

Summary
BAX is a pro-apoptotic BCL-2 family member that lies dormant in the cytosol until converted into a killer protein in response to cellular stress. Having recently identified the elusive trigger site for direct BAX activation, we now delineate by NMR and biochemical methods the essential allosteric conformational changes that transform ligand-triggered BAX into a fully activated monomer capable of propagating its own activation. Upon BAX engagement by a triggering BH3 helix, the unstructured loop between α-helices 1 and 2 is displaced, the carboxy terminal helix 9 is mobilized for membrane translocation, and the exposed BAX BH3 domain propagates the death signal through an auto-activating interaction with the trigger site of inactive BAX monomers. Our structure-activity analysis of this seminal apoptotic process reveals new pharmacologic opportunities to modulate cell death by interceding at key steps of the BAX activation pathway.
Introduction
BAX is a pro-apoptotic BCL-2 family protein that functions as a critical gateway to mitochondrial apoptosis (Wei et al., 2001) and was discovered based on its heterodimeric interaction with anti-apoptotic BCL-2 (Oltvai et al., 1993). Despite their striking structural similarities (Petros et al., 2001; Suzuki et al., 2000), BAX and BCL-2 have opposing functions. Whereas BCL-2 is a resident mitochondrial outermembrane protein that blocks BAX through protein interaction (Yin et al., 1994), BAX is a cytosolic protein that, when triggered by cellular stress signals, translocates to the mitochondria to form a putative homo-oligomeric pore, irreversibly damaging the mitochondria (Wei et al., 2001). The afferent signal transduction events that convert BAX from a cytosolic to a membrane-embedded mitochondrial protein distinguish BAX from its functional homologue, BAK, which is constitutively localized to the outer mitochondrial membrane. The BH3 domain of BAX confers its killing functionality, as documented by mutagenesis studies (Wang et al., 1998). The first structure of a BH3 death domain in complex with the surface hydrophobic pocket of an anti-apoptotic protein established a paradigm in which BCL-2 family survival proteins neutralize the death proteins through sequestration of pro-apoptotic BH3 α-helices (Sattler et al., 1997). Thus, the level of anti-apoptotic reserve at the outer mitochondrial membrane dictates the lethality of exposed BH3 death helices, establishing a rheostat for the cell's life and death decisions (Korsmeyer et al., 1993).
The structure-based paradigm for anti-apoptotic suppression of BAX dictates that its BH3 domain is trapped by the neutralizing BH3-binding pocket. Implicit in this model is the notion that the hydrophobic surface of the α-helical BAX BH3 domain becomes exposed for protein interaction. In inactive monomeric BAX, this BH3 interface is buried within the hydrophobic core of the protein (Suzuki et al., 2000). Thus, a major conformational change is required to convert BAX to its activated, BH3-exposed form. Once the quantity of activated BAX at the mitochondria exceeds the capacity of anti-apoptotic proteins to sequester the exposed BH3 domain, homo-oligomeric interactions predominate, leading to BAX-mediated mitochondrial apoptosis. Our current structural understanding of BAX is based on the NMR analysis of the inactive monomer (Suzuki et al., 2000) and its engagement by a triggering BH3 helix (Gavathiotis et al., 2008). Whereas a series of biochemical and cellular studies have demonstrated roles for the BAX amino terminus and carboxy terminus in its functional reorganization (Cartron et al., 2002; George et al., 2009; Goping et al., 1998; Hsu and Youle, 1997; Kim et al., 2009; Nechushtan et al., 1999; Schinzel et al., 2004), the explicit cascade of conformational events required for the initiation, propagation, and execution of BAX activity remains structurally undefined.
BID was the first protein classified as a BCL-2 family member based solely on the presence of a homologous BH3 domain, and was discovered as a result of its dual interactions with BCL-2 and BAX (Wang et al., 1996). The identification of a BID construct containing BH3 mutations that blocked BCL-2 interaction but retained BAX binding and killing activity led to the hypothesis that BID's BH3 domain could directly trigger BAX/BAK by a “hit and run” mechanism (Wang et al., 1996; Wei et al., 2000). Subsequent studies have provided substantial additional support for a mechanism by which BH3 domains of select BH3-only proteins can directly bind and activate BAX/BAK (Cartron et al., 2004a; Kim et al., 2006; Kim et al., 2009; Kuwana et al., 2005; Kuwana et al., 2002; Letai et al., 2002; Lovell et al., 2008; Walensky et al., 2006; Wei et al., 2000; Wei et al., 2001). The transient nature of these catalytic interactions, the consequent challenges in trapping them using experimental methods best suited for analyzing stable complexes, and the finding that genetic deletion of certain subsets of activating BH3-only proteins have yet to phenocopy Bax-/-Bak-/- mice, have continued to fuel a controversy regarding both the existence and physiologic relevance of direct BAX/BAK activation (Chen et al., 2005; Giam et al., 2008; Uren et al., 2007; Willis et al., 2007). Interestingly, a comprehensive Bim knock-in analysis, in which a variety of anti-apoptotic targeting BH3 domains were substituted singly and in combination for the native domain, documented that only BIM BH3 could sufficiently activate apoptosis to fully restore immune cell homeostasis in Bim-/- mice (Merino et al., 2009). These data suggest that BIM BH3 possesses pro-apoptotic functionality beyond that of pan-inhibition of anti-apoptotic BCL-2 family proteins. Indeed, BIM protein co-immunoprecipitates with BAX in a variety of experimental and cellular contexts (Harada et al., 2004; Lee et al., 2009; Merino et al., 2009; Willis et al., 2005).
To interrogate direct BH3-BAX interactions, we developed and applied stabilized alpha-helix of BCL-2 domains (SAHBs) based on hydrocarbon stapling of BH3 peptides (Bird et al., 2008; Walensky et al., 2004). These chemically-reinforced BH3 helices recapitulate the natural α-helical structure of BH3 domains as they exist in native BCL-2 family proteins such as BID and BAX (Chou et al., 1999; McDonnell et al., 1999; Suzuki et al., 2000), or when bound to their physiologic targets (Day et al., 2008; Hinds et al., 2007; Sattler et al., 1997). As predicted based on prior biochemical studies, BID and BIM SAHBs directly bound to BAX, but BAD SAHB did not (Walensky et al., 2006). Whereas SAHBs engaged anti-apoptotic targets such as BCL-XL with improved affinity compared to the corresponding unmodified BH3 peptides, consistent with the energetic benefit from prefolding, only the hydrocarbon-stapled BID and BIM BH3 peptides showed measurable binding interactions with full-length BAX (Walensky et al., 2006). This intriguing distinction between the binding of unmodified BH3 peptides to multidomain anti-apoptotic and pro-apoptotic targets provided the first clue that an important difference may exist between the BH3 binding sites on these homologous but functionally opposed classes of BCL-2 family proteins. Indeed, the biochemical consequences of these two types of BH3 binding interactions are starkly dissimilar. Whereas BH3 engagement of anti-apoptotic proteins is a static and inhibitory interaction, BH3 contact with pro-apoptotic BAX is a dynamic and stimulatory binding event.
Among the SAHBs tested for BAX binding activity, BIM SAHB displayed the strongest binding affinity (EC50, 24 nM) (Walensky et al., 2006) and was subsequently employed in nuclear magnetic resonance (NMR) spectroscopy studies designed to localize the BH3-binding site on BAX. However, chemical shift perturbation mapping of 15N-BAX upon BIM SAHB titration was precluded by the potency of BIM SAHB in binding and triggering BAX oligomerization in solution. To achieve a complex stable enough for short-term NMR acquisitions, we screened a series of BIM SAHBs of differential length and sequence composition, ultimately identifying a BIM SAHBA construct that, while still triggering BAX, exhibited sufficiently weak binding activity to monitor the complex (Gavathiotis et al., 2008). Heteronuclear single quantum coherence and paramagnetic relaxation enhancement (HSQC-PRE) NMR studies surprisingly localized the BH3-binding site on BAX to the intersection of helices α1 and α6, a geographically distinct region from the canonical BH3-binding pocket of anti-apoptotic proteins (Gavathiotis et al., 2008). Whereas the molecular topography of the BH3-binding sites are quite similar, the anti-apoptotic pocket has a comparatively deeper groove that may better facilitate α-helical folding of unstructured BH3 peptides upon binding. In contrast, the shallower site on BAX favors a prefolded BH3 α-helix as found in native BCL-2 proteins or their complexes. Having defined the binding site for initiation of BH3-triggered direct BAX activation and confirmed the functional specificity of the interaction by mutagenesis in vitro and in cells (Gavathiotis et al., 2008), we now apply structural and biochemical methodologies to investigate the conformational changes required for conversion of BH3-triggered BAX into a lethal, activated form.
