

Abstract
Photocatalytic oxidation of nucleic acid components on aqueous metal oxides (TiO2, α-FeOOH, and α-Fe2O3) has been studied. The oxidation of purine nucleotides results in the formation of the purine radical cations and sugar-phosphate radicals, whereas the oxidation of pyrimidine nucleotides other than thymine results in the oxidation of only the sugar-phosphate. The oxidation of the thymine (and to a far less extent for the 5-methylcytosine) derivatives results in deprotonation from the methyl group of the base. Some single stranded (ss) oligoribonucleotides and wild-type ss RNA were oxidized at purine sites. In contrast, double stranded (ds) oligoribonucleotides and DNA were not oxidized. These results account for observations suggesting that cellular ds DNA is not damaged by exposure to photoirradiated TiO2 nanoparticles inserted into the cell, whereas ss RNA is extensively damaged. The astrobiological import of our observations is that the rapid degradation of monomer nucleotides make them poor targets as biosignatures, whereas duplex DNA is a better target as it is resilient to oxidative diagenesis. Another import of our studies is that ds DNA (as opposed to ss RNA) appears to be optimized to withstand oxidative stress both due to the advantageous polymer morphology and the subtle details of its radical chemistry. This peculiarity may account for the preference for DNA over RNA as a “molecule of life” provided that metal oxides served as the template for synthesis of polynucleotides, as suggested by Orgel and others.
1. INTRODUCTION
The search for organic life on other planets in the Solar System, including Mars, is conceived as the search for terrestrial “biosignatures,” 1 that is the common biomolecules and metabolites and the products of their transformation in the environment (known as “diagenesis”). 2–7 As the solar radiation could be the source of energy for tentative life forms, the top soil is a possible habitat. The atmosphere of Mars transmits ultra-violet (UV) light mainly in the UVA region. 2,4,5 Martian soil is deficient in organic components in general, 2–6 which was rationalized as evidence for oxidative diagenesis occurring in the soil. 2–8 The submicron soil particles include perchlorates 8 and several light-absorbing metal oxides, such as titanium (IV) and iron (III) oxides; the latter oxides give Mars its characteristic red-brownish color. 9 Such oxides are semiconductors, and UV irradiation of their surfaces results in charge separation. The holes (electron deficiencies) that migrate to the surface can readily oxidize organic molecules adsorbed at the oxide surface. 3,4,10–13 Without the occurrence of catalysis, the photoreactions would require more energetic photons that have a much lower flux than the UVA radiation. 2–5 This brings the question, what types of biomarkers can exhibit long-term photostablity in such environment?
In our previous studies, 3,4 we demonstrated that carboxylated compounds (RCO2−) in general and natural amino acids in particular exhibit low resistance 12 to the above-mentioned catalytic photoreactions due to the occurrence of oxidative decarboxylation (photo-Kolbe reaction), 11 in which the photogenerated hole accepts an electron from RCO2− (which is linked to the surface in a bidentate fashion) yielding R• and CO2. Using electron paramagnetic resonance (EPR) spectroscopy, we demonstrated that this reaction (for aliphatic amino acids) and concurrent deprotonation (for aromatic amino acids) readily occur even at low temperature (50–100 K). 3 The same reaction occurred for short polypeptides, 3,4 however, for large peptides the efficiency of photocatalytic oxidation was quite low, as the hole on the oxide is not sufficiently energetic to react with the polymer chain or the aromatic residues in the hydrophobic core. 3,4
In the present study, we examine the photolytic stability of DNA, RNA, and related compounds on UV light exposed metal oxides, which is important not only in its astrobiological context, but also in its geochemical and biomedical contexts (see Section 4 and 5). These nucleic acids involve (2′-deoxy)ribose moieties. In ref. 13, the mechanisms for the photocatalytic oxidation of carbohydrates on the metal oxides have been examined. The main import of these studies for this work is that the TiO2 and α–FeOOH (but not α-Fe2O3) can readily oxidize chemisorbed D-ribose and 2′-deoxy-D-ribose and the corresponding 5′-sugar-phosphates. The reaction proceeds through the formation of the radical cations of the carbohydrates that deprotonate yielding the corresponding H loss neutral radicals (in the following designated as C(n′)•, where n′ is the corresponding carbon atom in the sugar residue, see Scheme 1. In bulk solutions, such neutral radicals tend to dehydrate. However, on the oxide surfaces this reaction is impeded by the bidentate oxo bridge formation between the vicinal hydroxyl groups and the metal. Instead, high yield of the formyl radical was observed. It was demonstrated that most of the formyl elimination involves the carbon-1 site and that the main two radicals formed by the catalytic photooxidation of the ribose and 2-deoxyribose are carbon -1 and -2 centered radicals, with the minor contribution from carbon-3 centered radicals. We interpret such preferences in the radical formation as the indication for the preferences in the modes of chemisorption for the corresponding molecules on the oxides.
Scheme 1.

Structural formulas and atom numbering for nucleobases and the general formula for the ribonucleosides (Z=Y =OH), ribonucleotides (Z=phosphate, Y=OH) and the corresponding 2-deoxyribo-derivatives (Y=H).
This study proceeds as follows. Photocatalytic oxidation of nucleic acids on aqueous metal oxides (TiO2, α-FeOOH, and α-Fe2O3) are examined in Section 3. Nucleosides and nucleotides are examined in Section 3.1, 2′-deoxynucleotides are examined in Section 3.2, and synthetic polynucleotides and wild-type RNA and DNA are examined in Section 3.3. For brevity, some tables and figures are supplied as supporting information; such materials have the designator “S”. In Section 4, we place these results in a broader context and in Section 5 we discuss the implications of our observations.
2. EXPERIMENTAL
Materials
Unless stated otherwise, all chemicals were obtained from Sigma-Aldrich (Milwaukee, WI) and used without further purification. Nucleosides (Scheme 1) are D-ribose substituted at the carbon-1′ with a pyrimidine (C and 5-Me-C for cytosine and 5-methylcytosine and U and T for uracil and 5-methyluracil (that is, thymine)) or a purine (A for adenine and G for guanine) nucleobase. The standard convention is followed for designating the nucleotides (5′-phosphate derivatives of the nucleosides): the middle latter M is for mono-, D is for di-, and T is for tri- phosphate; for example, ADP is adenosine-5′-diphosphate. The small letter “d” designates 2′-deoxynucleotides (as in dTMP).
d8-Deuterated (substituted at carbon-8) purine (2′-deoxy)ribonucleosides and ribonucleotides were synthesized by H/D exchange of the protiated compound in tetraethylamine/D2O for 1–3 days at 55 °C with the subsequent evaporation of the solvent in vacuum and repeated dissolution in D2O followed by the evaporation (4–5 cycles). 14–16 Employing Bruker Avance DMX 500 MHz NMR spectrometer (Bruker BioSpin, Billerica, MA), the degree of the H/D exchange was determined as 92 to 98% by integration. Other deuterated biochemicals were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA) and Omicron Biochemicals, Inc. (South Bend, IN). The 13CH3-labeled thymidine (dT) was obtained from Cambridge Isotope Laboratories (Andover, MA).
Anatase nanoparticles were synthesized by hydrolysis of TiCl4 as described in ref. 17 and stabilized in pH=2 solution. Hematite nanoparticles were prepared by hydrothermal oxidation of FeCl3 as described in ref. 3. These nanoparticles (30–50 nm) were stabilized at pH=4. The optical absorption spectra of these two solutions are given in Figure 1 in ref. 3. Goethite (α-FeOOH) particles were synthesized by hydrolysis of iron(III) nitrate at 50 °C for three days at pH=2. Goethite particles ranged in size from 50 nm to 0.2–1 μm.
Spectroscopy and simulations
The aqueous solutions/suspensions were irradiated using 6 ns laser pulses from the third harmonic (355 nm) of a Nd:YAG laser (Quantel Brilliant). N2-purged aqueous solutions and/or suspensions of the oxide particles containing the adsorbate were flash frozen at 77 K in O.D. 4 mm Suprasil tubes and subsequently irradiated using 355 nm light (15 mJ/pulse, 100–500 pulses) while immersed in liquid N2. The irradiated samples were transferred into the cryostat (Oxford Instruments model CF935) of a continuous-wave EPR spectrometer (Bruker model ESP300E operating at 9.45 GHz), and the radicals and spin centers on the metal oxides were detected in situ at 50–200 K. First-derivative EPR spectra were recorded at several temperatures, microwave power levels (typically, 0.2 mW), and field modulation amplitudes (typically, 2–5 G) at 100 KHz. The radicals on the oxide surface exhibit hindered rotation that results in poor averaging of the anisotropic part of hyperfine coupling tensors (hfcc) for magnetic nuclei, such as 14N and 1H, which complicated structural attribution and required selective deuteration of the photooxidized compounds. Another frequent complication is interference from trapped-electron centers on the oxides 3,4,10,13 which renders certain regions of the EPR spectra inaccessible to analysis. The trapped electron centers (surface and lattice TiIII ions) exhibit prominent resonance lines to the higher field of g=1.97 which are generally excluded from the spectra shown in Section 3.
Because some of the radicals reported in this work yield poorly-resolved EPR spectra, selective deuteration or 13C substitution were required to establish the radical identity. The deuteron, which is a spin-1 nucleus, has ~15% of the magnetic moment of the (spin-1/2) proton and, to a first approximation, the effect of such deuteration is nullifying the hfcc in the corresponding protons.
