

Abstract
Background
Activation of the protein tyrosine kinase c-Src (c-Src kinase) induced by the exposure to the environmental pollutant 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) has been shown in various cell types. Most previous works used Western blot analysis to detect the phosphorylation on the Tyr416 residue, which activates c-Src kinase.
Methods
Here we compared results of c-Src tyrosine phosphorylation via aryl hydrocarbon receptor (AhR)-dependent mechanisms from Western blot analysis with fluorescent resonance energy transfer (FRET) assay detecting c-Src activation after treatment with TCDD to activate AhR in two different human cell types.
Results
Western blot analyses show time-dependent phosphorylation of c-Src by TCDD in HepG2 and MCF-10A cells. Data from FRET assay visualized and quantified activation of c-Src kinase induced by TCDD in living cells of both cell types. The FRET efficiency decreased by 20%, 5 min after TCDD treatment and continued decreasing until the end of the experiment, 25 min after TCDD treatment. PP2, a c-Src specific inhibitor, suppressed both TCDD- and epidermal growth factor- (EGF) induced c-Src activation. In contrast, the AhR antagonist 3’-methoxy-4’nitroflavone (MNF) blocked only TCDD- but not EGF-induced activation of c-Src.
Conclusions
The current study shows that early activation of c-Src via EGF and AhR signaling pathways can be visualized in living cells using FRET assay which is in line with Western blot analysis.
General Significance
The FRET assay provides a useful tool to visualize and quantify c-Src kinase activation via AhR in living cells.
Keywords: AhR, COX-2, c-Src, EGF, FRET, TCDD
1. Introduction
c-Src kinase is the first discovered non-receptor protein tyrosine kinase. c-Src plays an important role in cell differentiation, proliferation, survival, cell adhesion, cell morphology, and motility [1; 2]. c-Src kinase is expressed ubiquitously and its activity is tightly regulated. Phosphorylation on the Tyr416 residue of c-Src kinase, which is located in the activation loop, promotes its kinase activity.
Rapid activation of c-Src kinase after TCDD treatment has been first reported in our lab in several different cell lines [3; 4; 5; 6]. Most previous work to detect the activation of c-Src kinase used specific antibody against phospho-c-Src (Tyr416) in Western blot analysis. However, in most cases, the results are only semi-quantified, which do not offer spatial resolution to resolve kinetics and cannot be visualized. Fluorescence resonance energy transfer (FRET) has been proven to be a powerful tool to visualize and quantify the signaling cascades in living cells with high spatiotemporal resolutions. A FRET-based biosensor has been developed in Dr. Tsien’s lab [7]. A plasmid encoding a c-Src kinase substrate peptide conjugated to YFP (yellow fluorescence protein) and CFP (cyan fluorescence protein) was generated and transfected into cells. When c-Src kinase inside the cell is activated, the tyrosine residue on the substrate peptide is phosphorylated and the conformational change of the substrate peptide increases the distance and changes the relative orientation between the YFP and CFP to generate a change of the fluorescence resonance energy transfer (FRET) effect. The yellow emission (527 nm) from YFP decreases and the cyan emission (476 nm) from CFP increases. As a result, the ratio of yellow/cyan emission decreases more drastically. The FRET-based biosensors provide a complementary approach to traditional biochemical assays for the analysis of c-Src kinase activity. FRET assay has been shown to be very useful method to detect and visualize the activation of c-Src kinase in living cells. The measurement is non-invasive and can be repeated with the same cells, allowing for determination of the time-course of kinase activation in live cells. Here we used this FRET-based biosensor to study the activation of c-Src kinase after TCDD exposure in two cell lines, the human hepatoma cell line HepG2 and the human mammary epithelial cell line MCF10A. The results show a rapid c-Src kinase activation in a Fret-based assay, which was confirmed