1.1 nM) and most selective (136-fold) PfPFT inhibitors.
QSAR Models
Quantitative structure–activity relationship (QSAR) models were generated to help determine the validity of both the proposed binding mode and the PfPFT active site homology model itself. Experimental data selected were the percentage inhibition of PfPFT at 5 nM inhibitor concentration. Given the often imprecise nature of percentage inhibition data and the unavailability of multiple assay results for any given compound, care was taken to ignore any model that suggested an accuracy of greater than 90% (i.e., r2 or q2 > 0.9). The corresponding theoretical data for the models were obtained as follows. For the first QSAR model, docked poses of each ligand constrained to our hypothesized binding mode were used for descriptor calculations, and the resultant GOLD-Score values were included as descriptors in the modeling. Nineteen samples were randomly chosen as a training set, leaving nine samples as a test set. Further details can be obtained by consulting the Experimental Methods. As shown in Figure 4, the training set has an r2 value of 0.86, suggesting there is very good correlation between the observed inhibition and the predicted inhibition of PfPFT at 5 nM inhibitor concentration. Indeed, a test set of the QSAR model was found to have a q2 value of 0.81, suggesting that this model may hold strong predictive power.
Figure 4.

QSAR model in which descriptors were calculated on the basis of docked poses of ligands constrained to our hypothesized binding mode: (blue diamond) training set, r2 = 0.86; (pink square) test set, q2 = 0.81.
In order to further test our hypothesis regarding the proposed binding mode of these molecules to PfPFT and of the homology model itself, a second QSAR model was prepared. For this model, descriptors were calculated on the basis of molecules that had been energy minimized rather than docked, with the exception of GOLD-Score derived values, which were still included but left unaltered. Also, the training and testing sets were deliberately chosen to be identical to those in the previous model rather than randomly chosen. Other than this, all operations were identical between the two models. The results of this second model are shown in Figure 5. While similar accuracy was achieved for the training set (r2 = 0.86), very little predictive power was seen with the test set (q2 = 0.37), suggesting that this model simply memorized, rather than learned, data from the training set. Also, it suggests that there is information content in the docked poses and thus that these docked poses are reasonable. Taken together, these QSAR models inform us that our hypothesis of the proposed binding mode is sound and that the homology model is a good approximation of the active site of PfPFT.
Figure 5.

QSAR model in which descriptors were calculated on the basis of ligands that had been energy-minimized: (blue diamond) training set, r2 = 0.86; (pink square) test set, q2 = 0.37.
Conclusions
We have synthesized several series of novel antimalarials incorporating a variety of scaffolds based on our previously reported ethylenediamine-derived inhibitors of PfPFT. These antimalarials were designed to allow exploration of the PfPFT active site and therein assess the validity of our predicted inhibitor binding mode and of the active site homology model itself. In turn, it was hoped that this garnered information would, in the near future, facilitate, and thereby accelerate, access to increasingly more-potent PfPFT inhibitors. Low energy docked conformations (GOLD), which were performed by loosely constraining our inhibitors to the docking pose we predicted in our previous publications,26,27 suggested which compounds would inhibit PfPFT well and which would not. Broadly speaking, biological evaluation of our compounds agreed with the computational docking studies, and from these data we developed two QSAR models that suggested the predicted binding mode for our inhibitors is reasonable and that the homology model we have used to design inhibitors is a good approximation of the PfPFT active site. In addition, our first QSAR model seems to have considerable predictive power and with suitable biological validation could therefore be used to design new inhibitors. Importantly, as well as proving particularly cytotoxic to cultured parasites (ED50 < 100 nM), some of our novel antimalarial PfPFTIs reported herein are among the most potent (IC50 ≈ 1 nM) and the most selective for parasitic PFT over mammalian PFT (up to 136-fold) currently reported in the literature.
Considering cost involved and speed and ease of synthesis, we conclude that the parent ethylenediamine-based inhibitors reported previously26,27 are our best antimalarials of this series thus far, and given the already high degree of selectivity over the mammalian isoform of PFT, future research should now be directed toward three goals. Inhibitor 1d is highly potent (IC50 = 1.2 nM), selective for parasitic over mammalian PFT (117-fold), and exhibits very good whole cell activity (ED50 = 88 nM (3D7), 54 nM (Kl)). Therefore, we first suggest further work is undertaken on this inhibitor to improve these ED50 values, which may be achieved by substituting the 3-methyl-3H-imidazole-4-sulfonyl group with the less basic and more hydrophobic pyridine-2-sulfonyl group,27 for example. Second, efforts should be made toward improving the limited metabolic stability of 1d reported by us previously.26,27 We showed how our inhibitors were quickly oxidized upon incubation with liver microsomes, probably by cytochrome P450, followed by loss of the aniline functionalized zinc-binding imidazole. It is likely that cytochrome P450-mediated inhibitor oxidation is initiated by loss of one electron of the aniline nitrogen lone pair, followed by abstraction of a hydrogen radical from the activated methylene group between the aniline and imidazole, ultimately leading to N-dealkylation of the imidazolylmethyl moiety. As described in the manuscript by Seto et al.,34 aniline N-dealkylation can only proceed when the aniline lone pair of electrons is oxidizable and when there is at least one hydrogen on the carbon directly attached to the aniline nitrogen. Thus, improved inhibitor metabolic stability may be achieved by (a) reducing electron density on the aniline nitrogen through incorporation of additional electron-withdrawing groups on the cyanophenyl ring and/or (b) replacing the methylene unit between the aniline nitrogen and the zinc-binding imidazole with a gem-dimethyl unit. Finally, our third goal should be the investigation of the activities of our antimalarials in drug-resistant strains of P. falciparum PFT (such as the Y837C strain19 that shows resistance to BMS-388891 and the G612A strain20 that shows resistance to BMS-339941). This research is essential not only to evaluate the potencies of our compounds in such strains but also, after preparing similar QSAR models for the mutant active sites as we did for the wild-type, to identify structural modifications that we may undertake to restore inhibitor potency where it may be needed. In this way, new antimalarial PfPFTIs that are active against drug-resistant strains of P. falciparum may be realized.
Experimental Methods
Ligand Docking Studies
Docking experiments were performed using the GOLD version 3.128 software package. Ligands were prepared for docking in InsightII.30 Each ligand was drawn as a two-dimensional representation and converted to three dimensions. Ligands were subsequently energy minimized with the cvff force field. The homology model was also prepared for use in InsightII, where each residue was protonated on the basis of calculated pKa values at a pH of 7.4.
Ligands were constrained in the active site of PfPFT at three points in order to maintain the binding mode hypothesized for lead compound 1a. A single atom of each imidazole and the cyanoaniline were chosen and constrained to occupy a region of space within 1 Å of that occupied by the equivalent atoms of docked 1a. This was performed using the substructure constraint functionality of GOLD. A small spring constant was used as a penalty function such that molecules could dock in alternative conformations, but these would be discouraged. GOLD was allowed to configure the optimal genetic algorithm settings for each ligand.
QSAR Modeling
Descriptors were calculated using the MOE software package.35 RECON TAE and RAD descriptors36 were calculated in addition to the standard assortment of 2D descriptors in MOE. Additional descriptors included energy measurements with the MMFF94x, Amber-99, and OPLS-AA force fields. Feature selection and modeling were performed using the Analyze software package.37 For further details, see Supporting Information.
Chemistry: General Methods
Solvents CH2Cl2, THF, CH3CN, and DMF were dried on an Innovative Technology SPS-400 dry solvent system. Anhydrous MeOH and DMSO were purchased from Sigma-Aldrich and used directly from their Sure-Seal bottles. Molecular sieves were activated by heating to 300 °C under vacuum overnight. All reactions were performed under an atmosphere of dry nitrogen in oven-dried glassware and were monitored for completeness by thin-layer chromatography (TLC) using silica gel (visualized by UV light or developed by treatment with KMnO4 stain or Hanessian’s stain). 1H and 13C NMR spectra were recorded on Bruker AM 400 MHz and Bruker AM 500 MHz spectrometers in either CDCl3, MeOH-d4 or DMSO-d6. Chemical shifts (δ) are reported in parts per million after calibration to residual isotopic solvent. Coupling constants (J) are reported in Hz. Mass spectrometry (MS) was performed using electrospray ionization on either a Varian MAT-CH-5 (HRMS) or a Waters Micromass ZQ (LRMS) instrument. Before biological testing, target molecules (2a–e, 3a–e, 4a, 4d, 5a, 5d, 6a–e, 7a–e, 8a–e, 9a–e, 10a, 10d, 11a, 11d), obtained as glassy films after silica gel flash column chromatographic purification (eluent CH2Cl2/MeOH/NH4OH, 192:7:1), were subjected to further purification by reversed-phase HPLC (rpHPLC). Analysis and purification by rpHPLC were performed using either Phenonenex Luna 5 µm C18 (2) 250 mm × 21 mm column run at 15 mL/min (preparative) or a Microsorb-MV 300 Å C18 250 mm × 4.6 mm column run at 1 mL/min (analytical), using gradient mixtures of (A) water with 0.1% TFA and (B) 10:1 acetonitrile/water with 0.1% TFA. Appropriate product fractions were pooled and lyophilized to dryness, affording the inhibitors as fluffy white powders as their TFA salts. Inhibitor purity was confirmed by analytical rpHPLC using linear gradients from 100% A to 100% B, with changing solvent composition of either (I) 4.5% or (II) 1.5% per minute after an initial 2 min of 100% A. For reporting HPLC data, percentage purity is given in parentheses after the retention time for each condition.
General Procedure A (Mitsunobu Reactions). Reaction of Primary and Secondary Alcohols with Secondary Sulfonamides
To a stirring solution of the alcohol (1 equiv) in THF (0.07 M) (occasionally, sonication and a little warming (40 °C) was required to achieve complete dissolution of the alcohol) was added the secondary sulfonamide (2.5 equiv) and PPh3 (3 equiv). After the mixture was stirred for 15 min at room temperature, DIAD (2.5 equiv) was added dropwise. For the acyclic, primary alcohols, reactions were typically complete within 1 h; for the less reactive, cyclic secondary alcohols and hindered primary alcohols, mixtures were left stirring overnight (16 h). All solvent was removed in vacuo.
General Procedure B (Sulfonamide Alkylations). Reaction of Secondary Sulfonamides with Benzylic and Alkyl Bromides
To a stirred solution of the secondary sulfonamide (1 equiv) in DMF (0.01 M for benzylic bromide or 0.1 M for alkyl bromide) was added Cs2CO3 (3 equiv). After 1 h, at room temperature, the bromide (or iodide) alkylating agent (1.1 equiv) was added dropwise. After 16 h, the reaction mixture was diluted with water and extracted into EtOAc (×3). The EtOAc extractions were combined and washed with 5% NaHCO3 (×3) and brine, dried (Na2SO4), filtered, and concentrated.
General Procedure C (Aniline Alkylation). Reaction of Secondary Anilines with 5-Chloromethyl-1-methyl-1H-imidazole•HCl (15)
The secondary aniline (1 equiv) was dissolved in DMF (0.07 M). Then the mixture was cooled to 0 °C. After 15 min, NaH (3 equiv) was added in one portion. After a further 15 min, 5-chloromethyl-1-methyl-1H-imidazole•HCl27 (15) (1.1 equiv) was added to the reaction mixture. The mixture was allowed to stir at 0 °C for 2–3 h, when TLC indicated the reaction was complete or had stalled. Upon quenching the reaction with brine (approximately 1 mL), the mixture was diluted with water and extracted with EtOAc (×3). The EtOAc extractions were combined and washed with 5% NaHCO3 (×3) and brine, dried (Na2SO4), filtered, and concentrated.
[N-Benzyl-N-3-{(4-cyanophenyl)(3-methyl-3H-imidazol-4-ylmethyl)amino}propyl]-1-methyl-1H-imidazole-4-sulfonamide (2a)
Primary alcohol 17 was coupled to secondary sulfonamide 19a on a 0.106 mmol scale via general procedure A. The crude residue was dry-loaded onto silica gel, then flash chromatographed (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to furnish 2a (51 mg, 96%): δH (400 MHz, MeOH-d4) 1.71 (quin, J = 7.0 Hz, 2H, CH2CH2CH2), 3.30–3.36 (obsc m, 4H, CH2CH2CH2), 3.83 (s, 3H, CH3(Im)), 3.89 (s, 3H, CH3(Im)), 4.34 (s, 2H, CH2Ph), 4.62 (s, 2H, CH2Im), 6.73 (d, J = 8.8 Hz, 2H, 2 CH (Ar)), 7.14 (s, 1H, CH (Im)), 7.31–7.43 (m, 5H, 5 CH (Ph)), 7.50 (d, J = 8.8 Hz, 2H, 2 CH (Ar)), 7.77 (s, 1H, CH (Im)), 7.84 (s, 1H, CH (Im)), 8.93 (s, 1H, CH (Im)); δC (100 MHz, MeOH-d4) 27.4, 34.3, 34.4, 45.8, 48.6, 49.3, 54.7, 99.8, 113.8, 118.8, 121.0, 126.6, 128.9, 129.7, 129.9, 133.1, 134.7, 137.7, 138.7, 139.3, 141.4, 151.8; HRMS (ES+) calcd for [C26H29N7O2S + H] 504.2182, found 504.2198; HPLC (I) tR = 15.69 min (100%), (II), tR = 27.81 min (100%).
[N-(2-Methylbenzyl)-N-3-{(4-cyanophenyl)(3-methyl-3H-imidazol-4-ylmethyl)amino}propyl]-1-methyl-1H-imidazole-4-sulfonamide (2b)
Primary alcohol 17 was coupled to secondary sulfonamide 19b on a 0.137 mmol scale via general procedure A. The crude residue was dry-loaded onto silica gel, then flash chromatographed (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to furnish 2b (64 mg, 90%): δH (400 MHz, MeOH-d4) 1.57 (quin, J = 7.3 Hz, 2H, CH2CH2CH2), 2.42 (s, 3H, CH3Ph), 3.21–3.27 (m, 4H, CH2-CH2CH2), 3.85 (s, 3H, CH3(Im)), 3.88 (s, 3H, CH3(Im)), 4.33 (s, 2H, CH2Ph), 4.60 (s, 2H, CH2Im), 6.70 (d, J = 8.8 Hz, 2H, 2 CH (Ar)), 7.11 (s, 1H, CH (Im)), 7.14–7.26 (m, 3H, 3 CH (Ph)), 7.31 (br d, J = 7.6 Hz, 1H, CH (Ph)), 7.49 (d, J = 8.8 Hz, 2H, 2 CH (Ar)), 7.78 (s, 1H, CH (Im)), 7.86 (s, 1H, CH (Im)), 8.93 (s, 1H, CH (Im)); δC (100 MHz, MeOH-d4) 19.5, 27.8, 34.3, 34.4, 45.3, 48.0, 49.2, 53.5, 99.8, 113.8, 118.8, 121.0, 126.7, 127.1, 129.3, 131.2, 131.7, 133.0, 134.7, 135.6, 137.7, 138.7, 138.8, 141.4, 151.7; HRMS (ES+) calcd for [C27H31N7O2S + H] 518.2338, found 518.2346; HPLC (I) tR = 15.98 min (100%), (II) tR = 28.75 min (100%).
