

Summary
Background and objective
A growing body of experimental evidences supports broad inhibitory and regulatory activity of plasminogen activator inhibitor 1 (PAI-1). The present study was designed to investigate whether PAI-1 inhibits FVIIa complexed with tissue factor (TF), a well known procoagulant risk factor.
Methods and results
The ability of PAI-1 to inhibit FVIIa-TF activity was evaluated in both clotting and factor X (FX) activation assays. PAI-1 and its complex with vitronectin inhibit (i) clotting activity of FVIIa-TF (PAI-1IC50, 817 and 125 nM, respectively), (ii) FVIIa-TF-mediated FX activation (PAI-1IC50, 260 and 50 nM, respectively), and (iii) FVIIa bound to TF expressed on the surface of stimulated endothelial cells (PAI-1IC50, 260 and 120 nM, respectively). The association rate constant (ka) for PAI-1 inhibition of FVIIa-TF was determined using a chromogenic assay. Ka for PAI-1 inhibition of FVIIa bound to relipidated TF is 3.3-fold higher than that for FVIIa bound to soluble TF (ka= 0.09 ± 0.01 and 0.027 ± 0.03 μM-1 min-1, respectively). Vitronectin increases ka for both soluble and relipidated TF by 3.5- and 30-fold, respectively (to 0.094 ± 0.020 and 2.7 ± 0.2 μM-1 min-1). However, only a 3.5-5.0- fold increase in the acylated FVIIa was observed on SDS PAGE in the presence of vitronectin for both relipidated and soluble TF, indicating fast formation of PAI-1/vitronectin/FVIIa/relipidated TF noncovalent complex.
Conclusions
Our results demonstrate potential anticoagulant activity of PAI-1 in the presence of vitronectin, which could contribute to regulation of hemostasis under pathological conditions such as severe sepsis, acute lung injury and pleural injury, where PAI-1 and TF are overexpressed.
Keywords: factor VIIa, plasminogen activator inhibitor-1, tissue factor
Introduction
The tissue factor (TF) pathway of coagulation plays a primary role in hemostasis, however the aberrant activation of TF-mediated coagulation leads to thrombus formation, the precipitating event in acute myocardial infarction, unstable angina and ischemic stroke [1]. TF-mediated coagulation also contributes to the pathogenesis of acute lung injury by promoting extravascular fibrin deposition [2]. Furthermore, TF initiates intrapleural fibrin deposition, which promotes pleural organization and loculation in a wide variety of fibrosing pleural diseases [3]. Thus, proper regulation of the TF pathway coagulation is critical for maintenance of hemostatic balance. TF-dependent coagulation is regulated primarily by tissue factor pathway inhibitor (TFPI) [4;5]. Antithrombin (AT) may function as an auxiliary physiological regulator of TF-mediated coagulation [6].
Plasminogen activator inhibitor-1 (PAI-1) is an important component of the plasminogen/plasmin system, as it is the primary inhibitor of tissue-type and urokinase-type plasminogen activator [7]. PAI-1 is also known to inhibit other plasma serine proteases, such as thrombin, factor XIIa [8;9] and activated protein C [10]. The presence of heparin and/or vitronectin markedly accelerates PAI-1-mediated inhibition of the above proteases [10;11]. It is well established that the abnormal expression of PAI-1 contributes to pathogenesis of many diseases, including lung injury, cardiovascular diseases and cancer [7]. Although PAI-1 circulates in plasma of healthy subjects in low quantities (6-80 ng/ml [12]), its concentration is elevated by 100 to 2000-fold in pathologic conditions [13;14]. The concentration of PAI-1 in plasma is increased approximately 100-fold in sepsis [13]. PAI-1 levels were shown to increase >2000 times that of normal plasma concentrations in coronary artery thrombi that formed in response to acute vascular injury [14].
Expression of both PAI-1 and TF are concurrently elevated in a number of diseases, including atherosclerosis and acute respiratory distress syndrome (ARDS) [14-17]. The resultant enhanced coagulation and depressed fibrinolysis is thought to predispose to alveolar fibrin deposition in acute lung injury, sepsis [3;18;19] and coronary thrombi in atherosclerosis [20;21]. However, it is also possible that increased expression of PAI-1 in vascular injury could be protective, particularly if it down-regulates TF-mediated coagulation. At present, there is no information as to whether PAI-1 can inhibit FVIIa or FVIIa bound to TF. In this study, we therefore investigated the ability of PAI-1 to modulate TF-FVIIa activity in vitro.
Material and Methods
Reagents
Recombinant human FVIIa (NovoSeven) was obtained from Novo Nordisk (Denmark). Human wild-type recombinant PAI-1 (PAI-1) was prepared and purified as described earlier [22]. SDS-PAGE analysis showed that PAI-1 was apparently homogenous. Analysis of PAI-1, by allowing it to react with one of its target proteases (tPA or UPA) and measuring the complex formation between PAI-1 and target proteinase by SDA-PAGE analysis, revealed that about 80 to 90% of PAI-1 is in active form (formed SDS-PAGE stable covalent complex with the target protease). Commercial rPAI-1 was obtained from (Molecular Innovations, MI). No significant differences were found between these two PAI-1 preparations on their ability to inhibit FVIIa-TF activity in various assays performed in the present study. Recombinant full-length TF was reconstituted into mixed phosphatidylcholine/phosphatidylserine (PC/PS) (80:20 w/w) vesicles as described previously [23]. Purified human plasma factor X and factor Xa were obtained from Enzyme Research Laboratories (South Bend, IN, USA). Antithrombin was from Kabi Pharmacia (Franklin, OH, USA). Vitronectin was obtained from either Promega (Madison, WI) or Molecular Innovations. Heparin was from American Pharmaceutical Partners (Schaumberg, IL). Factor VII-deficient plasma was obtained from George King Bio-Medical Inc (Overland Park, KS).