Results
The BH3 Trigger Converts BAX from a “Closed-loop” to an Activated “Open-loop” Conformation
NMR analysis of the BIM SAHB-BAX interaction identified significant changes in the loop residues located between α-helices 1 and 2 upon ligand binding (Gavathiotis et al., 2008). Comparison of the unbound and bound calculated model structures revealed that BAX optimally accommodates BIM SAHB at the trigger site by converting its loop from a “closed” to an “open” position (Fig. 1A), displacing the interactions of loop residues P43, E44, and L45 with α6 residues I133, R134, and M137, respectively (Fig. 1B). To test the hypothesis that conversion of BAX from a closed-loop to an open-loop conformation is required to initiate direct BAX activation, we engineered a BAX construct that covalently enforced the native interaction between the α1-α2 loop and the α6 helix of BAX through disulfide bond formation between installed cysteines at positions L45 and M137 (Fig. 1C). By enforcing the disulfide bond with 0.5 mM glutathione disulfide (GSSG) and reducing the covalent tether with 100 mM β-mercaptoethanol (BME), we established an experimental system that can toggle BAX between “tethered-loop” and “closed-loop” forms, designated BAX(L45C-M137C) and BAX(L45C, M137C) respectively (Supplementary Fig. 1A-1C).
Figure 1. BAX activation is initiated by ligand-induced displacement of the α1-α2 loop from a closed to an open conformation.
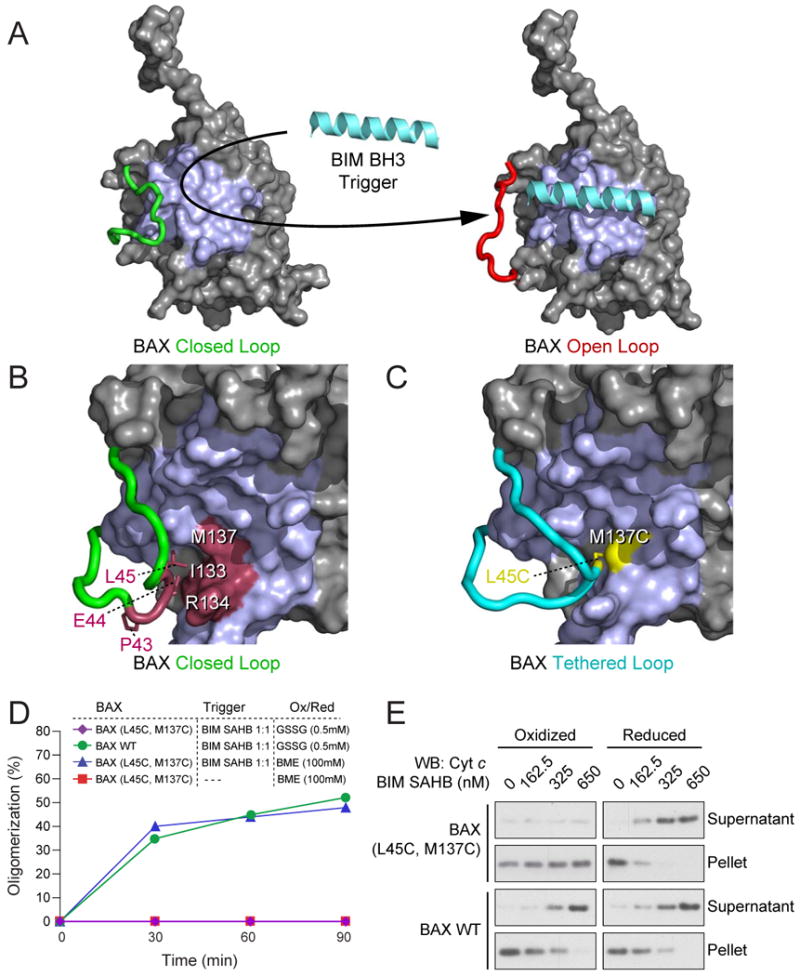
(A) NMR analysis of 15N-BAX upon BIM SAHB titration revealed conversion of BAX from a “closed-loop” to an “open-loop” conformation, implicating α1-α2 loop displacement as the initial conformational change upon BH3-triggered BAX activation.
(B) Noncovalent interactions between select α1-α2 loop and α6 residues reinforce the “closed-loop” conformation of inactive BAX (Suzuki et al., 2000).
(C) The L45 (α1-α2 loop) and M137 (α6) noncovalent interaction pair was replaced with cysteines to generate a disulfide linkage that covalently locks the α1-α2 loop in the closed conformation.
(D) BIM SAHB-induced BAX oligomerization was monitored by size-exclusion chromatography. Oxidized wild-type BAX monomer (0.5 mM GSSG) exhibited time-dependent oligomerization in response to equimolar BIM SAHB treatment, whereas oxidized BAX(L45C-M137C) monomer showed no response. The addition of 100 mM BME to BAX(L45C-M137C) restored BIM SAHB-induced oligomerization, but had no effect in the absence of triggering ligand.
(E) Cytochrome c release assays were performed using mitochondria isolated from Alb-creposBaxflox/-Bak−/− mice. Oxidized (0.5 mM GSSG) BAX(L45C-M137C) (100 nM) did not induce mitochondrial cytochrome c release in response to BIM SAHB treatment. However, the addition of BME (100 mM) to BAX(L45C-M137C) restored BIM SAHB-triggered cytochrome c release to wild-type levels. The addition of 0.5 mM GSSG or 100 mM BME to wild-type BAX had no independent effect on the induction of cytochrome c release by BIM SAHB.
See also Supplementary Figure 1.
To dissect the initiating events of BAX activation, we first tested whether the triggering ligand, BIM SAHB, could still bind to the tethered-loop form of BAX. Comparison of the chemical shift perturbation maps of 15N-BAX (Gavathiotis et al., 2008) and 15N-BAX(L45C-M137C) upon BIM SAHB titration revealed similar interaction profiles, documenting that ligand engagement of the α1/α6 trigger site can still occur when the BAX α1-α2 loop is covalently tethered (Supplementary Fig. 1D). This retention of binding activity is consistent with our tethering of the loop to the base of the trigger site so as to maintain access to the interaction surface (Fig. 1C). Despite preserving ligand binding, even excess BIM SAHB did not induce exposure of the 6A7 activation epitope (data not shown), representing a blockade of the characteristic N-terminal conformational change previously observed upon triggering wild-type BAX (Gavathiotis et al., 2008). We next conducted size-exclusion chromatography (SEC)-based BAX oligomerization analyses (Fig. 1D) and BAX-mediated mitochondrial cytochrome c release assays (Fig. 1E), performed as previously described in detail (Gavathiotis et al., 2008), to assess the functional impact of immobilizing the α1-α2 loop on direct activation of monomeric BAX. Strikingly, oxidized BAX(L45C-M137C) monomer showed no time- or dose-responsive activity upon BIM SAHB treatment (Fig. 1D, 1E, Supplementary Fig. 1E). To determine if the inactivity of BAX(L45C-M137C) derived from covalent lock-down of the α1-α2 loop, we repeated the experiments with reduced BAX(L45C, M137C) monomer and observed complete restoration of BIM SAHB-induced activation (Fig. 1D, 1E). Importantly, we confirmed that adding oxidizing or reducing agents to BAX did not independently inhibit or promote its activity. Oxidized wild-type BAX monomer was readily activated by BIM SAHB in both BAX oligomerization and BAX-mediated cytochrome c release assays (Fig. 1D, 1E). Likewise, the exposure of wild-type BAX or BAX(L45C-M137C) monomer to BME had no effect unless BIM SAHB was present (Fig. 1D, 1E). As previously documented, BIM SAHB had no independent effect on triggering mitochondrial cytochrome c release in the absence of BAX across a broad nanomolar dosing range (Gavathiotis et al., 2008) (Supplementary Fig. 1E). Taken together, these data demonstrate that initiation of BH3-triggered direct BAX activation requires ligand-induced conversion of BAX from a closed-loop to an open-loop conformation.
BAX Activation at the Amino Terminus Induces Mobilization of Carboxy Terminal Helix 9 for Mitochondrial Translocation
A longstanding challenge in studying BAX activation has been the inability to capture any structural information beyond that of the inactive monomer due to the catch 22 of trying to study a moving target using structural methods. In order to interrogate allosteric changes in BAX conformation induced by BIM SAHB binding, we sought to capture downstream events that lie at the cusp of protein oligomerization, which is reflected by global chemical shift changes followed by loss of NMR signal altogether. To overcome this obstacle, we examined dose-responsive changes induced by our weakly-binding BIM SAHB ligand at relatively short time points and also employed disulfide restraints within the BAX protein. Although in some cases only weak chemical shift changes can be captured using these approaches, the significance of the findings is supported by the topographical colocalization of ligand-induced changes to discrete functional regions of the BAX protein. Indeed, the derived NMR data provide important clues for dissecting the molecular mechanism of BAX activation and form the basis for hypotheses that can be tested and validated in a battery of biochemical experiments.
