

Abstract
Recent reports of increased tolerance to artemisinin derivatives—the last widely effective class of antimalarials — bolster the medical need for new treatments. The spirotetrahydro-β–carbolines, or spiroindolones, are a new class of fast-acting and potent schizonticidal drugs displaying low nanomolar potency against Plasmodium falciparum and Plasmodium vivax clinical isolates. Spiroindolones rapidly diminish protein synthesis in P. falciparum, an effect that is ablated in parasites bearing non-synonymous mutations in the gene encoding the P-type cation-transporter ATPase4 (PfATP4). The optimized spiroindolone NITD609 shows an acceptable safety profile and pharmacokinetic properties compatible with once-daily oral dosing; and demonstrates single-dose efficacy in a rodent malaria model. Collectively, these data demonstrate that NITD609 possesses a pharmacological profile suitable for a new drug candidate for the treatment of malaria.
Globally, 3.3 billion people are exposed to malaria, a devastating disease that causes over 800,000 deaths each year and kills more under five-year-olds than any other infectious agent (1). Fifty years ago, malaria had been eliminated from many areas of the world through effective antimalarial drug treatments, vector control interventions and disease prevention (2). However, the global spread of drug resistance resulted, by the 1980s, in a substantial increase in disease incidence and mortality. Today, some encouraging epidemiological data suggest that the introduction of new drugs (notably the artemisinin-based combination therapies or ACTs) may have reversed that trend (3). Derivatives of the endoperoxide artemisinin constitute the only antimalarial drugs that remain effective in all malaria-endemic regions, but recent reports suggest that decades of continuous use as monotherapies might have fostered the emergence of resistance (4–6). This realization has triggered a concerted search for new drugs that could be deployed if artemisinin resistance were to spread.
Many of the therapies currently in development utilize known antimalarial pharmacophores (e.g. aminoquinolines and/or peroxides) chemically modified to overcome the liabilities of their predecessors (7). While these compounds may prove to be important in the treatment of malaria, it would be preferable to discover novel chemotypes with a distinct mechanism of action (8). However, despite significant advances in our understanding of Plasmodium genome biology, the identification and validation of new drug targets has proven challenging (9).
To identify novel antimalarial leads, we and others have screened diverse chemical libraries using Plasmodium whole-cell proliferation assays with cultured intra-erythrocytic parasites (10–12). From a library of about 12,000 pure natural products and synthetic compounds with structural features found in natural products, our screen identified 275 primary hits with sub-micromolar activity against the most lethal human malarial parasite P. falciparum. We discarded those hits whose activity was not reconfirmed against multi-drug resistant parasites and/or that displayed some cytotoxicity against mammalian cells (more than 50% inhibition at 10 μM). Pharmacokinetic and physical properties were then determined for the remaining 17 compounds. From this, a synthetic compound related to the spiroazepineindole class, having a favorable pharmacological profile, stood out as an attractive chemical starting point for a medicinal chemistry lead optimization effort. Synthesis and evaluation of about 200 derivatives yielded the optimized spirotetrahydro-β–carboline (or spiroindolone) compound NITD609 (Fig. 1A). This compound is synthesized in eight steps including chiral separation of the active enantiomer, and is amenable to large-scale manufacturing. NITD609 has good drug-like attributes (see below) and displays physicochemical properties compatible with conventional tablet formulation.
Fig. 1.
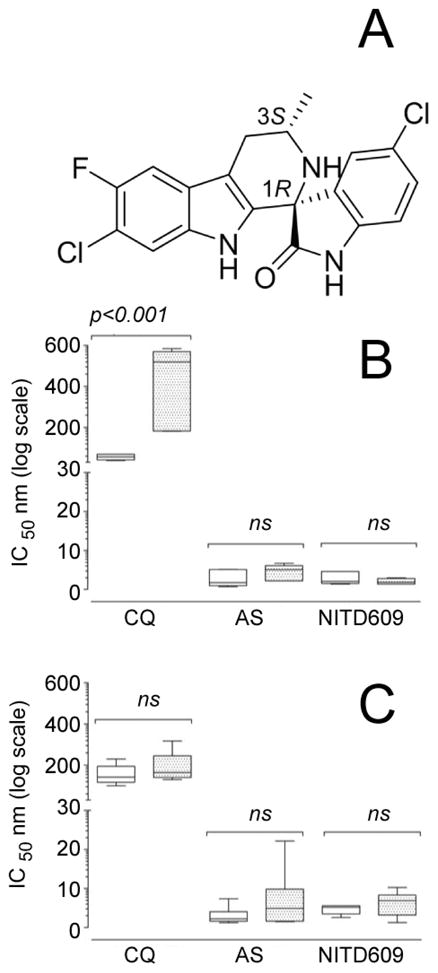
(A) Chemical structure of NITD609, showing the 1R,3S configuration that is essential for antimalarial activity. Key physicochemical properties are: solubility (pH 6.8) 39 μg/mL; logP (pH 7.4) 4.7; logD (pH 7.4) 4.6; pKa1 4.7; pKa2 10.7; polar surface area 56.92 Å2. (B) Ex vivo sensitivity of Plasmodium falciparum and (C) Plasmodium vivax (9 and 10 clinical isolates respectively) to NITD609 compared with the reference drugs chloroquine and artesunate. The antimalarial sensitivity of these two species was measured after exposing ring (unshaded boxes) and trophozoite stages (shaded boxes) to drug for 20 h. Data are shown as max–min box plots, with the solid internal line indicating median IC50 values. Inhibition of parasite growth was determined after 42 hours. Only chloroquine-treated P. vivax displayed a significant stage-specific sensitivity (p<0.001).