Cell Culture
Primary human umbilical vein endothelial cells (HUVEC) were purchased from Lonza (Walkersville, MD). Monolayers of HUVEC were grown to confluence at 37°C and 5% CO2 in a humidified incubator in EBM-2 basal media supplemented with 5% fetal bovine serum and growth supplements (Lonza). Endothelial cell passages between 3 and 8 were used in the present study. Human fibroblast cell line WI-38 was obtained from ATCC. Fibroblasts were cultured until confluence in Dulbecco's modified Eagle medium (Invitrogen, Carlsbad, CA) supplemented with 1% penicillin/streptomycin and 10% fetal bovine serum as described previously [24].
FVIIa clotting assay
FVIIa activity was measured in FVIIa specific clotting assay as described earlier [25] with minor modifications. Briefly, 50 μl of appropriately diluted aliquot from the reaction mixture was added to pre-warmed (3 min at 37°C) mixture of FVII-deficient plasma (100 μl) and soluble TF (100 μl of 100 nM, diluted in 2× rabbit brain cephalin, Sigma, St. Louis, MO). Clotting was initiated by the addition of 50 μl of CaCl2 (25 mM) warmed to 37°C and clot times were measured using COAG-MATE Xm coagulizer (Organon Technika, Durham, NC). Known concentrations of FVIIa were used to generate a standard reference curve.
FVIIa-TF activation of factor X
FVIIa (10 nM) complexed with relipidated TF (50 nM) in TBS (50 mM Tris-HCl, 0.15 M NaCl, pH 7.5) containing 1 mg/ml BSA and 5 mM CaCl2, was incubated with varying concentrations of PAI-1 (0 to 1.0 μM) for 1 h or with a fixed concentration of PAI-1 (1 μM) for varying time intervals (0 to 60 min) in the presence or absence of cofactors, heparin (10 U/ml) or vitronectin (1.0 μM) at 37°C. The reaction was stopped by removing an aliquot into TBS/BSA buffer containing EDTA (2 mM). FVIIa-TF activity in the sample, after diluting it appropriately, was determined in FX activation assay by adding FX (175 nM) and CaCl2 (5 mM) after which the amount of FXa generated was measured in a chromogenic assay using chromogenic substrate (Chromogenix S2765; diaPharma, West Chester, PA, USA) as previously described [26].
Measurement of FVIIa-TF activity on cell surfaces
Confluent monolayers of HUVEC were stimulated with TNFα (20 ng/ml) and IL1β (20 ng/ml) for 6 h. The cells were washed twice with buffer A (10 mM HEPES, 0.15 M NaCl, 4 mM KCl, 11 mM glucose, pH 7.5) and incubated with 10 nM of FVIIa for 30 min at 37°C in buffer B (buffer A containing 1 mg/ml BSA, and 5 mM CaCl2). Unbound FVIIa was removed, and the cells were washed twice with buffer B before exposing them to varying concentrations of PAI-1 ± vitronectin for 1 h. After 1 h of incubation, the cells were washed with buffer B to remove the inhibitor and cell surface FVIIa-TF activity was then determined by adding FX (175 nM) and measuring the amount of FXa generated in a chromogenic assay.
Measurements of second-order association rate constants for the reaction between PAI-1 and FVIIa
The values of the association rate constants (kinh) for inhibition of FVIIa-TF by PAI-1 were determined by tracing changes in absorbance at 405 nm (A405) for the reaction of FVIIa-TF (2.5 nM FVIIa and 5 nM relipidated TF or 25 nM sTF) with PAI-1 (0 -1.5μM) or PAI-1/vitronectin (equimolar concentrations of PAI-1 and vitronectin) complex in the presence of 0.5 mM of chromogenic tPA substrate (Centerchem, Inc.; Norwalk, CT). Changes in the A405 were measured using SpectraMax 96-well optical absorbance plate reader (Molecular Devices, Sunnyvale, CA). A single exponential equation (At = A∞ + A e-(kobs)t; where At is the A405 at time t, A∞ is the final A405; A and kobs, are amplitude and pseudo-first-order rate constants, respectively), for the changes in A405 was fit to the traced changes in absorbance using SigmaPlot 11.0 for Windows (SPSS Inc.). Values of the second order association rate constants for inhibition of FVIIa-TF by PAI-1 ± vitronectin were calculated from the slopes of the linear dependence of kobs on [serpin]/(1 + [S]/Km) (where Km is the Michaelis constant for the reaction between FVIIa (FVIIa-TF) and chromogenic substrate S using SigmaPlot.
Measurements of endpoint assay for the reaction between PAI-1 and FVIIa
FVIIa (10 nM) complexed with soluble TF (50 nM) was incubated with 0.1 or 0.2 μM PAI-1 or PAI-1/vitronectin complex at 37°C in 0.05M HEPES buffer, pH 7.4. Aliquots were withdrawn at different time points (0, 5, 10, 20 and 60 min). The reaction was stopped by 4-fold dilution of an aliquot with cold buffer in a 96 well plate on ice. Forty μl aliquots were withdrawn and diluted into 100 μl of the Hepes buffer containing 0.5 mM (final concentration) of chromogenic tPA substrate. Residual FVIIa amidolytic activity was measured as described above. In a control reaction, FVIIa-TF complex was incubated in the buffer without PAI-1 and analyzed in a similar manner. A logarithm of the residual FVIIa activity, which was calculated from slopes of dependences of A405 on time for controls and experiments, was plotted against time. A linear equation was fitted to the data.