[N-(Thiophen-3-ylmethyl)-N-3-{(4-cyanophenyl)(3-methyl-3H-imidazol-4-ylmethyl)amino}propyl]-1-methyl-1H-imidazole-4-sulfonamide (2c)
Primary alcohol 17 was coupled to secondary sulfonamide 19c on a 0.16 mmol scale via general procedure A. The crude residue was dry-loaded onto silica gel, then flash chromatographed (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to give 2c (80 mg, 98%): δH (400 MHz, MeOH-d4) 1.78 (quin, J = 7.2 Hz, 2H, CH2CH2CH2), 3.32–3.36 (obsc m, 2H, CH2CH2CH2NSO2), 3.37–3.41 (m, 2H, CH2CH2CH2NSO2), 3.83 (s, 3H, CH3(Im)), 3.91 (s, 3H, CH3(Im)), 4.36 (s, 2H, CH2thiophene), 4.68 (s, 2H, CH2Im), 6.79 (d, J = 8.8 Hz, 2H, 2 CH (Ar)), 7.13 (dd, J = 5.0, 1.2 Hz, 1H, CH (thiophene)), 7.18 (s, 1H, CH (Im)), 7.32–7.35 (m, 1H, CH (thiophene)), 7.41 (dd, J = 5.0, 2.8 Hz, 1H, CH (thiophene)), 7.53 (d, J = 8.8 Hz, 2H, 2 CH (Ar)), 7.74 (s, 1H, CH (Im)), 7.83 (s, 1H, CH (Im)), 8.93 (s, 1H, CH (Im)); δC (100 MHz, MeOH-d4) 27.3, 34.3, 34.4, 45.4, 48.2, 49.3, 49.4, 99.8, 113.8, 118.9, 121.0, 125.1, 126.5, 127.5, 129.2, 133.1, 134.7, 137.7, 139.4, 139.4, 141.3, 151.8; HRMS (ES+) calcd for [C24H27N7O2S2 + H] 510.1746, found 510.1761; HPLC (I) tR = 15.12 min (100%), (II) tR = 26.20 min (100%).
[N-(N-tert-Butoxycarbonylpiperidin-4-ylmethyl)-N-3-{(4-cyanophenyl)(3-methyl-3H-imidazol-4-ylmethyl)amino}propyl]-1-methyl-1H-imidazole-4-sulfonamide (2d)
Primary alcohol 17 was coupled to secondary sulfonamide 19d on a 0.24 mmol scale via general procedure A. The crude residue was dry-loaded onto silica gel, then flash chromatographed (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to afford 2d (132 mg, 90%): δH (400 MHz, MeOH-d4) 1.07 (qd, J = 12.3, 4.0 Hz, 2H, 2 CH (piperidinylmethyl)), 1.49 (s, 9H, C(CH3)3), 1.68–1.76 (m, 2H, 2 CH (piperidinylmethyl)), 1.79–1.90 (m, 1H, CH (piperidinylmethyl)), 1.99 (quin, J = 7.4 Hz, 2H, CH2CH2CH2), 2.64–2.80 (m, 2H, 2 CH (piperidinylmethyl)), 3.01 (d, J = 7.6 Hz, 2H, 2 CH (piperidinylmethyl)), 3.29 (t, J = 7.2 Hz, 2H, CH2CH2CH2NSO2), 3.61 (t, J = 7.2 Hz, 2H, CH2CH2CH2NSO2), 3.81 (s, 3H, CH3(Im)), 3.95 (s, 3H, CH3(Im)), 4.03–4.10 (m, 2H, 2 CH (piperidinylmethyl)), 4.87 (s, 2H, CH2Im), 6.96 (d, J = 8.8 Hz, 2H, 2 CH (Ar)), 7.30 (s, 1H, CH (Im)), 7.58 (d, J = 8.8 Hz, 2H, 2 CH (Ar)), 7.73 (s, 1H, CH (Im)), 7.79 (s, 1H, CH (Im)), 8.94 (s, 1H, CH (Im)); δC (100 MHz, MeOH-d4) 27.9, 28.7, 30.9, 34.2, 34.3, 36.5, 44.7 (br), 45.6, 48.9, 49.0, 56.3, 81.0, 100.0, 114.1, 119.0, 120.9, 126.5, 133.2, 134.8, 137.8, 139.3, 141.3, 151.9, 156.5; HRMS (ES+) calcd for [C30H42N8O4S + H] 611.3128, found 611.3129; HPLC (I) tR = 12.56 min (100%), (II) tR = 18.92 min (100%).
[N-{N-(2-Pyrimidinyl)-piperidin-4-ylmethyl}-N-3-{(4-cyanophenyl)(3-methyl-3H-imidazol-4-ylmethyl)amino}propyl]-1-methyl-1H-imidazole-4-sulfonamide (2e)
Primary alcohol 17 was coupled to secondary sulfonamide 19e on a 0.106 mmol scale via general procedure A. The crude residue was dry-loaded onto silica gel, then flash chromatographed (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to yield 2e (38 mg, 61%): δH (400 MHz, MeOH-d4) 1.17 (qd, J = 12.2, 4.0 Hz, 2H, 2 CH (piperidinylmethyl)), 1.78–1.86 (m, 2H, 2 CH (piperidinylmethyl)), 1.94–2.04 (m, 3H, CH2CH2CH2, CH (piperidinylmethyl)), 2.90–2.98 (m, 2H, 2 CH (piperidinylmethyl)), 3.04 (d, J = 7.6 Hz, 2H, 2 CH (piperidinylmethyl)), 3.26–3.31 (m, 2H, CH2CH2CH2NSO2), 3.59–3.66 (m, 2H, CH2CH2-CH2NSO2), 3.81 (s, 3H, CH3(Im)), 3.96 (s, 3H, CH3(Im)), 4.64–4.71 (m, 2H, 2 CH (piperidinylmethyl)), 4.88 (s, 2H, CH2Im), 6.66 (t, J = 5.1 Hz, 1H, CH (pyrimidine)), 6.97 (d, J = 9.2 Hz, 2H, 2 CH (Ar)), 7.31 (s, 1H, CH (Im)), 7.59 (d, J = 9.2 Hz, 2H, 2 CH (Ar)), 7.73 (s, 1H, CH (Im)), 7.80 (s, 1H, CH (Im)), 8.38 (d, J = 5.1 Hz, 2H, 2 CH (pyrimidine)), 8.95 (s, 1H, CH (Im)); δC (100 MHz, MeOH-d4) 27.9, 30.9, 34.2, 34.3, 36.6, 45.2, 45.6, 49.1, 49.3, 56.3, 100.1, 110.6, 114.1, 119.0, 120.9, 126.5, 133.2, 134.8, 137.8, 139.3, 141.3, 151.9, 158.7, 160.7; HRMS (ES+) calcd for [C29H31N10O2S + H] 589.2822, found 589.2831; HPLC (I) tR = 13.00 min (100%), (II) tR = 20.28 min (99.69%).
[N-Benzyl-N-{(4-cyanophenyl)(3-methyl-3H-imidazol-4-ylmethyl)amino}-2,2-dimethylethyl]-1-methyl-1H-imidazole-4-sulfonamide (3a)
The synthesis was as per general procedure A with alcohol 27 on a 0.037 mmol scale and secondary sulfonamide 19a. The mixture was heated to 45 °C for 18 h, reduced, and then dry-loaded onto silica gel and purified by flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to give 3a (12 mg, 65%): δH (400 MHz, CDCl3) 1.40 (s, 6H, C(CH3)2), 3.46 (s, 3H, CH3(Im)), 3.72 (s, 3H, CH3(Im)), 3.87 (s, 2H, CH2C(CH3)2), 4.47 (s, 2H, CH2Ph), 4.70 (s, 2H, CH2Im), 6.58 (s, 1H, CH(Im)), 6.60 (d, J = 8.3 Hz, 2H, 2 CH (Ar)), 7.27–7.38 (m, 7H, 2 CH (Im), 5 CH (Ph)), 7.45 (d, J = 8.3 Hz, 2H, 2 CH (Ar)), 7.47 (s, 1H, CH (Im)); δC (125 MHz, CDCl3) 26.9, 31.8, 33.9, 47.1, 50.8, 59.3, 65.6, 99.3, 113.7, 120.0, 123.5, 126.7, 127.4, 127.7, 128.6, 129.0, 133.2, 138.4, 138.6, 139.4, 143.1, 151.6; HRMS (ES+) calcd for [C28H31N6O2S + H] 518.2343, found 518.2338; HPLC (I) tR = 12.25 min (99.66%), (II) tR = 17.75 min (99.34%).
[N-(2-Methylbenzyl)-N-{(4-cyanophenyl)(3-methyl-3H-imidazol-4-ylmethyl)amino}-2,2-dimethylethyl]-1-methyl-1H-imidazole-4-sulfonamide (3b)
The synthesis was as per general procedure A with alcohol 27 on a 0.035 mmol scale and secondary sulfonamide 19b. The mixture was heated to 45 °C for 18 h, then reduced, and dry-loaded onto silica gel and purified by flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to give 3b (11 mg, 60%): δH (400 MHz, CDCl3) 1.40 (s, 6H, C(CH3)2), 2.23 (s, 3H, CH3Ph), 3.51 (s, 3H, CH3(Im)), 3.70 (s, 3H, CH3(Im)), 4.02 (s, 2H, CH2C(CH3)2), 4.56 (s, 2H, CH2Ph), 4.62 (s, 2H, CH2Im), 6.65 (s, 1H, CH (Im)), 6.72 (d, J = 9.0 Hz, 2H, 2 CH (Ar)), 7.10–7.39 (m, 6H, 2 CH (Im), 4 CH (Ar)), 7.45 (s, 1H, CH (Im)), 7.60 (d, J = 9.0 Hz, 2H, 2 CH (Ar)); δC (125 MHz, CDCl3) 18.9, 26.5, 31.6, 33.6, 47.0, 47.3, 58.9, 65.2, 99.2, 113.6, 119.7, 123.6, 125.8, 126.5, 126.6, 127.0, 128.8, 130.0, 133.0, 133.6, 136.8, 138.2, 138.7, 142.5, 151.4; HRMS (ES+) calcd for [C28H33N7O2S + H] 532.2416, found 532.2452; HPLC (I) tR = 12.80 min (95.01%), (II) tR = 19.70 min (95.06%).
[N-(Thiophen-3-ylmethyl)-N-{(4-cyanophenyl)(3-methyl-3H-imidazol-4-ylmethyl)amino}-2,2-dimethylethyl]-1-methyl-1H-imidazole-4-sulfonamide (3c)
The synthesis was as per general procedure A with alcohol 27 on a 0.088 mmol scale and secondary sulfonamide 19c. The mixture was heated to 45 °C for 18 h, reduced, and then dry-loaded onto silica gel and purified by flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to give 3c (25 mg, 55%): δH (400 MHz, CDCl3) 1.45 (s, 6H, C(CH3)2), 3.48 (s, 3H, CH3(Im)), 3.72 (s, 3H, CH3(Im)), 3.82 (s, 2H, CH2C(CH3)2), 4.46 (s, 2H, CH2thiophene), 4.67 (s, 2H, CH2Im), 6.61 (d, J = 8.5 Hz, 2H, Ar), 6.64 (s, 1H, CH (Im)), 7.22–7.37 (m, 7H, 2 CH (Im), 2 CH (Ar), 3 CH (thiophene)), 7.45 (s, 1H, CH (Im)); δC (125 MHz, CDCl3) 26.9, 31.8, 33.9, 46.0, 46.9, 59.2, 65.3, 99.3, 113.7, 120.0, 122.9, 123.4, 126.0, 126.8, 128.1, 128.8, 133.2, 138.4, 138.5, 140.7, 143.2, 151.6; HRMS (ES+) calcd for [C25H29N7O2S2 + H] 524.1904, found 524.1902; HPLC (I) tR = 12.56 min (100%), (II) tR = 18.70 min (100%).
[N-(N-tert-Butoxycarbonylpiperidin-4-ylmethyl)-N-{(4-cyanophenyl)(3-methyl-3H-imidazol-4-ylmethyl)amino}-2,2-dimethylethyl]-1-methyl-1H-imidazole-4-sulfonamide (3d)
The synthesis was as per general procedure A with alcohol 27 on a 0.217 mmol scale and secondary sulfonamide 19d. The mixture was heated to 45 °C for 18 h, reduced, and then dry-loaded onto silica gel and purified by flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to give 3d (68 mg, 50%): δH (400 MHz, CDCl3) 1.10 (qd, J = 12.3, 4.0 Hz, 2H, 2 CH (piperidinylmethyl)), 1.39 (s, 6H, C(CH3)2), 1.44 (s, 9H, C(CH3)3), 1.67–1.79 (m, 3H, 3 CH (piperidinylmethyl)), 2.58–2.67 (m, 2H, 2 CH (piperidinylmethyl)), 3.28–3.36 (m, 2H, 2 CH (piperidinylmethyl)), 3.53 (s, 3H, CH3(Im)), 3.72 (s, 3H, CH3(Im)), 3.85 (s, 2H, CH2(CH3)2), 4.05–4.15 (m, 2H, 2 CH (piperidinylmethyl)), 4.66 (s, 2H, CH2Im), 6.70 (s, 1H, CH (Im)), 6.92 (d, J = 9.0 Hz, 2H, 2 CH (Ar)), 7.35 (s, 1H, CH (Im)), 7.39 (s, 1H CH (Im)), 7.41 (s, 1H CH (Im)), 7.45 (d, J = 9.0 Hz, 2H, 2 CH (Ar)); δC (125 MHz, CDCl3) 14.4, 27.3, 28.7, 29.9, 30.4, 32.1, 34.2, 38.3, 47.1, 52.5, 58.1, 64.8, 79.6, 100.0, 114.1, 120.1, 123.4, 129.4, 133.7, 138.5, 144.0, 152.1, 155.0, 171.4; HRMS (ES+) calcd for [C31H44N8O4S + H] 625.2765, found 625.2720; HPLC (I) tR = 13.61 min (100%), (II) tR = 21.80 (100%).
[N-{N-(2-Pyrimidinyl)-piperidin-4-ylmethyl}-N-{(4-cyanophenyl)(3-methyl-3H-imidazol-4-ylmethyl)amino}-2,2-dimethylethyl]-1-methyl-1H-imidazole-4-sulfonamide (3e)
The synthesis was as per general procedure A with alcohol 27 on a 0.037 mmol scale and secondary sulfonamide 19e. The mixture was heated to 45 °C for 18 h, then dry-loaded onto silica gel and purified by flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 92:7:1) to give 3e (15 mg, 68% yield): δH (500 MHz, CDCl3) 1.17 (qd, J = 12.2, 4.0 Hz, 2H, 2 CH (piperidinylmethyl)), 1.41 (s, 6H, C(CH3)2), 1.81–2.02 (m, 3H, 3 CH (piperidinylmethyl)), 2.79 (m, 2H, 2 CH (piperidinylmethyl)), 3.33 (d, J = 7.0 Hz, 2H, 2 CH (piperidinylmethyl)), 3.54 (s, 3H, CH3(Im)), 3.71 (s, 3H, CH3(Im)), 3.87 (m, 2H, CH2C(CH3)2), 4.68 (s, 2H, CH2Im), 4.73–4.79 (m, 2H, 2 CH (piperidinylmethyl)), 6.42 (t, J = 4.9 Hz, 1H, CH (pyrimidine)), 6.69 (s, 1H, CH (Im)), 6.91 (d, J = 9.0 Hz, 2H, 2 CH (Ar)), 7.35 (s, 1H, CH (Im)), 7.39 (s, 1H, CH (Im)), 7.42 (s, 1H, CH (Im)), 7.44 (d, J = 9.0 Hz, 2H, 2 CH (Ar)), 8.27 (d, J = 4.9 Hz, 2H, 2 CH (pyrimidine)); δC (125 MHz, CDCl3) 27.4, 30.3, 32.1, 34.2, 38.6, 44.2, 47.1, 52.6, 58.3, 64.8, 99.9, 109.6, 114.1, 120.2, 123.5, 129.1, 133.7, 138.5, 139.0, 143.9, 152.1, 157.9, 161.8, 162.7; HRMS (ES+) calcd for [C30H38N10O2S + H] 603.2969, found 603.2978; HPLC (I) tR = 13.62 min (100%), (II) tR = 19.16 min (100%).