There is general agreement that a new antimalarial should ideally meet the following criteria: (i) display cidal activity against the parasite blood stages; (ii) be active against drug-resistant parasites, (iii) have no intrinsic safety liabilities (e.g. cytotoxicity, genotoxicity and/or cardiotoxicity), and finally (iv) have pharmacokinetic properties compatible with once-daily oral dosing. The data reported below demonstrate that NITD609 meets all of the criteria for a new antimalarial. We also provide insights into a mechanism of drug resistance involving the P-type cation-transporter ATPase4 (PfATP4).
Spiroindolones display fast and potent blood-schizonticidal activity against drug-resistant Plasmodium
Antimalarial blood-stage activity was evaluated in vitro against a panel of culture-adapted P. falciparum strains. NITD609 displayed low nanomolar IC50 values (range 0.5–1.4 nM; Table S1), with no evidence of diminished potency against drug-resistant strains (Table S1). This compound was also tested in ex vivo assays with fresh isolates of P. falciparum and P. vivax (13), collected from malaria patients on the Thai-Burmese border where drug resistance has been widely reported (14, 15). NITD609 was found to be as effective as artesunate, with potency in the low nanomolar range (IC50 values consistently <10 nM) against all P. falciparum (Fig. 1B) and P. vivax (Fig. 1C) isolates. NITD609 is also similar to artesunate in its ability to kill both mature trophozoite and immature P. vivax ring stages, in contrast to the trophozoite stage-specific activity observed with chloroquine (16). Regardless of their initial developmental stages, NITD609-treated parasites rapidly displayed morphological hallmarks of dying parasites, including pycnotic nuclei and abnormal digestive vacuoles and/or nuclear segmentation. Collectively, our in vitro and ex vivo data show that spiroindolones are potent against the intra-erythrocytic stages of the major human malarial pathogens P. falciparum and P. vivax, including a range of drug-resistant strains.
The rapid activity of artemisinin derivatives against all Plasmodium asexual erythrocytic stages is a key feature of their excellent therapeutic efficacy (6). To precisely determine which parasite blood stages are most sensitive to the spiroindolones, and to evaluate the time required for these drugs to act, we conducted in vitro drug sensitivity assays with synchronized parasites treated at ring, trophozoite and schizont stages for various durations (1, 6, 12 and 24 hours) prior to removal of drug and continuation of culture for 24 hours in the presence of [3H]-hypoxanthine. At a high concentration of NITD609 (≈100 × IC50 value), all stages (rings, trophozoites and schizonts) were similarly sensitive (Fig. S1). However, at low concentrations ≈(1 or 10 × IC50 value), schizonts were the most susceptible. These data suggest that the target is present in all asexual blood stages but might be particularly vulnerable in schizonts. With respect to drug action, while clearly significantly faster-acting than the former first-line antifolate agent pyrimethamine, NITD609 did not inhibit parasite growth as quickly as the artemisinin derivative artemether (Fig. S1). Strong inhibition was achieved with artemether treatment at 8 nM for only 6 hours, whereas similar activity was achieved with NITD609 at 1.6 nM for 24 hours.