Detection of PAI-1-FVIIa complexes
FVIIa (10 nM) complexed with relipidated TF (20 nM) was incubated with PAI-1 (1 μM) ± vitronectin (1 μM) or heparin (10 U/ml). At varying times, an aliquot was removed from the reaction mixture and added to SDS-PAGE sample buffer. The samples were subjected to SDS-PAGE and then transferred to PVDF membrane. The blots were probed with polyclonal anti-FVIIa antibody followed by horse radish-peroxidase-conjugated anti-rabbit IgG. The blots were developed by chemiluminescence. To evaluate the formation of FVIIa/PAI-1 complexes in a physiological fluid, 125I-FVIIa (10 nM) was added to a pleural fluid obtained from a patient with empyema (pre-existing specimen) spiked with PAI-1 (1 μM) and/or relipidated TF (20 nM). At the end of 60 min, SDS-PAGE sample buffer was added to the reaction mixture and the samples were subjected to SDS-PAGE, followed by autoradiography.
Results
PAI-1 Inhibition of FVIIa and FVIIa-TF coagulant activity
A clotting assay was employed to investigate whether or not PAI-1 and its complexes with heparin or vitronectin affect coagulation in plasma. FVIIa or FVIIa-TF complexes were incubated with PAI-1 (1 μM) for varying time periods in the absence or presence of cofactors, heparin and vitronectin. FVIIa activity remaining in the reaction mixtures at different intervals was measured in a FVIIa-specific clotting assay. PAI-1 alone or in the presence of heparin had no significant inhibitory effect on free FVIIa activity. In the presence of vitronectin, PAI-1 inhibited free FVIIa activity, albeit very slowly (∼15% inhibition in 2 h) (Fig. 1A). By contrast, PAI-1 readily inhibited FVIIa coagulant activity when FVIIa was in complex with TF (Fig. 1B). In the absence of any cofactor, PAI-1 inhibited 50% of FVIIa-TF activity within 85 min. Heparin increased the rate of PAI-1 inactivation of FVIIa-TF approximately 2-fold, whereas vitronectin enhanced the PAI-1 inactivation of FVIIa-TF by about 4 to 5-fold, inhibiting 50% of the FVIIa coagulant activity in less than 20 min (Fig. 1B). Heparin or vitronectin alone had no inhibitory effect on FVIIa-TF coagulant activity.
Fig. 1.

Time-dependent inhibition of free FVIIa or FVIIa complexed with relipidated TF by PAI-1. FVIIa (10 nM) (panel A) or FVIIa-TF complexes (FVIIa, 10 nM; relipidated TF, 50 nM) (panel B) were incubated with control buffer (□), heparin (10 U/ml) (△), vitronectin (1 μM) (inverted △), PAI-1 (1 μM) (○), PAI-1 (1 μM) + heparin (10 U/ml (▲), or PAI-1 (1 μM) + vitronectin (1 μM) (●). At different time intervals (0-120 min), aliquots were removed from the reaction mixtures and diluted in TBS/BSA buffer containing 2 mM EDTA. FVIIa coagulant activity was measured in the FVIIa specific clotting assay. FVIIa activity measured at 0 min time point was taken as 100%. Data represent the mean ± SEM (n =3).
Next, to compare the rate of PAI-1 inhibition of FVIIa-TF with a known serpin inhibitor of FVIIa-TF, i.e., antithrombin (AT), we examined the effect of equal concentrations of AT and PAI-1 on the inhibition of FVIIa-TF coagulant activity. In the absence of any cofactors, PAI-1 inhibited FVIIa-TF activity at a faster rate than AT. The time required for 50% inactivation of FVIIa-TF activity by PAI-1 was approximately 85 min. In this time frame, AT inhibited FVIIa-TF activity minimally (∼15%) as reported earlier [27] (Fig. 2A). In the presence of heparin, AT inhibited FVIIa-TF activity 25-times faster than PAI-1 (t50% inh. 2 min vs. 47 min). PAI-1 inhibited FVIIa-TF at a faster rate in the presence of vitronectin compared to incubation with heparin, yet this was 10 times slower than that observed with AT/heparin (Fig. 2B). To compare the efficiency of PAI-1 and PAI-1/vitronectin with AT/heparin inhibition of FVIIa-TF activity, we determined the loss of FVIIa coagulant activity in the presence of varying concentrations of PAI-1 ± vitronectin (1 μM) or AT ± heparin (10 U/ml). The IC50 values were the following: AT, > 5 μM; PAI-1, 817 nM; AT/heparin, 25 nM; PAI-1/vitronectin, 125 nM (Fig. 2C). Overall, these data illustrate that in the absence of any cofactors, PAI-1 is a more effective inhibitor than AT in inhibiting FVIIa-TF activity. However, in the presence of heparin, AT is a much more efficient inhibitor of FVIIa-TF activity compared to PAI-1.
Fig. 2.