in Western blot analysis of the phosphorylated form of src in both cell lines tested. The effect of TCDD on c-Src activation required activation of the aryl hydrocarbon receptor (AhR). TCDD is an environmental pollutant and the prototypical ligand of the transcription factor AhR. After ligand binding the AhR dimerizes with the aryl hydrocarbon receptor nuclear transporter (ARNT), and binds on dioxin responsive elements (DRE) and regulates the expression of a diversity of genes, which is well established for the induction of cytochrome P4501A1 (CYP1A1) in response to several environmental pollutants such as polyhalogenated aromatic hydrocarbons [8]. Furthermore, recent studies show that activation of AhR by TCDD also induces several proinflammatory genes like interleukin 8, IL-1β, CCL1 as well as cyclooxygenase 2 (COX-2) [9; 10; 11; 12]. COX-2, also known as prostaglandin H synthase 2 (PGHS-2), is the key enzyme in prostaglandin biosynthesis, and acts both as a dioxygenase and as a peroxidase. In contrast to the isoform COX-1, COX-2 is inducible by drugs and environmental toxicants [13]. The induction of COX-2 by TCDD has been shown to involve the activation of c-Src and to involve binding of the CAAT enhancer binding protein (C/EBP)β on the COX-2 promoter [12; 14; 15]. Activation of the AhR by omeprazole for instance, has been shown to require a signal transduction pathway that involves c-Src kinase and that this activation pathway is different from the mechanism exerted by high-affinity ligands [16; 17]. In summary, c-Src kinase activity seems to be a critical component in AhR signaling and gene regulation.
2. Material and Methods
2.1. Reagents and Antibodies
TCDD (>99% purity) was obtained from Dow Chemicals Co. (Midland, MI). Epidermal Growth Factor (EGF) was purchased from Sigma-Aldrich Co. (St. Louis, MO). 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine (PP2) was purchased from Calbiochem (San Diego, CA). 3’-methoxy-4’-nitroflavone (MNF) was a kind gift from Dr. Josef Abel (University of Duesseldorf, Germany). Rabbit polyclonal anti-c-Src and horseradish peroxidase-conjugated secondary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit polyclonal anti-phospho-Src (Tyr 416) was purchased from Cell Signaling Technology (Danvers, MA).
2.2. Cell Culture
HepG2 human hepatoma and MCF10A human mammary epithelial cells were purchased from the American Tissue Culture Collection (Manassas, VA). HepG2 cells were grown in MEM containing 10% fetal bovine serum (Sigma-Aldrich, St. Louis, MO), 100 units of penicillin and 100 μg/mL streptomycin. MCF10A cells were grown in DMEM/F12 containing 5% fetal bovine serum, 100 units of penicillin and 100 μg/mL streptomycin supplemented with 20 ng/mL EGF, 10 μg/mL insulin, 100 ng/mL cholera toxin and 500 ng/mL hydrocortisone. Cells were incubated at 37°C with 5% CO2 and medium was changed every two or three days.
2.3. Whole Cell Protein Extraction
Cells were rinsed twice with phosphate-buffered saline (PBS) and then lysed on ice with 200 μL cold RIPA buffer containing 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate, 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM sodium vanadate and protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO) for 30 min. The lysates were then centrifuged at 16,000 g at 4°C for 20 min and the supernatants were stored at −80°C as whole cell protein extracts. Protein concentrations were determined by the method of Bradford.
2.4. Western Blotting
Proteins were separated by 10% SDS-PAGE and then transferred to PVDF membrane. The PVDF membrane was blocked with 5% non-fat milk in TBST (10 mM Tris-HCl, pH 8.0, 150 mM NaCl and 0.05% Tween −20) for 1 hr at room temperature. The PVDF membrane was then incubated with primary antibody (1:500 dilution) in blocking buffer at 4°C for overnight. After incubation with horseradish peroxidase-conjugated secondary antibody (1:2000 dilution) in TBST for 3 hr at room temperature, blots were developed using SuperSignal West Pico detection kit (Pierce, Rockford, IL). All Western blots were repeated at least three times for each experiment to confirm the reproducibility of the results.