Although at least 12 hours of continuous drug exposure was required to reduce by 90% the incorporation of [3H]-hypoxanthine into parasite DNA (Fig. S1), a [35S]-radiolabelled methionine and cysteine ([35S]-Met/Cys) incorporation assay revealed that NITD609 effectively blocked protein synthesis in P. falciparum parasites within one hour (Fig. 2A). A similar effect was observed with the known protein translation inhibitors anisomycin (an inhibitor of peptidyl transferase) and cycloheximide (an inhibitor of translocation activities during polypeptide elongation). In contrast, the antimalarial drugs artemisinin and mefloquine only showed a nominal decrease in [35S]-Met/Cys incorporation within one hour. These data suggest that NITD609 has a mechanism of action different from artemisinin and mefloquine.
Fig. 2.

Spiroindolones rapidly diminish protein synthesis in the parasite. The rate of parasite protein synthesis was evaluated by monitoring [35S]-radiolabelled methionine and cysteine ([35S]-Met/Cys) incorporation into asynchronous cultures. Parasites were assayed for 1 hour in the presence of NITD609 (inverted triangle), anisomycin (diamond), cycloheximide (square), artemisinin (circle), or mefloquine (triangle), then extracted for radiographic measurements. Radiolabel incorporation was measured against inhibitor dosed over a five-log concentration range and percent incorporation was calculated by comparison to cultures assayed in the absence of inhibitor. Anisomycin and cycloheximide were included as positive controls. (A) Spiroindolone treatment rapidly diminishes protein synthesis in Dd2; however, this effect is mostly absent in (B) NITD609-RDd2 clone #2 except at very high concentration. 50% inhibition of [35S]-Met/Cys incorporation was observed with 3x and 78x IC50 of NITD609 on the NITD609-treated Dd2 wild type and NITD609-RDd2 drug-resistant clones respectively. Data are expressed in mean±SD and represent three independent experiments performed in triplicate. Similar losses of protein synthesis inhibition upon NITD609 treatment were observed in the resistant clones NITD609-RDd2 #1 and #3 respectively (see Fig. S2).
The spiroindolone NITD609 has a large selectivity index and displays an acceptable safety profile
Considering the malaria patient population—mostly young children and pregnant women—and the resource limitations in providing adequate medical supervision of treatment, new antimalarial drug candidates require a very good safety profile. To assess for intrinsic cytotoxic activity, we measured the concentration leading to 50% cell death (CC50) in vitro with cell lines of neural, renal, hepatic or monocytic origins. With NITD609, no significant cytotoxicity was observed (CC50 > 10 μM; Table S2). Given that NITD609 has an IC50 of ~1 nM against P. falciparum (Table S1), the cytotoxicity data establish a selectivity index (CC50/IC50) > 10,000. Multiple antimalarial drugs have cardiotoxicity liabilities due to hERG channel inhibition, which in extreme cases has resulted in their withdrawal (17, 18). hERG binding and patch clamp assays with NITD609 yielded IC50 values >30 μM, consistent with a very low risk of cardiotoxicity (Table S3). Using a miniaturized Ames assay, we also established that NITD609 lacked intrinsic mutagenic activity. Finally, we observed no significant binding with a panel of human G-protein coupled receptors, enzymes and ion channels (Table S4).
In agreement with these in vitro data, male rats tolerated an oral administration of NITD609 daily for 14 days at a dose yielding daily exposure (AUC0–24h) values between 29,400 and 56,500 ng*h/ml. This is equivalent to 10 to 20 times the daily exposure to a dose that reduced parasitemia by 99% in a malaria mouse model (ED99 = 5.3 mg/kg; see below). Under these conditions, no adverse events or significant histopathological findings were observed. Overall, these data show that NITD609 has an acceptable safety profile for a new antimalarial drug.
The clinical candidate NITD609 shows favorable pharmacokinetic properties and displays single dose cure efficacy in a malaria mouse model
Having shown that our lead spiroindolone NITD609 fulfills the first three criteria for a new antimalarial drug candidate, we determined its pharmacokinetic properties. Upon oral and intravenous administration in mice and rats, this compound displayed a moderate volume of distribution and a low total systemic clearance (Table S5). Orally administered NITD609 was well absorbed, and displayed a long half-life and excellent bioavailability. Based on our in vitro metabolic stability data, this favorable profile is likely to extend to humans, as the predicted metabolic clearance for this compound was low across several species (Table S6). Taken together, these data suggest that NITD609 displays favorable pharmacokinetic properties consistent with once-daily oral dosing. We tested this drug candidate through the oral route in a virulent P. berghei malaria mouse model (19, 20). The results of these experiments show that NITD609 displays efficacious doses (ED50/90/99 = 1.2, 2.7, and 5.3 mg/kg) that are lower than those of the reference antimalarial drugs chloroquine, artesunate and mefloquine (Table S7). A single oral dose of NITD609 at 100 mg/kg was found to completely clear P. berghei infection in all treated mice, and partial cure (50%) was achieved with a single oral dose at 30 mg/kg (Table 1). Three daily oral doses of 50 mg/kg allowed for complete cure and partial cure (90%) was achieved at 3×30 mg/kg (Table S8). At similar dosing regimens, none of the reference drugs tested could achieve even partial cure in this lethal infection model. To our knowledge, among the drug candidates currently in development, only the new-generation synthetic peroxide compounds (e.g. OZ439) have been reported to have this level of curative activity upon single-dose oral administration (7).