Whereas our initial NMR analysis of BAX upon BIM SAHB titration to a molar ratio of 1:1 showed little chemical shift perturbation of residues aside from those identified at the surface of the α1/α6 trigger site (Gavathiotis et al., 2008), we conducted a dose-responsive analysis to determine if allosteric changes became evident with higher concentrations of the ligand trigger. When the 1H-15N HSQC spectra were examined using 1:2, 1:4, and 1:6 ratios of BAX:BIM SAHB, dose-dependent cross-peak changes corresponding to multiple BAX α9 residues became evident (Figure 2A). Whereas α9 residue shifts were among the most numerous and significant changes aside from those at the trigger site, subtle but progressive changes were also observed for α2, α4, and α5 residues that lie within the hydrophobic core of BAX (Supplementary Fig. 2A). Based on these data, we hypothesized that direct engagement of BAX by BIM SAHB at the N-terminal trigger site induces reverberations within the very core of the protein, leading to allosteric sensing at the BAX C-terminus.
Figure 2. Release of α9 from its hydrophobic binding pocket is required for BH3-triggered BAX activation.
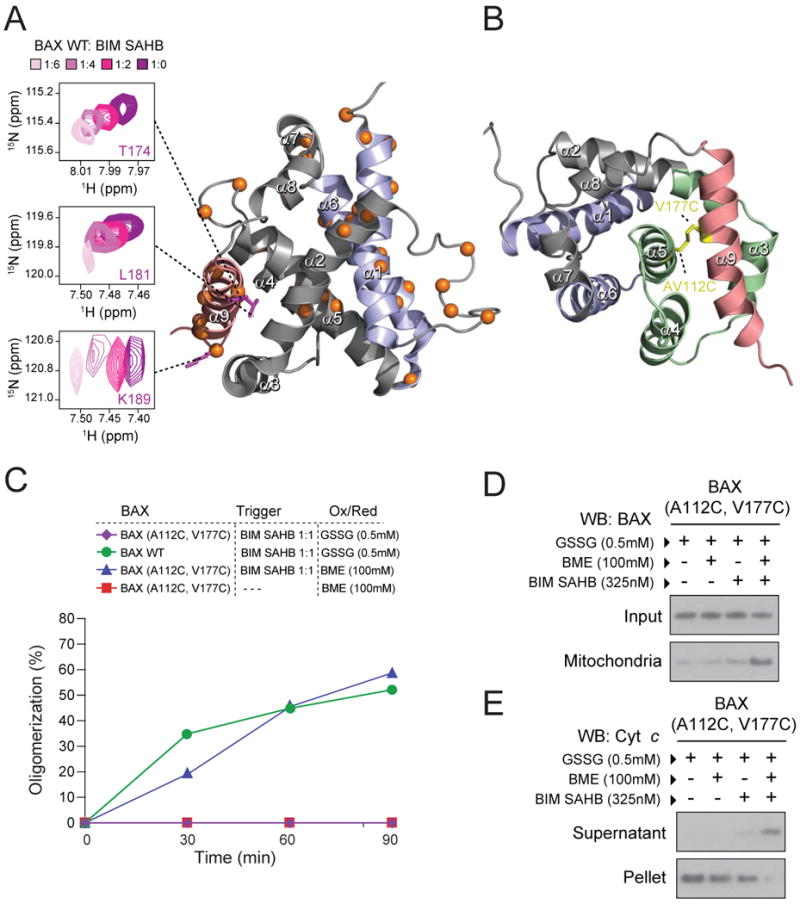
(A) The Cα atoms of 15N-BAX residues affected by BIM SAHB titration up to a ratio of 1:6 BAX:BIM SAHB are represented as orange spheres in the ribbon diagram. A side view of the BAX structure, rotated 90° from the α1/α6 trigger site (purple), demonstrates the series of α9 residues (red) with significant backbone amide chemical shift changes (calculated significance threshold ≥0.02 p.p.m.) The corresponding 1H-15N HSQC spectra demonstrate the dose-responsive chemical shift changes of BAX α9 residues (e.g. T174, L187, and K189) in response to BIM SAHB titration at 1:2, 1:4, and 1:6 ratios of BAX:BIM SAHB compared to unliganded BAX.
(B) BAX residues A112 of helix α5 and V177 of helix α9 were mutated to cysteines to generate the α9-tethered BAX(A112C-V177C) construct, in which α9 is covalently locked into its binding pocket (green).
(C) Oxidized BAX(A112-V117C) monomer (0.5 mM GSSG) was not activated by equimolar BIM SAHB (1:1), whereas wild-type BAX monomer exposed to oxidant underwent BIM SAHB-triggered oligomerization. The addition of BME to BAX(A112C-V117C) monomer completely restored BIM SAHB-induced oligomerization, an effect that was ligand-dependent, as the addition of BME alone did not induce activation.
(D) Upon exposure to isolated mitochondria, BAX(A112C,V117C) (100 nM) remained in the soluble fraction irrespective of redox status. BIM SAHB triggered mitochondrial translocation of reduced BAX(A112C,V117C), but not oxidized BAX(A112C-V117C).
(E) Correspondingly, BAX(A112C,V117C) (100 nM) induced mitochondrial cytochrome c release only when reduced with BME and treated with BIM SAHB.
See also Supplementary Figure 2.
To examine if BH3-triggering indeed causes release of α9 from its binding groove as a required step for regulated BAX translocation and functional insertion into the outer mitochondrial membrane (George et al., 2009; Goping et al., 1998; Kim et al., 2009; Suzuki et al., 2000), we generated a BAX construct in which α9 was covalently tethered to its binding pocket, which corresponds to the canonical BH3 binding groove of anti-apoptotic proteins (Fig. 2B). Specifically, we installed a disulfide bridge between Ala112 of α5 and Val177 of α9, establishing an experimental system that can toggle BAX between α9-tethered (covalent) and α9-closed (non-covalent) forms, designated BAX(A112C-V177C) and BAX(A112C, V177C), respectively. As was observed for BAX(L45C, M137C), comparison of the 1H-15N HSQC spectrum of the α9-tethered form of BAX with the reduced construct revealed a shift in cross-peaks for residues in the region of the released tether, which in this case corresponds to amino acids of the structurally defined α9-binding groove (Supplementary Fig. 2B). In functional studies, oxidized BAX(A112C-V177C) monomer showed no oligomerization in response to BIM SAHB treatment, but time-dependent activity was restored by reducing BAX(A112C-V177C) with BME (Fig. 2C). We again confirmed that BME treatment had no independent effect on the activation of BAX(A112C, V177C) monomer unless BIM SAHB was added (Fig. 2C). To examine the etiology of BAX(A112C-V177C) inactivity, we monitored the impact of reversible α9-tethering in the membrane environment by use of mitochondrial translocation (Fig. 2D) and cytochrome c release (Fig. 2E) assays. Whereas oxidized BAX(A112C-V177C) showed no translocation or release activity in response to BIM SAHB treatment, BIM SAHB-triggered translocation and cytochrome c release was restored by reducing BAX(A112C-V177C) with BME.
To further dissect the intermediate state defined by BIM SAHB-induced N-terminal activation in the context of C-terminal blockade, we exposed oxidized BAX(A112C-V177C) to BIM SAHB, followed by SEC-based repurification and functional assessment of triggered BAX(A112C-V177C). The repurified BAX(A112C-V177C) retained 6A7 positivity (Supplementary Fig. 2C), but was unable to induce cytochrome c release (Supplementary Fig. 2D). However, exposure to BME alone restored the capacity of this semi-activated form of BAX to induce cytochrome c release (Supplementary Fig. 2D). These data highlight that N-terminal conformationally-activated BAX(A112C-V177C) is stable to reisolation and can complete the activation process once the C-terminus is liberated by BME treatment. Thus, once BAX is directly triggered, allosteric release of the α9 helix is required for mitochondrial translocation and functional activation.
BAX BH3 is Exposed in Response to Direct BAX Activation
Exposure of the hydrophobic surface of the BAX BH3 helix is believed to be an essential component of the BAX death signal. Although it is known that the BAX BH3 domain is required for functional BAX homo-oligomerization (Wang et al., 1998) and that BAX can undergo auto-activation to propel homo-oligomerization (Tan et al., 2006), the mechanistic role of the BAX BH3 domain in this process is unknown. To examine whether BAX engagement by the BIM SAHB trigger also induces allosteric changes in the BAX BH3 domain, we employed our oxidized BAX(A112C-V177C) construct, in which α-9 is locked in place to prevent ligand-triggered oligomerization. In addition to the previously characterized changes induced by ligand binding at the trigger site, NMR analysis of 15N-BAX(A112C-V177C) upon BIM SAHB titration also revealed numerous weak but reproducible chemical shift changes that explicitly colocalized to the topographic region of the BAX BH3 domain (Fig. 3A). We sought to confirm the BAX BH3 NMR findings by repeating the analysis with a P168G BAX construct, which bears a mutation in the α8-α9 loop and was previously shown to impair BAX activity by disrupting α9 release (Schinzel et al., 2004). The inability of BAX(P168G) to oligomerize (Supplementary Fig. 3A) or induce mitochondrial cytochrome c release (Supplementary Fig. 3B) in response to BIM SAHB under the conditions employed for wild-type BAX activation, deemed this construct especially useful for further exploring ligand-induced changes in BAX BH3 by NMR. NMR analysis of 15N-BAX(P168G) upon BIM SAHB titration recapitulated the BAX BH3 findings of the α9-tethered construct and displayed chemical shift changes of comparatively greater magnitude (Fig. 3B). Based on these NMR data, we hypothesize that engagement of the trigger site also causes allosteric sensing at the BAX BH3 domain as part of the incipient global conformational change that defines the transformation of BAX from an inactive to an activated monomer.