2-[Benzyl(1-methyl-1H-imidazole-4-sulfonyl)amino]-N-(4-cyanophenyl)-N-(3-methyl-3H-imidazol-4-ylmethyl)acetamide (4a)
Compound 32 was coupled to secondary sulfonamide 19a as per general procedure A on a 0.164 mmol scale. After workup, the crude residue was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to furnish 4a (43 mg, 52%): δH (500 MHz, CDCl3) 3.60 (s, 3H, CH3(Im)), 3.70 (s, 2H, CH2CO), 3.76 (s, 3H, CH3(Im)), 4.60 (s, 2H, CH2Ph), 4.85 (s, 2H, CH2Im), 6.59 (br s, 1H, CH (Im)), 6.95 (d, J = 8.5 Hz, 2H, 2 CH (Ar)), 7.19–7.24 (m, 2H, 2 CH (Ph)), 7.27–7.31 (m, 3H, 3 CH (Ph)), 7.43 (br s, 1H, CH (Im)), 7.45 (s, 1H, CH (Im)), 7.46 (s, 1H, CH (Im)), 7.58 (d, J = 8.5 Hz, 2H, 2 CH (Ar)); δC (125 MHz, CDCl3) 31.7, 33.9, 41.5, 47.6, 51.6, 112.6, 117.6, 123.9, 126.1, 127.9, 128.5, 128.6, 129.3, 130.4, 133.6, 135.2, 138.9, 139.1, 140.1, 143.6, 167.1; HRMS (ES+) calcd for [C25H25N7O3S + H] 504.1818, found 504.1830; HPLC (I) tR = 11.48 min (100%), (II) tR = 15.67 min (100%).
2-[(N-tert-Butoxycarbonylpiperidin-4-ylmethyl)(1-methyl-1H-imidazole-4-sulfonyl)amino]-N-(4-cyanophenyl)-N-(3-methyl-3H-imidazol-4-ylmethyl)acetamide (4d)
Compound 32 was coupled to secondary sulfonamide 19d as per general procedure A on a 0.164 mmol scale. After workup, the crude residue was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to furnish 4b (20 mg, 20%): δH (500 MHz, CDCl3) 0.99 (m, 2H, 2 CH (piperidinylmethyl)), 1.44 (s, 9H, C(CH3)3), 1.59–1.75 (m, 3H, 3 CH (piperidinylmethyl)), 2.54–2.68 (m, 2H, 2 CH (piperidinylmethyl)), 3.07–3.19 (m, 2H, 2 CH (piperidinylmethyl)), 3.61 (s, 3H, CH3(Im)), 3.71 (s, 3H, CH3(Im)), 3.73–3.81 (m, 2H, CH2CO), 3.95–4.08 (m, 2H, CHCH2N (piperidinylmethyl)), 4.81–4.94 (m, 2H, CH2Im), 6.59 (s, 1H, CH (Im)), 7.22 (d, J = 8.5 Hz, 2H, 2 CH (Ar)), 7.33 (app s, 2H, 2 CH (Im)), 7.43 (s, 1H, CH (Im)), 7.69 (d, J = 8.5 Hz, 2H, 2 CH (Ar)); δC (125 MHz, CDCl3) 28.4, 29.6, 31.9, 34.0, 35.3, 41.7, 43.5 (br), 50.2, 55.1, 79.5, 112.8, 117.6, 123.6, 126.1, 129.7, 130.4, 133.9, 138.8, 139.1, 140.0, 143.8, 154.6, 167.5; HRMS (ES+) calcd for [C29H38N8O5S + H] 611.2764, found 611.2769; HPLC (I) tR = 12.44 min (99.53%), (II) tR = 17.97 min (98.67%).
3-[Benzyl-(1-methyl-1H-imidazole-4-sulfonyl)amino]-N-(4-cyanophenyl)-N-(3-methyl-3H-imidazol-4-ylmethyl)propanamide (5a)
5a was prepared as per general procedure B with 35 and benzyl bromide on a 0.0845 mmol scale. The crude residue was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to give 5a (42 mg, 96%): δH (500 MHz, CDCl3) 2.12 (m, 2H, CH2CH2CO), 3.47 (t, J = 7.3 Hz, 2H, CH2CH2CO), 3.53 (s, 3H, CH3(Im)), 3.72 (s, 3H, CH3(Im)), 4.31 (s, 2H, CH2Ph), 4.78 (s, 2H, CH2Im), 6.53 (s, 1H, CH (Im)), 6.68 (d, J = 8.3 Hz, 2H, 2 CH (Ar)), 7.19–7.25 (m, 5H, 5 CH (Ph)), 7.37 (s, 1H, CH (Im)), 7.38 (br s, 1H, CH (Im)), 7.41 (s, 1H, CH (Im)), 7.58 (d, J = 8.3 Hz, 2H, 2 CH (Ar)); δC (125 MHz, CDCl3) 31.7, 34.9, 35.2, 40.9, 45.3, 53.8, 112.3, 117.7, 124.2, 126.5, 127.7, 128.3, 128.4, 129.3, 129.7, 130.2, 133.6, 136.8, 138.9, 139.7, 144.5, 169.7; HRMS (ES+) calcd for [C26H27N7O3S + H] 518.1974, found 518.1994; HPLC (I) tR = 11.82 min (98.91%), (II) tR = 16.38 min (99.01%).
3-[(N-tert-Butoxycarbonylpiperidin-4-ylmethyl)(1-methyl-1H-imidazole-4-sulfonyl)amino]-N-(4-cyanophenyl)-N-(3-methyl-3H-imidazol-4-ylmethyl)propanamide (5d)
5d was prepared as per general procedure B with 35 and N-tert-butoxycarbonylpiperidin-4-ylmethyl bromide (1.5 equiv) in DMF (0.1 M) on a 0.0986 mmol scale. After the mixture was stirred at room temperature for 36 h, byproduct began to form. So the reaction mixture was worked up and then the crude residue was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to give 5d (30 mg, 50% (73% brsm)): δH (500 MHz, CDCl3) 1.03 (qd, J = 12.2, 4.0 Hz, 2H, 2 CH (piperidinylmethyl)), 1.43 (s, 9H, C(CH3)3), 1.59 (br app d, J = 12.2 Hz, 2H, 2 CH (piperidinylmethyl)), 1.77 (m, 1H, CH (piperidinylmethyl)), 2.46 (m, 2H, CH2CH2CO), 2.63 (m, 2H, 2 CH (piperidinylmethyl)), 2.92 (m, 2H, 2 CH (piperidinylmethyl)), 3.50 (m, 2H, 2 CH (piperidinylmethyl)), 3.59 (s, 3H, CH3(Im)), 3.71 (s, 3H, CH3(Im)), 4.05 (m, 2H, CHCH2N (piperidinylmethyl)), 4.90 (s, 2H, CH2Im), 6.61 (s, 1H, CH (Im)), 7.13 (d, J = 8.0 Hz, 2H, 2 CH (Ar)), 7.32 (s, 1H, CH (Im)), 7.33 (s, 1H, CH (Im)), 7.42 (s, 1H, CH (Im)), 7.68 (d, J = 8.3 Hz, 2H, 2 CH (Ar)); δC (125 MHz, CDCl3) 28.4, 29.7, 31.8, 33.9, 34.9, 35.1, 41.2, 43.4 (br), 46.0, 55.3, 79.4, 112.5, 117.7, 124.2, 126.5, 129.5, 130.3, 133.7, 138.7, 139.0, 139.5, 144.6, 154.7, 170.0; HRMS (ES+) calcd for [C30H40N8O5S + H] 625.2921, found 625.2923; HPLC (I) tR = 12.74 min (98.83%), (II) tR = 19.28 min (99.47%).
(±)-[N-Benzyl-N-{cis-2-[(4-cyanophenyl)(3-methyl-3H-imidazol-4-ylmethyl)amino]cyclopentyl}]-1-methyl-1H-imidazole-4-sulfonamide (6a)
The synthesis was as per general procedure B with 42 and benzyl bromide on a 0.0273 mmol scale. The crude residue was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to afford 6a (8 mg, 55%): δH (400 MHz, CDCl3) 1.37–1.50 (m, 1H, CH (cyclopentyl)), 1.84–1.96 (m, 2H, 2 CH (cyclopentyl)), 2.04–2.19 (m, 2H, 2 CH (cyclopentyl)), 2.49–2.56 (m, 1H, CH (cyclopentyl)), 3.55 (s, 3H, CH3(Im)), 3.67 (s, 3H, CH3(Im)), 3.88 (d, J = 16.4 Hz, 1H, CHaPh), 4.14 (m, 1H, CHN (cyclopentyl)), 4.20 (d, J = 18.0 Hz, 1H, CHaIm), 4.25 (m, 1H, CHN (cyclopentyl)), 4.52 (d, J = 16.4 Hz, 1H, CHbPh), 4.75 (d, J = 18.0 Hz, 1H, CHbIm), 6.52 (s, 1H, CH (Im)), 6.68 (d, J = 9.2 Hz, 2H, 2 CH (Ar)), 6.97–7.01 (m, 2H, 2 CH (Ph)), 7.15–7.20 (m, 3H, 3 CH (Ph)), 7.25 (s, 1H, CH (Im)), 7.37 (s, 1H, CH (Im)), 7.43 (d, J = 9.2 Hz, 2H, 2 CH (Ar)), 7.47 (s, 1H, CH (Im)); δC (125 MHz, CDCl3) 21.1, 28.4, 28.6, 31.6, 33.9, 41.6, 52.3, 61.2, 61.8, 98.8, 113.2, 120.3, 124.7, 127.0, 127.4, 127.9, 128.3, 128.4, 133.2, 136.7, 137.8, 138.9, 140.6, 151.4; HRMS (ES+) calcd for [C28H31N7O2S + H] 530.2338, found 530.2350; HPLC (I) tR = 12.73 min (100%), (II) tR = 18.89 min (99.35%).
(±)-[N-(2-Methylbenzyl)-N-{cis-2-[(4-cyanophenyl)(3-methyl-3H-imidazol-4-ylmethyl)amino]cyclopentyl}]-1-methyl-1H-imidazole-4-sulfonamide (6b)
The synthesis was as per general procedure B with 42 and 2-methylbenzyl bromide on a 0.116 mmol scale. The crude residue was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to afford 6b (14 mg, 22%): δH (500 MHz, CDCl3) 1.38–1.48 (m, 1H, CH (cyclopentyl)), 1.86–1.99 (m, 5H, CH3Ph, 2 CH (cyclopentyl)), 2.14–2.26 (m, 2H, 2 CH (cyclopentyl)), 2.67–2.76 (m, 1H, CH (cyclopentyl)), 3.59 (s, 3H, CH3(Im)), 3.62 (s, 3H, CH3(Im)), 4.05 (d, J = 17.5 Hz, 1H, CHaPh), 4.09–4.15 (m, 1H, CHN (cyclopentyl)), 4.26 (m, 1H, CHN (cyclopentyl)), 4.31 (d, J = 18.0 Hz, 1H, CHaIm), 4.42 (d, J = 17.5 Hz, 1H, CHbPh), 4.90 (d, J = 18.0 Hz, 1H, CHbIm), 6.55 (s, 1H, CH (Im)), 6.69 (d, J = 9.0 Hz, 2H, 2 CH (Ar)), 6.95–7.01 (m, 3H, 3 CH (Ph)), 7.07 (td, J = 7.3, 1.5 Hz, 1H, CH (Ph)), 7.16 (s, 1H, CH (Im)), 7.25 (s, 1H, CH (Im)), 7.43 (d, J = 9.0 Hz, 2H, 2 CH (Ar)), 7.45 (s, 1H, CH (Im)); δC (125 MHz, CDCl3) 18.8, 21.2, 28.6, 28.7, 31.6, 33.8, 41.7, 50.6, 61.5, 62.2, 98.8, 112.9, 120.3, 124.5, 125.5, 126.9, 127.1, 127.3, 128.4, 130.2, 133.4, 134.2, 135.7, 137.8, 138.7, 140.8, 151.5; HRMS (ES+) calcd for [C29H33N7O2S + H] 544.2495, found 544.2509; HPLC (I) tR = 12.77 min (100%), (II) tR = 19.02 min (100%).
(±)-[N-(Thiophen-3-ylmethyl)-N-{cis-2-[(4-cyanophenyl)(3-methyl-3H-imidazol-4-ylmethyl)amino]cyclopentyl}]-1-methyl-1H-imidazole-4-sulfonamide (6c)
The synthesis was as per general procedure B with 42 and thiophen-3-ylmethyl bromide (prepared by employing a standard bromination procedure of thiophen-3-ylmethanol with PPh3Br2; the bromide darkened on standing at room temperature, but 1H NMR of the material after 1 month suggested no decomposition had occurred) on a 0.121 mmol scale. The crude residue was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to afford 6c (35 mg, 54%): δH (500 MHz, CDCl3) 1.43–1.53 (m, 1H, CH (cyclopentyl)), 1.85–1.96 (m, 2H, 2 CH (cyclopentyl)), 2.04–2.13 (m, 2H, 2 CH (cyclopentyl)), 2.41–2.50 (m, 1H, CH (cyclopentyl)), 3.57 (s, 3H, CH3(Im)), 3.66 (s, 3H, CH3(Im)), 3.96 (d, J = 16.5 Hz, 1H, CHathiophene), 4.18 (m, 1H, CHN (cyclopentyl)), 4.27–4.34 (m, 2H, CHaIm, CHN (cyclopentyl)), 4.43 (d, J = 16.5 Hz, 1H, CHbthiophene), 4.74 (d, J = 18.0 Hz, 1H, CHbIm), 6.53 (s, 1H, CH (Im)), 6.70 (dd, J = 5.0, 1.5 Hz, 1H, CH (thiophene)), 6.72 (d, J = 9.0 Hz, 2H, 2 CH (Ar)), 6.90–6.93 (m, 1H, CH (thiophene)), 7.11–7.13 (dd, J = 5.0, 2.8 Hz, 1H, CH (thiophene)), 7.22 (s, 1H, CH (Im)), 7.39 (s, 1H, CH (Im)), 7.43 (d, J = 9.0 Hz, 2H, 2 CH (Ar)), 7.44 (s, 1H, CH (Im)); δC (125 MHz, CDCl3) 21.0, 28.2, 28.3, 31.5, 33.9, 41.8, 47.4, 60.7, 61.8, 98.8, 113.2, 120.4, 123.0, 124.5, 125.7, 127.5, 127.7, 128.2, 133.2, 137.9, 138.0, 138.8, 140.8, 151.5; HRMS (ES+) calcd for [C26H29N7O2S2 + H] 536.1902, found 536.1915; HPLC (I) tR = 11.08 min (100%), (II) tR = 18.85 min (98.38%).