Table 1.
In vivo efficacy data in the P. berghei rodent malaria model.
| Compound | 1 × 10 mg/kg oral | 1 × 30 mg/kg oral | 1 × 100 mg/kg oral | |||||
|---|---|---|---|---|---|---|---|---|
| Activity (%) | Survival (%) | Activity (%) | Survival (days) | Cure (%) | Activity (%) | Survival (days) | Cure (%) | |
| NIT609* | 99.6 | 13.3 | 99.6 | 24.1 | 50 | 99.2 | 30 | 100 |
| Artesunate# | 70 | 7.3 | 89 | 7.2 | -- | 97 | 6.7 | -- |
| Artemether# | 81 | 6.2 | 97 | 6.9 | -- | 99 | 7.6 | -- |
| Chloroquine# | 99.6 | 8 | 99.7 | 8.7 | -- | >99.9 | 12 | -- |
| Mefloquine# | 95 | 15.2 | 98 | 18.2 | -- | 89 | 28 | -- |
Survival of control animals: 6–7 days
Cure = no parasite present at day 30
0.5% MCM/0.1% Solutol HS15 formulation (n=10 mice)
7% Tween/3% Ethanol formulation (n≥10 mice)
Mutations in the P-type cation-transporter ATPase4 (PfATP4) confer low-level drug-resistance to spiroindolones
The rapid development of drug resistance has plagued malaria control programs in almost all endemic regions. In vitro selection of resistance has proven to be a powerful predictor of the molecular determinants employed by parasites in field settings (21, 22). To evaluate the potential for drug resistance and gain insight into the mechanism of action (MOA), we applied drug pressure to a cultured clone of Dd2—a multidrug-resistant parasite strain believed to have a higher propensity to mutate (23) (Fig. S3A). Six independent cultures were exposed to incrementally increasing sub-lethal concentrations of two compounds—NITD609 and the less potent derivative NITD678 (Fig. S3B and C). Following 3–4 months of constant drug pressure, the IC50 values had increased 7–24 fold (attaining a mean of 3–11nM) for NITD609-selected parasites, and 7–11 fold (mean of 162–241 nM) for NITD678-selected parasites (Fig. S3D and E; Table S9). The relatively high number of passages required to yield drug-resistant parasites, together with the low level of resistance that was achieved, suggest that spiroindolones are not readily prone to select for high-level resistance in vitro. Subsequent passaging of drug-selected parasites in drug-free media for four months showed no evidence of revertants, providing evidence that resistance was stable (Fig. S4). None of the selected mutants showed cross-resistance to a panel of antimalarial agents with diverse modes of action, including artemisinin and mefloquine (Table S10).
To determine the molecular basis of in vitro resistance, we prepared genomic DNA (gDNA) from each of the six drug-resistant clones. gDNA samples were then fragmented, labeled and hybridized to a high-density tiling array that contains ~6 million single-stranded 25-mer probes complementary to the P. falciparum genome. We compared the hybridization data for each haploid clone, using software that identifies regions on the array that show a loss or gain of hybridization relative to the non-resistant parental reference line (24), and that calculates a probability of a genomic change based on the number of consecutive probes showing a hybridization difference. The microarray covers approximately 90% of coding regions and 60% of non-coding regions, with probes spaced every 2 to 3 bases. Sequence coverage is limited only by the high AT content of P. falciparum that causes some 25-mer sequences to be represented more than once throughout the genome, rendering those non-informative (25). Former studies showed that this genome-tiling analysis can identify 90% of the differences that distinguish two strains in unique regions of the genome (24). Detectable genomic changes include single nucleotide polymorphisms (SNPs), insertion/deletion events and copy number variations (CNVs). Using a permissive p-value cutoff of 1×10−5, we identified 7–95 genomic differences, localized to within 2–3 nucleotides, in each resistant clone. A similar comparison between two highly diverged strains such as Dd2 and 3D7 would yield >13,000 genomic differences (24). Using a stricter cutoff (p < 10−10) that should give fewer false positives at the expense of more false negatives, we found 27 total differences amongst all six mutants. Seven of these mapped to a single gene, pfatp4 (PFL0590c; Fig. 3A and 3C), with the remainder found largely in randomly assorted subtelomeric or intergenic regions. Inspection of the hybridization patterns showed that one strain carried a copy number variant that encompassed the pfatp4 locus (Fig. 3B). These data strongly suggest that treatment with spiroindolones specifically selects for mutations in pfatp4.