A comparison between AT and PAI-1 inhibition of TF-FVIIa. FVIIa (10 nM) and relipidated TF (50 nM) were incubated with AT or PAI-1 (1 μM) (panels A and B) in the absence of their cofactors (panel A) or presence of the cofactors, heparin (10 U/ml) or vitronectin (1 μM) (panel B). At varying time intervals, an aliquot was removed from the reaction mixture and residual FVIIa activity was determined as described in Fig. 1. (C) FVIIa (10 nM) and relipidated TF (50 nM) were incubated with varying concentrations of AT or PAI-1 (0 to 1 μM) in the absence or presence of heparin (10 U/ml) or vitronectin (1 μM) for 1 h and the residual FVIIa coagulant activity was determined as described in Materials and Methods. Symbols denote, buffer, (□); PAI-1, (○); AT, (◇) in panel A; buffer, (□); AT, (◇); AT + heparin (◆); PAI-1 + heparin, (▲); PAI-1 + vitronectin, (●) in panels B and C. FVIIa activity measured at 0 min or with no inhibitor was taken as 100%. Data shown in the figure represent mean ± SEM (n=3).
We next investigated the ability of PAI-1 to inhibit the proteolytic activity of FVIIa-TF in a factor X activation assay. Consistent with data obtained in clotting activity assays, PAI-1 inhibited FVIIa-TF catalyzed activation of FX in a dose-dependent manner (Fig. 3A). Heparin enhanced the inhibitory effect of PAI-1 by two-fold whereas vitronectin enhanced the inhibitory effect by 5-fold. The PAI-1IC50 values for inhibition of FVIIa-TF proteolytic activity were as follows: PAI-1, 260 nM; PAI-1/heparin, 100 nM; and PAI-1/vitronectin, 50 nM. Time-dependent studies of PAI-1 inhibition of FVIIa-TF activity studies confirmed that PAI-1 inhibited FVIIa-TF at a much faster rate in the presence of vitronectin (Fig. 3B).
Fig. 3.

Inhibition of FVIIa-TF proteolytic activity by PAI-1. (A) FVIIa (10 nM) and relipidated TF (50 nM) were incubated with varying concentrations of PAI-1 in the absence (○) or presence of vitronectin (1 μM) (●) or heparin (10 U/ml) (▲) for 1 h. At the end 1 h, residual FVIIa-TF proteolytic activity was measured in FX activation assay as described in Materials and Methods. (B) FVIIa (10 nM) and relipidated TF (50 nM) were incubated for varying times with PAI-1 (1 μM) in the absence (○) or presence of vitronectin (1 μM) (●) or heparin (10 U/ml) (▲) and the residual FVIIa-TF activity was measured in FX activation assay. Data shown in the figure represent the mean ± SEM (n = 3).
Kinetics of FVIIa/PAI-1 complex formation
We have used continuous chromogenic assay to determine second order rate constants for PAI-1 ± vitronectin inhibition of FVIIa in the presence of relipidated or soluble TF (Fig. 4). Vitronectin potentiated the inactivation of FVIIa bound to relipidated TF by 30 fold (Fig. 4 A, B, C and Table 1). In contrast, the increase in the kinh for the reaction of PAI-1 with FVIIa-soluble TF was only 3.5 fold (Fig. 4D, Table 1). The rate of inactivation of FVIIa complexed with relipidated TF by PAI-1 in the absence of vitronectin was also 3.3 fold higher than that for FVIIa-soluble TF and equal to kinh for the reaction of with the FVIIa-soluble TF with PAI-1/vitronectin (Table 1). The results of the end point assay of the effect of vitronectin on the reaction between PAI-1 and FVIIa-soluble TF complex (Fig. 4E) agreed with the data obtained with the continuous assay (4D).
Fig. 4.

Effects of vitronectin on inhibition of FVIIa-TF by PAI-1. Varying concentrations of PAI-1 alone (0 -1.0 μM) (A) or PAI-1 with equimolar concentration of vitronectin (0-0.11 μM) (B) were added to FVIIa (2.5 nM) complexed with relipidated TF (5 nM) in the presence of 0.5 mM chromogenic substrate. FVIIa amidolytic activity was detected via accumulation of 4-nitraniline as an increase in the A405. (C) The values of kobs were calculated by fitting a single exponential equation to the traces observed in panels A and B. The values of kobs were plotted versus an effective inhibitory concentration of PAI-1 or its complex with vitronectin ([PAI-1]/(1+ [S]/Km); where S is substrate, and Km is the Michaelis constant for hydrolysis of S by FVIIa-TF. Solid lines represent best fits of the linear equation. Open symbols depict PAI-1 and closed symbols denote PAI-1 + vitronectin. (D) Same as in panel C except that the values of kobs were calculated by fitting a single exponential equation to the traces observed for inhibition of FVIIa bound to soluble TF (not shown). (E) Inactivation of FVIIa (10 nM) complexed with soluble TF (50 nM) by 0.1 (○, ●) and 0.2 μM (△, ▲) PAI-1 with (filled symbols) or without (empty symbols) vitronectin measured by an end point assay as described under Material and Methods. Residual amidolytic activity of FVIIa was plotted against time in semilogarithmic scale. Solid lines represent the best fit of the linear equation to the data.
Table I. Values of kinha (μM-1 min-1) for the reaction between PAI-1 (PAI-1/vitronectin) and FVIIa complexed with soluble TF or relipidated TF.
| TF | PAI-1 (kinh, μM-1 min-1) | PAI-1/vitronectin (kinh, μM-1 min-1) |
|---|---|---|
| Soluble TF | 0.027 ± 0.003 | 0.094 ± 0.02 |
| Relipidated TF | 0.09 ± 0.01 | 2.70 ± 0.20 |
the values of kinh were calculated from the slopes of linear dependencies of kobs on concentration of PAI-1 (PAI-1/vitronectin) (Fig. 4C and 4D) as described under Materials and Methods.