(±)-[N-(N-tert-Butoxycarbonylpiperidin-4-ylmethyl)-N-{cis-2-[(4-cyanophenyl)(3-methyl-3H-imidazol-4-ylmethyl)amino]cyclopentyl}]-1-methyl-1H-imidazole-4-sulfonamide (6d)
The synthesis was as per general procedure B with 42 and N-tert-butoxycarbonylpiperidin-4-ylmethyl iodide (prepared by Finkelstein transformation on N-tert-butoxycarbonylpiperidin-4-ylmethyl bromide with sodium iodide in acetone) (1.5 equiv) on a 0.116 mmol scale in DMF (0.1 M), and the mixture was stirred for 7 days at room temperature. After the usual workup, the crude residue was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to yield 6d (60 mg, 81%): δH (500 MHz, CDCl3) 0.62–0.72 (m, 1H, CH (piperidinylmethyl)), 0.81–0.89 (m, 1H, CH (piperidinylmethyl)), 1.24–1.31 (m, 2H, 2 CH (piperidinylmethyl)), 1.41 (s, 9H, C(CH3)3), 1.42–1.61 (m, 3H, 2 CH (piperidinylmethyl), CH (cyclopentyl)), 1.85–1.95 (m, 2H, 2 CH (cyclopentyl)), 2.06–2.23 (m, 2H, 2 CH (cyclopentyl)), 2.25–2.35 (m, 1H, CH (piperidinylmethyl)), 2.38–2.49 (m, 1H, CH (cyclopentyl)), 2.73–2.81 (m, 1H, CH (piperidinylmethyl)), 2.86–2.99 (m, 1H, CH (piperidinylmethyl)), 3.64 (s, 3H, CH3(Im)), 3.73 (s, 3H, CH3(Im)), 3.81–3.97 (m, 2H, CHCH2N (piperidinylmethyl)), 4.13 (m, 1H, CHN (cyclopentyl)), 4.24 (m, 1H, CHN (cyclopentyl)), 4.57 (d, J = 18.0 Hz, 1H, CHaIm), 5.06 (br d, J = 18.0 Hz, 1H, CHbIm), 6.56 (s, 1H, CH (Im)), 6.81 (d, J = 9.0 Hz, 2H, 2 CH (Ar)), 7.38 (s, 1H, CH (Im)), 7.41–7.46 (m, 4H, 2 CH (Im), 2 CH (Ar)); δC (125 MHz, CDCl3) 20.9, 28.0, 28.2, 28.4, 29.9, 31.6, 34.0, 35.1, 42.3, 43.4 (br), 55.9, 61.8, 62.1, 79.4, 99.1, 113.3, 120.1, 124.1, 127.5, 128.0, 133.3, 138.0, 138.8, 141.4, 151.5, 154.6; HRMS (ES+) calcd for [C32H44N8O4S + H] 637.3284, found 637.3288; HPLC (I) tR = 13.28 min (100%), (II) tR = 20.60 min (100%).
(±)-[N-{N-(2-Pyrimidinyl)-piperidin-4-ylmethyl}-N-{cis-2-[(4-cyanophenyl)(3-methyl-3H-imidazol-4-ylmethyl)amino]cyclopentyl}]-1-methyl-1H-imidazole-4-sulfonamide (6e)
The synthesis was as per general procedure B with 42 and N-(2-pyrimidinyl)-piperidin-4-ylmethyl iodide (prepared by the reaction of PPh3Br2 on N-(2-pyrimidinyl)-piperidin-4-ylmethanol to give N-(2-pyrimidinyl)-piperidin-4-ylmethyl bromide, followed by Finkesltein halide exchange with NaI) (1.5 equiv) on a 0.118 mmol scale in DMF (0.1 M), and the mixture was stirred for 7 days at room temperature. The crude residue was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to furnish 6e (57 mg, 79%): δH (500 MHz, CDCl3) 0.76 (qd, J = 12.5, 4.0 Hz, 1H, CH (piperidinylmethyl)), 0.94 (qd, J = 12.5, 4.0 Hz, 1H, CH (piperidinylmethyl)), 1.35–1.41 (m, 1H, 1 CH (cyclopentyl)), 1.48–1.57 (m, 2H, 2 CH (piperidinylmethyl)), 1.68–1.77 (m, 1H, CH (piperidinylmethyl)), 1.87–1.97 (m, 2H, 2 CH (cyclopentyl)), 2.09–2.20 (m, 2H, 2 CH (cyclopentyl)), 2.39 (td, J = 12.5, 2.5 Hz, 1H, CH (piperidinylmethyl)), 2.44–2.52 (m, 2H, CH (cyclopentyl), CH (piperidinylmethyl)), 2.80 (dd, J = 14.5, 7.0 Hz, 1H, CH (piperidinylmethyl)), 2.97 (dd, J = 14.5, 7.5 Hz, 1H, CH (piperidinylmethyl)), 3.66 (s, 3H, CH3(Im)), 3.73 (s, 3H, CH3(Im)), 4.17 (m, 1H, CHN (cyclopentyl)), 4.26 (m, 1H, CHN (cyclopentyl)), 4.53–4.63 (m, 3H, CHaIm, CHCH2N (piperidinylmethyl)), 5.02 (br d, J = 17.5 Hz, 1H, CHbIm), 6.42 (t, J = 4.9 Hz, 1H, CH (pyrimidine), 6.59 (s, 1H, CH (Im)), 6.82 (d, J = 9.5 Hz, 2H, 2 CH (Ar)), 7.38 (s, 1H, CH (Im)), 7.44–7.47 (m, 3H, CH (Im), 2 CH (Ar)), 7.49 (s, 1H, CH (Im)), 8.25 (d, J = 4.9 Hz, 2H, 2 CH (pyrimidine)); δC (125 MHz, CDCl3) 21.0, 28.1, 28.4, 29.8, 31.7, 34.0, 35.4, 42.3, 43.5, 55.7, 61.9, 62.7, 99.2, 109.4, 113.3, 120.1, 124.0, 127.0, 128.2, 133.4, 137.9, 138.7, 141.6, 151.5, 157.7, 161.4; HRMS (ES+) calcd for [C31H38N10O2S + H] 615.2978, found 615.2999; HPLC (I) tR = 13.12 min (99.94%), (II) tR = 19.98 min (99.39%).
(±)-[N-Benzyl-N-{trans-2-[(4-cyanophenyl)(3-methyl-3H-imidazol-4-ylmethyl)amino]cyclopentyl}]-1-methyl-1H-imidazole-4-sulfonamide (7a)
The synthesis was as per general procedure B with 45 and benzyl bromide on a 0.0228 mmol scale. The crude residue was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to afford 7a (10 mg, 85%): δH (400 MHz, CDCl3) 1.19–1.29 (m, 1H, CH (cyclopentyl)), 1.54–1.64 (m, 2H, 2 CH (cyclopentyl)), 1.65–1.79 (m, 2H, 2 CH (cyclopentyl)), 1.83–1.91 (m, 1H, CH (cyclopentyl)), 3.60 (s, 3H, CH3(Im)), 3.73 (s, 3H, CH3(Im)), 3.87 (m, 1H, CHN (cyclopentyl)), 4.04 (d, J = 16.4 Hz, 1H, CHaPh), 4.41 (d, J = 17.8 Hz, 1H, CHaIm), 4.61–4.72 (m, 2H, CHbPh, CHN (cyclopentyl)), 4.80 (d, J = 17.8 Hz, 1H, CHbIm), 6.33 (d, J = 8.8 Hz, 2H, 2 CH (Ar)), 6.60 (s, 1H, CH (Im)), 6.94–7.02 (m, 3H, 3 CH (Ph)), 7.20–7.25 (m, 4H, 2 CH (Ph), 2 CH (Ar)), 7.39 (s, 1H, CH (Im)), 7.47 (s, 1H, CH (Im)), 7.45 (s, 1H, CH (Im)); δC (125 MHz, CDCl3) 19.3, 24.1, 26.2, 32.1, 34.0, 39.6, 48.1, 60.0, 61.1, 98.6, 113.1, 120.3, 124.2, 127.0, 127.3 (2), 128.2, 128.6, 133.0, 137.1, 137.9, 139.0, 140.2, 151.2; HRMS (ES+) calcd for [C28H31N7O2S + H] 530.2338, found 530.2353; HPLC (I) tR = 12.98 min (98.35%), (II) tR= 19.76 min (98.05%)
(±)-[N-(2-Methylbenzyl)-N-{trans-2-[(4-cyanophenyl)(3-methyl-3H-imidazol-4-ylmethyl)amino]cyclopentyl}]-1-methyl-1H-imidazole-4-sulfonamide (7b)
The synthesis was as per general procedure B with 45 and 2-methylbenzyl bromide on a 0.082 mmol scale. The crude residue was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to afford 7b (37 mg, 83%): δH (500 MHz, CDCl3) 1.20–1.29 (m, 1H, CH (cyclo-pentyl)), 1.55–1.66 (m, 2H, 2 CH (cyclopentyl)), 1.76–1.89 (m, 3H, 3 CH (cyclopentyl)), 2.21 (s, 3H, CH3Ph), 3.58 (s, 3H, CH3(Im)), 3.76 (s, 3H, CH3(Im)), 3.90 (m, 1H, CHN (cyclopentyl)), 4.23 (d, J= 16.0 Hz, 1H, CHaPh), 4.35 (d, J = 18.0 Hz, 1H, CHaIm), 4.57 (d, J = 16.0 Hz, 1H, CHbPh), 4.62 (m, 1H, CHN (cyclopentyl)), 4.75 (d, J = 18.0 Hz, 1H, CHbIm), 6.40 (d, J = 9.0 Hz, 2H, 2 CH (Ar)), 6.61 (s, 1H, CH (Im)), 6.83–6.97 (m, 3H, 3 CH (Ph)), 7.25–7.30 (m, 3H, 2 CH (Ar), CH (Ph)), 7.41 (s, 1H, CH (Im)), 7.43 (s, 1H, CH (Im)), 7.50 (s, 1H, CH (Im)); δC (125 MHz, CDCl3) 19.2, 19.6, 24.2, 26.4, 31.9, 34.0, 39.5, 46.3, 59.1, 61.2, 98.6, 113.2, 120.4, 124.3, 125.7, 127.3, 128.0, 128.3, 128.4, 130.4, 133.1, 134.6, 135.5, 138.2, 139.0, 140.4, 151.4; HRMS (ES+) calcd for [C29H33N7O2S + H] 544.2495, found 544.2501; HPLC (I) tR = 12.91 min (100%), (II) tR = 19.69 min (99.61%).
(±)-[N-(Thiophen-3-ylmethyl)-N-{trans-2-[(4-cyanophenyl)(3-methyl-3H-imidazol-4-ylmethyl)amino]cyclopentyl}]-1-methyl-1H-imidazole-4-sulfonamide (7c)
The synthesis was as per general procedure B with 45 and thiophen-3-ylmethyl bromide on a 0.0888 mmol scale. The crude residue was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to afford 7c (36 mg, 75%): δH (500 MHz, CDCl3) 1.27–1.35 (m, 1H, CH (cyclopentyl)), 1.57–1.69 (m, 3H, 3 CH (cyclopentyl)), 1.75–1.82 (m, 1H, CH (cyclopentyl)), 1.88–1.95 (m, 1H, CH (cyclopentyl)), 3.56 (s, 3H, CH3(Im)), 3.71 (s, 3H, CH3(Im)), 4.02 (m, 1H, CHN (cyclopentyl)), 4.14 (d, J = 16.0 Hz, 1H, CHathiophene), 4.44 (d, J = 18.0 Hz, 1H, CHaIm), 4.55 (d, J = 16.0 Hz, 1H, CHbthiophene), 4.63 (m, 1H, CHN (cyclopentyl)), 4.76 (d, J = 18.0 Hz, 1H, CHbIm), 6.57 (d, J = 9.0 Hz, 2H, 2 CH (Ar)), 6.60 (s, 1H, CH (Im)), 6.92 (dd, J = 5.3, 1.5 Hz, 1H, CH (thiophene)), 6.97 (dd, J = 5.3, 3.0 Hz, 1H, CH (thiophene)), 7.00–7.02 (m, 1H, CH (thiophene)), 7.30 (d, J = 9.0 Hz, 2H, 2 CH (Ar)), 7.34 (s, 1H, CH (Im)), 7.41 (s, 1H, CH (Im)), 7.44 (s, 1H, CH (Im)); δC (125 MHz, CDCl3) 19.4, 24.7, 26.3, 31.8, 33.9, 39.8, 43.5, 60.0, 61.3, 98.8, 113.3, 120.3, 122.4, 124.1, 126.0, 127.2, 128.0, 128.2, 133.1, 138.2, 138.5, 138.9, 140.4, 151.4; HRMS (ES+) calcd for [C26H29N7O2S2 + H] 536.1902, found 536.1912; HPLC (I) tR = 12.93 min (100%), (II) tR = 19.55 min (100%).
(±)-[N-(N-tert-Butoxycarbonylpiperidin-4-ylmethyl)-N-{trans-2-[(4-cyanophenyl)(3-methyl-3H-imidazol-4-ylmethyl)amino]cyclopentyl}]-1-methyl-1H-imidazole-4-sulfonamide (7d)
The synthesis was as per general procedure B with 45 and N-tert-butoxycarbonylpiperidin-4-ylmethyl bromide (1.5 equiv) on a 0.0774 mmol scale in DMF (0.1 M), and the mixture was stirred for 7 days at room temperature. The crude residue was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to afford 7d (41 mg, 83%): δH (500 MHz, CDCl3) 1.00–1.09 (m, 1H, CH (piperidinylmethyl)), 1.24–1.35 (m, 2H, CH (cyclopentyl), CH (piperidinylmethyl)), 1.37–1.46 (m, 11H, C(CH3)3, and 2 CH (piperidinylmethyl)), 1.51–1.65 (m, 5H, 3 CH (cyclopentyl), 2 CH (piperidinylmethyl)), 2.00–2.08 (m, 2H, 2 CH (cyclopentyl)), 2.17–2.26 (m, 1H, CH (piperidinylmethyl)), 2.68–2.80 (m, 1H, CH (piperidinylmethyl)), 3.23 (dd, J = 15.0, 9.0 Hz, 1H, CH (piperidinylmethyl)), 3.65 (s, 3H, CH3(Im)), 3.75 (s, 3H, CH3(Im)), 3.89–4.00 (m, 3H, CHCH2N (piperidinylmethyl), CHN (cyclopentyl)), 4.63 (m, 1H, CHN (cyclopentyl)), 4.72 (d, J = 18.0 Hz, 1H, CHaIm), 5.06 (d, J = 18.0 Hz, 1H, CHbIm), 6.71 (s, 1H, CH (Im)), 6.78 (d, J = 9.0 Hz, 2H, 2 CH (Ar)), 7.42 (s, 1H, CH (Im)), 7.46 (s, 1H, CH (Im)), 7.48 (d, J = 9.0 Hz, 2H, 2 CH (Ar)), 7.60 (s, 1H, CH (Im)); δC (125 MHz, CDCl3) 18.5, 23.0, 25.8, 28.4, 30.2, 32.0, 34.0, 36.8, 39.5, 43.7 (br), 49.6, 60.0, 61.5, 79.3, 99.5, 113.1, 119.9, 124.1, 127.7, 128.2, 133.6, 138.2, 139.0, 140.1, 151.6, 154.5; HRMS (ES+) calcd for [C32H44N8O4S + H] 637.3284, found 637.3292; HPLC (I) tR = 13.45 min (100%), (II) tR = 21.09 min (100%).