Calculations of the geometry and magnetic parameters of the radicals were carried out using density functional theory (DFT) with a B3LYP functional 18 and 6–31G(d,p) basis set with polarization functions from Gaussian 98. 19
3. RESULTS
3.1. Nucleotides and nucleosides
As discussed in refs. 3, 4, and 17, the holes on α-FeOOH and TiO2 are sufficiently energetic to oxidize both the (2′-deoxy) ribose and the corresponding 5′-phosphate derivatives. The nucleobases that attached in the β-position to the carbon-1′ of the sugar (phosphate) unit can serve either as an electron donor or an electron acceptor 20–23 (see also references 1 to 17 in ref. 16 for reviews). In the gas phase, the theoretically calculated adiabatic ionization potentials of the free U, T, C, A, and G nucleobases are 9.32, 8.87, 8.68, 8.26, and 7.77 eV, respectively, 24a,25; these values agree well with the corresponding experimental values 24b and microhydration, H bonding, and stacking do not change this ordering. 25 Thus, the two purines are easiest to oxidize, whereas the pyrimidines are more difficult to oxidize (Schemes 2 and 3). Oxidation by hydroxyl radicals results in the formation of the corresponding hydroxyl adducts at carbon-4 for A and carbon-5 for G, whereas fast one-electron oxidizers, such as SO4 −•, generate the corresponding radical cations, A+• and G+•. 20 At low pH, these radical cations are stable to deprotonation; e.g., G+• has pKa of 3.9. 26 Such one-electron oxidation occurs both in free purine bases and their derivatives, including ribonucleotides and 2′-dexoyribonucleotides (see Scheme 3 for the guanine). Recent work has shown that the purine cations in nucleosides or in DNA are easily photoconverted to sugar (H atom loss) radicals at carbons-1′, -3′, and -5′ via a proton-coupled hole transfer process; 27 – 29 however, in highly polymerized salmon sperm ds DNA, only 1′ radical is produced. 29 Thus, purine radical cation excited states can play an important role in sugar radical formation (resulting in strand breaks).
Scheme 2.

Redox reactions of cytosine-based derivatives, including the radicals generated by oxidation of cytidine (see also ref 27a.)
Scheme 3.

Oxidation pathways for guanine (see also ref. 27a)
In contrast, the radical cations of pyrimidines exhibit complex chemistry. 30–41 Oxidized (free) pyrimidines (such as cytidine (1) in Scheme 2) deprotonate from their nitrogen-1 site, yielding the corresponding neutral N-centered radicals (radical 2 and, to a lesser degree, radical 3. 30,42 As a minor reaction pathway, these radical cations 35–39 also react with water and deprotonate, yielding hydroxyl adducts at the carbon-6 sites for thymine (Scheme 4), e.g., 21,39 the carbon-5 site for cytosine (radical 4 in Scheme 2), and the carbon-5 and -6 sites for the uracil. 42 In N(1)-substituted pyrimidines, N(1)-C(1′) scission also occurs, as suggested by product analyses and spin trapping, 21,22,32,33,44 but the corresponding N-centered radicals 2 was not observed by EPR. For ribonucleotides, the U+•, C+•, and 5-methyl-C+• yield C(1′)• radical 6, as shown in Scheme 2 (some studies suggest the formation of other ribose-centered radicals). 33,34,44 On the other hand, H-atom abstraction by the hydroxyl radical results in the formation of sugar radicals at all of the carbon sites in the sugar moiety, but the preferred site for the H-atom abstraction is at the carbon-2′ site. 45 We note here that the C(1′)• radical undergoes C(1′)-O bond (ring) scission to yield radical 7 or undergoes a 1,2 H-shift to yield radical 8. The latter eliminates water and loses the free pyrimidine base (or, in the oligonucleotides, the phosphate group) yielding radical 9. The latter reactions cause irreversible strand breaks. 21–23 Analogous internal H abstraction causing the elimination of uracil has been reported for uridine and polyuridine in γ-radiolysis of aqueous solutions at room temperature; 46 product analyses indicating the formation of sugar phosphate radicals 7 and 9 are corroborated by spin-trapping studies. 44 For 2′-deoxycytidine, in addition to deprotonation from the sugar the dC+• radical cation can also deprotonate from the amine group yielding a dC-C(4)-•NH radical that undergoes internal proton transfer to yield =N• radical 3. 30,33 For oxidized uridine, dU+•, there is also the formation of a C(6)-OH adduct radical (through the concerted hydrolysis/deprotonation) analogous to radical 4. 42,44
Scheme 4.

Redox reactions of thymine and thymidine.
In 2′-deoxyribonucleotides cation radicals, deprotonation from the ribose is less facile and the hydroxyl adducts are observed, with the hydroxyl addition at carbon-5 for dC and carbon-5 and -6 for dU. For dT (structure 14 in Scheme 4), the main product is the allyl-dT (dU-C(5)-CH2•) radical 15 that is formed by deprotonation of the radical cation 16 from the methyl group; 35,37,40,43 the hydroxyl adduct at carbon-6 (radical 17) is generated as a byproduct. 39 EPR studies of frozen glass matrices 34 and crystals 43,47 indicate that oxidation of the cytidine and 2′-deoxycytidine (and the corresponding 5′-phosphates) produces primarily dehydrated radical 9 formed with unaltered base release. Such ribose-centered radicals are formed not only under direct attack of the oxidizing radicals (which may involve the ribose rather than the purine moiety), 21 but also through biphotonic ionization of the cytidine and 5-methylcytidine. 34,36,37 For 5-methylated cytosine and cytidine, the formation of allyl-C radical analogous to allyl-dT has been reported; 37,40 such radicals (known as 3αH) 43 are also observed from 5-methylcytosine impurity in cytosine and cytidine monocrystals, as this methylation reduces the ionization potential by 140–290 meV. 43 In aqueous solutions that do not contain electron scavengers, photoionization and radiolysis generate hydrated electrons that yield radical anions and their protonated forms, as shown in Schemes 2 and 4. The resulting chemistry is complex, with a number of species generated simultaneously.
Given that the oxides deeply trap photogenerated electrons, the photochemistry of nucleic acids on the titanium (IV) and iron (III) oxides should be oxidative, and the radicals are less prone to secondary reactions, such as the loss of water. In this section we consider the results for ribonucleotides; the 2′-deoxyribonucleotides are examined in the next section. The parent nucleosides and 2′-deoxynucleosides of the purines have prohibitively low solubilities in acidic solutions, and we did not observe photooxidation for such compounds.
As shown in Figures 1a and 1b, the EPR spectra for nucleotide mono-, di-, and tri- phosphates on photoirradiated TiO2 are dominated by a relatively narrow resonance line whose appearance does not change with the extent of 5′-phosphorylation. In addition to this line, there is a line of the formyl radical, suggesting the damage to the ribose ring. The strongest EPR signals are observed for purines, as can be expected given their ease of oxidation (Figure 1a). The proof of the identity of this radical as G+• is provided by d8-substitution in guanine (Figure 2). Upon this substitution, the resonance line narrows due to the elimination of a 8.9 G isotropic hfcc for H(8) in h8-G+• (see simulations in Figure 1S). The line narrowing reveals the shoulders (indicated by arrow in Figure 2). This feature likely forms a ribose-centered radical. In contrast, d8-substitution in adenine does not result in a significant change of the spectral envelope (Figure 2). This is consistent with previous experimental and computational results for h8-A+• that indicate weak coupling of the unpaired electron density to the H(8) proton (~6 G) and strong coupling constants in the NH2 protons (~7.3 G). 48 The simulated EPR spectra for h8-A+• and d8-A+• are shown in Figure 1S. In accordance with the previous reports, 48 warming of the photoirradiated purine 5′-phosphates to 140 K (when the matrix softens) did not result in significant changes in their EPR spectrum (Figure 2S).
Figure 1.

EPR spectra of (a) purine and (b) pyrimidine 5′-mono, di-, and triphosphates of ribonucleotides on photoirradiated aqueous TiO2 at 50 K (355 nm). The narrow low-field line is from the formyl radical.
Figure 2.

As Figure 1, for isotopomers of purine ribonucleotide-5-monophosphates. The deuteration sites are indicated in the plot labels; the atom numbering corresponds to Scheme 1. The arrow indicates a line of sugar-phosphate radical.
Importantly, the EPR spectra observed for the cytidine and uridine (and their 5′-phosphates) are very similar (Figure 1b), despite the structural differences. The EPR spectra observed for these pyrimidines are not fully consistent with the spectra simulated and/or observed for their radical cations and anions (including the corresponding deprotonated and protonated forms, respectively), the hydroxyl adduct radicals at carbon-5 and -6 positions (that have large hfcc’s, ~20 G, in α– and β–protons) and the N(1)- radicals. The photooxidation of the pyrimidine nucleotides and nucleosides appear to have resulted either in direct oxidation of the ribose moiety at the surface or the secondary reactions of the radical cations, as explained above.