2.5. Fluorescence Resonance Energy Transfer Assay
The Src reporter plasmid was a kind gift from Dr. Roger Tsien (University of California, San Diego, CA). Cells were transfected with the Src reporter plasmid using Effectene (Qiagen, Valencia, CA) following the manufacture’s guide. Before FRET assay, cells were transferred into Hank’s balanced salt solution with 20 mM HEPES (pH 7.4) and 2 g/L D-glucose. Fluorescence imaging was carried out with a fully automatic Olympus microscope (IX-81) driven by MetaMorph. Fluorescence emission was detected with a Hamamatsu HQ CCD camera. A spectrograph (Acton SpectraPro 2150i) was attached to the microscope’s exit port to support spectral imaging. For each cell, two images were taken: one with CFP excitation and one with YFP excitation. FRET efficiency was calculated from enhanced YFP emission using a previously described method [18].
3. Results
3.1. Enhanced phosphorylation on the Tyr416 residue of c-Src Kinase after TCDD Treatment in HepG2 and MCF10A cells
The activation c-Src kinase after TCDD treatment was first verified in HepG2 and MCF10A cells through the Western blot using a specific antibody capable of reacting with a phosphorylated (Tyr 416), and therefore an activated form of c-Src kinase. In HepG2 cells, c-Src kinase was found to be activated as early as 5 min after TCDD treatment, manifested by the enhanced phosphorylation on the the Tyr416 residues (Figure 1A). The activation of c-Src kinase became significant 15 min after TCDD treatment and stayed activated 30 min after TCDD treatment. Two chemical inhibitors, PP2 (c-Src kinase inhibitor) and MNF (AhR antagonist) can both inhibit the ability of TCDD to activate c-Src kinase as expected (Figure 1B). In MCF10A cells, the temporal pattern of TCDD to activate c-Src kinase is very similar to that in HepG2 cells (Figure 1C).
Figure 1.
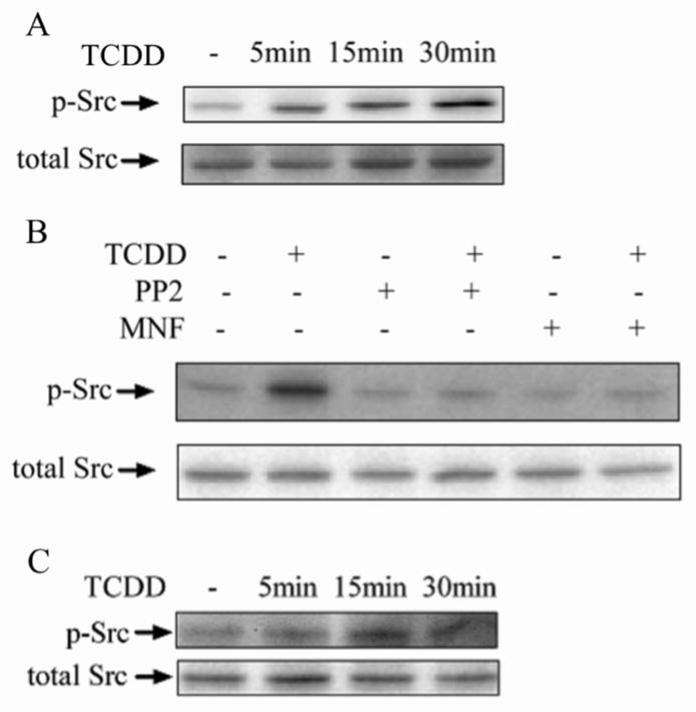
Action of TCDD to activate c-Src kinase by enhancing the phosphorylation on the Tyr416 residue. (A) HepG2 cells were treated with 10 nM TCDD for different time periods as indicated. (B) HepG2 cells were preincubated with 2 μM PP2 or 10 μM MNF for 30 min followed by treatment of 10 nM TCDD for 30 min. (C) MCF10A cells were treated with 10 nM TCDD for different time periods as indicated. The activation of c-Src kinase was judged by the phosphorylation on the Tyr416 residue.