(±)-[N-{N-(2-Pyrimidinyl)-piperidin-4-ylmethyl}-N-{trans-2-[(4-cyanophenyl)(3-methyl-3H-imidazol-4-ylmethyl)amino]cyclopentyl}]-1-methyl-1H-imidazole-4-sulfonamide (7e)
The synthesis was as per general procedure B with 45 and N-(2-pyrimidinyl)-piperidin-4-ylmethyl bromide (1.5 equiv) on a 0.0799 mmol scale in DMF (0.1 M), and the mixture was stirred for 7 days at room temperature. The crude residue was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to afford 7e (38 mg, 78%): δH (500 MHz, CDCl3) 0.88 (qd, J = 12.2, 4.0 Hz, 1H, CH (piperidinylmethyl)), 1.01 (qd, J = 12.3, 4.0 Hz, 1H, CH (piperidinylmethyl)), 1.25–1.34 (m, 1H, 1 CH (cyclopentyl)), 1.38–1.48 (m, 2H, 2 CH (piperidinylmethyl)), 1.52–1.78 (m, 4H, 3 CH (cyclopentyl), CH (piperidinylmethyl)), 1.98–2.09 (m, 2H, 2 CH (cyclopentyl)), 2.28 (br t, J = 12.0, 1H, CH (piperidinylmethyl)), 2.40 (br t, J = 12.0 Hz, 1H, CH (piperidinylmethyl)), 2.74 (dd, J = 14.8, 6.0 Hz, 1H, CH (piperidinylmethyl)), 3.24 (dd, J = 14.8, 8.5 Hz, 1H, CH (piperidinylmethyl)), 3.61 (s, 3H, CH3(Im)), 3.74 (s, 3H, CH3(Im)), 3.97 (m, 1H, CHN (cyclopentyl)), 4.47–4.54 (m, 2H, CHCH2N (piperidinylmethyl)), 4.63 (m, 1H, CHN (cyclopentyl)), 4.73 (d, J = 17.5 Hz, 1H, CHaIm), 5.05 (d, J = 17.5 Hz, 1H, CHbIm), 6.41 (t, J = 4.9 Hz, 1H, CH (pyrimidine), 6.59 (s, 1H, CH (Im)), 6.78 (d, J = 8.5 Hz, 2H, 2 CH (Ar)), 7.41 (s, 1H, CH (Im)), 7.43–7.49 (m, 4H, 2 CH (Im), 2 CH (Ar)), 8.25 (d, J = 4.9 Hz, 2H, 2 CH (pyrimidine)); δC (125 MHz, CDCl3) 18.6, 23.1, 25.8, 29.9, 31.8, 33.9, 37.0, 39.5, 43.5, 49.7, 60.1, 61.6, 99.4, 109.3, 113.2, 119.9, 124.1, 128.2, 128.4, 133.6, 138.4, 138.9, 140.1, 151.8, 157.6, 161.3; HRMS (ES+) calcd for [C31H38N10O2S + H] 615.2978, found 615.2990; HPLC (I) tR = 12.91 min (99.70%), (II) tR = 19.32 min (99.65%).
(±)-[N-Benzyl-N-{cis-3-[(4-cyanophenyl)(3-methyl-3H-imidazol-4-ylmethyl)amino]cyclopentyl}]-1-methyl-1H-imidazole-4-sulfonamide (8a)
The synthesis was as per general procedure A with alcohol 55 and sulfonamide 19a on a 0.102 mmol scale. The crude residue was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to furnish 8a (38 mg, 71%): δH (500 MHz, CDCl3) 1.45–1.55 (m, 1H, CH (cyclopentyl)), 1.62–1.86 (m, 4H, 4 CH (cyclopentyl)), 1.92–1.98 (m, 1H, CH (cyclopentyl)), 3.42 (s, 3H, CH3(Im)), 3.65 (s, 3H, CH3(Im)), 4.02–4.17 (m, 4H, CH2Ph, 2 CHN (cyclopentyl)), 4.30 (d, J = 16.5 Hz, 1H, CHaIm), 4.41 (d, J = 16.5 Hz, 1H, CHbIm), 6.52–6.58 (m, 3H, 2 CH (Ar), CH (Im)), 7.14–7.27 (m, 6H, 5 CH (Ph), CH (Im)), 7.30 (d, J = 8.5 Hz, 2H, 2 CH (Ar)), 7.33 (s, 1H, CH (Im)), 7.39 (s, 1H, CH (Im)); δC (125 MHz, CDCl3) 26.8, 28.0, 31.5, 33.6, 33.9, 41.6, 49.5, 56.4, 57.3, 99.2, 113.5, 120.0, 123.8, 127.1, 127.2, 127.3, 128.0, 128.4, 133.3, 138.2, 138.4, 138.9, 140.7, 151.9; HRMS (ES+) calcd for [C28H31N7O2S + H] 530.2338, found 530.2357; HPLC (I) tR = 12.28 min (100%), (II) tR = 18.26 min (100%).
(±)-[N-(2-Methylbenzyl)-N-{cis-3-[(4-cyanophenyl)(3-methyl-3H-imidazol-4-ylmethyl)amino]cyclopentyl}]-1-methyl-1H-imidazole-4-sulfonamide (8b)
The synthesis was as per general procedure A with alcohol 55 and sulfonamide 19b on a 0.102 mmol scale. The crude residue was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to yield 8b (36 mg, 65%): δH (500 MHz, CDCl3) 1.51–1.58 (m, 1H, CH (cyclopentyl)), 1.66–1.75 (m, 1H, CH (cyclopentyl)), 1.75–1.93 (m, 3H, 3 CH (cyclopentyl)), 2.03–2.08 (m, 1H, CH (cyclopentyl)), 2.26 (s, 3H, CH3Ph), 3.46 (s, 3H, CH3(Im)), 3.73 (s, 3H, CH3(Im)), 4.13 (m, 1H, CHN (cyclopentyl)), 4.17–4.24 (m, 3H, CH2Ph, CHN (cyclopentyl)), 4.34 (d, J = 16.5 Hz, 1H, CHaIm), 4.46 (d, J = 16.5 Hz, 1H, CHbIm), 6.60–6.64 (m, 3H, CH (Im), 2 CH (Ar)), 7.08–7.11 (m, 1H, CH (Ph)), 7.13–7.17 (m, 2H, 2 CH (Ph)), 7.28 (s, 1H, CH (Im)), 7.36–7.40 (m, 3H, 2 CH (Ar), CH (Im)), 7.41–7.44 (m, 1H, CH (Ph)), 7.45 (s, 1H, CH (Im)); δC (125 MHz, CDCl3) 19.2, 26.9, 27.8, 31.5, 33.5, 34.0, 41.7, 47.4, 56.4, 57.2, 99.3, 113.5, 120.1, 123.9, 126.0, 127.2, 127.7, 128.0, 128.1, 130.2, 133.4, 135.2, 135.8, 138.3, 138.9, 140.7, 151.9; HRMS (ES+) calcd for [C29H33N7O2S + H] 544.2495, found 544.2495; HPLC (I) tR = 12.41 min (98.55%), (II) tR = 17.99 min (98.89%).
(±)-[N-(Thiophen-3-ylmethyl)-N-{cis-3-[(4-cyanophenyl)(3-methyl-3H-imidazol-4-ylmethyl)amino]cyclopentyl}]-1-methyl-1H-imidazole-4-sulfonamide (8c)
The synthesis was as per general procedure A with alcohol 55 and sulfonamide 19c on a 0.102 mmol scale. The crude residue was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to afford 8c (43 mg, 79%): δH (500 MHz, CDCl3) 1.53–1.62 (m, 1H, CH (cyclopentyl)), 1.74–1.86 (m, 3H, 3 CH (cyclopentyl)), 1.87–1.93 (m, 1H, CH (cyclopentyl)), 2.01–2.06 (m, 1H, CH (cyclopentyl)), 3.54 (s, 3H, CH3(Im)), 3.71 (s, 3H, CH3(Im)), 4.11–4.19 (m, 2H, 2 CHN (cyclopentyl)), 4.25 (AB quartet, J = 17.8 Hz, 2H, CH2Ph), 4.37 (d, J = 16.4 Hz, 1H, CHaIm), 4.45 (d, J = 16.4 Hz, 1H, CHbIm), 6.61–6.61 (m, 3H, 2 CH (Ar), CH (Im)), 7.04 (dd, J = 5.0, 1.2 Hz, 1H, CH (thiophene)), 7.05–7.07 (m, 1H, CH (thiophene)), 7.22 (dd, J = 5.0, 3.0 Hz, 1H, CH (thiophene)), 7.25 (s, 1H, CH (Im)), 7.38 (d, J = 9.0 Hz, 2H, 2 CH (Ar)), 7.41 (s, 1H, CH (Im)), 7.42 (s, 1H, CH (Im)); δC (125 MHz, CDCl3) 26.9, 28.1, 31.5, 33.6, 33.9, 41.8, 45.2, 56.5, 57.2, 99.3, 113.5, 120.0, 122.0, 123.7, 126.0, 127.2, 127.4, 128.1, 133.4, 138.3, 138.8, 139.7, 141.0, 152.0; HRMS (ES+) calcd for [C26H29N7O2S2 + H] 536.1902, found 536.1910; HPLC (I) tR = 12.09 min (100%), (II) tR = 17.62 min (100%).
(±)-[N-(N-tert-Butoxycarbonylpiperidin-4-ylmethyl)-N-{cis-3-[(4-cyanophenyl)-(3-methyl-3H-imidazol-4-ylmethyl)amino]cyclopentyl}]-1-methyl-1H-imidazole-4-sulfonamide (8d)
The synthesis was as per general procedure A with alcohol 55 and sulfonamide 19d on a 0.102 mmol scale. The crude residue was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to furnish 8d (40 mg, 62%): δH (500 MHz, CDCl3) 0.92–0.98 (m, 1H, CH (piperidinylmethyl)), 1.04 (qd, J = 12.5, 4.0 Hz, 1 H, CH (piperidinylmethyl)), 1.45 (s, 9H, C(CH3)3), 1.65–1.77 (m, 3H, 2 CH (piperidinylmethyl), CH (cyclopentyl)), 1.79–1.91 (m, 4H, 3 CH (cyclopentyl), CH (piperidinylmethyl)), 1.94–2.06 (m, 2H, 2 CH (cyclopentyl)), 2.58–2.69 (m, 2H, 2 CH (piperidinylmethyl)), 2.87–2.98 (m, 1H, CH (piperidinylmethyl)), 2.99–3.10 (m, 1H, CH (piperidinylmethyl)), 3.62 (s, 3H, CH3(Im)), 3.72 (s, 3H, CH3(Im)), 3.98 (m, 1H, CHN (cyclopentyl)), 4.03–4.14 (m, 2H, CHCH2N (piperidinylmethyl)), 4.18 (m, 1H, CHN (cyclopentyl)), 4.42 (AB quartet, J = 17.5 Hz, 2H, CH2Im), 6.67 (s, 1H, CH (Im)), 6.69 (d, J = 9.0 Hz, 2H, 2 CH (Ar)), 7.20 (s, 1H, CH (Im)), 7.39 (s, 1H, CH (Im)), 7.41 (d, J = 9.0 Hz, 2 CH (Ar)), 7.44 (s, 1H, CH (Im)); δC (125 MHz, CDCl3) 26.9, 28.2, 28.4, 29.9, 31.6, 33.5, 33.9, 36.7, 41.9, 43.6 (br), 52.5, 56.3, 58.3, 79.3, 99.4, 113.6, 120.0, 123.7, 128.1, 128.3, 133.4, 138.3, 138.7, 140.7, 152.0, 154.7; HRMS (ES+) calcd for [C32H44N8O4S + H] 637.3284, found 637.3283; HPLC (I) tR = 12.89 min (99.52%), (II) tR = 20.14 min (99.30%).
(±)-[N-{N-(2-Pyrimidinyl)-piperidin-4-ylmethyl}-N-{cis-3-[(4-cyanophenyl)(3-methyl-3H-imidazol-4-ylmethyl)amino]cyclopentyl}]-1-methyl-1H-imidazole-4-sulfonamide (8e)
The synthesis was as per general procedure A with alcohol 55 (0.203 mmol, 1 equiv) and sulfonamide 21 (1.5 equiv), with 2 equiv of PPh3 and 1.5 equiv of DIAD. The crude residue was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to furnish (±)-[N-tert-butoxycarbonyl-N-{cis-3-[(4-cyanophenyl)(3-methyl-3H-imidazol-4-ylmethyl)amino]cyclopentyl}]-1-methyl-1H-imidazole-4-sulfonamide as a white powder (108 mg, 99%): δH (500 MHz, CDCl3) 1.37 (s, 9H, C(CH3)3), 1.90–1.96 (m, 1H, CH (cyclopentyl)), 1.98–2.06 (m, 1H, CH (cyclopentyl)), 2.09–2.17 (m, 1H, CH (cyclopentyl)), 2.25–2.41 (m, 3H, 3 CH (cyclopentyl)), 3.66 (s, 3H, CH3(Im)), 3.76 (s, 3H, CH3(Im)), 4.28 (m, 1H, CHN (cyclopentyl)), 4.50 (AB quartet, J = 17.5 Hz, 2H, CH2Im), 4.95 (m, 1H, CHN (cyclopentyl)), 6.72–6.79 (m, 3H, CH (Im) 2 CH (Ar)), 7.41–7.50 (m, 4H, 2 CH (Im), 2 CH (Ar)), 7.59 (s, 1H, CH (Im)); δC (125 MHz, CDCl3) 27.5, 27.7, 28.1, 31.3, 32.7, 33.9, 41.1, 56.1, 57.5, 83.8, 98.5, 113.1, 120.1, 124.9, 127.6, 128.3, 133.2, 138.0, 138.4, 140.0, 150.6, 152.0; HRMS (ES+) calcd for [C26H33N7O4S + H] 540.2393, found 540.2391. The material (108 mg, 0.201 mmol) was redissolved in a 1:1 mixture of CH2Cl2/TFA (7 mL). After the mixture was stirred for 3 h at room temperature, TLC indicated the reaction was complete, and so all solvent was removed in vacuo. The residue was dry-loaded onto silica gel and purified by flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 92:7: 1) to give (±)-[N-{cis-3-[(4-cyanophenyl)(3-methyl-3H-imidazol-4-ylmethyl)amino]cyclopentyl}]-1-methyl-1H-imidazole-4-sulfonamide as a glassy film (88 mg, 96%): δH (500 MHz, MeOH-d4) 1.55–1.61 (m, 1H, CH (cyclopentyl)), 1.64–1.72 (m, 2H, 2 CH (cyclopentyl)), 1.81–1.89 (m, 1H, CH (cyclopentyl)), 1.93–1.99 (m, 1H, CH (cyclopentyl)), 2.08–2.13 (m, 1H, CH (cyclopentyl)), 3.56 (m, 1H, CHNHSO2), 3.69 (s, 3H, CH3(Im)), 3.75 (s, 3H, CH3(Im)), 4.26 (m, 1H, CHN (cyclopentyl)), 4.46 (AB quartet, J = 18.0 Hz, 2H, CH2Im), 6.57 (s, 1H, CH (Im)), 6.68 (d, J = 9.3 Hz, 2H, 2 CH (Ar)), 7.41 (d, J = 9.3 Hz, 2H, 2 CH (Ar)), 7.47 (s, 1H, CH (Im)), 7.50 (s, 1H, CH (Im)), 7.52 (s, 1H, CH (Im)); δC (125 MHz, CDCl3) 26.3, 30.3, 30.5, 32.9, 35.7, 40.6, 51.7, 56.8, 97.5, 112.7, 119.5, 123.9, 125.7, 128.5, 132.7, 137.5, 138.9, 139.2, 151.7; HRMS (ES+) calcd for [C21H25N7O2S + H] 440.1869, found 440.1881. Finally, (±)-[N-{cis-3-[(4-cyanophenyl)(3-methyl-3H-imidazol-4-ylmethyl)amino]cyclopentyl}]-1-methyl-1H-imidazole-4-sulfonamide (0.096 mmol) and N-(2-pyrimidinyl)-4-iodomethylpiperidine (1.5 equiv) were reacted together as per general procedure B in DMF (0.1 M), and the mixture was stirred for 3 days at room temperature. After the usual workup, the crude residue was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 92:7:1) to yield the title compound 8e (50 mg, 85%): δH (500 MHz, CDCl3) 1.04 (qd, J = 12.3, 4.0 Hz, 1H, CH (piperidinylmethyl)), 1.11 (qd, J = 12.3, 4.0 Hz, 1H, CH (piperidinylmethyl)), 1.65–1.72 (m, 1H, 1 CH (cyclopentyl)), 1.78–2.09 (m, 8H, 5 CH (cyclopentyl), 3 CH (piperidinylmethyl)), 2.77–2.85 (m, 2H, 2 CH (piperidinylmethyl)), 2.95 (dd, J = 14.5, 7.0 Hz, 1H, CH (piperidinylmethyl)), 3.07 (dd, J = 14.5, 7.5 Hz, 1H, CH (piperidinylmethyl)), 3.59 (s, 3H, CH3(Im)), 3.72 (s, 3H, CH3(Im)), 4.04 (m, 1H, CHN (cyclopentyl)), 4.18 (m, 1H, CHN (cyclopentyl)), 4.43 (AB quartet, J = 17.5 Hz, 2H, CH2Im), 4.73–4.80 (m, 2H, CHCH2N (piperidinylmethyl)), 6.44 (t, J = 4.8 Hz, 1H, CH (pyrimidine), 6.67 (s, 1H, CH (Im)), 6.69 (d, J = 9.0 Hz, 2H, 2 CH (Ar)), 7.20 (s, 1H, CH (Im)), 7.39 (s, 1H, CH (Im)), 7.40–7.44 (m, 3H, CH (Im), 2 CH (Ar)) 8.28 (d, J = 4.8 Hz, 2H, 2 CH (pyrimidine)); δC (125 MHz, CDCl3) 26.9, 28.3, 29.9, 31.6, 33.5, 33.9, 37.1, 41.9, 43.7, 52.6, 56.3, 58.3, 99.6, 109.4, 113.6, 120.0, 123.7, 128.2, 128.3, 133.5, 138.4, 138.7, 140.9, 152.1, 157.7, 161.5; HRMS (ES+) calcd for [C31H38N10O2S + H] 615.2978, found 615.3008; HPLC (I) tR = 12.56 min (99.21%), (II) tR = 19.15 min (99.37%).