That sugar-centered radicals are indeed formed in the course of the photoreaction is supported by Figure 3 (an enlarged, annotated version of this figure is given in Figure 3S), in which we compared the EPR spectra for 5,5″-h2- and 5′,5″-d2-cytidine and CMP (the bottom traces). The EPR spectra observed for CMP and 5,5″-h2 -cytidine are very similar (Figure 4), whereas the EPR spectrum for 5′,5″-d2-cytidine has a narrow component that can only be from the 5′,5″-d2-C(5′)• (indicated with arrows in Figure 3S). A similar transformation is observed for AMP and 5′,5″-d2-AMP, as demonstrated in Figure 2. The formation of ribose-centered radicals is also suggested by the comparison of pyrimidine nucleosides, 2′-deoxynucleosides, and nucleotide monophosphates in Figure 4: the nucleosides and nucleotides have very similar spectra, while the 2′-deoxynucleosides have rather different spectra indicative of the involvement of the 2′-deoxy- C(1′)• or C(3′)• radicals (as explained in Section 3.2). However, when the cytidine is methylated at carbon-5, the spectrum changes considerably as compared to cytosine. As shown below, this is due to the formation of allyl-C/dC radical via deprotonation of methyl group in the 5-methyl- cytosine radical cation.
Figure 3.
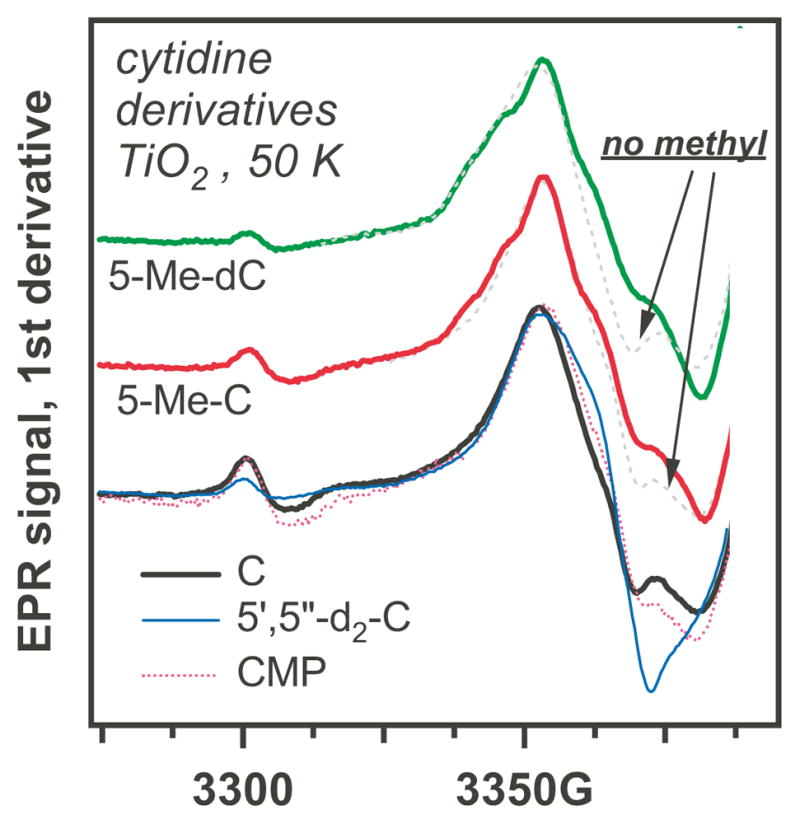
EPR spectra from cytidine (C) and 2′-deoxycytidine (dC) derivatives on photoirradiated TiO2 at 50 K. The upper two traces compare 5-methyl (solid lines) and 5-H derivatives (broken lines). The bottom trace compares cytidine, 5′,5″-d-cytidine and cytidine-5′-monophosphate. The annotated, enlarged version of this figure is given in the Supplement (Figure 3S). The spectra for 5-methyl derivative indicate the presence of allyl-C/dC radical (see Table 1S for magnetic parameters and Figure 11S for simulations).
Figure 4.

EPR spectra from pyrimidine (X=C, U; Scheme 1) ribonucleosides (X, dotted lines), 2′-deoxyribonucleosides (dX, solid lines), and the corresponding ribonucleotide-5′-monophosphates (XMP, dash dot lines) on photoirradiated aqueous TiO2 at 50 K. The arrows indicate spectral regions where nucleotides and 2′-deoxynucleotides have different spectral features.
On iron (III) oxides, the photooxidation was observed only for nucleotides (no photooxidation was observed for the nucleosides) and only on α-FeOOH, with the single exception of GxP which was oxidized by α-Fe2O3 (see below).
3.2. 2′-Deoxynucleotides
While oxidation of the purine bases in ribonucleotides and 2′-deoxyribonucleotides produces spectroscopically identical radical cations (e.g., G+• and dG+•), the direct or secondary (for purines) oxidation of the corresponding sugar moieties should yield different EPR patterns, as the sugars are different. This is certainly true if the main locus of the H loss (following the oxidation of the nucleobase) in the cytidine and uridine (and their 2′-deoxy analogs) is at carbon-1′, as suggested by previous research (Section 3.1). In other words, the difference between the EPR spectra of the two classes of nucleotides can be interpreted as evidence for the formation of neutral carbon-centered sugar radicals.
In Figure 5, we compare the EPR spectra obtained by photooxidation of dGMP and GMP and dCMP and CMP, respectively. It is clear from Figure 5 that in addition to dG+•, the dGMP yields radicals with a wider EPR spectrum. This transformation parallels Figure 2 in ref. 13, illustrating similar spectral transformations for 2-deoxyribose vs. ribose due to the presence of carbon-1′ to -3′ centered radicals. The resonance lines from the sugar radicals are also observed for d8-dGMP in which the d8-dG+• radical has a narrower line than h8-dG+• and so interferes less with the observation of other radicals. The considerable differences between C/dC and U/dU, respectively, are also seen in Figure 4. These results indicate that the sugar moiety is oxidized regardless of the 2′-OH substitution. In contrast to the ease of the photooxidation of (d)GMP on TiO2, no photooxidation was observed in solutions containing guanosine, adenosine, their 2′-deoxy analogs, nor with dAMP, suggesting weak chemisorption of these compounds by the TiO2 surface.
Figure 5.

As Figures 3 and 4, for 2′-deoxyribonucleotides (C, T, G, and d8-G; see Scheme 1). The dotted lines are for the corresponding ribonucleotides. The regions of spectral difference are indicated by arrows.
Thymine (Schemes 1 and 4), which is a DNA-specific pyrimidine, presents an exceptional case (Figures 5 and 6). In radiolysis of DNA, the radical anion dT−• is a major radical product. 20,31 The oxidation of this nucleobase results in the formation of the allyl-dT radical by deprotonation of T+• from the 5-methyl group (Scheme 4). This radical has been observed by transient absorption - pulse radiolysis of aqueous solutions 21,45 and EPR in γ-irradiated aqueous salt glasses containing thymine, which is readily oxidized by strongly oxidative radicals, such as Cl2−•. 35,36 The products of the reaction are (i) the C(6)-OH• adduct, 21 (ii) dT+•, 35, 36,38,39 and (iii) radical 15 (Figure 4S). 35,37 The latter can also be observed at 200 K in irradiated aqueous BeF2 glasses. 40 Typically, the low-temperature EPR spectra of irradiated samples are dominated by dT−• doublet whereas 35,40 the EPR spectrum from the allyl-dT gradually appears on annealing the sample to 210 K where the anion decays. This is a result of methyl deprotonation of the cation radical (whose ESR spectrum is broad and not as readily observed at low temperature). 38,40
Figure 6.
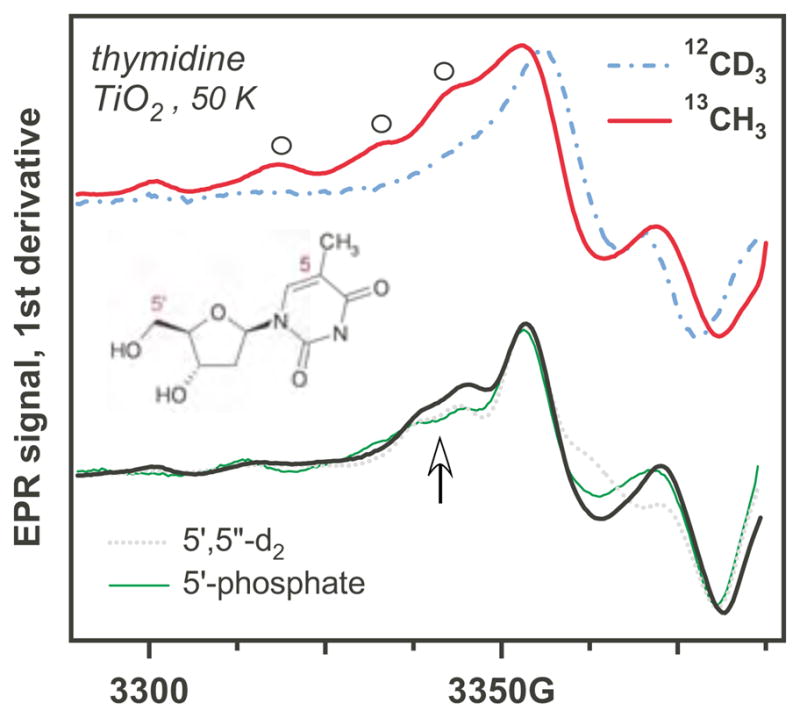
EPR spectra for photooxidation of thymine derivatives on aqueous TiO2. The upper traces are for the methyl isotopomers (Schemes 1 and 4). The bold lines are for the 2′-deoxy-β-thymidine, the dash-dot lines are for the thymine, other derivatives are indicated in the plot. Open circles indicate the resonance lines also indicated in Figure 9S.