To further investigate the effect of vitronectin on the reaction between PAI-1 and FVIIa-relipidated TF, the rate of accumulation of the acylated FVIIa, which is stable under SDS-PAGE conditions, was estimated. First, FVIIa, in the presence or absence of relipidated TF, was incubated with PAI-1 ± vitronectin. Serial subsamples removed from the reaction mixtures were subjected to SDS-PAGE followed by immunoblotting with anti-FVIIa antibodies. As shown in Fig. 5A, in the presence of TF a significant amount of FVIIa formed an inhibitory complex with PAI-1 with the molecular weight of about 100 kDa as early as the first time point (i.e. 5 min). The complex formation progressively increased with increasing incubation time. Vitronectin enhanced the rate of FVIIa/PAI-1 complex formation by approximately 3 to 5 fold, which agreed with the results of the clotting and FX activation assays (Fig. 1 and 2). At the end of a 30 min incubation period, most of the FVIIa was present as a FVIIa/PAI-1 complex (Fig. 5). While the rate of formation of the final inhibitory complex due to the reaction of PAI-1 with FVIIa-soluble TF was several times slower than that for relipidated TF, vitronectin potentiated it 4 to 5 times (data not shown). These results support the data observed in an amidolytic assay with soluble TF (Fig. 4D and 4E, Table 1). Also as expected (Fig. 1 and 2), in the absence of TF, no FVIIa-PAI-1 complexes were detected regardless of whether or not the reaction mixture contained vitronectin or heparin (Fig. 5A). Next, we examined whether PAI-1 forms a stable covalent complex with FVIIa in pleural fluid. As shown in Fig. 5B, 125I-FVIIa added to the pleural fluid spiked with TF and PAI-1 formed a covalent complex with PAI-1. In the absence of exogenously added TF, no FVIIa/PAI-1 complex was detected on SDS-PAGE analysis. We observed similar data when unlabeled FVIIa was added to the pleural fluid spiked with TF and PAI-1 and the complex formation was recognized by western blot analysis using either anti-FVII or anti-PAI-1 antibodies (data not shown).
Fig. 5.

FVIIa bound to TF forms complex with PAI-1. (A) FVIIa (10 nM) in the presence or absence of relipidated TF (20 nM) was incubated with PAI-1 (1 μM) in the presence or absence of vitronectin (1 μM) or heparin (10 U/ml) in buffer B. At varying time intervals, an aliquot was removed from the reaction mixture and added to SDS-PAGE sample buffer. The samples were subjected to SDS-PAGE and immunoblotted with anti-FVII antibodies. (B) Radiolabeled FVIIa (10nM) in the presence and absence of relipidated TF (20nM) was incubated with PAI-1 or control vehicle in pleural fluid for 60 min. Samples were subjected to SDS-PAGE and the gel was dried and exposed on X-ray film.
PAI-1 inhibition of FVIIa bound to cell surface TF
To investigate the effect of PAI-1 on the catalytic activity of FVIIa bound to TF expressed on cell surfaces, we employed two different cell model systems, human endothelial cells (HUVEC) and fibroblasts (WI-38 cells). Monolayers of HUVEC stimulated with TNFα and IL1β for 6 h to induce TF expression were incubated with FVIIa (10 nM) in Ca2+-containing buffer (buffer B) for 30 min at 37°C to allow FVIIa binding to TF at the cell surface. Thereafter, the cells were washed twice in buffer B to remove unbound FVIIa, after which the cells were incubated with varying concentrations of PAI-1 in presence or absence of vitronectin for 1 h. At the end of 1 h, the cells were washed to remove PAI-1, FX (10 μg/ml) was added to the cells and the amount of FXa generated was measured. As shown in Fig. 6A, PAI-1 inhibited cell surface TF bound FVIIa-catalyzed activation of FX in a dose-dependent manner with a half-maximal inhibition at 260 nM. Addition of vitronectin with PAI-1 decreased the half-maximal inhibition to 120 nM. PAI-1 also inhibited FVIIa bound to TF that is constitutively expressed on fibroblasts in a very similar fashion, but addition of vitronectin along with PAI-1 did not further enhance PAI-1 inhibition of FVIIa-TF formed on cell surfaces of fibroblasts (Fig. 6B).
Fig. 6.

Inhibition of FVIIa bound to cell surface TF by PAI-1. Monolayers of stimulated HUVEC (A) or WI-38 fibroblasts (B) were incubated with FVIIa (10 nM) for 30 min to allow FVIIa bound to cell surface TF. Thereafter, free FVIIa was removed and the monolayers were incubated with varying concentrations of PAI-1 in the absence (○) or presence of vitronectin (1 μM) (●) for 1 h. At the end of 1 h, the supernatant media was removed and FVIIa-TF activity remained at the cell surface was determined by adding FX (175 nM) and measuring the rate of FXa generation. The rate of FX activation observed in the absence of PAI-1, vitronectin was taken as 100%. Data represent the mean ± SEM (n= 4 wells).
Discussion
Blood coagulation is initiated following disruption of intact endothelial surfaces that exposes TF to FVIIa in flowing blood. The FVIIa-TF complex then activates FIX and FX to initiate the TF-dependent pathway of blood coagulation. TF-dependent blood coagulation plays a primary role in hemostasis after tissue injury. However, aberrant activation of TF-dependent coagulation contributes to the pathogenesis of diseases characterized by thrombotic events or extravascular fibrin deposition, including acute myocardial infarction and diverse forms of acute lung injury [20;28;29]. Therefore, the proper regulation of TF expression is critical not only for maintenance of hemostatic balance but for preservation of normal organ function and general health. TFPI is the major regulator of FVIIa-TF-induced coagulation [5;30]. Although AT also has been shown to inhibit TF-FVIIa activity, the regulation of pathophysiologic TF-dependent coagulation remains unclear at this time [31]. The data presented herein show that PAI-1, an essential inhibitor of the fibrinolytic system, also inhibits the activity of FVIIa-TF, the key initiator of coagulation.