3.2. Activation of c-Src Kinase after TCDD Treatment in HepG2 cells
Using FRET assay, the activation of c-Src kinase by TCDD in HepG2 cells was visualized in living cells for the first time (Figure 2A). We quantified FRET efficiency using a method as previously described [18]. Assessment of the FRET signal from the plasma membrane clearly showed decrease FRET efficiency after EGF and TCDD treatment for MCF10A as well as HepG2, which clearly indicated the activation of c-Src kinase in both cell types. The effect of PP2 and MNF to abolish the ability of TCDD to activate c-Src kinase is shown in figure 2B using FRET assay. TCDD, as well as EGF, a well-known c-Src kinase activator, both reduced the FRET efficiency significantly. However, pretreatment of PP2 or MNF abolished TCDD’s ability to reduce the FRET efficiency, which indicates that TCDD activates c-Src kinase in an AhR-dependent manner. The temporal patter of the activation of c-Src kinase by TCDD measured by FRET assay was similar to data obtained by Western blot (Figure 2C). The FRET efficiency decreased by 20%, 5 min after TCDD treatment and continued decreasing until the end of this experiment, 25 min after TCDD treatment. The results show that the FRET assay is sensitive and data obtained by FRET assay can be easily quantified and visualized.
Figure 2.

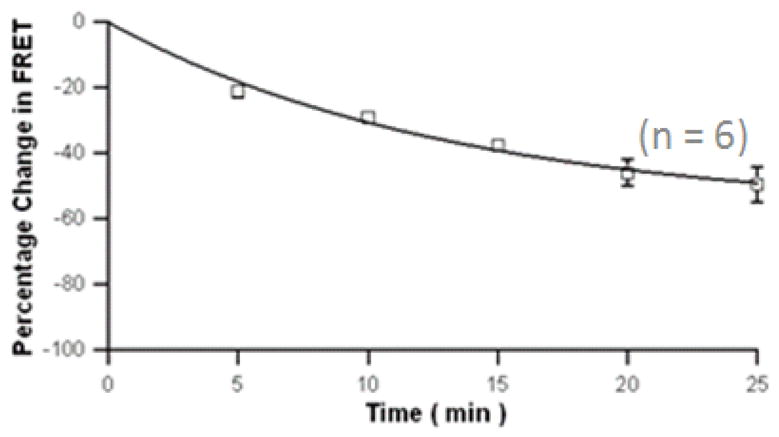
Activation of c-Src kinase after TCDD exposure in HepG2 measured by FRET assay. (A) Fluorescence images of a representative single HepG2 cell expressing CFP- and YFP- tagged of the plasmid encoding a c-Src kinase substrate peptide was observed with CFP or YFP excitation. HepG2 cells were treated with 10 nM TCDD or 1,4-dioxane (as control) for 30 min. The cyan emission from CFP (left), the yellow emission from YFP (middle), and the overlap of the two (right) are shown. (B) Treatment with EGF and TCDD did decrease the FRET signal. HepG2 cells were preincubated with 2 μM PP2 or 10 μM MNF for 30 min followed by treatment of 1 ng/ml EGF or 10 nM TCDD for 30 min. The number of cells measured for each treatment was labeled above each bar. FRET efficiencies were averaged values and error bars represent SEM. **significantly different p<0.001 (C) Time course of TCDD treatment for HepG2 cells. A decreased FRET efficiency was observed. HepG2 cells were treated with 10 nM TCDD for different time periods as indicated. The percentage change of FRET efficiency is shown.
3.3. Activation of c-Src Kinase after TCDD Treatment in HepG2 and MCF10A cells
In MCF10A cells, the activation of c-Src kinase is very similar to that in HepG2 cells. TCDD, as well as EGF activated c-Src kinase shown by the significant reduction of d the FRET efficiency (Figure 3A). The activation of c-Src kinase by TCDD also started as early as 5 min after exposure and sustained over the time period of 30 min after TCDD treatment (Figure 3B).
Figure 3.