On TiO2, the EPR spectra observed for dTMP and dT (Figures 5 and 6) reveal a characteristic doublet that may be confused for that observed for dT−• in the bulk. 40 However, this identification cannot be correct. CD3-substitution in dT (Figure 6) results in a narrow, structureless resonance line. This transformation cannot be accounted for by dT−•, as in this radical anion, the hfcc on the methyl protons is negligible (~0.3 G); the largest hfcc is for H(6) (~15 G). e.g., 31 On the other hand, the observed EPR spectrum is also inconsistent with that of dT+• as observed by Sevilla and co-workers in aqueous glasses. 35–39 While the radical cation has large hfcc in the methyl protons and N(1) (~20.4 G and 11.8 G, respectively), the appearance of the spectra for the CH3- and CD3- isotopomers of the radical cation is completely different from the one observed. The N(1) centered radical (for thymine) and the C(6) hydroxyl adduct can also be excluded, for similar reasons. On the other hand, the allyl-dT radical 15 would rationalize the observed features. Indeed, (i) this is the known major product of dT oxidation; (ii) it has large proton hfcc’s in the methyl and H(6) (Table 1S); it accounts not only for the central doublet, but also for the side lines observed in Figures 6 (see arrow) and 4S. That the latter are unlikely to be from the ribose radicals is suggested by Figure 6 in which we plotted together the EPR spectra obtained from 5,5″-h2-dT and 5′,5″-d2-dT. It is apparent from this spectrum that 5′,5″-d2-substitution in the 2′-deoxyribose had no effect on these shoulders, although there is some effect of this substitution at the center of the spectrum which is evidence for some C(5′)• radical. To support the identification of the allyl-dT radical, we examined 13C-methylated dT (Figure 6). The DFT calculations indicate that the allyl-dT radical would have a large hfcc’s for the methylene carbon-13 in this radical (Table 1S) The EPR spectrum indeed exhibits prominent resonance lines from the 13C splittings indicated by open circles.
To ascertain the identification of allyl-dT radical we simulated the EPR spectra first assuming the planar geometry of the nucleobase residue and an axial g-tensor (Figure 4S and Table 1S). While the overall shape of the spectrum compared favorably to experimental spectrum and the reference spectrum of the allyl-T as observed in γ-irradiate solution of (dT)6 in 8 M sodium perchlorate in D2O, the outer resonance lines were too widely spaced and the amplitude of the middle resonance was unrealistically large. These deviations are indicative of conformational strain in the radical adsorbed at the oxide surface. To simulate these effects we studied the dependencies of the hfcc’s and simulated EPR spectra on the rotation of methylene group (Figures 5S and 6S) and out-of-plane C(6)-H bending (Figures 7S and 8S). Both of these distortions have the desired effect on the simulated EPR spectra (Figures 6S and 8S) through the decrease in isotropic and anisotrpoic hyperfine coupling constants in the H(6) proton. The estimated degree of strain is ~ 0.2–0.3 eV, which is fairly typical for matrix interactions. Figure 9S illustrates how the spectra for the three methyl group isotopomers of dT can be accounted for assuming a nonplanar allyl-dT radical with the angular distribution of methylene group rotation shown in Figure 5S (which corresponds to the mean angle of 17.5°). In this calculation, we assumed isotropic g-tensor, while the g-tensor of a planar allyl-T/dT radical is known to be axial. e.g., 40 Unfortunately, it is difficult to predict how the g-tensor might change in the distorted radicals. The simulations shown in Figures 4S and 9S demonstrate how the “split” low field resonance line can be accounted for by the slight axiality of the g-tensor. We conclude that all of the features observed in the EPR spectra for the isotopomers of dT on TiO2 can be consistently accounted for by a nonplanar allyl-dT radical adsorbed at the oxide surface.
These considerations suggest that the spectrum for photoirradiated dT on TiO2 is comprised mainly of the resonance lines of the allyl-dT with some ribose-centered radicals at carbon-5′. In the previously studied (homogeneous) systems, the presence of radical 15 (generated via hole scavenging) became apparent only after the complete decay of the dT−• or scavenging of the precursor electron to the radical anion. In TiO2 photooxidation (due to deep trapping of the electrons), this radical is observed without interference from dT−• even at low temperature, and the appearance of the EPR spectra does not change upon warming of the sample to 150 K (e.g., Figure 10S).
Revisiting Figures 3 and 3S in the view of these results, we observe that 5-methylation of the cytosine and 2′-deoxycytidine leads to the appearance of extra features (indicated by open circles in Figure 3S) that are not observed for the unmethylated species. The positions of these extra resonance lines (that account for 15–17% of the spin-1/2 species, by integration) are similar to those in the allyl-T/dT radical in Figure 4S and 9S and simulations shown in Figure 11S using the calculated magnetic parameters for the planar allyl-C/dC radical (given in Table 1S) are supportive of this attribution. While 5-methylation of uracil results in deprotonation of the radical cation from the methyl group, the methylation in cytosine is less efficient in diverting oxidative damage from the sugar phosphate backbone to the methyl group on the base.
While the oxidation of dT and dTMP was observed on TiO2; no oxidation of the dT was observed on α-FeOOH and α-Fe2O3 and only dTMP was oxidized on α-FeOOH (Figure 12S). This may be the result of weaker chemisorption on the iron (III) oxide surfaces and the decreased oxidative potential. There was also no evidence for oxidation of the purine (2′-deoxy)nucleosides on these iron oxides. dCMP and dGMP were both oxidized on α-FeOOH, whereas only the GxP and dGMP were oxidized by α-Fe2O3 (Figures 13S and 14S).
3.4. Oligonucleotides, RNA, and DNA
While all natural nucleotides are readily oxidized on TiO2 and some nucleotides are oxidized by α-FeOOH and even α-Fe2O3, no oxidation was observed for poly-C (pC), pU, the pA•pU duplexes and DNA, even at high concentration of these compounds. The EPR spectra revealed only the characteristic lines of the oxygen hole centers of the corresponding oxides. In contrast, photoirradiated pA on TiO2 and α-FeOOH revealed a spectrum that was similar to that of AMP (Figure 7). Conversely, baker’s yeast RNA (at 50 g/L) yielded EPR spectra that were very similar to those from GxP (Figure 8a). We remind that the (d)GxP nucleotide derivatives were the only ones that were photooxidized on all three oxides (Section 3.2). RNA was also oxidized on all of the oxides (Figure 8b), and the spectra suggest that in all three cases the photooxidation resulted in the formation of G+•.
Figure 7.
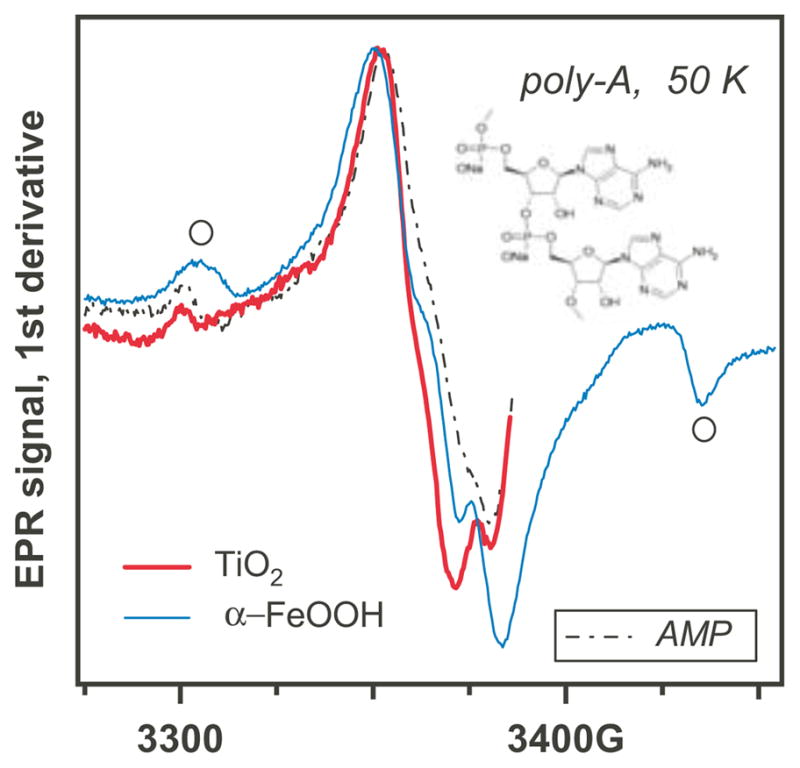
EPR spectra for photooxidation of pA on aqueous TiO2 and α-FeOOH; the spectrum of AMP on TiO2 is shown for comparison (dash-dot trace). The features indicated with open circles in the α–FeOOH trace are from the oxide matrix.
Figure 8.

EPR spectra from photooxidation of baker’s yeast ss RNA on metal oxides. (a) Comparison of spectra from ss RNA and GMP. (b) Comparison of EPR spectra obtained on different oxides. The features indicated with open circles in the α–FeOOH trace are from the oxide matrix.