In addition to inhibiting tPA and uPA, PAI-1 also inhibits other serine proteases involved in blood coagulation and fibrinolysis, such as thrombin, APC and plasmin, albeit with low efficiency [32-35]. In the presence of cofactors, either vitronectin and/or heparin, PAI-1 becomes an efficient inhibitor of thrombin [36-38] and APC [10]. However, these cofactors do not convert PAI-1 to a general serine protease inhibitor as no inhibition of FXa by PAI-1 was detected irrespective of the presence or absence of these cofactors [39]. Thus, the present observation that PAI-1 inhibits FVIIa and that vitronectin enhances this inhibition reflects specific inhibition of FVIIa by PAI-1 and not a generic inhibition of serine proteases by serpins. As observed earlier with AT inhibition of FVIIa [27;40], PAI-1 inhibits FVIIa complexed with TF but not free FVIIa. Earlier studies have suggested that TF induces allosteric modifications in FVIIa and stabilizes the active conformation of FVIIa [41]. Such stabilization may be necessary for PAI-1 to inactivate FVIIa. Earlier studies from our laboratory showed that AT-mediated inhibition of FVIIa-TF was strictly dependent on the presence of heparin [27]. In contrast to AT, PAI-1 inhibition of FVIIa-TF does not require the presence of heparin. The inhibition of FVIIa complexed with relipidated TF by PAI-1 is improved slightly by the presence of heparin.
The simplest mechanism-based inhibition of proteinase (E) by serpin (I) (Scheme 1) includes reversible formation of the Michaelis-like complex (EI) and final inhibitory complex (E-I). While both EI and E-I possess no enzymatic activity only acylated enzyme E-I is stable under conditions of SDS PAGE. Although vitronectin accelerated formation of FVIIa/PAI-1 complex in the presence of either soluble or relipidated TF, the corresponding rate of accumulation of E-I (Scheme 1) was always higher with relipidated TF. At present the precise reason for the substantial difference between PAI-1 inhibition of FVIIa bound to soluble TF and relipidated TF is unknown, but a more favorable conformational change in FVIIa bound to relipidated TF could be a possible reason. Vitronectin significantly enhanced kinh for the reaction of FVIIa bound to relipidated TF with PAI-1 (Table 1) most likely via increasing the affinity of PAI-1 to FVIIa complexed with relipidated TF (an increase in the rate of formation of EI (k1; Scheme I)). It is unlikely that liposomes are directly contributes to the cofactor effect of vitronectin since the addition of liposomes to soluble TF did not alter the observed enhancement of the FVIIa inhibition by vitronectin (data not shown). Therefore, overall, our data suggest that in the presence of vitronectin, PAI-1 rapidly forms a catalytically inactive Michaelis-like complex EI with FVIIa bound to relipidated TF, which slowly progresses to the final inhibitory complex E-I (Scheme 1). Such an inhibitory mechanism could explain observed differences in PAI-1 ± vitronectin inhibition of FVIIa-TF in different assays. The loss of FVIIa activity in amidolytic assay (Fig. 4A and B) reflects formation of both EI and E-I (Scheme 1). In contrast, the proteolytic activity assays, where sample are diluted many fold, and SDS-PAGE analysis measure the terminal stable inhibitor complex and not the intermediate. Earlier studies established that the active form of PAI-1 is spontaneously converted to inactive latent form with a half-life 0.5-1 h under physiological conditions and that vitronectin binding increases half life of active PAI-1 (see rev[42]). Vitronectin binding to PAI-1 was also shown to induce a conformational change in the reactive center of PAI-1, thus making it more accessible for the catalytic center of target protease [43]. However, given that vitronectin primarily affects affinity of PAI-1 to FVIIa bound to relipidated TF, it is likely that exosite interactions between the proteinase and serpin (in complex with vitronectin) outside of the active site govern the observed effects of vitronectin.
Scheme 1.
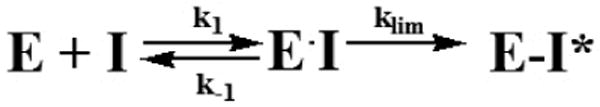
Studies performed using a stimulated endothelial and fibroblasts cell model system confirmed that PAI-1 is capable of inhibiting FVIIa bound to TF at the cell surface. However, in contrast to the data obtained with FVIIa bound to relipidated or soluble TF, vitronectin either had a modest (2-fold increase) or no effect on PAI-1 inhibition of FVIIa bound to cell surface TF. While it is unclear why vitronectin has a negligible cofactor effect on PAI-1 inhibition of FVIIa bound to cell surface TF, our results clearly indicate that the effects of vitronectin on serpin mechanism strongly depend on the microenvironment.