(±)-[N-Benzyl-N-{trans-3-[(4-cyanophenyl)(3-methyl-3H-imidazol-4-ylmethyl)amino]cyclopentyl}]-1-methyl-1H-imidazole-4-sulfonamide (9a)
The synthesis was as per general procedure A with alcohol 60 and sulfonamide 19a on a 0.102 mmol scale. The crude residue was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to furnish 9a (29 mg, 54%): δH (500 MHz, CDCl3) 1.31–1.39 (m, 1H, CH (cyclopentyl)), 1.46–1.54 (m, 1H, CH (cyclopentyl)), 1.69–1.76 (m, 1H, CH (cyclopentyl)), 1.76–1.84 (m, 2H, 2 CH (cyclopentyl)), 1.91–1.97 (m, 1H, CH (cyclopentyl)), 3.56 (s, 3H, CH3(Im)), 3.63 (s, 3H, CH3(Im)), 4.14 (m, 1H, CHN (cyclopentyl)), 4.24 (s, 2H, CH2Ph), 4.26 (d, J = 16.8 Hz, 1H, CHaIm), 4.43 (d, J = 16.8 Hz, 1H, CHbIm), 4.49 (m, 1H, CHN (cyclopentyl)), 6.48 (d, J = 9.0 Hz, 2H, 2 CH (Ar)), 6.54 (s, 1H, CH (Im)), 7.20–7.23 (m, 2H, CH (Ph), CH (Im)), 7.25–7.31 (m, 5H, 2 CH (Ar), 2 CH (Ph), CH (Im)), 7.32–7.36 (m, 2H, CH (Im), CH (Ph)), 7.38 (s, 1H, CH (Im)); δC (125 MHz, CDCl3) 29.0, 29.1, 30.9, 31.6, 33.9, 41.7, 48.1, 56.9, 57.9, 99.1, 113.3, 120.1, 123.7, 127.1, 127.3, 127.9, 128.2, 128.5, 133.3, 138.2, 138.3, 138.9, 140.5, 151.8; HRMS (ES+) calcd for [C28H31N7O2S + H] 530.2338, found 530.2349; HPLC (I) tR = 12.21 min (100%), (II) tR = 18.09 min (99.84%).
(±)-[N-(2-Methylbenzyl)-N-{trans-3-[(4-cyanophenyl)(3-methyl-3H-imidazol-4-ylmethyl)amino]cyclopentyl}]-1-methyl-1H-imidazole-4-sulfonamide (9b)
The synthesis was as per general procedure A with alcohol 60 and sulfonamide 19b on a 0.136 mmol scale. The crude residue was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to give 9b (36 mg, 49%): δH (500 MHz, CDCl3) 1.37–1.53 (m, 2H, 2 CH (cyclopentyl)), 1.78–1.85 (m, 1H, CH (cyclopentyl)), 1.86–1.92 (m, 2H, 2 CH (cyclopentyl)), 1.97–2.02 (m, 1H, CH (cyclopentyl)), 2.32 (s, 3H, CH3Ph), 3.63 (s, 3H, CH3(Im)), 3.69 (s, 3H, CH3(Im)), 4.17 (m, 1H, CHN (cyclopentyl)), 4.27–4.31 (m, 3H, CH2Ph, CHaIm), 4.44 (d, J = 16.5 Hz, 1H, CHbIm), 4.58 (m, 1H, CHN (cyclopentyl)), 6.53 (d, J = 9.0 Hz, 2H, 2 CH (Ar)), 6.61 (s, 1H, CH (Im)), 7.12–7.15 (m, 1H, CH (Ph)), 7.16–7.22 (m, 2H, 2 CH (Ph)), 7.28 (s, 1H, CH (Im)), 7.34–7.37 (m, 3H, 2 CH (Ar), CH (Im)), 7.44 (s, 1H, CH (Im)), 7.48–7.51 (m, 1H, CH (Ph)); δC (125 MHz, CDCl3) 19.3, 28.9, 29.0, 31.5, 31.6, 33.9, 41.8, 45.9, 57.0, 57.9, 99.2, 113.3, 120.1, 123.8, 126.1, 127.1, 127.4, 128.0, 128.2, 130.1, 133.4, 134.9, 135.8, 138.3, 138.9, 140.4, 151.8; HRMS (ES+) calcd for [C29H33N7O2S + H] 544.2495, found 544.2503; HPLC (I) tR = 12.34 min (98.09%), (II) tR = 18.02 min (98.07%).
(±)-[N-(Thiophen-3-ylmethyl)-N-{trans-3-[(4-cyanophenyl)(3-methyl-3H-imidazol-4-ylmethyl)amino]cyclopentyl}]-1-methyl-1H-imidazole-4-sulfonamide (9c)
The synthesis was as per general procedure A with alcohol 60 and sulfonamide 19c on a 0.136 mmol scale. The crude residue was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to furnish 9c (47 mg, 65%): δH (500 MHz, CDCl3) 1.40–1.48 (m, 1H, CH (cyclopentyl)), 1.58–1.67 (m, 1H, CH (cyclopentyl)), 1.77–1.83 (m, 1H, CH (cyclopentyl)), 1.86–1.92 (m, 2H, 2 CH (cyclopentyl)), 2.00–2.06 (m, 1H, CH (cyclopentyl)), 3.62 (s, 3H, CH3(Im)), 3.68 (s, 3H, CH3(Im)), 4.25 (m, 1H, CHN (cyclopentyl)), 4.31 (s, 2H, CH2thiophene), 4.32 (d, J = 16.5 Hz, 1H, CHaIm), 4.46 (d, J = 16.5 Hz, 1H, CHbIm), 4.52 (m, 1H, CHN (cyclopentyl)), 6.58 (d, J = 9.1 Hz, 2H, 2 CH (Ar)), 6.61 (s, 1H, CH (Im)), 7.09 (dd, J = 5.0, 1.2 Hz, 1H, CH (thiophene)), 7.12–7.14 (m, 1H, CH (thiophene)), 7.26 (s, 1H, CH (Im)), 7.28 (dd, J = 5.0, 3.0 Hz, 1H, CH (thiophene)), 7.35 (s, 1H, CH (Im)), 7.37 (d, J = 9.1 Hz, 2H, 2 CH (Ar)), 7.43 (s, 1H, CH (Im)); δC (125 MHz, CDCl3) 29.0, 29.1, 31.6, 31.8, 33.9, 41.8, 43.9, 57.0, 57.9, 99.2, 113.3, 120.1, 122.1, 123.7, 126.0, 127.2, 128.0, 128.1, 133.4, 138.3, 138.8, 139.6, 140.6, 151.8; HRMS (ES+) calcd for [C26H29N7O2S2 + H] 536.1902, found 536.1911; HPLC (I) tR = 12.05 min (98.16%), (II) tR = 17.43 min (98.44%).
(±)-[N-(N-tert-Butoxycarbonylpiperidin-4-ylmethyl)-N-{trans-3-[(4-cyanophenyl)(3-methyl-3H-imidazol-4-ylmethyl)amino]cyclopentyl}]-1-methyl-1H-imidazole-4-sulfonamide (9d)
The synthesis was as per general procedure A with alcohol 60 and sulfonamide 19d on a 0.136 mmol scale. The crude residue was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to afford 9d (27 mg, 32%): δH (500 MHz, CDCl3) 1.00–1.13 (m, 2H, 2 CH (piperidinylmethyl)), 1.40–1.52 (s, 11H, C(CH3)3, CH (piperidinylmethyl), CH (cyclopentyl)), 1.56–1.65 (m, 1H, CH (cyclopentyl)), 1.70–1.84 (m, 3H, 2 CH (piperidinylmethyl), CH (cyclopentyl)), 1.87–2.01 (m, 2H, 2 CH (cyclopentyl)), 2.12–2.19 (m, 1H, CH (cyclopentyl)), 2.61–2.75 (m, 3H, 3 CH (piperidinylmethyl)), 3.02–3.11 (m, 1H, CH (piperidinylmethyl)), 3.67 (s, 3H, CH3(Im)), 3.68 (s, 3H, CH3(Im)), 4.04–4.19 (m, 2H, CHCH2N (piperidinylmethyl)), 4.31–4.37 (m, 3H, CH2Im, CHN (cyclopentyl)), 4.42 (m, 1H, CHN (cyclopentyl)), 6.62 (s, 1H, CH (Im)), 6.66 (d, J = 9.3 Hz, 2H, 2 CH (Ar)), 7.24 (s, 1H, CH (Im)), 7.25 (s, 1H, CH (Im)), 7.41 (d, J = 9.3 Hz, 2H, 2 CH (Ar)), 7.47 (s, 1H, CH (Im)); δC (125 MHz, CDCl3) 28.4, 29.0, 29.8, 30.2, 31.4, 33.9, 34.1, 36.9, 41.1, 43.7 (br), 49.9, 56.2, 58.7, 79.7, 100.7, 113.2, 118.6, 119.5, 124.7, 132.4, 133.8, 136.0, 138.1, 139.2, 150.9, 154.9; HRMS (ES+) calcd for [C32H44N8O4S + H] 637.3284, found 637.3293; HPLC (I) tR = 12.97 min (98.72%), (II) tR = 20.52 min (99.01%).
(±)-[N-{N-(2-Pyrimidinyl)-piperidin-4-ylmethyl}-N-{trans-3-[(4-cyanophenyl)(3-methyl-3H-imidazol-4-ylmethyl)amino]cyclopentyl}]-1-methyl-1H-imidazole-4-sulfonamide (9e)
The synthesis was as per general procedure A with alcohol 60 (0.203 mmol, 1 equiv) and sulfonamide 21 (1.5 equiv), with 2 equiv of PPh3 and 1.5 equiv of DIAD. The crude residue was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to furnish (±)-[N-tert-butoxycarbonyl-N-{2-trans-[(4-cyanophenyl)(3-methyl-3H-imidazol-4-ylmethyl)amino]cyclopentyl}]-1-methyl-1H-imidazole-4-sulfonamide as a white powder (90 mg, 83%): δH (500 MHz, CDCl3) 1.42 (s, 9H, C(CH3)3), 1.51–1.61 (m, 1H, CH (cyclopentyl)), 1.92–1.99 (m, 1H, CH (cyclopentyl)), 2.09–2.18 (m, 2H, 2 CH (cyclopentyl)), 2.20–2.28 (m, 1H, CH (cyclopentyl)), 2.43–2.49 (m, 1H, 1 CH (cyclopentyl)), 3.65 (s, 3H, CH3(Im)), 3.76 (s, 3H, CH3(Im)), 4.38 (AB quartet, J = 17.0 Hz, 2H, CH2Im), 4.72 (m, 1H, CHN (cyclopentyl)), 5.03 (m, 1H, CHN (cyclopentyl)), 6.72–6.77 (m, 3H, CH (Im), 2 CH (Ar)), 7.41 (d, J = 9.0 Hz, 2H, 2 CH (Ar)), 7.45 (s, 1H, CH (Im)), 7.51 (s, 1H, CH (Im)), 7.52 (s, 1H, CH (Im)); δC (125 MHz, CDCl3) 28.1, 30.0, 30.8, 31.6, 32.2, 34.0, 41.4, 56.4, 57.7, 84.3, 98.8, 113.3, 120.2, 125.0, 127.6, 128.0, 133.3, 138.5, 139.1, 140.1, 150.5, 152.2; HRMS (ES+) calcd for [C26H33N7O4S + H] 540.2393, found 540.2396. The material (85 mg, 0.158 mmol) was redissolved in a 1:1 mixture of CH2Cl2/TFA (5 mL). After the mixture was stirred for 3 h at room temperature, TLC indicated the reaction was complete, and so all solvent was removed in vacuo. The residue was dry-loaded onto silica gel and purified by flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 92:7:1) to give (±)-[N-{2-trans-[(4-cyanophenyl)(3-methyl-3H-imidazol-4-ylmethyl)amino]cyclopentyl}]-1-methyl-1H-imidazole-4-sulfonamide as a glassy film (68 mg, 99%): δH (500 MHz, CDCl3) 1.39–1.47 (m, 1H, CH (cyclopentyl)), 1.57–1.70 (m, 2H, 2 CH (cyclopentyl)), 1.90–2.06 (m, 3H, 3 CH (cyclopentyl)), 3.58 (s, 3H, CH3(Im)), 3.68 (s, 3H, CH3(Im)), 3.78 (m, 1H, CHNHSO2), 4.27 (s, 2H, CH2Im), 4.40 (m, 1H, CHN (cyclopentyl)), 6.56 (s, 1H, CH (Im)), 6.61 (d, J = 9.0 Hz, 2H, 2 CH (Ar)), 7.20 (s, 1H, CH (Im)), 7.28 (d, J = 9.0 Hz, 2H, 2 CH (Ar)), 7.40 (s, 1H, CH (Im)), 7.43 (s, 1H, CH (Im)), 7.47 (s, 1H, CH (Im)); δC (125 MHz, CDCl3) 27.9, 31.6, 32.7, 34.0, 35.7, 42.6, 53.0, 57.2, 98.6, 113.4, 120.2, 123.9, 127.6, 128.2, 133.2, 138.3, 139.0, 140.5, 151.9; HRMS (ES+) calcd for [C21H25N7O2S + H] 440.1869, found 440.1882. Finally, (±)-[N-{trans-3-[(4-cyanophenyl)(3-methyl-3H-imidazol-4-ylmethyl)amino]cyclopentyl}]-1-methyl-1H-imidazole-4-sulfonamide (0.095 mmol) and N-(2-pyrimidinyl)-piperidin-4-ylmethyl iodide (1.5 equiv) were reacted together as per general procedure B in DMF (0.1 M), and the mixture was stirred for 3 days at room temperature. After the usual workup, the crude residue was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 92:7:1) to yield the title compound 9e (49 mg, 85%): δH (500 MHz, CDCl3) 1.08–1.19 (m, 2H, 2 CH (piperidinylmethyl)), 1.41–1.51 (m, 1H, 1 CH (cyclopentyl)), 1.57–1.65 (m, 1H, CH (cyclopentyl)), 1.69–1.93 (m, 3H, 2 CH (piperidinylmethyl), CH (cyclopentyl)), 1.97–2.02 (m, 1H, CH (cyclopentyl)), 2.04–2.15 (m, 2H, 2 CH (cyclopentyl)), 2.74 (dd, J = 14.8, 8.1 Hz, 1H, CH (piperidinylmethyl)), 2.80–2.87 (m, 2H, 2 CH (piperidinylmethyl)), 2.97 (dd, J = 14.5, 7.5 Hz, 1H, CH (piperidinylmethyl)), 3.08 (dd, J = 14.8, 6.8 Hz, 1H, CH (piperidinylmethyl)), 3.67 (s, 3H, CH3), 3.68 (s, 3H, CH3), 4.29–4.39 (m, 3H, CHN (cyclopentyl), CH2Im), 4.45 (m, 1H, CHN (cyclopentyl)), 4.76–4.81 (m, 2H, CHCH2N (piperidinylmethyl)), 6.43 (t, J = 4.8 Hz, 1H, CH (pyrimidine), 6.60 (s, 1H, CH (Im)), 6.65 (d, J = 9.0 Hz, 2H, 2 CH (Ar)), 7.26 (m, 2H, 2 CH (Im)), 7.38 (d, J = 8.9 Hz, 2H, 2 CH (Ar)), 7.41 (s, 1H, CH (Im)), 8.28 (d, J = 4.8 Hz, 2H, 2 CH (pyrimidine)); δC (125 MHz, CDCl3) 26.6, 29.5, 29.9, 31.0, 31.7, 33.9, 37.4, 41.8, 43.8, 50.4, 57.7, 58.6, 99.2, 109.4, 113.3, 120.0, 123.8, 127.8, 128.3, 133.1, 138.4, 138.8, 139.8, 151.7, 157.8, 161.5; HRMS (ES+) calcd for [C31H38N10O2S + H] 615.2978, found 615.2959; HPLC (I) tR = 12.59 min (100%), (II) tR = 19.17 min (100%).