4. DISCUSSION
4.1. Monomers
Summarizing our EPR observations in Section 3 and ref. 13, photoirradiated TiO2 and α–FeOOH oxides readily oxidize D-ribose (and other carbohydrates and polyols) and ribose-5-phosphate, as well as the corresponding 2-deoxy compounds. The mediation of the charge transfer by the nucleobases is not required for such oxidation, and the resulting species are H loss radicals that are stable on the oxide surface. The photooxidation of (2′-deoxy)ribonucleosides was less efficient (especially for purines) due to poor chemisorption at the oxide surface: the substitution at the carbon-1′ decreased the sorption at the oxide surface by elimination of the anchoring hydroxyl group that is critically involved in the chemisorption of ribose (Section 3.2). The chemisorption is improved upon phosphorylation at carbon-5′. The 2′-deoxyribonucleotides exhibited poorer sorption, suggesting the critical involvement of the C(2′)OH groups in the ribonucleotides; nevertheless, dGMP and dTMP were readily oxidized on α–FeOOH and TiO2. The oxidation of purine (2′-deoxy) nucleotides resulted in the formation of the corresponding radical cations and, to a lesser extent, ribose-centered radicals, including the C(5′)• radicals that can undergo subsequent phosphate loss (resulting in a strand break). Among the DNA bases, guanine is the easiest base to oxidize. 20,23–25 With the exception of the thymine derivatives (that yield radical 15), the oxidation of pyrimidine (2′-deoxy)nucleotides resulted in the formation of sugar-centered radicals. Overall, the photochemistry found in this work is entirely oxidative and the radical products are similar to those observed in bulk solutions containing strongly oxidizing radicals, but with the advantage that these photooxidation reactions do not have the complications due to electron scavenging and H atom and hydroxyl adduct formation. The resulting radicals are more stable on the oxide surfaces than in the bulk.
Our results agree with the picture of surface interactions emerging from studies of chemisorption on oxide surfaces. Recently, Cleaves et al. studied the adsorption of nucleic acid components on rutile. 49 Free nucleobases were found to adsorb weakly at low pH, with A and C showing greater affinity than U due to the interactions involving their free amino group. By contrast, all ribonucleosides exhibited similar absorption isotherms and the surface density at saturation (0.2–0.3 molecules per nm2), while the corresponding 2′-deoxyribonucleotides showed 3–5 times lower adsorptivity. This suggests that (2′-deoxy)nucleosides are adsorbed through the hydroxyl groups in the sugar moiety. Phosphorylation increased the adsorption 3–6 times, suggesting additional anchoring involving the phosphate group; still, the 2′-deoxyribonucleotides showed lower adsorptivity than the ribonucleotides. It is inferred that β-nucleosides and nucleotides adsorb in such a way that the nucleobase points away from the oxide surface and the molecules are bridged through their 2′- and 3′-hydroxyl and 5′-phosphate groups. Exposing the 3′-hydroxyl group to the oxide surface primes it for “template polymerization” that is known to occur on iron oxides. 50 The occurrence of such polymerization led Schwartz and Orgel 51 and Holm 50 (among many others) to suggest such surfaces to be cradles of prebiotic evolution: the mineral surfaces selectively concentrated organic molecules and catalyzed their reactions. The iron (III) oxides in banded-iron formations are closely associated with the oldest microfossils (> 3.8 Gya).50
Our study shows a possible problem with such scenarios: photocatalytic activity at such oxide surfaces may result in the oxidation and breakdown of organic molecules, including the components of nucleic acids. On the other hand, as discussed in Section 5, some otherwise mysterious features of DNA chemistry can be rationalized as a means of minimizing the undesired chemical effects of precisely such damage.
4.2 Nucleic acids
Photooxidation of monomer nucleotides and 2′-deoxynucleotides can be contrasted with photolysis and radiolysis of DNA and model oligoribonucleotides. In radiolysis of aqueous solutions of DNA, the primary species are dG+• (35%), dT−• (25%, structure 18 in Scheme 4) and protonated dC−• (25%, structure 5 in Scheme 2). Additionally, 15% of the radical species originate from the sugar-phosphate backbone, such as C(1′,3′,5′)• and dephosphorylated C(3′)•. These radicals are generated through direct ionization as well as the indirect effect via reactions of hydroxyl radicals (46%) and H atoms and solvated electrons (54%). The hydroxyl radicals add to or oxidize all nucleobases in DNA. For holes generated in DNA, subsequent electron transfer along the duplex (either by tunneling or thermally assisted electron hopping) rapidly relocates the hole on guanine bases in dGn steps. 16,23,52,53 Conversely, pyrimidines have the highest electron affinity and so C and T account for the majority of the electron centers. 20,23 Strand breaks in the DNA are caused by damage to the sugar-phosphate backbone rather than the redox reactions of the nucleobases 22,23 (although, as discussed in section 4.1, oxidation of dC and dU does lead to the formation of sugar-centered radicals, Scheme 2) and excited states of purine cation radicals also lead to sugar radical formation. 20,27–29 While guanine is the primary locus of oxidative damage (Scheme 2), this oxidation has a radioprotective effect: in γ-irradiated, H217O-hydrated ds DNA, the dG+• cation (radical cation 10 in Scheme 4) hydrolyses and deprotonates, yielding the dG-C(8)-17OH• radical 11 that can react and stepwise donate electrons to nearby dG+• radicals, yielding 8-oxo-dG (13) and subsequently the 8-oxo-dG+• radical 12. In this way, radical 12 serves as a “sink” for the oxidation, minimizing oxidative damage to the DNA. 20
In contrast, the photolysis of DNA (requiring short-wave UVC and UVB light) results in [2+2] pyrimidine dimerization, 54 with photohydration of the pyrimidine bases being a minor side reaction. In a cell, the UVA light can excite various pigments, some of which have sufficiently energetic excited states to oxidize purine nucleobases. 54 From the standpoint of chemistry, photocatalysis on metal oxides is closer to radiolysis than photolysis. The important difference is that while radiolysis results both in oxidation and reduction, photocatalysis results in oxidative chemistry, as the negative charge is trapped by the metal oxide particles.
The preference for the oxidation of guanine sites in single strand (ss) and double strand (ds) oligo(2′-deoxy)ribonucleotides is well known from previous studies. 20,23,52,53 For example, the studies of Shafirovich and co-workers established the preference for oxidation of dGn sites by the CO3 −• radical and the excited state of the riboflavin. 52a The free energy of electron transfer increases in the order of ..G.., ..GG.., and ..GGG.. sequences, the latter two steps exhibiting 52 and 77 meV higher free energy gain than isolated dG bases. 52b It is known from such studies that the photooxidation proceeds in two stages: hole injection into the DNA duplex occurring primarily at the dGn-sites is followed by hole hopping towards the dGn sequences with the lowest ionization potentials. Once the positive charge is trapped, this is followed by chemical trapping (the slower reaction of dG+• with water). 16,20,52a For highly oxidative radicals like SO4−• and CO3−•, the oxidation is very exergonic and the dGn sites are oxidized randomly, whereas for weaker oxidizers, there are sequence-specific preferences: e.g., for oxidation by riboflavin, the oxidized moiety is always located 5′ to guanine in a …GG… sequence. 15,52,53 Shortly after the initial injection (regardless of the initial injection sites), the hole localizes at the dGn sites with the lowest ionization potential. The oxidized dG+• undergoes further transformations (Scheme 3). In particular, visible light excitation of dG+• results in the internal charge transfer, leading to formation of the C(1′,3′,5′)• radicals. 27–29 From the standpoint of EPR spectroscopy, there is no difference between the isolated (d)G+• radicals and the radical cations in the (d)Gn sequences in DNA and RNA, i.e., the electron density is not shared between several G residues; rather, it is localized on the residue that has the lowest ionization potential. From the EPR spectra alone (unless selective d8-deuteration is done at specific loci) 16 it is not possible to determine which at (d)Gn sequences are hosting the observed (d)G+• radical.
Majima and co-workers studied hole injection to dGn sequences in short DNA duplexes that were either unmodified or modified with catechol at the 5′ terminus (this modification is used to improve surface binding). 55 Only a small fraction of such duplexes (< 15%) were bound to the surface regardless of the modification; the photoinduced oxidation proceeded both through the hole injection to DNA bound to the surface and photogenerated hydroxyl radicals that migrated from the surface to the bulk and oxidized DNA in the bulk. 55 Rajh and co-workers 56,57,58 studied DNA duplexes bound to TiO2 through a dopamine moiety conjugated to the carboxy-derivatized terminal dT base (cdT). The EPR spectra obtained at 10 K exhibited resonance lines from the cdT+•, whereas spectra observed above 80 K were from the dG+• radicals located further out. 57 The extent of photoreduction of the adsorbed Ag+ cations (serving as a measure of negative charge stored on the nanoparticle) exponentially decreased with the overall length of the GG-terminated duplex, suggesting that the yield of the oxidation was determined by the efficiency of the forward electron transfer: the further was the locus of the electron donating GG site, the lower was the probability of electron transfer to this site. 58 For example, for the GG sites located 7 nm from the surface the reduction of Ag+ was only ~ 1/3 of that at 1 nm (in the GG-interspersed (TA)n duplexes of the same overall length). Including single-G hopping sites into the GG-terminate duplexes increased the efficiency of charge separation, providing “stepping stones” for the charge transfer. Importantly, in both studies, the DNA was chemically modified in such a way that the straight DNA duplexes were protruding from the surface (the authors call this mode of adsorption “tentacle binding”). 56,57,58
Wamer et al. 59 studied native DNA and RNA in UVA-irradiated TiO2 suspensions (aiming to use the TiO2 nanoparticles for photodynamic therapy). Oxidative damage resulting in the formation of 8-OH-dG was observed in both cases (in an aqueous solution) and attributed to reactions of hydroxyl radicals occurring in the bulk. Mysteriously, the photoirradiation of cells impregnated with TiO2 resulted in considerable damage to the RNA but not the DNA. As the cell has many hydroxyl radical scavengers present in a much higher concentration than nucleic acids and the rate of photoproduction of hydroxyl was low, the likely mechanism for photooxidation was direct hole injection from the embedded TiO2 nanoparticles to the chemisorbed nucleic acids. This heretofore unexplained result can be interpreted as indication that chemisorbed RNA was oxidized, whereas DNA was not. This result seemingly contradicts the studies of Majima and co-workers 55 and Rajh and co-workers 57,58 in which the charge separation on the TiO2 surfaces dressed by DNA “tentacles” was observed. On the other hand, these observations are fully consistent with our EPR results: while the individual nucleotides and 2′-deoxynucleotides, single-stranded sodium poly(adenosine-5′-monophosphate), and RNA were readily photooxidized on TiO2, the duplexes (including DNA) were not oxidized. As both RNA and DNA involve multiple (d)Gn sites, why can ss polynucleotides be oxidized whereas ds polynucleotides and DNA cannot be oxidized?