Figure 3. The BAX BH3 helix is exposed upon ligand-triggered direct BAX activation.
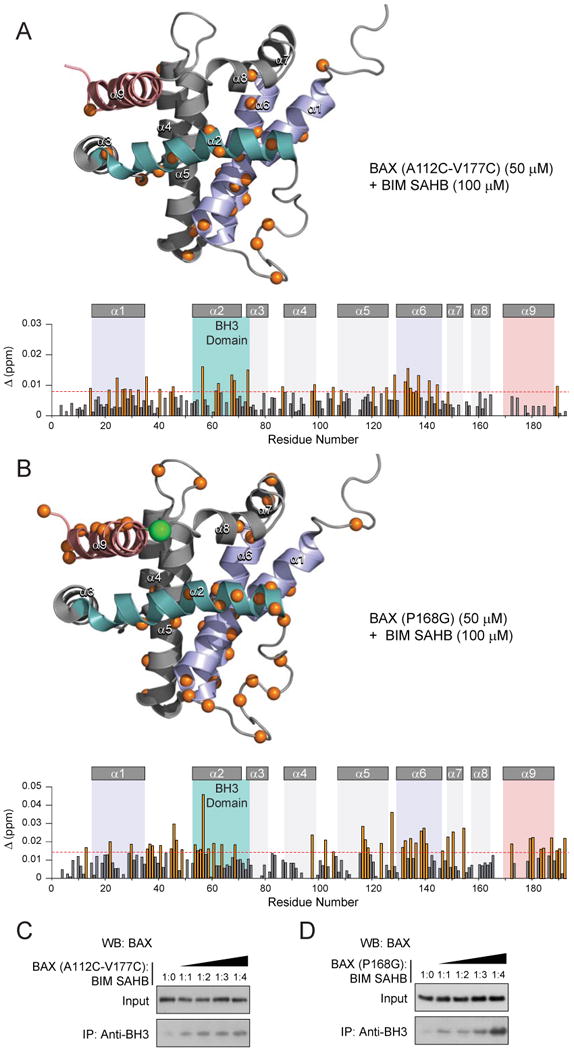
(A, B) Measured chemical shift changes of 15N-BAX(A112C-V117C) (A) and 15N-BAX(P168G) (B) upon BIM SAHB titration up to a ratio of 1:2 BAX:BIM SAHB are plotted as a function of BAX residue number. The weighted average chemical shift difference Δ at the indicated molar ratio was calculated as in p.p.m, as previously reported (Gavathiotis et al., 2008). The absence of a bar indicates no chemical shift difference, or the presence of a proline or residue that is overlapped or not assigned. The significance threshold for backbone amide chemical shift changes was calculated based on the average chemical shift across all residues plus the standard deviation, as previously reported (Gavathiotis et al., 2008) and in accordance with standard methods (Marintchev et al., 2007). Residues with significant backbone amide chemical shift change are concentrated in the region of the trigger site (α1, α1-α2 loop, α6) and BH3 domain (α2). BIM SAHB-induced chemical shift of BAX BH3 residue T56, which lies on the inward-facing surface of the BH3 helix in direct contact with α1, becomes even more prominent than previously observed for wild-type BAX(Gavathiotis et al., 2008), in addition to new changes for BH3 residues E61, C62, G67, D68, and N73 of 15N-BAX(A112C-V177C) and D53, A54, S55, K58, S60, L63, and D68 of 15N-BAX(P168G). Whereas no α9 chemical shift changes are observed when α9 is covalently tethered, the point mutant construct displays allosteric sensing at the C-terminus in a manner similar to wild-type BAX treated with higher doses of BIM SAHB (Fig. 2A). Cα atoms of affected residues are represented as orange spheres in the ribbon diagram and orange bars in the plot, based on a calculated significance threshold of ≥0.008 p.p.m. and ≥0.015 for chemical shift changes of 15N-BAX(A112C-V177C) and 15N-BAX(P168G) (50 μM), respectively, upon addition of BIM SAHB (100 μm). The site of P168G mutagenesis is indicated by a green sphere in the ribbon diagram.
(C, D) BIM SAHB triggered dose-responsive exposure of the BAX BH3 domain (residues 53-71) as monitored by immunoprecipitation of BAX(A112C-V117C) (C) and BAX(P168G) (D) using a BAX BH3 antibody and anti-BAX western analysis.
See also Supplementary Figure 3.
To determine if the BAX BH3 domain becomes exposed upon ligand-induced direct BAX activation, we incubated wild-type BAX with escalating doses of BIM SAHB in the presence of an anti-BAX BH3 antibody, but observed little to no immunoprecipitated BAX by western analysis (data not shown). We reasoned that the kinetics of BH3 exposure and rapid engagement in propagation and oligomerization interactions may preclude trapping the BAX BH3 with an antibody. Therefore, in accordance with our NMR approach, we performed the experiment using α9-tethered BAX(A112C-V177C) in order to prevent downstream oligomerization. In contrast to α1-α2 loop-tethered BAX(L45C-M137C) but similar to wild-type BAX (Gavathiotis et al., 2008), BIM SAHB induced dose-responsive exposure of the N-terminal 6A7 activation epitope of BAX(A112C-V177C) (Supplementary Fig. 3C). However, by engaging the N-terminal trigger site while preventing α9-release, we were now able to detect dose-responsive exposure of the BAX BH3 domain as well (Fig. 3C). We likewise observe dose-responsive anti-BAX BH3 immunoprecipitation using the BAX(P168G) construct (Fig. 3D). Thus, direct engagement of the BAX trigger site induces allosteric exposure of the BAX BH3 helix, which is an essential driver of BAX lethality (Wang et al., 1998).
BAX BH3 Propagates the Death Signal Through an Autoactivating Interaction with the BAX Trigger Site
Once exposed, what is the functional role of the BAX BH3 helix in promoting BAX activation? Interestingly, sequence alignment of the BIM and BAX BH3 domains revealed striking conservation in a consecutive group of amino acids implicated in complementary binding interactions between BIM BH3 and BAX at the trigger site, namely the core BH3 sequences LRRIGDE of BIM and LKRIGDE of BAX (Fig. 4A). Based on the calculated model structure of BIM BH3 and BAX, the charged residues R153, D157, and E158 of BIM exhibit electrostatic interactions with amino acids E131, R134, and K21 of BAX, respectively (Gavathiotis et al., 2008). The corresponding charged residues K64, D68, and E69 of BAX could enforce these same electrostatic pairings. To test the hypothesis that BAX BH3 itself propagates BAX activation through direct interaction at the trigger site, we synthesized a BAX SAHB for structural and biochemical analysis. We localized the hydrocarbon staple away from the core LKRIGDE sequence to preserve the key charged residues K64, D68, and E69 for protein interaction, and designated the peptide BAX SAHBB in accordance with the nomenclature of BIM SAHBB, which contains the analogous staple position (Fig. 4A) (Gavathiotis et al., 2008). NMR analysis of 15N-BAX upon BAX SAHB titration indeed revealed shifts of those cross-peaks corresponding to residues of the α1/α6 trigger site (Fig. 4B).
Figure 4. BAX BH3 propagates BAX activation by engaging the α1/α6 trigger site.
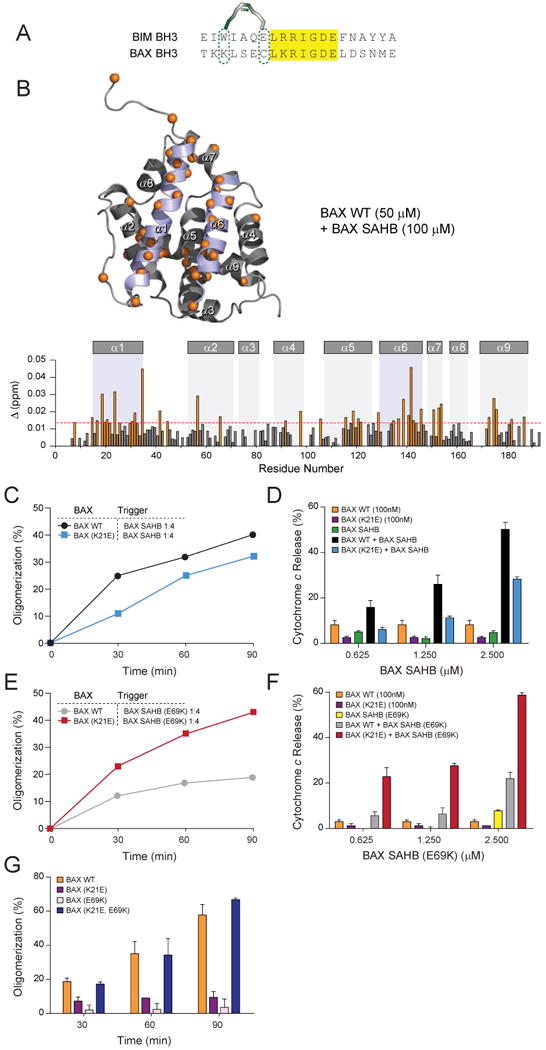
(A) Sequence alignment of the BIM and BAX BH3 domains revealed striking amino acid identity of core sequences implicated in BIM SAHB binding to the BAX trigger site.