Activation of c-Src kinase after TCDD exposure in MCF10A measured by FRET assay. (A) MCF10A cells were treated with 1 ng/mL EGF or 10 nM TCDD for 30 min. The number of cells measured for each treatment was labeled above each bar. FRET signals were assessed selectively from the plasma membrane. FRET efficiencies were averaged values and error bars represent SEM. **significantly different p<0.001 (B) Time course of TCDD treatment for MCF10A cells. A decreased FRET was observed. MCF10A cells were treated with 10 nM TCDD for various time points as indicated. The percentage change of FRET efficiency is shown.
4. Discussion
c-Src kinase is expressed ubiquitously and its activity is tightly regulated. There are two phosphorylation sites in c-Src kinase, which are critical in regulating its activity. One site is the Tyr 527 residue. Under basal conditions in vivo, 90–95% of c-Src kinase is phosphorylated at Tyr 527 [19]. Phosphorylation on this site makes it bind to its own SH2 domain and this intramolecular association stabilizes the inactive form of the enzyme. On the other hand, phosphorylation on another site, the Tyr 416 residue, which is located in the activation loop, promotes its kinase activity [1]. Rapid activation of c-Src kinase after TCDD treatment has been first reported in our lab in several different cell lines [3; 4; 5; 6]. Interestingly, c-Src kinase contributes to the eventual toxic manifestations of TCDD such as wasting syndrome since certain toxic endpoints were less pronounced in c-Src KO mice compared to the C57BL/6 wild type [12; 20; 21]. The importance of c-Src kinase activity for AhR signaling has also been demonstrated for various other effects. For instance, Src kinase activity was completely essential for TCDD-mediated adhesion restoration, sustained Erk activation and suppression of peroxisome proliferator-activated receptor (PPAR) gamma1 [22]. c-Src kinase activity is also critically involved in the induction mechanism of COX-2 mediated by TCDD [12] as well as TCDD-induced adipocyte differentiation as shown in mouse embryonic fibroblasts (MEFs) derived from c-Src-deficient mice [21]. Moreover, compounds like omeprazole, Benzimidazoles or thiabendazole mediate induction of CYP1A1 differently from AhR ligands such as TCDD [16; 17], which implicates c-Src tyrosine kinase activity. Together, the literature shows that c-Src kinase is playing a critical role in especially non-classical AhR-mediated activation of various genes, and therefore might be indispensable to exert AhR-dependent effects including non-classical AhR ligands, which became the focus of recent studies to explore the physiological function of the AhR regulated via natural compounds [23].
The activation of c-Src kinase after TCDD exposure can be detected by Western blot using anti-phospho-c-Src (Tyr 416) antibody or measuring the c-Src protein located to the plasma membrane because c-Src kinase is translocated to the plasma membrane from cytosol after activation [24], but this method has limitations. The sensitivity of these methods largely depends on the quality of the antibody and the results provide only semi-quantitative data. An improved method would be helpful to study the mechanism of the activation of c-Src kinase by TCDD. Our study shows that in two cell lines including HepG2 and MCF10A, the new method using FRET assay to detect early and AhR-dependent activation of c-Src kinase which was confirmed using traditional Western blot method. However, the FRET assay is more sensitive and can be accurately quantified. Moreover, it can visualize the spatial or temporal pattern of the activation of c-Src kinase in living cells, which provide more details of the process of c-Src kinase activation and might help to fully understand the exact mechanism how TCDD activates c-Src kinase. In summary, we presented here experimental evidence for a regulatory role of AhR during activation of c-Src in living cells.
Research Highlights.
Activation of c-Src kinase by TCDD in living cells
FRET assay to detect early and AhR-dependent activation of c-Src kinase
Regulatory role of AhR and cPLA2 during activation of c-Src in living cells.