The crucial difference is the polymer morphology. As suggested by our results, the TiO2 can oxidize not only all of the nucleobases, but even the bare ribose units; the energetics may be a consideration for the α-Fe2O3 but not the α-FeOOH and TiO2. As wild-type RNA and DNA include multiple (d)Gn sequences, the low abundance of such sequences cannot be the reason. Furthermore, charge separation readily occurred when the DNA duplexes were chemically linked to the oxide surface. 55–58 These observations suggest that ds oligoribonucleotides and oligoribo(2′-deoxy)ribonucleotides, including DNA duplexes, are not chemisorbed by the oxide surface in a fashion conducive to hole injection, whereas ss oligoribonucleotides and RNA are.
This rationale is not surprising in view of the results discussed in Sections 3 and 4.1, suggesting that the ribose hydroxyl groups are crucially involved in anchoring of the nucleotides (Section 4.1). In duplex DNA, the conformation of the polymer chains is rigid, the 3′ and 5′ hydroxyl groups are involved in the formation of the polymer backbone while the 1′ hydroxyl and 2′ hydroxyl groups of the ribose are missing; i.e. the binding can only be through the negatively charged phosphate groups and the 2′-OH groups (for RNA). While the unsubstituted HPO42− strongly binds to TiO2 at pH=2.3 (the binding constant is 4×104 M−1) 60 in a bidentate fashion, disubstitution of the phosphate results in negligible adsorption. Connor and McQuillan 60 explain this through the changeover from a bidentate to monodentate mode of adsorption. For this reason, adsorption of the ss and ds DNA duplexes at the oxide surfaces should be inefficient. The same relates to rigid ds RNA duplexes, in which the 2′-OH groups are hidden inside the double helix. By contrast, the ss RNA is open and flexible (as at least some of the nucleobases are not paired) and has an exposed 2′-hydroxyl group in every ribose unit, which allows the adsorption through several hydroxyl groups. This places the Gn sites next to the surface and the electron transfer becomes facile.
5. CONCLUSION
In addition to mechanistic insight and photodynamic therapy 56,59 aspects discussed in the previous sections, our studies have impact on two broad areas of interest:
(1) Photostability of biomolecules in the terrestrial and extraterrestrial environments
The surfaces of planets, including Mars, are most accessible for exploration by space probes, 1 but the abundance of the tentative biosignatures at the surface depends on the rates of generation and decomposition. The first rate is unknown, while the decomposition rate can be estimated. 3–6 The biosignatures that can accumulate in the soil are biomolecules that are most difficult to degrade – as opposed to the biomolecules which are most abundant in the living organisms. Thus, the most appealing target biomolecules (or products of their diagenesis) are precisely those that are most stable on the light-exposed soil and yet recognizably biotic in their origin. Instead, current emphasis has been placed on the simplest (rather than the most durable) biosignatures, such as α–amino acids and nucleotides. 1 The choice of such targets is dictated by many considerations, including the ease of remote detection. On the other hand, the chemical stability of amino acids on light exposed martian soil (regolith) is rather poor, 3,4,6 and the steady-state concentration of such molecules might be prohibitively low.
In the present study, DNA and RNA subunit monomers (nucleosides, etc.) on the oxide particles are found to be far more susceptible to photolytic damage than the corresponding polymers. There is a striking similarity as to how the polymerization affects the photooxidation of α-amino acids and ribonucleotides on metal oxide surfaces: 3,4 the monomers are strongly bound to the surfaces and readily oxidized, whereas the polymers have fewer exposed anchoring groups and so are more resistant to oxidation. In the case of nucleic acids, the major factor is the chain morphology: ss RNA can attach to the oxide surfaces through multiple 2′-hydroxyl groups and in this way donate the electron from the guanine placed close to the oxide surface, while the ds DNA and RNA either are not chemisorbed at all or adsorbed in such a way that the electron transfer is suppressed. The individual ribonucleotides and 2′-deoxyribonucleotides are easily oxidized, and this oxidation involves not only their bases (which is a reaction that might be reversible) but also the sugar phosphate moieties (resulting in the irreversible damage). It appears that the monomer α–amino acids or nucleotides make rather poor choices as biosignatures on the light exposed martian regolith due to the ease of their photocatalytic oxidation: the steady-state concentration of such “simple” biosignatures (even if these are being generated by the tentative martian “bacteria”) should be quite low. On the other hand, peptides (especially coiled ones) 3,4 and polynucleotides (especially, in their duplex forms) are more resilient to such photoreactions and so may more readily accumulate in the soil. These observations argue for reconsideration of the strategy for search of organic life on Mars: a greater emphasis should be placed on the search of macromolecules rather than monomers and small metabolites.
(2) Prebiotic evolution and the origin of the “DNA world”
An interesting corollary of our study is that ds DNA seems to be optimized to withstand long-term oxidative damage, on more than one level. That nature has preferred DNA over RNA is a long standing puzzle in molecular biology, especially in the view of the “RNA world” scenarios of prebiotic evolution. 61 Our study (supported by the previous work on radiolysis of DNA) 15,16,20–23 points to the crucial difference that heretofore escaped the attention of much of the scientific community: that DNA has a higher resistance to oxidative stress. For example, the methylation of uracil at carbon-5 in dT provides a site for deprotonation at the nucleobase, whereas in dU, the radical cation oxidizes the sugar-phosphate, resulting in a strand break. Conversely, the 2′-hydroxyl group in ribose not only makes various reactions involving hydroxyl groups possible (e.g., transesterification), 62 but it also exposes the sugar moiety to attack by oxidizing radicals (and surface holes), resulting in a strand break. While the 5-methylation of uracil in 2′-deoxythymine protects the ribose from the oxidative damage on the oxides, for 5-Me-C and 5-Me-dC, this protective action is inefficient and >80% of the damage is channeled to the sugar phosphate. Thus, our study provides a possible explanation why the methylation of uracil was preferred over the methylation of cytidine, provided that the DNA structure had evolved to withstand a long-term oxidative damage.
Thus rationalized features of the DNA chemistry may appear surprising in the view of relatively low radiation background, anoxic conditions, and low levels of the UVC and UVB light ground exposure on the primitive earth. 63 However, such oxidation-protective features become understandable by assuming, following the hypothesis of Orgel and Schwartz, 51 that mineral surfaces, including transition metal oxides, such as iron (III) oxides, served as a matrix and a catalyst for prebiotic evolution. Reduction of hydrous ferric oxide is still used by dissimilatory iron-reducing soil and sediment bacteria for terminating the respiration chain. 64 The importance of such ubiquitous minerals as terminal electron acceptors was even higher on the anoxic earth, which may explain why the early life (including the early photosynthetic life) could have become dependent on such oxides. While the iron (III) oxides can serve as efficient catalysts and terminal electron acceptors, these oxides are phototoxic, which necessitates a degree of protection against the irreversible structural damage to organic biomolecules on or near the light exposed surface. This phototoxicity may account for the preference of DNA over RNA, and our study demonstrates how such “chemical” protection of the DNA is accomplished at several levels of organization: from the morphology (counteracting DNA binding to the oxide surface) to the minute details of radical (ion) chemistry resulting into channeling the oxidation damage away from the sugar phosphate backbone (Scheme 2) into oxidized guanine which acts as a multiple hole sink (Scheme 3). While this scenario is speculative, it provides a natural explanation for the preference of DNA over RNA under the prebiotic conditions without invoking the workings of modern cellular machinery.
Supplementary Material
Acknowledgments
IAS thanks S. D. Chemerisov, T. Rajh, N. Dimitrijevic, H. J. Cleaves, and D. Catling for useful discussions. This work was supported by the Office of Science, Division of Chemical Sciences, US-DOE under contract No. DE-AC-02-06CH11357 and the NASA Planetary Division Mars Fundamental Research grant No. NNH08AI65I. MDS and AA acknowledge support from NCI RO1 CA045424.
Work performed under the auspices of the Office of Science, Division of Chemical Science, US-DOE under contracts Nos. DE-AC02-06CH11357 and NASA’s Mars Fundamental Research Program grant NNH08AI65I.