(B) Measured chemical shift changes of 15N-BAX upon BAX SAHB titration up to a ratio of 1:2 BAX:BAX SAHB are plotted as a function of BAX residue number. Residues with significant backbone amide chemical shift change are concentrated at the trigger site (α1, α1-α2 loop, α6), with notable allosteric sensing again observed, for example, at internal residues T56 of the BH3 domain and T174 of α9. Cα atoms of affected residues are represented as orange spheres in the ribbon diagram and orange bars in the plot, based on a calculated significance threshold of ≥0.014 p.p.m. for chemical shift changes of 15N-BAX (50 μM) upon addition of BAX SAHB (100 μM).
(C, D) BAX SAHB induced BAX oligomerization and BAX-mediated cytochrome c release, an effect that was impaired by K21E mutagenesis of the BAX trigger site.
(E, F) E69K mutagenesis of BAX SAHB impaired ligand-induced BAX oligomerization and BAX-mediated cytochrome c release, but restored BAX K21E activity to wild-type levels.
(G) The capacity of native BAX BH3 to propagate BAX activation through intermonomeric interaction at the trigger site was explored by complementary mutagenesis. Heat-initiated BAX oligomerization was impaired by K21E mutagenesis of the trigger site or E69K mutagenesis of the BH3 domain, but fully restored by complementary K21E and E69K mutagenesis.
Error bars represent the mean +/- s.d. for experiments performed in at least triplicate.
See also Supplementary Figure 3.
To explicitly link the BAX SAHB interaction with functional activation of BAX, we performed BAX oligomerization and BAX-mediated cytochrome c release assays. Like BIM SAHB, BAX SAHB induced time-responsive BAX oligomerization as assessed by SEC (Fig. 4C). In addition, BAX SAHB dose-responsively triggered BAX-mediated mitochondrial cytochrome c release (Fig. 4D), but had no effect in the absence of added recombinant BAX (Supplementary Fig. 4A). The specificity of this functional interaction was explored by mutagenesis (Supplementary Fig. 4B). We previously demonstrated that a K21E mutation, which reverses the polarity of an electrostatic residue at the BAX trigger site, impairs BIM SAHB-induced BAX activation in vitro and in cells (Gavathiotis et al., 2008). Here, we find that BAX SAHB-induced BAX(K21E) activation is likewise blunted in both the oligomerization and cytochrome c release assays (Fig. 4C, 4D). Whereas the complementary reverse polarity mutant BAX SAHB(E69K) is a poor activator of wild-type BAX, it effectively reverses the impairment of BAX(K21E) (Fig. 4E, 4F), underscoring both the specificity and functional importance of direct BAX BH3 interaction at the trigger site. Finally, to extend our observations to an exclusively protein-based analysis of native BAX BH3-mediated propagation, we compared the efficiency of BAX, BAX(K21E), BAX(E69K), and BAX(K21E, E69K) constructs to undergo auto-activation in response to heat (Pagliari et al., 2005). In this experimental context, initiation of BAX activation is not imposed by exogenous ligand but is instead driven by an intermonomeric trigger once the protein is sufficiently destabilized by heat (Supplementary Fig. 4C). We find that single residue mutagenesis of the BAX BH3 domain or the trigger site impaired heat-induced BAX activation, but complementary K21E, E69K mutagenesis within the BAX monomer restored the oligomerization response to wild-type levels (Fig. 4G). Taken together, these data indicate that exposure of the BAX BH3 domain propagates the BAX activation pathway through a triggering interaction between the BH3 helix of activated BAX and the trigger site of inactive BAX monomers. Thus, consistent with the “hit and run” model for direct BAX activation (Perez and White, 2000; Tan et al., 2006; Wei et al., 2000), the triggering BH3-only interaction catalyzes the activation process, which is then propelled by BAX BH3/trigger site auto-activating interactions (Fig. 5).
Figure 5. BH3-triggered structural reorganization drives the BAX activation pathway.

Cytosolic BAX activation is initiated by BIM BH3 engagement of the α1/α6 trigger site. A series of discrete structural changes ensue, including α1-α2 loop displacement, 6A7 epitope exposure, BAX BH3 exposure, and α9 release for mitochondrial translocation. BAX propagates its activation through triggering interactions between the exposed BAX BH3 domain of fully activated monomers and the α1/α6 binding site of inactive monomers. BAX assembles into a structurally undefined homo-oligomeric pore that promotes apoptosis by releasing mitochondrial factors such as cytochrome c.
Discussion
BAX is a meticulously regulated death-inducing protein that is deployed in response to cellular stress, but can be intercepted and neutralized by survival proteins. Because the relative influence of BCL-2 family pro-apoptotic and anti-apoptotic proteins can dictate cellular fate in health and disease, the activation and inhibitory mechanisms that govern their functions remain an area of intensive investigation. It is well established that functionally activated BAX, as defined by its mitochondrial localization and availability of its helical BH3 domain for interaction, can be blocked by anti-apoptotic sequestration and then derepressed through displacement of the BAX BH3 by other BH3-domain containing proteins (Willis et al., 2005; Willis et al., 2007). However, a major gap in understanding of the BAX activation pathway relates to the mechanism by which inactive, cytosolic BAX becomes activated in the first place and what structural changes underlie its conversion to a fully activated monomer capable of propagating its own activation.
Here we apply a combination of structural and biochemical approaches to identify and validate key interactions and conformational changes that drive BAX activation (Fig. 5). First, we distinguish the critical binding event between the BIM BH3 ligand and the BAX trigger site from the initial conformational change, which involves displacement of the α1-α2 loop from a closed to an open configuration. This N-terminal initiation event, which coincides with exposure of the N-terminal 6A7 epitope (Gavathiotis et al., 2008), unleashes rapid structural changes that culminate in functional BAX oligomerization. Along this continuum, we detect allosteric changes at the C-terminal α9 helix, which when covalently restrained in its binding pocket, prevents mitochondrial translocation and functional BAX activation. By arresting monomeric BAX activation at an intermediate stage, the α9-tether enabled us to identify allosteric changes at the BAX BH3 domain, which becomes exposed upon BIM SAHB engagement of the N-terminal trigger site. BAX BH3 shares striking sequence identity with BIM BH3 at key interacting positions, suggesting that once exposed, the BAX BH3 helix can subserve the function of BIM BH3 in triggering BAX activation through engagement of the α1/α6 binding site. Indeed, BAX SAHB directly binds the trigger site and functionally activates BAX, with the specificity of interaction confirmed by complementary mutagenesis. Thus, once catalyzed by a triggering BH3-only interaction, BAX can propagate the death signal through auto-activating interactions between its own BH3 domain and trigger site.
A common theme that emerges from the study of BAX activation is the critical role of N- and C-terminal structural elements in regulating exposure of the protein's toxic, hydrophobic core. Whether by direct BH3-triggered activation (Cartron et al., 2004a; Desagher et al., 1999; Gavathiotis et al., 2008; Kim et al., 2006; Kim et al., 2009; Kuwana et al., 2002; Letai et al., 2002; Walensky et al., 2006), activating mutations (Zhou et al., 2007) or deletions (Fu et al., 2009), heat (Pagliari et al., 2005), changes in pH (Cartron et al., 2004b; Khaled et al., 1999), or engagement with other known (Chipuk et al., 2004) or undiscovered interactors, any modality that unzips the inactive conformation of BAX to expose its inner hydrophobic surfaces will trigger BAX activation. Indeed, our structural and biochemical studies document that ligand engagement of the trigger site sets in motion a ripple effect involving displacement of the α1-α2 loop, followed by allosteric changes at key functional domains such as BAX BH3 and α9, ultimately resulting in a major conformational reorganization that effectively exposes previously buried hydrophobic surfaces. Conversely, covalent lock-down of the α1-α2 loop or α9 regulatory sites structurally and functionally restrains BAX. Importantly, whereas α1-α2 loop tethering prevents BIM SAHB-induced N-terminal 6A7 epitope exposure and all downstream activation features, locking α9 in place does not preclude ligand-induced 6A7 and BH3 epitope exposure but instead prevents terminal activation events, including mitochondrial membrane targeting and functional oligomer formation. Once activated by exogenous triggering interactions and self-propagated by intermonomeric interactions, the execution phase of BAX activation is ultimately dependent upon homo-oligomerization within the membrane environment (Lovell et al., 2008; Schlesinger and Saito, 2006). Mutagenesis of the hydrophobic BAX BH3 interface (Wang et al., 1998) and other key residues within the hydrophobic core (George et al., 2007; Meijerink et al., 1998) abrogate homo-oligomerization and toxicity, indicating that once monomeric BAX is activated, oligomer formation is predominantly driven by hydrophobic contacts within the membrane (Annis et al., 2005; Lucken-Ardjomande et al., 2008). The structural nature of the oligomeric interactions is currently unknown, although several models for BAX/BAK homo-oligomerization have been proposed based upon the structure of an N- and C-terminally truncated BAK homodimer (Moldoveanu et al., 2006), BAK mutagenesis studies (Dewson et al., 2009; Dewson et al., 2008), and a BAX crosslinking analysis (Zhang et al., 2010). Intriguingly, these studies implicate the region of the BAX trigger site as a critical protein interaction surface for oligomer formation.