Fig. 3.

Genomic tiling arrays identified shared mutations in the pfatp4 gene (PFL0590c) in all drug-resistant parasites. (A) Distinct pairs of single nucleotide polymorphisms (SNPs) in pfatp4 were detected in NITD678-RDd2 clones #2 (blue) and #3 (orange). P-values were calculated for all probes covering pfatp4 and flanking regions; a spike in the p-value reflects a difference in hybridization between the parental clone and the drug-selected clone, a hallmark of a SNP. Direct sequencing of pfatp4 from each clone confirmed that these SNPs cause non-synonymous changes in the coding region, indicated by the red boxes. The resulting change in the primary sequence is given next to each SNP. (B) A 120kb copy number variation covering 37 genes in chromosome 12 was detected in the genome of NITD678-RDd2 clone #1. The pfatp4 gene (asterisk) was contained within this amplification. Direct sequencing of pfatp4 from this clone identified an additional non-synonymous SNP at amino acid position 223 (G223R). This mutation was continuously observed throughout numerous sequencing reads of sub-cloned pfatp4 PCR products of NITD678-RDd2 clone#1, suggesting that the mutation occurred before the amplification event and, thus, resides in all pfatp4 gene copies in the genome. (C) The three NITD609-RDd2 clones showed no evidence of copy number variants; however, each clone contained non-synonymous SNPs in pfatp4 (clone#1, blue; clone#2, orange; clone#3, red).
Sequencing of the entire pfatp4 gene from the different resistant strains revealed eleven non-synonymous mutations (Table S9), with at least one in every clone. Nine of these were considered true genomic alterations for the thresholds used with the microarray analysis (p <10−5). One exception resulted from a SNP lying within the copy number variant (NITD678-RDd2 clone#1). The second exception (NITD609-RDd2 clone#1) was the result of an emergent mixed population that had been cultured for several weeks after cloning in order to obtain enough DNA for hybridization. Sequencing of different PCR products from this clone showed that some fragments contained three mutations (Ile398Phe, Pro990Arg, and Asp1247Tyr) in pfatp4 while others harbored only two mutations (Ile398Phe and Pro990Arg). The probability of 11 non-synonymous mutations occurring in pfatp4 by chance is extremely unlikely. Sequencing of 14 different isolates of P. falciparum from different continents (26, 27) revealed only 7 non-synonymous SNPs and 6 synonymous SNPs for pfatp4 (from a total of ~32,000 SNPs), placing the gene at about the 70th percentile in terms of diversity. In contrast, none of the genes with the highest number of non-synonymous SNPs from sequencing field isolates (26, 27) showed any differences in our tiling array analysis. Thus our data suggest a strong selective pressure on this single gene.
In order to confirm that drug resistance was conferred directly by these mutations, the full-length pfatp4 gene was amplified from either wild-type or resistant lines, and cloned into an expression vector that allows for site-specific integration in transgenic parasites (Fig. S5). Following integrase-mediated recombination (28) to stably introduce these genes into parasites, transgenic lines were evaluated for inhibition of parasite growth as a function of drug concentration. Lines expressing mutant PfATP4 harboring either the single Asp1247Tyr (D1247Y) or double Ile398Phe/Pro990Arg (I398F/P990R) mutations showed an increase in IC50 values relative to the parental line (Fig. 4A and 4B; Table S11). This effect was enhanced when the I398F/P990R double mutant was placed under the control of the strong P. falciparum calmodulin (PF14_0323) promoter, conferring a 5-fold increase in the IC50 value against NITD609. The reduced level of resistance compared to the original drug-selected mutants can be attributed to the co-expression of variant pfatp4 and the endogenous wild-type allele in our transfected lines. Artemisinin (Fig. 4C) and mefloquine (Fig. 4D) displayed equivalent potency against the wild-type and transgenic strains (Table S11), confirming that resistance was specific to the spiroindolones.
Fig. 4.