Footnotes
Supporting Information Available: A PDF file containing Table 1S and Figures 1S to 14S with captions. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Hoehler TM, Westall F. Astrobiology. 2010;9:859–867. doi: 10.1089/ast.2010.0527.see also Goals I and IV in Mars Science Goals, Objectives, Investigations, and Priorities: 2008 Mars Exploration Program Analysis Group. Johnson JR, editor. 2008 September 15; http://mepag.jpl.nasa.gov/reports/index.html.
- 2.Benner SA, Devine KG, Matveeva LN, Powell DH. Proc Natl Acad Sci. 2000;97:2425. doi: 10.1073/pnas.040539497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shkrob IA, Chemerisov SD. J Phys Chem C. 2009;113:17138. [Google Scholar]
- 4.Shkrob IA, Chemerisov SD, Marin TW. Astrobiology. 2010;10:425. doi: 10.1089/ast.2009.0433. [DOI] [PubMed] [Google Scholar]
- 5.ten Kate IL. Astrobiology. 2010;10:589. doi: 10.1089/ast.2010.0498. [DOI] [PubMed] [Google Scholar]
- 6.Garry JRC, Ten Kate IL, Martins Z, Nornberg P, Ehrenfruend P. Meteoritics Planet Sci. 2006;41:391. [Google Scholar]; Ten Kate IL, Garry JRC, Peeters Z, Foing B, Ehrenfreund Planet Space Sci. 2006;54:296. [Google Scholar]; Ten Kate IL, Garry JRC, Peeters Z, Quinn R, Foing BH, Ehrenfreund Meteoritics Planet Sci. 2005;40:1185. [Google Scholar]; Ten Kate IL, Ruiterkamp I, Botta O, Lehmann B, Hernandez Gomez, Boudin N, Foing BH, Ehrenfreund P. Int J Astrobiol. 2003;1:387. [Google Scholar]; Ehrenfreund P, Bernstein MP, Dworkin JP, Sanford SA, Allamandola LJ. ApJ. 2001;550:L95. [Google Scholar]
- 7.Shuerger AC, Clark BC. Space Sci Rev. 2008;135:233. [Google Scholar]
- 8.Hecht MH, Kounaves SP, Quinn RC, West SJ, Youn SMM, Ming DW, Catling DC, Clark BC, Boynton WV, Hoffman J, DeFlores LP, Gospodinova K, Kapit J, Smith PH. Science. 2009;325:64. doi: 10.1126/science.1172466. [DOI] [PubMed] [Google Scholar]; Catling DC, Claire MW, Zahle KJ, Quinn RC, Clark BC, Hecht MH, Kounaves S. J Geophys Res. 2010;115:E00E11. [Google Scholar]
- 9.See references 5 to 8 in ref. 3 for the reviews of soil composition and chemistry.
- 10.Rajh T, Poluektov OG, Thurnauer MC. In: Chemical Physics of Nanostructured Semiconductors. Kokorin AI, editor. NOVA Science Publ., Inc; New York: 2003. [Google Scholar]; Chiesa M, Giamello E, Che M. Chem Rev. 2010;110:1320. doi: 10.1021/cr800366v. [DOI] [PubMed] [Google Scholar]
- 11.Nosaka Y, Koenuma K, Ushida K, Kira A. Langmuir. 1996;12:736. [Google Scholar]; Kraetler B, Jaeger CD, Bard A. J Am Chem Soc. 1978;100:2239, 4904, 5985. [Google Scholar]; Chemseddine A, Boehm HP. J Mol Catal. 1990;60:295. [Google Scholar]; Bideau M, Claudel B, Faure L, Rachimoellah M. J Photochem. 1987;39:107. [Google Scholar]
- 12.Tran TH, Nosaka AY, Nosaka Y. J Phys Chem B. 2006;110:25525. doi: 10.1021/jp065255z. [DOI] [PubMed] [Google Scholar]
- 13.Shkrob IA, Marin TW, Chemerisov SD, Sevilla MD. J Phys Chem C. 2010 (submitted, see “Review Only” supplemental) jp-2010–10612s. [Google Scholar]
- 14.Huang X, Yu P, LeProust E, Gao X. Nucleic Acid Res. 1997;25:4758. doi: 10.1093/nar/25.23.4758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adhikary A, Kumar A, Becker D, Sevilla MD. J Phys Chem B. 2006;110:24171. doi: 10.1021/jp064361y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adhikary A, Khanduri D, Sevilla MD. J Am Chem Soc. 2009;131:8614. doi: 10.1021/ja9014869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shkrob IA, Sauer MC. J Phys Chem B. 2004;108:12497. [Google Scholar]; Shkrob IA, Sauer MC, Gosztola D. J Phys Chem B. 2004;108:12512. [Google Scholar]
- 18.Becke AD. Phys Rev A. 1988;38:3098. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]; Lee C, Yang W, Parr RG. Phys Rev B. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 19.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Zakrzewski VG, Montgomery JA, Jr, Stratmann RE, Burant JC, Dapprich S, Millam JM, Daniels AD, Kudin KN, Strain MC, Farkas O, Tomasi J, Barone V, Cossi M, Cammi R, Mennucci B, Pomelli C, Adamo C, Clifford S, Ochterski J, Petersson GA, Ayala PY, Cui Q, Morokuma K, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Cioslowski J, Ortiz JV, Baboul AG, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Gomperts R, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Gonzalez C, Challacombe M, Gill PMW, Johnson BG, Chen W, Wong MW, Andres JL, Head- Gordon M, Replogle ES, Pople JA. Gaussian 98, rev. A.1. Gaussian, Inc; Pittsburgh, PA: 1998. [Google Scholar]
- 20.Becker D, Adhikary A, Sevilla MD. In: Recent Trends in Radiation Chemistry. Wishart JF, Rao Rao BSM, editors. Word Scientific; Singapore: 2010. p. 509. [Google Scholar]; Becker D, Adhikary A, Sevilla MD. In: Charge Migration in DNA: Physics, Chemistry and Biology Perspectives. Chakraborty T, editor. Springer-Verlag; Germany, Heidelberg: 2007. p. 139. [Google Scholar]; Kumar A, Sevilla MD. In: Radical and Radical Ion Reactivity in Nucleic Acid Chemistry. Greenberg MM, editor. John Wiley & Sons, Inc; New Jersey: 2009. p. 1. [Google Scholar]
- 21.von Sonntag C. In: Recent Trends in Radiation Chemistry. Wishart JF, Rao Rao BSM, editors. Word Scientific; Singapore: 2010. p. 543. [Google Scholar]
- 22.von Sonntag C, Schuchmann H-P. Radiation Chemistry: Present Status and Future Trends. Elsevier Science; Amsterdam, The Netherlands: 2010. p. 513. [Google Scholar]
- 23.O’Neill P. Radiation Chemistry: Present Status and Future Trends. Elsevier Science; Amsterdam, The Netherlands: 2010. p. 585. [Google Scholar]
- 24.(a) Roca-Sanjuan D, Rubio M, Merchan M, Serrano-Andres L. J Chem Phys. 2006;125:084302. doi: 10.1063/1.2336217. [DOI] [PubMed] [Google Scholar]; (b) Orlov VM, Smirnov AN, Varshavsky YM. Tetrahedron Lett. 1976;48:4377. [Google Scholar]; Hush NS, Cheung AS. Chem Phys Lett. 1975;34:11. [Google Scholar]; Kumar A, Sevilla MD. In: Radiation Induced Molecular Phenomena in Nucleic Acid: A Comprehensive Theoretical and Experimental Analysis. Shukla MK, Leszczynski J, editors. Springer-Verlag; Germany, Heidelberg: 2008. p. 577. [Google Scholar]
- 25.Close DM. J Phys Chem A. 2010;114:1860. doi: 10.1021/jp906963f. [DOI] [PubMed] [Google Scholar]; Close DM. J Phys Chem A. 2008;112:12702. doi: 10.1021/jp807265y. [DOI] [PubMed] [Google Scholar]; Close DM, Øhman KT. J Phys Chem A. 2008;112:11207. doi: 10.1021/jp805308p. [DOI] [PubMed] [Google Scholar]; Close DM, Crespo-Hernández CE, Gorb L, Leszczynski J. J Phys Chem A. 2008;112:4405. doi: 10.1021/jp711157b. [DOI] [PubMed] [Google Scholar]; Close DM, Crespo-Hernández CE, Gorb L, Leszczynski J. J Phys Chem A. 2005;109:9279. doi: 10.1021/jp053235b. [DOI] [PubMed] [Google Scholar]
- 26.e.g., Steenken S. Free Radical Res Commun. 1992;16:349. doi: 10.3109/10715769209049187.Steenken S. Chem Rev. 1989;89:503.Steenken S, Candeias LP. J Am Chem Soc. 1989;111:1094.