Each step along the BAX activation pathway provides a potential opportunity for therapeutic intervention in diseases of premature cell death or unchecked cellular survival. Pharmacologic approaches that stimulate the trigger site, displace the α1-α2 loop, release α9, and/or expose the BAX BH3 carry the potential to induce apoptosis through BAX activation. Whereas unstressed cells may have sufficient anti-apoptotic reserve to constitutively bind and sequester activated BAX, diseased cells whose anti-apoptotic reserve is taxed by tonic death signaling may be unable to withstand further BAX-mediated mitochondrial assault. Conversely, targeted therapies that block the BAX trigger site, protect the α1-α2 loop from displacement, reinforce the interaction between α9 and its binding pocket, and/or block BAX BH3 exposure may serve to protect cells from unwanted cell death. Another pivotal opportunity for potential pharmacologic intervention may derive from modulating key interactions of the BAX homo-oligomer itself, an elusive death channel whose biological structure and defining interactions await discovery.
Experimental Procedures
SAHB Synthesis and Characterization
Hydrocarbon-stapled peptides corresponding to the BH3 domains of BIM and BAX were synthesized, purified, and characterized according to our established methods (Bird et al., 2008). NMR and biochemical studies employed (1) BIM SAHBA, an N-acetylated, C-amidated 20-mer: Ac-145EIWIAQELRXIGDXFNAYYA164-CONH2, (2) BAX SAHBB, an N-acetylated, C-amidated 20-mer: Ac-56TKXLSEXLKRIGDELDSNBE75-CONH2, and (3) BAX SAHBB(E69K), an N-acetylated, C-amidated 20-mer: Ac-56TKXLSEXLKRIGDKLDSNBE75-CONH2. The letter “X” represents the non-natural amino acid (S)-2-(((9H-fluoren-9-yl)methoxy)carbonylamino)-2-methyl-hept-6-enoic acid that is substituted at i, i+4 positions for olefin metathesis and the letter “B” represents norleucine, a conservative substitution for methionine that lacks sulfur in order to preserve full activity of the Grubb's ruthenium catalyst. All peptides were purified by liquid chromatography-mass spectrometry to >95% purity and quantified by amino acid analysis.
BAX Preparation
Recombinant BAX and its point mutants were produced as previously described (Suzuki et al., 2000; Walensky et al., 2006). Point mutations were generated by PCR-based site-directed mutagenesis followed by DNA sequencing to verify the construct. BAX(L45C-M137C) and BAX(A112C-V177C) were maintained in 0.5 mM glutathione disulfide (GSSG) to preserve the engineered disulfide bond. Reduction of the disulfide tethers was achieved by the addition of 100 mM β-mercaptoethanol (BME) for 30 min.
NMR Samples and Spectroscopy
Uniformly 15N-labeled full-length human BAX was generated as previously described (Gavathiotis et al., 2008; Suzuki et al., 2000). Protein samples were prepared in 25 mM sodium acetate, 50 mM NaCl solution at pH 6.0 in 5% D2O. SAHBs (6 mM stock), GSSG (100 mM stock) and BME (7.1 M stock) were titrated into a solution of 50 μM BAX to achieve the indicated concentrations or molar ratios. Correlation 1H-15N HSQC spectra (Grzesiek and BAX, 1993) were acquired at 25°C on a Bruker 800 MHz NMR spectrometer equipped with a cryogenic probe, processed using NMRPipe (Delaglio et al., 1995), and analyzed with NMRView (Johnson, 2004). The weighted average chemical shift difference Δ at the indicated molar ratio was calculated as in p.p.m. The absence of a bar indicates no chemical shift difference, or the presence of a proline or residue that is overlapped or not assigned. BAX cross-peak assignments were applied as previously reported (Suzuki et al., 2000) and assignments for BAX(L45C, M137C), BAX(A112C,V177C), and BAX(P168G) were determined by comparison with the wild-type BAX spectra. Mutagenized residues and any residues in their immediate vicinity that sustained large chemical shifts were left unassigned. The significance threshold for backbone amide chemical shift changes was calculated based on the average chemical shift across all residues plus the standard deviation, in accordance with standard methods (Marintchev et al., 2007).
Structure Modeling
Energy-minimized structures of BAX(L45C-M137C) and BAX(A112C-V177C) were generating by cysteine mutagenesis of the BAX solution structure (PDB ID# 1F16) (Suzuki et al., 2000) followed by implementation of a conjugate gradient energy minimization protocol using the molecular dynamics software GROMACS (Van Der Spoel et al., 2005). Structures were analyzed using WHATCHECK (Hooft et al., 1996) and PYMOL (DeLano, 2002).
Mass Spectrometry Analysis
BAX protein was oxidized with 0.5 mM GSSG and free cysteine residues derivatized with iodoacetamide. Following SDS-PAGE electrophoresis, the coomassie-stained gel band was excised, in-gel digested with trypsin (Shevchenko et al., 1996), and the generated peptide mixture subjected to nano-LC-MS/MS using a hybrid linear ion trap/FT-ICR mass spectrometer (LTQ FT, Thermo Electron) as described (Haas et al., 2006). MS/MS spectra were assigned by searching all possible pairs of cysteine-containing tryptic peptides using a custom algorithm and matched to the peptide shown. High mass accuracy MS/MS unambiguously confirmed covalent modification of cysteines with multiple matching b- and y-type ions.
BAX Oligomerization Assay
SAHB was added to a 200 μL solution (20 mM Hepes/KOH pH 7.2, 150 mM KCl, 0.5% CHAPS) containing size exclusion chromatography (SEC)-purified, monomeric BAX with the indicated additive (ie. 0.5 mM GSSG, 100 mM BME) at the indicated BAX:SAHB ratio. The mixtures and BAX monomer alone were incubated at 30°C for the indicated durations and then subjected to analysis by SEC using an SD75 column and 20 mM Hepes/KOH pH 7.2, 150 mM KCl running buffer. The monomeric and oligomeric fractions elute at ∼11.5-12.0 min and ∼6.5-7.5 min, respectively. Protein standards (GE Healthcare) were used to calibrate the molecular weights of gel filtration peaks. For heat-induced oligomerization, BAX constructs (7.5 μM) in 200μL solution (20 mM Hepes/KOH pH 7.2, 150 mM KCl, 0.2% n-octyl-β-D-glucoside) were incubated at 37°C for the indicated durations and subjected to analysis by SEC. Replicates were performed using at least two independent preparations of freshly SEC-purified monomeric BAX protein.
BAX Conformational Change Assays
BIM SAHB was added to a 50 μL PBS solution containing monomeric BAX (0.5 μM) at a ratio of 1:0, 1:1, 1:2, 1:3 and 1:4 BAX:BIM SAHB. The mixtures and a BAX monomer sample were incubated at room temperature for 15 minutes and then added to a 3% BSA in PBS solution (250 μL) containing 3 μL of 6A7 (sc-23959, Santa Cruz Biotechnology) or anti-BH3 (AP1302a, Abgent) antibody for 1 hour incubation at 4°C. Additionally, 5 μL of each input sample (10%) was mixed with 20 μL of SDS-sample buffer to measure baseline BAX levels across specimens. Preclarified sepharose beads (30 μL) were added to the BAX:BIM SAHB and BAX monomer solutions for an additional 1 hour incubation at 4°C. The sepharose beads were spun down, washed 3 times with 1 mL of 3% BSA in PBS solution, resuspended in 50 μL of SDS-sample buffer, and boiled at 95 °C for 2 minutes. Samples were separated on 10% SDS-PAGE Bis-Tris gel, blotted on a PVDF membrane, and western analysis performed using the rabbit polyclonal N20 anti-BAX antibody (sc493, Santa Cruz Biotechnology) and chemiluminescence-based detection (PerkinElmer).