Introduction of mutant pfatp4 into Dd2attB parasites decreases susceptiblity to spiroindolones. pfatp4 transgenes harboring mutations identified in either NITD609-RDd2 clone #1 (I398F/P990R) or clone #3 (D1247Y) were individually introduced into the parental Dd2 background to evaluate the ability of the mutant protein to protect against spiroindolone activity. As a control, wild-type pfatp4 was also introduced. Expression of pfatp4 was regulated by either the P. berghei EF1α promoter (PbEF1α) or the stronger P. falciparum calmodulin promoter (PfCam). IC50 values were determined for (A) NITD609, (B) NITD678, (C) artemisinin, and (D) mefloquine. IC50 values are shown as means±stdev and were derived from three independent experiments performed in quadruplicate with the SYBR Green-based cell proliferation assay (12). Statistical significance was calculated using a two-tailed unpaired t test, comparing transgenic pfatp4 lines to the Dd2attB parental line: *p<0.0001. (E, F) Localization of PfATP4 to the parasite plasma membrane. Transgenic parasites co-expressing PfATP4-GFP and an ER marker, mRFP-PfSec12 were labeled with Hoechst 33382 to visualize the nucleus. PfATP4-GFP was observed at the parasite plasma membrane in (E) early schizont (two nuclei) and (F) late-segmented schizont parasites. Bar = 5 μm.
Molecular characterization of PfATP4
The pfatp4 gene product is annotated as a cation-transporting P-type ATPase (PfATP4) (29–31). This family of ATP-consuming transporters can be inhibited by chemically diverse compounds including thapsigargin, cyclopiazonic acid and lansoprazole, and as such constitutes an attractive set of drug targets (reviewed in (32)). P-type ATPases are ubiquitous in eukaryotic organisms and those involved in divalent cation transport appear to maintain a conserved structural mechanism for ion translocation (33). PfATP4 shows significant homology to Saccharomyces cerevisiae PMR1, a P-type ATPase required for high-affinity Ca2+ and Mn2+ transport. The human ortholog (hSPCA1) is associated with Hailey-Hailey disease, an acantholytic skin condition. The structural elucidation of a related rabbit SERCA pump (34), which shares 30% amino acid identity with PfATP4, enabled us to generate a homology model (Fig. 5). This localizes most of the eight resistance-associated mutations to transmembrane domains. The transmembrane region is predicted to act as a funnel to translocate cations across biological membranes, and includes eight conserved residues required for cation coordination (35, 36). Importantly, none of those residues were altered in our resistant lines. We note that a number of P-type ATPase inhibitors, including cyclopiazonic acid and thapsigargin, bind to the transmembrane region (37–39). However, no cross-resistance to either inhibitor was observed in our spiroindolone-selected mutant parasite lines (Table S10).
Fig 5.

Resistance-associated SNPs map to the predicted transmembrane region of PfATP4. A homology model of PfATP4 was generated in SWISS-MODEL based on the crystal structure of the rabbit SERCA pump. Amino acid alignment analysis by EMBOSS (41) revealed 30% identity and 48% similarity between these proteins. Residues corresponding to resistance-associated mutations are indicated in red for NITD609-RDd2 and in green for NITD678-RDd2. These mutations mapped to the putative transmembrane helices. The sites of divalent cation entry and exit are indicated as Xx2+.
To characterize PfATP4 further, we generated a PfATP4-GFP fusion that was co-transfected into Dd2 parasites along with mRFP-PfSec12, an endoplasmic reticulum (ER) marker (40). Live cell imaging of transgenic parasites localized PfATP4-GFP to the parasite plasma membrane throughout the intraerythrocytic lifecycle (Fig. 4E–F and Fig. S6). In late-stage segmented schizonts, this fusion protein was invaginated and surrounded the developing daughter merozoites, confirming a parasite plasma membrane distribution rather than delivery to the surrounding parasitophorous vacuole (Fig. 4F). This finding is supported by earlier immunofluorescence data that localized PfATP4 at or near the plasma membrane (30).
Several possibilities may explain how mutations in PfATP4 could confer resistance to spiroindolones. First, the protein could play a role in drug transport; however it shows no homology to known transporters. An alternative hypothesis could be that the PfATP4 mutations attenuate the spiroindolone-induced disruption of cellular homeostasis through an indirect mechanism that remains to be determined. Finally, PfATP4 may be the actual spiroindolones drug target since cation-transporting ATPases are chemically-validated drug targets (32). It is difficult to distinguish between these possibilities as little is known about the molecular function of PfATP4. We have been unable to reproduce results suggesting that PfATP4 plays a role in calcium transport (31) (Fig S7). And, it is unlikely that the protein regulates calcium transport of the endoplasmic reticulum calcium stores, given it’s localization to the plasma membrane. It is possible nonetheless that the protein regulates the trafficking of other cations. Although further functional characterization of PfATP4 is clearly warranted, the mutations we identified will likely be useful molecular markers of drug resistance once NITD609 enters clinical trials.