(±)-[N-Benzyl-N-{cis-4-[(4-cyanophenyl)(3-methyl-3H-imidazol-4-ylmethyl)amino]cyclohexyl}] 1-methyl-1H-imidazole-4-sulfonamide (10a)
The synthesis was as per general procedure C with 64a on a 0.149 mmol scale. After workup, the crude residue was dry-loaded onto silica gel, then purified by flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to afford 10a (52 mg, 65% (or 92% brsm)): δH (400 MHz, CDCl3) 1.55–1.67 (m, 8H, 2 NCH2CH2N), 3.35 (s, 3H, CH3(Im)), 3.65 (m, 1H, CHN (cyclohexyl)), 3.73 (s, 3H, CH3(Im)), 3.77 (m, 1H, CHN (cyclopentyl)), 4.17 (s, 2H, CH2Ph), 4.43 (s, 2H, CH2Im), 6.58 (s, 1H, CH (Im)), 6.67 (d, J = 9.0 Hz, 2H, CH (Ar)), 7.23–7.44 (m, 10H, CH (Im), 5 CH (Ph), 2 CH (Ar), 2 CH (Im)); δC (125 MHz, CDCl3) 27.0, 27.2, 31.1, 33.7, 42.3, 48.9, 52.1, 56.3, 94.9, 97.5, 101.0, 116.5, 119.5, 123.6, 126.9, 127.2, 128.0, 128.3, 132.9, 138.6, 138.7, 140.9, 151.6; HPLC (I) tR = 12.59 min (96.06%), (II) tR = 18.98 min (95.85%); HRMS (ES+) calcd for [C29H33N7O2S + H] 544.2507, found 544.2495.
(±)-[N-(N-tert-Butoxycarbonylpiperidin-4-ylmethyl)-N-{cis-4-[(4-cyanophenyl)(3-methyl-3H-imidazol-4-ylmethyl)amino]cyclohexyl}]-1-methyl-1H-imidazole-4-sulfonamide (10d)
The synthesis was as per general procedure C with 64d on a 0.108 mmol scale. After workup, the crude residue was dry-loaded onto silica gel, then purified by flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to furnish 10d (48 mg, 69% (or 94% brsm)): δH (400 MHz, CDCl3) 0.94–1.03 (m, 2H, 2 CH (piperidinylmethyl)), 1.46 (s, 9H, C(CH3)3), 1.56–1.85 (m, 9H, 2 NCH2CH2N (cyclohexyl), CH (piperidinylmethyl)), 2.00–2.10 (m, 2H, 2 CH (piperidinylmethyl)), 2.58–2.70 (m, 2H, 2 CH (piperidinylmethyl)), 2.87–3.01 (m, 2H, 2 CH (piperidinylmethyl)), 3.49 (s, 3H, CH3Im), 3.63 (m, 1H, CHN (cyclohexyl)), 3.72 (s, 3H, CH3(Im)), 3.81 (m, 1 H, CHN (cyclohexyl)), 4.03–4.16 (m, 2H, 2 CH (piperidinylmethyl)), 4.48 (s, 2H, CH2Im), 6.66 (s, 1H, CH (Im)), 6.78 (d, J = 9.0 Hz, 2H, 2 CH (Ar)), 7.32 (s, 1H, CH (Im)), 7.37 (s, 1H, CH (Im)), 7.39 (s, 1H, CH (Im)), 7.45 (d, J = 9.0 Hz, 2H, CH(Ar)); δC (125 MHz, CDCl3) 27.9, 28.2, 28.7, 29.7, 30.3, 31.7, 34.2, 36.9, 43.6, 51.3, 52.1, 58.3, 79.6, 101.9, 117.5, 119.9, 124.0, 127.8, 129.0, 133.5, 138.6, 139.0, 141.5, 152.4, 155.0; HRMS (ES+) calcd for [C33H46N8O4S + H] 651.3420, found 651.3455; HPLC (I) tR = 13.42 min (100%), (II) tR = 21.42 min (100%).
(±)-[N-Benzyl-N-{trans-4-[(4-cyanophenyl)(3-methyl-3H-imidazol-4-ylmethyl)amino]cyclohexyl}]-1-methyl-1H-imidazole-4-sulfonamide (11a)
Compound 66 was benzylated on the sulfonamide NH as per general procedure B on a 0.139 mmol scale with benzyl bromide. The sample was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 292:7:1 1:2) to furnish (±)-[N-benzyl-N-{trans-4-(4-cyanophenylamino)cyclohexyl}]-1-methyl-1H-imidazole-4-sulfonamide as a colorless film (61 mg, 98%): δH (500 MHz, CDCl3) 1.17 (qd, J = 12.5, 3.0 Hz, 2H, 2 CH (cyclohexyl)), 1.41 (qd, J = 12.5, 3.0 Hz, 2H, 2 CH (cyclohexyl)), 1.76–1.82 (m, 2H, 2 CH (cyclohexyl)), 1.96–2.02 (m, 2H, 2 CH (cyclohexyl)), 3.02 (tt, J = 11.0, 3.5 Hz, 1H, CHNSO2), 3.69 (s, 3H, CH3(Im)), 3.91 (tt, J = 12.5 Hz, 3.5 Hz, 1H, CHNHAr), 4.43 (s, 2H, CH2Ph), 6.42 (d, J = 8.0 Hz, 2H, 2 CH (Ar)), 7.21–7.32 (m, 5H, 5 CH (Ph)), 7.33 (s, 1H, CH (Im)), 7.39 (d, J = 8.0 Hz, 2H, 2 CH (Ar)), 7.44 (s, 1H, CH (Im)); δC (125 MHz, CDCl3) 30.0, 32.2, 33.9, 47.8, 50.6, 57.7, 98.3, 112.4, 120.4, 123.8, 127.2, 127.5, 128.3, 133.5, 138.8, 138.9, 141.2, 150.2; HRMS (ES+) calcd for [C24H27N5O2S + H] 450.1964, found 450.1972. The secondary aniline NH was then alkylated with imidazole 15 as per general procedure C on a 0.107 mmol scale, but the mixture was allowed to stir for 12 h, with the temperature gradually warming from 0 °C to room temperature, after which time the reaction appeared to have stalled. After usual workup, the crude residue was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to give the title compound 11a (38 mg, 65% (or 97% brsm)): δH (500 MHz, CDCl3) 1.41–1.53 (m, 4H, 4 CH (cyclohexyl)), 1.80–1.86 (m, 4H, 4 CH (cyclohexyl)), 3.55 (m, 1H, CHNSO2), 3.61 (s, 3H, CH3(Im)), 3.71 (s, 3H, CH3(Im)), 3.95 (m, 1H, CHNAr), 4.29 (s, 2H, CH2Im), 4.42 (s, 2H, CH2Ph), 6.59 (d, J = 9.5 Hz, 2H, 2 CH (Ar)), 6.62 (br s, 1H, CH (Im)), 7.24–7.44 (m, 10H, 5 CH (Ph), 2 CH (Ar), 3 CH (Im)); δC (125 MHz, CDCl3) 29.1, 30.4, 31.6, 33.9, 40.7, 47.6, 56.3, 57.5, 98.9, 112.9, 120.1, 123.7, 127.3, 127.4, 128.1, 127.9, 128.3, 133.4, 138.2, 138.6, 138.8, 141.2, 151.2; HRMS (ES+) calcd for [C29H33N7O2S + H] 544.2495, found 544.2502; HPLC (I) tR = 11.97 (100%), (II) tR = 17.04 (99.37%).
(±)-[N-(N-tert-Butoxycarbonylpiperidin-4-ylmethyl)-N-{trans-4-[(4-cyanophenyl)(3-methyl-3H-imidazol-4-ylmethyl)amino]cyclohexyl}]-1-methyl-1H-imidazole-4-sulfonamide (11d)
Compound 66 was alkylated on the sulfonamide NH as per general procedure B on a 0.139 mmol scale and with N-tert-butoxycarbonyl-4-bromom-ethylpiperidine in DMF (0.1 M). After 4 days, the reaction was worked up, then purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 292:7:1) to furnish the (±)-[N-(N-tert-butoxycarbonylpiperidin-4-ylmethyl)-N-{4-(4-trans-cyanophenylamino)cyclohexyl}]-1-methyl-1H-imidazole-4-sulfonamide as a colorless film (77 mg, 100%): δH (500 MHz, CDCl3) 1.07 (qd, J = 12.3, 4.3 Hz, 2H, 2 CH (piperidinylmethyl)), 1.25 (qd, J = 12.3, 3.0 Hz, 2H, 2 CH (cyclohexyl)), 1.44 (s, 9H, C(CH3)3), 1.56 (qd, J = 12.2, 3.0 Hz, 2H, 2 CH (cyclohexyl)), 1.72–1.79 (m, 2H, 2 CH (cyclohexyl)), 1.81–1.92 (m, 3H, 3 CH (piperidinylmethyl)), 2.07–2.13 (m, 2H, 2 CH (cyclohexyl)), 2.61–2.71 (m, 2H, 2 CH (piperidinylmethyl)), 3.01–3.08 (m, 2H, 2 CH (piperidinylmethyl), 3.18 (tt, J = 11.5, 3.5 Hz, 1H, CHNSO2), 3.73 (s, 3H, CH3(Im)), 3.77 (qd, J = 12.0, 3.5 Hz, 1H, CHNH), 4.06–4.15 (m, 2H, 2 CH (piperidinylmethyl)), 6.50 (d, J = 8.8 Hz, 2H, 2 CH (Ar)), 7.36 (d, J = 8.8 Hz, 2H, 2 CH (Ar)), 7.39 (s, 1H, CH (Im)), 7.44 (s, 1H, CH (Im)); δC (125 MHz, CDCl3) 28.4, 30.0, 30.2, 32.2, 33.9, 36.9, 43.4 (br), 50.2, 50.7, 57.7, 78.6, 98.3, 112.4, 120.4, 123.6, 134.6, 139.8, 141.0, 150.2, 154.7; HRMS (ES+) calcd for [C28H40N6O4S + H] 557.2910, found 557.2931. The secondary aniline NH was then alkylated with imidazole 15 as per general procedure C on a 0.0937 mmol scale, but the mixture was allowed to stir for 12 h, with the temperature gradually warming from 0 °C to room temperature. After usual workup, the crude residue was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to furnish the target molecule 11d (37 mg, 61% (or 95% brsm)): δH (500 MHz, CDCl3) 1.07 (qd, J = 12.3, 4.3 Hz, 2H, 2 CH (piperidinylmethyl)), 1.44 (s, 9H, C(CH3)3), 1.51–1.61 (m, 4H, 4 CH (cyclohexyl)), 1.72–1.80 (m, 2H, 2 CH (cyclohexyl)), 1.82–1.95 (m, 5H, 3 CH (piperidinylmethyl), 2 CH (cyclohexyl)), 2.60–2.71 (m, 2H, 2 CH (piperidinylmethyl)), 2.97–3.03 (m, 2H, 2 CH (piperidinylmethyl)), 3.63 (s, 3H, CH3(Im)), 3.68 (m, 1H, CHNSO2), 3.72 (s, 3H, CH3(Im)), 3.80 (m, 1H, CHNAr), 4.05–4.17 (m, 2H, 2 CH (piperidinylmethyl)), 4.36 (s, 2H, CH2Im), 6.63–6.69 (m, 3H, 2 CH (Ar), CH (Im)), 7.37 (s, 1H, Im), 7.40 (d, J = 9.0 Hz, 2H, 2 CH (Ar)), 7.42 (s, 1H, CH (Im)), 7.45 (br s, 1H, CH (Im)); δC (125 MHz, CDCl3) 28.2, 29.1, 30.0, 30.5 (br), 31.8, 33.9, 36.8, 40.8, 43.4 (br), 50.0, 56.4, 57.5, 79.5, 99.1, 113.0, 120.1, 123.5, 127.9, 128.2, 133.5, 138.3, 138.9, 140.9, 151.2, 154.7; HRMS (ES+) calcd for [C33H46N8O4S + H] 651.3441, found 651.3446; HPLC (I) tR = 13.18 (100%), (II) tR = 20.66 (99.92%).
Biological Assays. Plasmodium Strains
The P. falciparum strains used in this study were 3D7 (The Netherlands, [airport-associated malaria], chloroquine-sensitive) provided by Dr. Pradipsinh Rathod from the University of Washington and K1 (Thailand, chloroquine-resistant, pyrimethamine-resistant) obtained from the MR4 Unit of the American Type Culture Collection (ATCC, Manassas, VA).