Mitochondrial Cytochrome c Release Assay
Mitochondrial cytochrome c release assays were performed on Alb-creposBaxflox/-Bak−/− mitochondria as described (Pitter et al., 2008; Walensky et al., 2006). Mitochondria were treated with the indicated concentrations of SAHB and oxidized (0.5 mM GSSG) or reduced (100 mM BME) BAX protein (100 nM), singly and in combination, and incubated at room temperature for 40 min. The supernatants were isolated by centrifugation at 5,500×g for 10 minutes and the mitochondrial pellets solubilized in 1% Triton X-100/PBS. Mitochondrial supernatant and pellet fractions were separated by 10% NuPAGE (Invitrogen) gels and analyzed by immunoblotting with anti-cytochrome c antibody (7H8.2C12, BD Pharmingen).
Mitochondrial Translocation Assay
Mitochondria were treated with the indicated concentrations of SAHB and oxidized (0.5 mM GSSG) or reduced (100 mM BME) BAX protein, singly and in combination, and incubated at room temperature for 30 min. The mitochondria were pelleted at 5,500×g for 10 minutes, resuspended and washed with 0.1 M sodium carbonate pH 11.5 for 30 min, centrifuged at 13,000×g for 10 min at 4°C, and then resuspended in 20 μl SDS-sample buffer for analysis by 10% NuPAGE (Invitrogen) gel electrophoresis and western blotting with N20 anti-BAX antibody.
Supplementary Material
Acknowledgments
We thank E. Smith for editorial and graphics assistance, N. Tjandra and M. Suzuki for BAX NMR assignments and helpful discussions, C. Turner of the MIT/Harvard Center for Magnetic Resonance and S. Pochapsky of the Brandeis University Nuclear Magnetic Resonance Facility for technical advice, and S. Gygi and J. Mintseris of Harvard Medical School for mass spectrometry analysis. This work was supported by NIH grant 2R01CA050239, an American Society of Hematology Junior Faculty Scholar Award, and a grant from the William Lawrence Children's Foundation to L.D.W., and an American Heart Association Founders Affiliate Scientist Development Grant to E.G. We also thank the LaTorre family for their generous support of D.E.R.
Footnotes
Supplemental Information: Supplemental Information includes four figures and figure legends.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Annis MG, Soucie EL, Dlugosz PJ, Cruz-Aguado JA, Penn LZ, Leber B, Andrews DW. Bax forms multispanning monomers that oligomerize to permeabilize membranes during apoptosis. Embo J. 2005;24:2096–2103. doi: 10.1038/sj.emboj.7600675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird GH, Bernal F, Pitter K, Walensky LD. Synthesis and biophysical characterization of stabilized alpha-helices of BCL-2 domains. Methods Enzymol. 2008;446:369–386. doi: 10.1016/S0076-6879(08)01622-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartron PF, Gallenne T, Bougras G, Gautier F, Manero F, Vusio P, Meflah K, Vallette FM, Juin P. The first alpha helix of Bax plays a necessary role in its ligand-induced activation by the BH3-only proteins Bid and PUMA. Mol Cell. 2004a;16:807–818. doi: 10.1016/j.molcel.2004.10.028. [DOI] [PubMed] [Google Scholar]
- Cartron PF, Moreau C, Oliver L, Mayat E, Meflah K, Vallette FM. Involvement of the N-terminus of Bax in its intracellular localization and function. FEBS Lett. 2002;512:95–100. doi: 10.1016/s0014-5793(02)02227-5. [DOI] [PubMed] [Google Scholar]
- Cartron PF, Oliver L, Mayat E, Meflah K, Vallette FM. Impact of pH on Bax alpha conformation, oligomerisation and mitochondrial integration. FEBS Lett. 2004b;578:41–46. doi: 10.1016/j.febslet.2004.10.080. [DOI] [PubMed] [Google Scholar]
- Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM, Huang DC. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, Green DR. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004;303:1010–1014. doi: 10.1126/science.1092734. [DOI] [PubMed] [Google Scholar]
- Chou JJ, Li H, Salvesen GS, Yuan J, Wagner G. Solution structure of BID, an intracellular amplifier of apoptotic signaling. Cell. 1999;96:615–624. doi: 10.1016/s0092-8674(00)80572-3. [DOI] [PubMed] [Google Scholar]
- Day CL, Smits C, Fan FC, Lee EF, Fairlie WD, Hinds MG. Structure of the BH3 domains from the p53-inducible BH3-only proteins Noxa and Puma in complex with Mcl-1. J Mol Biol. 2008;380:958–971. doi: 10.1016/j.jmb.2008.05.071. [DOI] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL Molecular Graphics System. San Carlos: DeLano Scientific; 2002. http://www.pymol.org. edn. [Google Scholar]
- Desagher S, Osen-Sand A, Nichols A, Eskes R, Montessuit S, Lauper S, Maundrell K, Antonsson B, Martinou JC. Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J Cell Biol. 1999;144:891–901. doi: 10.1083/jcb.144.5.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewson G, Kratina T, Czabotar P, Day CL, Adams JM, Kluck RM. Bak activation for apoptosis involves oligomerization of dimers via their alpha6 helices. Mol Cell. 2009;36:696–703. doi: 10.1016/j.molcel.2009.11.008. [DOI] [PubMed] [Google Scholar]
- Dewson G, Kratina T, Sim HW, Puthalakath H, Adams JM, Colman PM, Kluck RM. To trigger apoptosis, Bak exposes its BH3 domain and homodimerizes via BH3:groove interactions. Mol Cell. 2008;30:369–380. doi: 10.1016/j.molcel.2008.04.005. [DOI] [PubMed] [Google Scholar]
- Fu NY, Sukumaran SK, Kerk SY, Yu VC. Baxbeta: a constitutively active human Bax isoform that is under tight regulatory control by the proteasomal degradation mechanism. Mol Cell. 2009;33:15–29. doi: 10.1016/j.molcel.2008.11.025. [DOI] [PubMed] [Google Scholar]
- Gavathiotis E, Suzuki M, Davis ML, Pitter K, Bird GH, Katz SG, Tu HC, Kim H, Cheng EH, Tjandra N, Walensky LD. BAX activation is initiated at a novel interaction site. Nature. 2008;455:1076–1081. doi: 10.1038/nature07396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George NM, Evans JJ, Luo X. A three-helix homo-oligomerization domain containing BH3 and BH1 is responsible for the apoptotic activity of Bax. Genes Dev. 2007;21:1937–1948. doi: 10.1101/gad.1553607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George NM, Targy N, Evans JJ, Zhang L, Luo X. Bax contains two functional mitochondrial targeting sequences and translocates to mitochondria in a conformational change- and homo-oligomerization-driven process. J Biol Chem. 2009;285:1384–1392. doi: 10.1074/jbc.M109.049924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giam M, Huang DC, Bouillet P. BH3-only proteins and their roles in programmed cell death. Oncogene. 2008;27 1:S128–136. doi: 10.1038/onc.2009.50. [DOI] [PubMed] [Google Scholar]
- Goping IS, Gross A, Lavoie JN, Nguyen M, Jemmerson R, Roth K, Korsmeyer SJ, Shore GC. Regulated targeting of BAX to mitochondria. J Cell Biol. 1998;143:207–215. doi: 10.1083/jcb.143.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas W, Faherty BK, Gerber SA, Elias JE, Beausoleil SA, Bakalarski CE, Li X, Villen J, Gygi SP. Optimization and use of peptide mass measurement accuracy in shotgun proteomics. Mol Cell Proteomics. 2006;5:1326–1337. doi: 10.1074/mcp.M500339-MCP200. [DOI] [PubMed] [Google Scholar]
- Harada H, Quearry B, Ruiz-Vela A, Korsmeyer SJ. Survival factor-induced extracellular signal-regulated kinase phosphorylates BIM, inhibiting its association with BAX and proapoptotic activity. Proc Natl Acad Sci U S A. 2004;101:15313–15317. doi: 10.1073/pnas.0406837101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinds MG, Smits C, Fredericks-Short R, Risk JM, Bailey M, Huang DC, Day CL. Bim, Bad and Bmf: intrinsically unstructured BH3-only proteins that undergo a localized conformational change upon binding to prosurvival Bcl-2 targets. Cell Death Differ. 2007;14:128–136. doi: 10.1038/sj.cdd.4401934. [DOI] [PubMed] [Google Scholar]
- Hooft RW, Vriend G, Sander C, Abola EE. Errors in protein structures. Nature. 1996;381:272. doi: 10.1038/381272a0. [DOI] [PubMed] [Google Scholar]
- Hsu YT, Youle RJ. Nonionic detergents induce dimerization among members of the Bcl-2 family. J Biol Chem. 1997;272:13829–13834. doi: 10.1074/jbc.272.21.13829. [DOI] [PubMed] [Google Scholar]
- Johnson BA. Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol Biol. 2004;278:313–352. doi: 10.1385/1-59259-809-9:313. [DOI] [PubMed] [Google Scholar]