Conclusions
These studies define the spiroindolones as a new antimalarial chemotype that acts through a novel mechanism of action. In contrast to mefloquine and artemisinin, these compounds rapidly suppress protein synthesis in the parasite. Our genome-wide investigations revealed a specific mechanism of resistance mediated by the P-type ATPase PfATP4 and demonstrates at single-base resolution how a small eukaryotic genome adapts to sub-lethal drug pressure. Our lead compound NITD609 displays outstanding antimalarial activity, and meets all of the criteria required for a new antimalarial drug candidate. Further safety and pharmacological preclinical evaluation is currently ongoing to support the initiation of human clinical trials.
Supplementary Material
Acknowledgments
We thank Thomas A. Smith (SwissTPH) for the statistical analysis of the stage and rate of action studies. We thank Srinivasa Rao (NITD) and Martin Traebert (Novartis-preclinical safety) respectively for the cytotoxicity and hERG inhibition data. We also thank Margaret Weaver (Novartis-preclinical safety) for the interpretation of the preclinical toxicology data. The team would like to acknowledge Dr. Pete Schultz who had the vision to see the potential of these experiments in the exploratory phase. In addition, we extend our gratitude to Dr. Rich T. Eastman for providing guidance to help establish the in vitro drug selection protocol and for providing the cloned Dd2 parasites. Finally, the scientific expertise provided by Drs. Selina E.R. Bopp and Shailendra K. Sharma and their helpful discussions throughout this project were greatly appreciated. Funding for the PfATP4 transfectants was provided to the Fidock laboratory in part by the Medicines for Malaria Venture (MMV08/0015; PI D. Fidock). SMRU is sponsored by The Wellcome Trust of Great Britain, as part of the Oxford Tropical Medicine Research Programme of Wellcome Trust-Mahidol University. Laurent Renia laboratory is supported by a core grant from the Singapore Immunology Network, A*STAR. This work was supported by a grant from the Medicines for Malaria Venture and a translational research grant (WT078285) from the Wellcome Trust to the Novartis Institute for Tropical Diseases, the Genomics Institute of the Novartis Research Foundation and the Swiss Tropical Institute and by grants to EAW from the W. M. Keck Foundation and NIH (R01AI059472).
Footnotes
One-sentence summary
We describe the pharmacological profile of a new antimalarial drug candidate—the spiroindolone NITD609—which through a novel mechanism of action rapidly clears a Plasmodium infection upon administration of a single oral dose in a malaria mouse model.
References
- 1.WHO. World Malaria Report. 2009 http://www.who.int/malaria/world_malaria_report_2009/en/
- 2.Greenwood BM, et al. J Clin Invest. 2008;118:1266. doi: 10.1172/JCI33996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eastman RT, Fidock DA. Nat Rev Microbiol. 2009;7:864. doi: 10.1038/nrmicro2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dondorp AM, et al. N Engl J Med. 2009;361:455. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Noedl H, et al. N Engl J Med. 2008;359:2619. doi: 10.1056/NEJMc0805011. [DOI] [PubMed] [Google Scholar]
- 6.White NJ. Science. 2008;320:330. doi: 10.1126/science.1155165. [DOI] [PubMed] [Google Scholar]
- 7.Olliaro P, Wells TN. Clin Pharmacol Ther. 2009;85:584. doi: 10.1038/clpt.2009.51. [DOI] [PubMed] [Google Scholar]
- 8.Wells TNC, Alonso PL, Gutteridge WE. Nat Rev Drug Discov. 2009;8:879. doi: 10.1038/nrd2972. [DOI] [PubMed] [Google Scholar]
- 9.Winzeler EA. Nature. 2008;455:751. doi: 10.1038/nature07361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gamo FJ, et al. Nature. 465:305. doi: 10.1038/nature09107. [DOI] [PubMed] [Google Scholar]