Acknowledgments
We like to thank Dr. Roger Tsien (University of California, San Diego, CA) for providing us with critical c-Src reporter plasmid. Funding was provided by the National Institute of Environmental Health grant R21ES15846 and Grant-in-Aid from the American Heart Association (C.F.A.V.), and National Eye Institute grant REY016754A and AHA grant 0665201Y (J.Z.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Brown MT, Cooper JA. Regulation, substrates and functions of src. Biochim Biophys Acta. 1996;1287:121–49. doi: 10.1016/0304-419x(96)00003-0. [DOI] [PubMed] [Google Scholar]
- 2.Frame MC. Src in cancer: deregulation and consequences for cell behaviour. Biochim Biophys Acta. 2002;1602:114–30. doi: 10.1016/s0304-419x(02)00040-9. [DOI] [PubMed] [Google Scholar]
- 3.Blankenship A, Matsumura F. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-induced activation of a protein tyrosine kinase, pp60src, in murine hepatic cytosol using a cell-free system. Mol Pharmacol. 1997;52:667–75. doi: 10.1124/mol.52.4.667. [DOI] [PubMed] [Google Scholar]
- 4.Bombick DW, Matsumura F. 2,3,7,8-Tetrachlorodibenzo-p-dioxin causes elevation of the levels of the protein tyrosine kinase pp60c-src. J Biochem Toxicol. 1987;2:141–54. doi: 10.1002/jbt.2570020207. [DOI] [PubMed] [Google Scholar]
- 5.Dong B, Matsumura F. Roles of cytosolic phospholipase A2 and Src kinase in the early action of 2,3,7,8-tetrachlorodibenzo-p-dioxin through a nongenomic pathway in MCF10A cells. Mol Pharmacol. 2008;74:255–63. doi: 10.1124/mol.107.044669. [DOI] [PubMed] [Google Scholar]
- 6.Park S, Dong B, Matsumura F. Rapid activation of c-Src kinase by dioxin is mediated by the Cdc37-HSP90 complex as part of Ah receptor signaling in MCF10A cells. Biochemistry. 2007;46:899–908. doi: 10.1021/bi061925f. [DOI] [PubMed] [Google Scholar]
- 7.Ting AY, Kain KH, Klemke RL, Tsien RY. Genetically encoded fluorescent reporters of protein tyrosine kinase activities in living cells. Proc Natl Acad Sci U S A. 2001;98:15003–8. doi: 10.1073/pnas.211564598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Denison MS, Nagy SR. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu Rev Pharmacol Toxicol. 2003;43:309–34. doi: 10.1146/annurev.pharmtox.43.100901.135828. [DOI] [PubMed] [Google Scholar]
- 9.N’Diaye M, Le Ferrec E, Lagadic-Gossmann D, Corre S, Gilot D, Lecureur V, Monteiro P, Rauch C, Galibert MD, Fardel O. Aryl hydrocarbon receptor - and calcium-dependent induction of the chemokine CCL1 by the environmental contaminant benzo[a]pyrene. J Biol Chem. 2006;281:19906–15. doi: 10.1074/jbc.M601192200. [DOI] [PubMed] [Google Scholar]
- 10.Vogel CF, Sciullo E, Wong P, Kuzmicky P, Kado N, Matsumura F. Induction of proinflammatory cytokines and C-reactive protein in human macrophage cell line U937 exposed to air pollution particulates. Environ Health Perspect. 2005;113:1536–41. doi: 10.1289/ehp.8094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vogel CF, Sciullo E, Li W, Wong P, Lazennec G, Matsumura F. RelB, a new partner of aryl hydrocarbon receptor-mediated transcription. Mol Endocrinol. 2007;21:2941–55. doi: 10.1210/me.2007-0211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vogel C, Boerboom AM, Baechle C, El-Bahay C, Kahl R, Degen GH, Abel J. Regulation of prostaglandin endoperoxide H synthase-2 induction by dioxin in rat hepatocytes: possible c-Src-mediated pathway. Carcinogenesis. 2000;21:2267–74. doi: 10.1093/carcin/21.12.2267. [DOI] [PubMed] [Google Scholar]