- 27.(a) Kumar A, Sevilla MD. Chem Rev. 2010 doi: 10.1021/cr100023g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kumar A, Sevilla MD. J Phys Chem B. 2009;113:13374. doi: 10.1021/jp9058593. [DOI] [PMC free article] [PubMed] [Google Scholar]; Khanduri D, Collins S, Kumar A, Adhikary A, Sevilla MD. J Phys Chem B. 2008;112:2168. doi: 10.1021/jp077429y. [DOI] [PubMed] [Google Scholar]; Becker D, Sevilla MD. Electron Spin Resonance. 2008;21:33. [Google Scholar]; Adhikary A, Collins S, Khanduri D, Sevilla MD. J Phys Chem B. 2007;111:7415. doi: 10.1021/jp071107c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adhikary A, Khanduri D, Kumar A, sevilla MD. J Phys Chem B. 2008;112:15844. doi: 10.1021/jp808139e. [DOI] [PMC free article] [PubMed] [Google Scholar]; Adhikary A, Becker D, Collins S, Koppen J, Sevilla MD. Nucleic Acids Res. 2006;34:1501. doi: 10.1093/nar/gkl026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adhikary A, Malkhasian AYS, Collins S, Koppen J, Becker D, Sevilla MD. 2005;33:5553. doi: 10.1093/nar/gki857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Naumov S, Hildenbrand K, von Sonntag C. J Chem Soc Perkin Trans. 2001;2:1648. [Google Scholar]
- 31.Lu JM, Geimer J, Naumov S, Beckert D. Phys Chem Chem Phys. 2001;3:952. [Google Scholar]
- 32.Wagner JR, Cadet J. Acc Chem Res. 2010;43:564. doi: 10.1021/ar9002637. [DOI] [PubMed] [Google Scholar]
- 33.Aravindakumar CT, Schuchmann MN, Rao BSM, von Sonntag J, von Sonntag C. Org Biomol Chem. 2003;1:401. doi: 10.1039/b209626a. [DOI] [PubMed] [Google Scholar]
- 34.Malone ME, Cullis PM, Symons MCR, Parker AW. J Phys Chem. 1995;99:9299. [Google Scholar]
- 35.Sevilla MD. J Phys Chem. 1971;75:626. doi: 10.1021/j100675a005. [DOI] [PubMed] [Google Scholar]
- 36.Sevilla MD, Van Paemel C, Nichols C. J Phys Chem. 1972;76:3571. doi: 10.1021/j100668a012. [DOI] [PubMed] [Google Scholar]
- 37.Sevilla MD, Van Paemel C, Zorman G. J Phys Chem. 1972;76:3577. doi: 10.1021/j100668a013. [DOI] [PubMed] [Google Scholar]
- 38.Sevilla MD. J Phys Chem. 1976;80:1898. [Google Scholar]
- 39.Sevilla MD, Suryanarayana D, Morehouse KM. J Phys Chem. 1981;85:1027. [Google Scholar]
- 40.Ohlmann J, Huttermann J. Int J Radiat Biol. 1993;63:427. doi: 10.1080/09553009314550581. [DOI] [PubMed] [Google Scholar]
- 41.Wang W, Razskazovskii Y, Sevilla MD. Int J Radiat Biol. 1997;71:387. doi: 10.1080/095530097144003. [DOI] [PubMed] [Google Scholar]
- 42.Neta P. Radiat Res. 1972;49:1. [PubMed] [Google Scholar]; Nicolau C, McMillan M, Norman RO. Biochim Biophys Acta. 1969;174:413. doi: 10.1016/0005-2787(69)90272-x. [DOI] [PubMed] [Google Scholar]
- 43.Close D. J Phys Chem B. 2003;107:864. [Google Scholar]
- 44.Ho WF, Gilbert BC, Davies MJ. J Chem Soc, Perkin Trans. 1997;2:2533. [Google Scholar]
- 45.von Sonntag C. Free-radical-induced DNA Damage and Its Repair. Springer-Verlag; Germany, Heidelberg: 2006. p. 213. [Google Scholar]; Dedon PC. Chem Res Toxicol. 2008;21:206. doi: 10.1021/tx700283c. [DOI] [PubMed] [Google Scholar]; Pogozelski WK, Tullius TD. Chem Rev. 1998;98:1089. doi: 10.1021/cr960437i. [DOI] [PubMed] [Google Scholar]; Colson AO, Sevilla MD. J Phys Chem. 1995;99:3867. [Google Scholar]
- 46.Lemaire DGE, Bothe E, Schulte-Frohlinde D. Int J Radiat Biol. 1984;45:351. doi: 10.1080/09553008414550491. [DOI] [PubMed] [Google Scholar]; Bothe E, Schulte-Frohlinde D. Z Naturforsch C. 1982;37:1191. doi: 10.1515/znc-1982-11-1222. [DOI] [PubMed] [Google Scholar]; Lemaire DGE, Bothe E, Schulte-Frohlinde D. Int J Radiat Biol. 1987;51:319. doi: 10.1080/09553008714550791. [DOI] [PubMed] [Google Scholar]; Deeble DJ, von Sonntag C. Int J radiat Biol. 1984;46:247. doi: 10.1080/09553008414551371. [DOI] [PubMed] [Google Scholar]; Deeble DJ, Schultz D, von Sonntag C. Int J radiat Biol. 1986;49:915. doi: 10.1080/09553008514553151. [DOI] [PubMed] [Google Scholar]
- 47.Hole EO, Nelson WH, Sagstuen E, Close DM. Radiat Res. 1992;129:119.Krivokapic A, Hole EO, Sagstuen E. Radiat Res. 2003;160:340. doi: 10.1667/rr3048.and refrences 3 to 14 therein.
- 48.Adhikary A, Kumar A, Khanduri D, Sevilla MD. J Am Chem Soc. 2008;130:10282. doi: 10.1021/ja802122s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cleaves HJ, Jonsson CM, Jonsson CL, Sverjensky DA, Hazen RA. Astrobiology. 2010;10:311. doi: 10.1089/ast.2009.0397. [DOI] [PubMed] [Google Scholar]
- 50.Holm NG, Ertem G, Ferris JP. Origins Life Evol Biosphere. 1993;23:195. doi: 10.1007/BF01581839. [DOI] [PubMed] [Google Scholar]
- 51.Schwartz AW, Orgel LE. Science. 1985;228:585. doi: 10.1126/science.228.4699.585. [DOI] [PubMed] [Google Scholar]; Schwartz AW, Orgel LE. J Mol Evol. 1985;21:299. doi: 10.1007/BF02102362. [DOI] [PubMed] [Google Scholar]; Visscher J, Schwartz AW. J Mol Evol. 1988;28:3. doi: 10.1007/BF02143492. [DOI] [PubMed] [Google Scholar]; Ferris JP, Hill AR, Liu R, Orgel LE. Nature. 1996;381:59. doi: 10.1038/381059a0. [DOI] [PubMed] [Google Scholar]; Ferris JP. In: Encyclopedia of Molecular Cell Biology and Molecular Medicine. Meyers RA, editor. Weinheim, Germany: Wiley-VCH Verlag; 2005. p. 1. [Google Scholar]
- 52.(a) Lee YA, Durandin A, Dedon PC, Geacintov NE, Shafirovich V. J Phys Chem B. 2008;112:1834. doi: 10.1021/jp076777x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lewis FD, Liu X, Liu J, Miller S, Hayes RT, Wasielewski MR. Nature. 2000;406:51. doi: 10.1038/35017524. [DOI] [PubMed] [Google Scholar]; Lewis FD, Liu J, Zuo X, Hayes RT, Wasielewski MR. J Am Chem Soc. 2003;125:4850. doi: 10.1021/ja029390a. [DOI] [PubMed] [Google Scholar]
- 53.Kino K, Saito I. J Am Chem Soc. 1998;120:7373. [Google Scholar]; Ito K, Inoue S, Yamamoto K, Kawanishi K. J Biol Chem. 1993;268:13221. [PubMed] [Google Scholar]
- 54.Desnous C, Guillaume D, Clivio P. Chem Rev. 2010;110:1213. doi: 10.1021/cr0781972. [DOI] [PubMed] [Google Scholar]
- 55.Tachikawa T, Asanoi Y, Kawai K, Tojo S, Sugimoto A, Fujitsuka M, Majima T. Chem Eur J. 2008;14:1492. doi: 10.1002/chem.200701030. [DOI] [PubMed] [Google Scholar]
- 56.Paunesku T, Rajh T, Wiederrecht G, Maser J, Vogt S, Stojićević N, Protić M, Lai B, Oryhon J, Thurnauer M, Woloschak G. Nat Mater. 2003;2:343. doi: 10.1038/nmat875. [DOI] [PubMed] [Google Scholar]
- 57.Rajh T, Saponjic Z, Liu J, Dimitrijevic NM, Scherer NF, Vega-Arroyo M, Zapol P, Curtiss LA, Thurnauer MC. Nano Lett. 2004;4:1017. [Google Scholar]
- 58.Liu J, de la Garza L, Zhang L, Dimitrijevic NM, Zuo X, Tiede DM, Rajh T. Chem Phys. 2007;339:154. [Google Scholar]
- 59.Wamer WG, Yin JJ, Wei RR. Free Rad Biol Med. 1997;23:851. doi: 10.1016/s0891-5849(97)00068-3. [DOI] [PubMed] [Google Scholar]
- 60.Connor PA, McQuillan AJ. Langmuir. 1999;15:2916. [Google Scholar]
- 61.Gesteland Cech TR, Atkins JF., editors. RNA world. Cold Spring Harbor Laboratory Press; Woodbury, NY: 2006. p. 1. [Google Scholar]
- 62.Emilisson GM, Nakamura S, Roth A, Breaker RR. RNA. 2003;9:907. doi: 10.1261/rna.5680603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.e.g., Cleaves HJ, Miller SL. Proc Natl Acad Sci USA. 1998;95:7260. doi: 10.1073/pnas.95.13.7260.
- 64.e.g., Frederickson JK, Zachara JM, Kennedy DW, Dong H, Onstott TC, Hinman NW, Li SM. Geochim Cosmochim Acta. 1998;62:3239.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.