The biological significance of PAI-1 inhibition of FVIIa-TF at present is unknown. TFPI is the most relevant inhibitor of FVIIa-TF in vivo [4]. TFPI-FXa inhibits FVIIa-TF at about three orders of magnitude faster (0.6 to >1 × 109 M-1 min-1) [44-46] than PAI-1 inhibition of FVIIa-TF. Therefore, it is unlikely that PAI-1, which circulates in plasma at a similar or lower concentration (60 to 80 ng/ml) than TFPI (100 ng/ml), inhibits FVIIa-TF under normal physiological conditions. In general, the physiological relevance of interaction between serine proteases and serpins is determined by a number of criteria, including the (1) presence of the target protease, inhibitors and cofactors in the same compartment, (2) concentration of the reactants in such a compartment, and (3) rate of inhibition [47]. Expression of both PAI-1 and TF are concurrently elevated in a number of diseases, including atherosclerosis and acute respiratory distress syndrome (ARDS) [14-17]. TF, PAI-1, and vitronectin were also found to be localized in atherosclerotic human vessel wall [48;49]. Leakage of circulating blood through damaged vessel walls or local synthesis of FVII [50] in atheroma may lead to formation of FVIIa-TF complexes in atherosclerotic vessel wall [48;49]. Similarly, bronchoalveolar lavage and pleural fluids from ARDS and other patients are shown to contain PAI-1, TF and FVIIa [16;17;51]. In some disease settings, PAI-1 concentrations are elevated by 100 to 2000-fold, exceeding the concentration of TFPI by 2 to 3-orders of magnitude [13;14;16;52]. Under this scenario, despite PAI-1, compared to TFPI, being a poor inhibitor of FVIIa-TF, the relative efficiency of PAI-1 to TFPI for inhibition FVIIa-TF could reach approximately 0.1 to 1. Thus, the regulation of FVIIa-TF activity by PAI-1 could be a relevant mechanism in the context of atherogenesis, sepsis and diseases characterized by elevated expression of PAI-1. Here it may pertinent to note that PAI-1 may also regulate FVIIa-TF pathway indirectly as the end product of the fibrinolytic system, plasmin, can activate FVII [53].
In summary, the present study documents for the first time that PAI-1 is an inhibitor of FVIIa-TF. Although PAI-1 inhibits FVIIa-TF with low efficiency, PAI-1 inhibition of the FVIIa-TF pathway of coagulation may have pathophysiological significance in disease conditions such as ARDS or atherogenesis, which are characterized by aberrant fibrin formation and increased expression of both TF and PAI-1.
Acknowledgments
The study was supported in part by grants HL58869 (LVMR), HL65500 (UP) and PO-1 HL076406 (SI) from National Institutes of Health.
Abbreviations used are
- AT
antithrombin
- FVIIa
activated factor VII
- PAI-1
plasminogen activator inhibitor-1
- TF
tissue factor
Footnotes
Addendum
P. Sen performed the majority of experiments described in the manuscript. A.A. Komissarov and G. Florova performed the kinetic studies. P. Sen, A.A. Komissarov and G. Florova analyzed the data and prepared figures included in the manuscript. All authors contributed to the research design and preparation of the manuscript at various stages of the study. L.V.M. Rao designed the research, analyzed the data and wrote the manuscript.
Disclosure of conflicts of interests
The authors state they have no conflict of interest.
References
- 1.Semeraro N, Colucci M. Tissue factor in health and disease. Thromb Haemost. 1997;78:759–64. [PubMed] [Google Scholar]
- 2.Idell S. Coagulation, fibrinolysis, and fibrin deposition in acute lung injury. Crit Care Med. 2003;31:S213–S220. doi: 10.1097/01.CCM.0000057846.21303.AB. [DOI] [PubMed] [Google Scholar]
- 3.Mutsaers SE, Prele CM, Brody AR, Idell S. Pathogenesis of pleural fibrosis. Respirology. 2004;9:428–40. doi: 10.1111/j.1440-1843.2004.00633.x. [DOI] [PubMed] [Google Scholar]
- 4.Broze GJ. The role of tissue factor pathway inhibitor in a revised coagulation cascade. Semin Hematol. 1992;29:159–69. [PubMed] [Google Scholar]
- 5.Rapaport SI, Rao LVM. Initiation and regulation of tissue factor-dependent blood coagulation. Arterioscler Thromb. 1992;12:1111–21. doi: 10.1161/01.atv.12.10.1111. [DOI] [PubMed] [Google Scholar]
- 6.Rao LVM, Nordfang O, Hoang AD, Pendurthi UR. Mechanism of antithrombin III inhibition of VIIa/tissue factor activity on cell surfaces. Comparison with tissue factor pathway inhibitor/factor Xa induced inhibition of factor VIIa/tissue factor activity. Blood. 1995;85:121–9. [PubMed] [Google Scholar]
- 7.Gils A, Declerck PJ. Plasminogen activator inhibitor-1. Curr Med Chem. 2004;11:2323–34. doi: 10.2174/0929867043364595. [DOI] [PubMed] [Google Scholar]
- 8.Ehrlich HJ, Gebbink RK, Keiher J, Linders M, Preissner KT, Pannekoek H. Alternation of serpin specificity by a protein cofactor vitronectin endows plasminogen activator inhibitor 1 with thrombin inhibitory properties. J Biol Chem. 1990;265:13029–35. [PubMed] [Google Scholar]