- 11.Guiguemde WA, et al. Nature. 465:311. doi: 10.1038/nature09099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Plouffe D, et al. Proc Natl Acad Sci U S A. 2008;105:9059. doi: 10.1073/pnas.0802982105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Russell BM, et al. Antimicrob Agents Chemother. 2003;47:170. doi: 10.1128/AAC.47.1.170-173.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guthmann JP, et al. Trop Med Int Health. 2008;13:91. doi: 10.1111/j.1365-3156.2007.01978.x. [DOI] [PubMed] [Google Scholar]
- 15.White NJ. J Antimicrob Chemother. 1992;30:571. doi: 10.1093/jac/30.5.571. [DOI] [PubMed] [Google Scholar]
- 16.Sharrock WW, et al. Malar J. 2008;7:94. doi: 10.1186/1475-2875-7-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Traebert M, Dumotier B. Expert Opin Drug Saf. 2005;4:421. doi: 10.1517/14740338.4.3.421. [DOI] [PubMed] [Google Scholar]
- 18.White NJ. Lancet Infect Dis. 2007;7:549. doi: 10.1016/S1473-3099(07)70187-1. [DOI] [PubMed] [Google Scholar]
- 19.Fidock DA, Rosenthal PJ, Croft SL, Brun R, Nwaka S. Nat Rev Drug Discov. 2004;3:509. doi: 10.1038/nrd1416. [DOI] [PubMed] [Google Scholar]
- 20.Vennerstrom JL, et al. Nature. 2004;430:900. doi: 10.1038/nature02779. [DOI] [PubMed] [Google Scholar]
- 21.Hayton K, Su XZ. Curr Genet. 2008;54:223. doi: 10.1007/s00294-008-0214-x. [DOI] [PubMed] [Google Scholar]
- 22.Nzila A, Mwai L. J Antimicrob Chemother. 2010;65:390. doi: 10.1093/jac/dkp449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rathod PK, McErlean T, Lee PC. Proc Natl Acad Sci U S A. 1997;94:9389. doi: 10.1073/pnas.94.17.9389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dharia NV, et al. Genome Biol. 2009;10:R21. doi: 10.1186/gb-2009-10-2-r21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gardner MJ, et al. Nature. 2002;419:498. doi: 10.1038/nature01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Volkman SK, et al. Nat Genet. 2007;39:113. doi: 10.1038/ng1930. [DOI] [PubMed] [Google Scholar]
- 27.Jeffares DC, et al. Nat Genet. 2007;39:120. doi: 10.1038/ng1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nkrumah LJ, et al. Nat Methods. 2006;3:615. doi: 10.1038/nmeth904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trottein F, Thompson J, Cowman AF. Gene. 1995;158:133. doi: 10.1016/0378-1119(95)00158-3. [DOI] [PubMed] [Google Scholar]
- 30.Dyer M, Jackson M, McWhinney C, Zhao G, Mikkelsen R. Mol Biochem Parasitol. 1996;78:1. doi: 10.1016/s0166-6851(96)02593-5. [DOI] [PubMed] [Google Scholar]
- 31.Krishna S, et al. J Biol Chem. 2001;276:10782. doi: 10.1074/jbc.M010554200. [DOI] [PubMed] [Google Scholar]
- 32.Yatime L, et al. Biochim Biophys Acta. 2009;1787:207. doi: 10.1016/j.bbabio.2008.12.019. [DOI] [PubMed] [Google Scholar]
- 33.Kuhlbrandt W. Nat Rev Mol Cell Biol. 2004;5:282. doi: 10.1038/nrm1354. [DOI] [PubMed] [Google Scholar]
- 34.Jensen AM, Sorensen TL, Olesen C, Moller JV, Nissen P. EMBO J. 2006;25:2305. doi: 10.1038/sj.emboj.7601135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clarke DM, Loo TW, Inesi G, MacLennan DH. Nature. 1989;339:476. doi: 10.1038/339476a0. [DOI] [PubMed] [Google Scholar]
- 36.Toyoshima C, Nakasako M, Nomura H, Ogawa H. Nature. 2000;405:647. doi: 10.1038/35015017. [DOI] [PubMed] [Google Scholar]
- 37.Toyoshima C, Nomura H. Nature. 2002;418:605. doi: 10.1038/nature00944. [DOI] [PubMed] [Google Scholar]
- 38.Moncoq K, Trieber CA, Young HS. J Biol Chem. 2007;282:9748. doi: 10.1074/jbc.M611653200. [DOI] [PubMed] [Google Scholar]
- 39.Takahashi M, Kondou Y, Toyoshima C. Proc Natl Acad Sci U S A. 2007;104:5800. doi: 10.1073/pnas.0700979104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee MC, Moura PA, Miller EA, Fidock DA. Mol Microbiol. 2008;68:1535. doi: 10.1111/j.1365-2958.2008.06250.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rice P, Longden I, Bleasby A. Trends Genet. 2000;16:276. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.