- 13.Vogel C. Prostaglandin H synthases and their importance in chemical toxicity. Curr Drug Metab. 2000;1:391–404. doi: 10.2174/1389200003338884. [DOI] [PubMed] [Google Scholar]
- 14.Fritsche E, Schafer C, Calles C, Bernsmann T, Bernshausen T, Wurm M, Hubenthal U, Cline JE, Hajimiragha H, Schroeder P, Klotz LO, Rannug A, Furst P, Hanenberg H, Abel J, Krutmann J. Lightening up the UV response by identification of the arylhydrocarbon receptor as a cytoplasmatic target for ultraviolet B radiation. Proc Natl Acad Sci U S A. 2007;104:8851–6. doi: 10.1073/pnas.0701764104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vogel CF, Sciullo E, Park S, Liedtke C, Trautwein C, Matsumura F. Dioxin increases C/EBPbeta transcription by activating cAMP/protein kinase A. J Biol Chem. 2004;279:8886–94. doi: 10.1074/jbc.M310190200. [DOI] [PubMed] [Google Scholar]
- 16.Lemaire G, Delescluse C, Pralavorio M, Ledirac N, Lesca P, Rahmani R. The role of protein tyrosine kinases in CYP1A1 induction by omeprazole and thiabendazole in rat hepatocytes. Life Sci. 2004;74:2265–78. doi: 10.1016/j.lfs.2003.09.056. [DOI] [PubMed] [Google Scholar]
- 17.Backlund M, Ingelman-Sundberg M. Regulation of aryl hydrocarbon receptor signal transduction by protein tyrosine kinases. Cell Signal. 2005;17:39–48. doi: 10.1016/j.cellsig.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 18.Cheng W, Yang F, Takanishi CL, Zheng J. Thermosensitive TRPV channel subunits coassemble into heteromeric channels with intermediate conductance and gating properties. J Gen Physiol. 2007;129:191–207. doi: 10.1085/jgp.200709731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zheng XM, Resnick RJ, Shalloway D. A phosphotyrosine displacement mechanism for activation of Src by PTPalpha. Embo J. 2000;19:964–78. doi: 10.1093/emboj/19.5.964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dunlap DY, Ikeda I, Nagashima H, Vogel CF, Matsumura F. Effects of src-deficiency on the expression of in vivo toxicity of TCDD in a strain of c-src knockout mice procured through six generations of backcrossings to C57BL/6 mice. Toxicology. 2002;172:125–41. doi: 10.1016/s0300-483x(02)00006-9. [DOI] [PubMed] [Google Scholar]
- 21.Vogel CF, Matsumura F. Interaction of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) with induced adipocyte differentiation in mouse embryonic fibroblasts (MEFs) involves tyrosine kinase c-Src. Biochem Pharmacol. 2003;66:1231–44. doi: 10.1016/s0006-2952(03)00404-0. [DOI] [PubMed] [Google Scholar]
- 22.Liu X, Jefcoate C. 2,3,7,8-tetrachlorodibenzo-p-dioxin and epidermal growth factor cooperatively suppress peroxisome proliferator-activated receptor-gamma1 stimulation and restore focal adhesion complexes during adipogenesis: selective contributions of Src, Rho, and Erk distinguish these overlapping processes in C3H10T1/2 cells. Mol Pharmacol. 2006;70:1902–15. doi: 10.1124/mol.106.026534. [DOI] [PubMed] [Google Scholar]
- 23.Kawajiri K, Kobayashi Y, Ohtake F, Ikuta T, Matsushima Y, Mimura J, Pettersson S, Pollenz RS, Sakaki T, Hirokawa T, Akiyama T, Kurosumi M, Poellinger L, Kato S, Fujii-Kuriyama Y. Aryl hydrocarbon receptor suppresses intestinal carcinogenesis in ApcMin/+ mice with natural ligands. Proc Natl Acad Sci U S A. 2009;106:13481–6. doi: 10.1073/pnas.0902132106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kohle C, Gschaidmeier H, Lauth D, Topell S, Zitzer H, Bock KW. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD)-mediated membrane translocation of c-Src protein kinase in liver WB-F344 cells. Arch Toxicol. 1999;73:152–8. doi: 10.1007/s002040050600. [DOI] [PubMed] [Google Scholar]