- Khaled AR, Kim K, Hofmeister R, Muegge K, Durum SK. Withdrawal of IL-7 induces Bax translocation from cytosol to mitochondria through a rise in intracellular pH. Proc Natl Acad Sci U S A. 1999;96:14476–14481. doi: 10.1073/pnas.96.25.14476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Rafiuddin-Shah M, Tu HC, Jeffers JR, Zambetti GP, Hsieh JJ, Cheng EH. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell Biol. 2006;8:1348–1358. doi: 10.1038/ncb1499. [DOI] [PubMed] [Google Scholar]
- Kim H, Tu HC, Ren D, Takeuchi O, Jeffers JR, Zambetti GP, Hsieh JJD, Cheng EHY. Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol Cell. 2009;36:487–499. doi: 10.1016/j.molcel.2009.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsmeyer SJ, Shutter JR, Veis DJ, Merry DE, Oltvai ZN. Bcl-2/Bax: a rheostat that regulates an anti-oxidant pathway and cell death. Semin Cancer Biol. 1993;4:327–332. [PubMed] [Google Scholar]
- Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR, Newmeyer DD. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell. 2005;17:525–535. doi: 10.1016/j.molcel.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR, Newmeyer DD. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111:331–342. doi: 10.1016/s0092-8674(02)01036-x. [DOI] [PubMed] [Google Scholar]
- Lee EF, Fedorova A, Zobel K, Boyle MJ, Yang H, Perugini MA, Colman PM, Huang DC, Deshayes K, Fairlie WD. Novel Bcl-2 homology-3 domain-like sequences identified from screening randomized peptide libraries for inhibitors of the pro-survival Bcl-2 proteins. J Biol Chem. 2009;284:31315–31326. doi: 10.1074/jbc.M109.048009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2:183–192. doi: 10.1016/s1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- Lovell JF, Billen LP, Bindner S, Shamas-Din A, Fradin C, Leber B, Andrews DW. Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell. 2008;135:1074–1084. doi: 10.1016/j.cell.2008.11.010. [DOI] [PubMed] [Google Scholar]
- Lucken-Ardjomande S, Montessuit S, Martinou JC. Contributions to Bax insertion and oligomerization of lipids of the mitochondrial outer membrane. Cell Death Differ. 2008;15:929–937. doi: 10.1038/cdd.2008.9. [DOI] [PubMed] [Google Scholar]
- Marintchev A, Frueh D, Wagner G. NMR methods for studying protein-protein interactions involved in translation initiation. Methods Enzymol. 2007;430:283–331. doi: 10.1016/S0076-6879(07)30012-8. [DOI] [PubMed] [Google Scholar]
- McDonnell JM, Fushman D, Milliman CL, Korsmeyer SJ, Cowburn D. Solution structure of the proapoptotic molecule BID: a structural basis for apoptotic agonists and antagonists. Cell. 1999;96:625–634. doi: 10.1016/s0092-8674(00)80573-5. [DOI] [PubMed] [Google Scholar]
- Meijerink JP, Mensink EJ, Wang K, Sedlak TW, Sloetjes AW, de Witte T, Waksman G, Korsmeyer SJ. Hematopoietic malignancies demonstrate loss-of-function mutations of BAX. Blood. 1998;91:2991–2997. [PubMed] [Google Scholar]
- Merino D, Giam M, Hughes PD, Siggs OM, Heger K, O'Reilly LA, Adams JM, Strasser A, Lee EF, Fairlie WD, Bouillet P. The role of BH3-only protein Bim extends beyond inhibiting Bcl-2-like prosurvival proteins. J Cell Biol. 2009;186:355–362. doi: 10.1083/jcb.200905153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldoveanu T, Liu Q, Tocilj A, Watson M, Shore G, Gehring K. The X-ray structure of a BAK homodimer reveals an inhibitory zinc binding site. Mol Cell. 2006;24:677–688. doi: 10.1016/j.molcel.2006.10.014. [DOI] [PubMed] [Google Scholar]
- Nechushtan A, Smith CL, Hsu YT, Youle RJ. Conformation of the Bax C-terminus regulates subcellular location and cell death. Embo J. 1999;18:2330–2341. doi: 10.1093/emboj/18.9.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- Pagliari LJ, Kuwana T, Bonzon C, Newmeyer DD, Tu S, Beere HM, Green DR. The multidomain proapoptotic molecules Bax and Bak are directly activated by heat. Proc Natl Acad Sci U S A. 2005;102:17975–17980. doi: 10.1073/pnas.0506712102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez D, White E. TNF-alpha signals apoptosis through a bid-dependent conformational change in Bax that is inhibited by E1B 19K. Mol Cell. 2000;6:53–63. [PubMed] [Google Scholar]
- Petros AM, Medek A, Nettesheim DG, Kim DH, Yoon HS, Swift K, Matayoshi ED, Oltersdorf T, Fesik SW. Solution structure of the antiapoptotic protein bcl-2. Proc Natl Acad Sci U S A. 2001;98:3012–3017. doi: 10.1073/pnas.041619798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitter K, Bernal F, LaBelle JL, Walensky LD. Chapter 23 Dissection of the BCL-2 Family Signaling Network with Stabilized alpha-Helices of BCL-2 Domains. Methods Enzymol. 2008;446:387–408. doi: 10.1016/S0076-6879(08)01623-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler M, Liang H, Nettesheim D, Meadows RP, Harlan JE, Eberstadt M, Yoon HS, Shuker SB, Chang BS, Minn AJ, et al. Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science. 1997;275:983–986. doi: 10.1126/science.275.5302.983. [DOI] [PubMed] [Google Scholar]
- Schinzel A, Kaufmann T, Schuler M, Martinalbo J, Grubb D, Borner C. Conformational control of Bax localization and apoptotic activity by Pro168. J Cell Biol. 2004;164:1021–1032. doi: 10.1083/jcb.200309013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlesinger PH, Saito M. The Bax pore in liposomes, Biophysics. Cell Death Differ. 2006;13:1403–1408. doi: 10.1038/sj.cdd.4401991. [DOI] [PubMed] [Google Scholar]
- Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Youle RJ, Tjandra N. Structure of Bax: coregulation of dimer formation and intracellular localization. Cell. 2000;103:645–654. doi: 10.1016/s0092-8674(00)00167-7. [DOI] [PubMed] [Google Scholar]
- Tan C, Dlugosz PJ, Peng J, Zhang Z, Lapolla SM, Plafker SM, Andrews DW, Lin J. Auto-activation of the apoptosis protein Bax increases mitochondrial membrane permeability and is inhibited by Bcl-2. J Biol Chem. 2006;281:14764–14775. doi: 10.1074/jbc.M602374200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uren RT, Dewson G, Chen L, Coyne SC, Huang DC, Adams JM, Kluck RM. Mitochondrial permeabilization relies on BH3 ligands engaging multiple prosurvival Bcl-2 relatives, not Bak. J Cell Biol. 2007;177:277–287. doi: 10.1083/jcb.200606065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, Wagner G, Verdine GL, Korsmeyer SJ. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science. 2004;305:1466–1470. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walensky LD, Pitter K, Morash J, Oh KJ, Barbuto S, Fisher J, Smith E, Verdine GL, Korsmeyer SJ. A stapled BID BH3 helix directly binds and activates BAX. Mol Cell. 2006;24:199–210. doi: 10.1016/j.molcel.2006.08.020. [DOI] [PubMed] [Google Scholar]
- Wang K, Gross A, Waksman G, Korsmeyer SJ. Mutagenesis of the BH3 domain of BAX identifies residues critical for dimerization and killing. Mol Cell Biol. 1998;18:6083–6089. doi: 10.1128/mcb.18.10.6083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Yin XM, Chao DT, Milliman CL, Korsmeyer SJ. BID: a novel BH3 domain-only death agonist. Genes Dev. 1996;10:2859–2869. doi: 10.1101/gad.10.22.2859. [DOI] [PubMed] [Google Scholar]
- Wei MC, Lindsten T, Mootha VK, Weiler S, Gross A, Ashiya M, Thompson CB, Korsmeyer SJ. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev. 2000;14:2060–2071. [PMC free article] [PubMed] [Google Scholar]
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, Adams JM, Huang DC. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005;19:1294–1305. doi: 10.1101/gad.1304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis SN, Fletcher JI, Kaufmann T, van Delft MF, Chen L, Czabotar PE, Ierino H, Lee EF, Fairlie WD, Bouillet P, et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science. 2007;315:856–859. doi: 10.1126/science.1133289. [DOI] [PubMed] [Google Scholar]
- Yin XM, Oltvai ZN, Korsmeyer SJ. BH1 and BH2 domains of Bcl-2 are required for inhibition of apoptosis and heterodimerization with Bax. Nature. 1994;369:321–323. doi: 10.1038/369321a0. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Zhu W, Lapolla SM, Miao Y, Shao Y, Falcone M, Boreham D, McFarlane N, Ding J, Johnson AE, et al. Bax forms an oligomer via separate, yet interdependent, surfaces. J Biol Chem. 2010;285:17614–17627. doi: 10.1074/jbc.M110.113456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Hou Q, Hansen JL, Hsu YT. Complete activation of Bax by a single site mutation. Oncogene. 2007;26:7092–7102. doi: 10.1038/sj.onc.1210517. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.