P. falciparum Culture
Strains of P. falciparum were sustained in vitro on the basis of experimental techniques as described by Trager and Jensen.38 Cultures were maintained in RPMI-1640 (Sigma, St. Louis, MO) with 2 mM l-glutamine, 25 mM HEPES, 33 mM NaHCO3, 20 µg/mL gentamicin sulfate, and 20% (v/v) heat-inactivated human plasma type A+ (RP-20P). Type A+ erythrocytes were obtained from laboratory donors, washed three times with RPMI, resuspended in 50% RPMI, and stored at 4 °C. Parasites were grown in 10 mL of a 2% hematocrit/RP-20P (v/v) in 50 mL flasks under a 5% CO2, 5% O2, and 90% N2 atmosphere.
P. falciparum ED50 Determination
An amount of 1 µL of P. falciparum PFT inhibitor (PfPFTI) dissolved in DMSO was added to each well of a 96–well plate followed by the addition of 200 µL of P. falciparum culture at parasitemia and hematocrit of 0.5%. Plates were flushed with 5% CO2, 5% O2, and 90% N2 and then incubated at 37 °C for 48 h. [8-3H]Hypoxanthine (0.3 µCi, 20 Ci/mmol, American Radiolabeled Chemicals) in 30 µL of RP-20P was added to cultures and incubated for an additional 24 h. Cells were harvested onto filter mats by a Multiharvester (Skatron, Sunnyvale, CA), and the radioactivity incorporated into the parasites was counted on a β-scintillation counter. The background level detected with uninfected erythrocytes was subtracted from the data. The 3H-incorporation into infected RBCs with 1 µL of DMSO vehicle alone represents 100% malaria growth. ED50 values were determined by linear regression analysis of the plots of 3H-hypoxanthine incorporation versus concentration of compound.
PfPFT and Rat PFT IC50 Determination
The PFT assay used to determine the IC50 values (inhibitor concentration that causes 50% enzyme inhibition) of the compounds is based on a PFT [3H] scintillation proximity assay (SPA) (TRKQ7010 Amersham Bio-sciences Corp., Piscataway, NJ). Assays were carried out in assay buffer (pH 7.5, 50 mM HEPES, 30 mM MgCl2, 20 mM KCl, 5 mM DTT, 0.01% Triton X-100), 1 µm human lamin-B carboxyterminus sequence peptide (biotin-YRASNRSCAIM), and 1 µCi [3H]farnesylpyrophosphate (Amersham specific activity 15–20 Ci/mM) in a total volume of 50 µL which included 1 µL of PfPFT inhibitor solution in DMSO and 5 µL of partially purified PfPFT.17 Assays in the absence of PfPFT inhibitor and PfPFT were included as positive and negative controls, respectively. Reaction mixtures were incubated at 37 °C for 60 min and terminated by addition of 70 µL of assay STOP solution and 5 µL of SPA beads. The assay mixture was incubated at room temperature for 30 min. The assay was counted on a plate Chameleon 425-104 multilabel counter (Hidex Oy Turku, Finland). IC50 values were calculated using linear regression analysis of the plots of the amount of radioprenylation versus the concentration of compound. For ratPFT IC50 determination, 0.01 µm human lamin-B carboxy-terminus sequence peptide (biotin-YRASNRSCAIM) was used. The assay was incubated at 37 °C for 15 min; otherwise, experimental conditions were the same as for PfPFT IC50 determination.
Supplementary Material
Acknowledgment
Financial support for this work was provided by the Medicines for Malaria Venture (MMV) and the National Institutes of Health (Grants CA67771 and AI54384). We thank David Floyd, Lou Lombardo, and David Williams from Bristol-Myers Squibb for helpful suggestions.
Footnotes
Abbreviations: PFT, protein farnesyltransferase; P. falciparum, Plasmodium falciparum; PfPFT, P. falciparum protein farnesyltransferase; FPP, farnesyl pyrophosphate; ED50, effective dose that inhibits 50% of P. falciparum proliferation; IC50, inhibitor concentration that inhibits 50% of PFT enzyme activity; rt, room temperature.
Supporting Information Available: GOLD low energy docked pictures of compounds 2a–5a, 7a, 8a, 10a, and 11a, experimental procedures and full characterizations of compounds 14–64d, and HPLC traces of two independent conditions for all target molecules (2a–9e, 10a, 10d, 11a, 11d). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Sachs J, Malaney P. The economic and social burden of malaria. Nature. 2002;415:680–685. doi: 10.1038/415680a. [DOI] [PubMed] [Google Scholar]
- 2. www.mmv.org.
- 3.Snow RW, Trape JF, Marsh K. The past, present and future of childhood malaria mortality in Africa. Trends Parasitol. 2001;17:593–597. doi: 10.1016/s1471-4922(01)02031-1. [DOI] [PubMed] [Google Scholar]
- 4.Olliaro PL, Bloland PB. Clinical and public health implications of antimalarial drug resistance. Antimalar. Chemother. 2001:65–83. [Google Scholar]
- 5.Wiesner J, Ortmann R, Jomaa H, Schlitzer M. New antimalarial drugs! Angew. Chem., Int. Ed. 2003;42:5274–5293. doi: 10.1002/anie.200200569. [DOI] [PubMed] [Google Scholar]
- 6.May J, Meyer CG. Chemoresistance in falciparum malaria. Trends Parasitol. 2003;19:432–435. doi: 10.1016/j.pt.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 7.Ridley RG. Medical need, scientific opportunity and the drive for antimalarial drugs. Nature. 2002;415:686–693. doi: 10.1038/415686a. [DOI] [PubMed] [Google Scholar]
- 8.Greenwood B, Mutabingwa T. Malaria in 2002. Nature. 2002;415:670–672. doi: 10.1038/415670a. [DOI] [PubMed] [Google Scholar]
- 9.Chakrabarti D, Azam T, DelVecchio C, Qiu L, Park Y-I, Allen CM. Protein prenyl transferase activities of Plasmodium falciparum. Mol. Biochem. Parasitol. 1998;94:175–184. doi: 10.1016/s0166-6851(98)00065-6. [DOI] [PubMed] [Google Scholar]
- 10.(a) Nallan L, Bauer KD, Bendale P, Rivas K, Yokoyama K, Hornéy CP, Pendyala PR, Floyd D, Lombardo LJ, Williams DF, Hamilton A, Sebti S, Windsor WT, Weber PC, Buckner FS, Chakrabarti D, Gelb MH, van Voorhis WC. Protein farnesyltransferase inhibitors exhibit potent antimalarial activity. J. Med. Chem. 2005;48:3704–3713. doi: 10.1021/jm0491039. [DOI] [PubMed] [Google Scholar]; (b) Bendale P, Olepu S, Suryadevara PK, Bulbule V, Rivas K, Nallan L, Smart B, Yokoyama K, Ankala S, Pendyala PR, Floyd D, Lombardo LJ, Williams DK, Buckner FS, Chakrabarti D, Verlinde CLMJ, van Voorhis WC, Gel MH. Second generation tetrahydroquinoline-based protein farnesyltransferase inhibitors as antimalarials. J. Med. Chem. 2007;50:4585–4605. doi: 10.1021/jm0703340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clough N, Wilson RJM. Antibiotics and plasmodial plastid organelle. Antimalar. Chemother. 2001:265–286. [Google Scholar]
- 12.Loyevsky M, Gordeuk VR. Iron chelators. Antimalar. Chemother. 2001:307–324. [Google Scholar]
- 13.Gelb MH, van Voorhis WC, Buckner FS, Yokoyama K, Eastman R, Carpenter EP, Panethymitaki C, Brown KA, Smith DF. Protein farnesyl and N -myristoyl transferases: piggy-back medicinal chemistry targets for the development of antitrypanosomatid and antimalarial therapeutics. Mol. Biochem. Parasitol. 2003;126:155–163. doi: 10.1016/s0166-6851(02)00282-7. [DOI] [PubMed] [Google Scholar]
- 14.(a) Wiesner J, Kettler K, Sakowski J, Ortmann R, Katzin AM, Kimura EA, Silber K, Klebe G, Jomaa H, Schlitzer M. Farnesyltransferase inhibitors inhibit the growth of malaria parasites in vitro and in vivo. Angew. Chem., Int. Ed. 2004;43:251–254. doi: 10.1002/anie.200351169. [DOI] [PubMed] [Google Scholar]; (b) Schlitzer M. Structure based design of benzophenone-based non-thiol farnesyltransferase inhibitors. Curr. Pharm. Des. 2002;8:1713–1722. doi: 10.2174/1381612023394061. [DOI] [PubMed] [Google Scholar]
- 15.Sebti SM, Hamilton AD. Farnesyltransferase and geranylgeranyltransferase-I inhibitors and cancer therapy: lessons from mechanism and bench-to-bedside translational studies. Oncogene. 2000;19:6584–6593. doi: 10.1038/sj.onc.1204146. [DOI] [PubMed] [Google Scholar]
- 16.Bell IM. Inhibitors of farnesyltransferase: a rational approach to cancer chemotherapy. J. Med. Chem. 2004;47:1870–1878. doi: 10.1021/jm0305467. [DOI] [PubMed] [Google Scholar]
- 17.Strickland CL, Windsor WT, Syto R, Wang L, Bond R, Wu Z, Schwartz J, Le HV, Beese LS, Weber PC. Crystal structure of farnesyl protein transferase complexed with a CaaX peptide and farnesyl diphosphate analog. Biochemistry. 1998;37:16601–16611. doi: 10.1021/bi981197z. [DOI] [PubMed] [Google Scholar]
- 18.Chakrabarti D, Da Silva T, Barger J, Paquette S, Patel H, Patterson S, Allen CM. Protein farnesyltransferase and protein prenylation in Plasmodium falciparum. J. Biol. Chem. 2002;277:42066–42073. doi: 10.1074/jbc.M202860200. [DOI] [PubMed] [Google Scholar]
- 19.Eastman RT, White J, Hucke O, Bauer K, Yokoyama K, Nallan L, Chakrabarti D, Verlinde CL, Gelb MH, Rathod PK, van Voorhis WC. Resistance to a protein farnesyltransferase inhibitor in Plasmodium falciparum. J. Biol. Chem. 2005;280:13554–13559. doi: 10.1074/jbc.M413556200. [DOI] [PubMed] [Google Scholar]
- 20.Eastman RT, White J, Hucke O, Yokoyama K, Verlinde CLMJ, Hast MA, Beese LS, Gelb MH, Rathod PK, van Voorhis WC. Resistance mutations at the lipid substrate binding site of Plasmodium falciparum protein farnesyltransferase. Mol. Biochem. Parasitol. 2007;152:66–71. doi: 10.1016/j.molbiopara.2006.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ohkanda J, Lockman JW, Yokoyama K, Gelb MH, Croft SL, Kendrick H, Harrell MI, Feagin JE, Blaskovich MA, Sebti SM, Hamilton AD. Peptidomimetic inhibitors of protein farnesyltransferase show potent antimalarial activity. Bioorg. Med. Chem. Lett. 2001;11:761–764. doi: 10.1016/s0960-894x(01)00055-5. [DOI] [PubMed] [Google Scholar]
- 22.Ohkanda J, Knowles DB, Blaskovich MA, Sebti SM, Hamilton AD. Inhibitors of protein farnesyltransferase as novel anticancer agents. Curr. Top. Med. Chem. 2002;2:303–323. doi: 10.2174/1568026023394281. [DOI] [PubMed] [Google Scholar]
- 23.Carrico D, Ohkanda J, Kendrick H, Yokoyama K, Blaskovich MA, Bucher CJ, Buckner FS, van Voorhis WC, Chakrabarti D, Croft SL, Gelb MH, Sebti SM, Hamilton AD. In vitro and in vivo antimalarial activity of peptidomimetic protein farnesyltransferase inhibitors with improved membrane permeability. Bioorg. Med. Chem. 2004;12:6517–6526. doi: 10.1016/j.bmc.2004.09.020. [DOI] [PubMed] [Google Scholar]
- 24.van Voorhis WC, Rivas KL, Bendale P, Nallan L, Horney C, Barrett LK, Bauer KD, Smart BP, Ankala S, Hucke O, Verlinde CLMJ, Chakrabarti D, Strickland C, Yokoyama K, Buckner FS, Hamilton AD, Williams DK, Lombardo LJ, Floyd D, Gelb MH. Efficacy, pharmacokinetics, and metabolism of tetrahydroquinoline inhibitors of Plasmodium falciparum protein farnesyltransferase. Antimicrob. Agents Chemother. 2007;51:3659–3671. doi: 10.1128/AAC.00246-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. www.plasmodb.org.
- 26.Glenn MP, Chang S-Y, Hucke O, Verlinde CL, Rivas K, Hornéy C, Yokoyama K, Buckner FS, Pendyala PR, Chakrabarti D, Gelb M, van Voorhis WC, Sebti SM, Hamilton AD. Structurally simple farnesyltransferase inhibitors arrest the growth of malaria parasites. Angew. Chem. Int. Ed. 2005;44:4903–4906. doi: 10.1002/anie.200500674. [DOI] [PubMed] [Google Scholar]
- 27.Glenn MP, Chang S-Y, Hornéy C, Rivas K, Yokoyama K, Pusateri EE, Fletcher S, Cummings CG, Buckner FS, Pendyala PR, Chakrabarti D, Sebti SM, Gelb M, van Voorhis WC, Hamilton AD. Structurally simple, potent Plasmodium selective farnesyltransferase inhibitors that arrest the growth of malaria parasites. J. Med. Chem. 2006;49:5710–5727. doi: 10.1021/jm060081v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 29.Reissig HU, Zimmer R. Donor–acceptor-substituted cyclopropane derivatives and their application in organic synthesis. Chem. ReV. 2003;103:1151–1196. doi: 10.1021/cr010016n. [DOI] [PubMed] [Google Scholar]
- 30.InsightII. San Diego, CA: Accelrys Inc; 2000. http://www.accelry-s.com. [Google Scholar]
- 31.Unpublished results.
- 32.Thorpe-Ingold effect, or the “gem-dialkyl effect”: replacement of a C–CH2–C group with a C–CR2–C group causes a compression of the C–C–C bond angle at the now-quarternary carbon.
- 33.Long SB, Hancock PJ, Kral AM, Hellinga HW, Beese LS. The crystal structure of human proteinfarnesyltransferase reveals the basis for inhibition by CaaX tetrapeptides and their mimetics. Proc. Natl. Acad. Sci. U.S.A. 2001;98:12948–12953. doi: 10.1073/pnas.241407898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seto Y, Guengerich FP. Partitioning between N-dealkylation and N-oxygenation in the oxidation of N,N -dialkylarylamines catalyzed by cytochrome P450 2B1. J. Biol. Chem. 1993;268:9986–9997. [PubMed] [Google Scholar]
- 35.Molecular Operating EnVironment. Montreal, Quebec, Canada: Chemical Computing Group; 2007. version 2007.11. [Google Scholar]
- 36.Sukumar N, Breneman CM. QTAIM in Drug Discovery and Protein Modeling. In: Matta CF, Boyd RJ, editors. The Quantum Theory of Atoms in Molecules: From Solid State to DNA and Drug Design; Weinheim, Germany: Wiley-VCH; 2007. [Google Scholar]
- 37.Embrechts MJ. KernelPLS(Analyze) Troy, NY: Rensselaer Polytechnic Institute; 2001. version 6.36. [Google Scholar]
- 38.Trager W, Jensen JB. Human malaria parasites in continuous culture. Science. 1976;193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.