- 9.Keijer J, Linders M, Wegman JJ, Ehrlich HJ, Mertens K, Pannekoek H. On the target specificity of plasminogen activator inhibitor 1: the role of heparin, vitronectin, and the reactive site. Blood. 1991;78:1254–61. [PubMed] [Google Scholar]
- 10.Rezaie AR. Vitronectin functions as a cofactor for rapid inhibition of activated protein C by plasminogen activator inhibitor-1. Implications for the mechanism of profibrinolytic action of activated protein C. J Biol Chem. 2001;276:15567–70. doi: 10.1074/jbc.C100123200. [DOI] [PubMed] [Google Scholar]
- 11.Huber K. Plasminogen activator inhibitor type-1 (part one): basic mechanisms, regulation, and role for thromboembolic disease. J Thromb Thrombolysis. 2001;11:183–93. doi: 10.1023/a:1011955018052. [DOI] [PubMed] [Google Scholar]
- 12.Zeerleder S, Schroeder V, Hack CE, Kohler HP, Wuillemin WA. TAFI and PAI-1 levels in human sepsis. Thromb Res. 2006;118:205–12. doi: 10.1016/j.thromres.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 13.Fay WP, Murphy JG, Owen WG. High concentrations of active plasminogen activator inhibitor-1 in porcine coronary artery thrombi. Arterioscler Thromb Vasc Biol. 1996;16:1277–84. doi: 10.1161/01.atv.16.10.1277. [DOI] [PubMed] [Google Scholar]
- 14.Thiruvikraman SV, Guha A, Roboz J, Taubman MB, Nemerson Y, Fallon JT. In situ localization of tissue factor in human atherosclerotic plaques by binding of digoxigenin-labeled factors VIIa and X. Lab Invest. 1996;75:451–61. [PubMed] [Google Scholar]
- 15.Idell S, Girard W, Koenig KB, Mclarty J, Fair DS. Abnormalities of pathways of fibrin turnover in the human pleural space. Am Rev Respir Dis. 1991;144:187–94. doi: 10.1164/ajrccm/144.1.187. [DOI] [PubMed] [Google Scholar]
- 16.Idell S, James KK, Levin EG, Schwartz BS, Manchanda N, Maunder RJ, Martin TR, Mclarty J, Fair DS. Local abnormalities in coagulation and fibrinolytic pathways predispose to alveolar fibrin deposition in the adult respiratory distress syndrome. J Clin Invest N Y. 1989;84:695–705. doi: 10.1172/JCI114217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vervloet MG, Thijs LG, Hack CE. Derangements of coagulation and fibrinolysis in critically ill patients with sepsis and septic shock. Semin Thromb Hemost. 1998;24:33–44. doi: 10.1055/s-2007-995821. [DOI] [PubMed] [Google Scholar]
- 18.Welty-wolf KE, Carraway MS, Ortel TL, Piantadosi CA. Coagulation and inflammation in acute lung injury. Thromb Haemost. 2002;88:17–25. [PubMed] [Google Scholar]
- 19.Taubman MB, Fallon JT, Schecter AD, Giesen P, Mendlowitz M, Fyfe BS, Marmur JD, Nemerson Y. Tissue factor in the pathogenesis of atherosclerosis. Thromb Haemost. 1997;78:200–4. [PubMed] [Google Scholar]
- 20.Vaughan DE. PAI-1 and atherothrombosis. J Thromb Haemost. 2005;3:1879–83. doi: 10.1111/j.1538-7836.2005.01420.x. [DOI] [PubMed] [Google Scholar]
- 21.Komissarov AA, Declerck PJ, Shore JD. Mechanisms of conversion of plasminogen activator inhibitor 1 from a suicide inhibitor to a substrate by monoclonal antibodies. J Biol Chem. 2002;277:43858–65. doi: 10.1074/jbc.M204110200. [DOI] [PubMed] [Google Scholar]
- 22.Rao LVM, Williams T, Rapaport SI. Studies of the activation of factor VII bound to tissue factor. Blood. 1996;87:3738–48. [PubMed] [Google Scholar]
- 23.Morrissey JH, Macik BG, Neuenschwander PF, Comp PC. Quantitation of activated factor VII levels in plasma using a tissue factor mutant selectively deficient in promoting factor VII activation. Blood. 1993;81:734–44. [PubMed] [Google Scholar]
- 24.Pendurthi UR, Williams JT, Rao LVM. Acidic and basic fibroblast growth factors suppress transcriptional activation of tissue factor and other inflammatory genes in endothelial cells. Arterioscler Thromb Vasc Biol. 1997;17:940–6. doi: 10.1161/01.atv.17.5.940. [DOI] [PubMed] [Google Scholar]
- 25.Rao LVM, Rapaport SI, Hoang AD. Binding of factor VIIa to tissue factor permits rapid antithrombin III/heparin inhibition of factor VIIa. Blood. 1993;81:2600–7. [PubMed] [Google Scholar]
- 26.Ardissino D, Merlini PA, Arlens R, Coppola R, Bramucci E, Lucreziotti S, Repetto A, Fetiveau R, Mannucci PM. Tissue factor in human coronary atherosclerotic plaques. Clinica Chimica Acta. 2000;291:235–40. doi: 10.1016/s0009-8981(99)00231-4. [DOI] [PubMed] [Google Scholar]
- 27.Marmur JD, Thiruvikraman SV, Fyfe BS, Guha A, Sharma SK, Ambrose JA, Fallon JT, Nemerson Y, Taubman MB. Identification of active tissue factor in human coronary atheroma. Circulation. 1996;94:1226–32. doi: 10.1161/01.cir.94.6.1226. [DOI] [PubMed] [Google Scholar]
- 28.Broze GJ., Jr Tissue factor pathway inhibitor and the current concept of blood coagulation. Blood Coagul Fibrinolysis. 1995;6:S7–S13. doi: 10.1097/00001721-199506001-00002. [DOI] [PubMed] [Google Scholar]
- 29.Rapaport SI, Rao LVM. The tissue factor pathway: How it has become a “prima ballerina”. Thromb Haemost. 1995;74:7–17. [PubMed] [Google Scholar]
- 30.Sakata Y, Curriden S, Lawrence D, Griffin JH, Loskutoff DJ. Activated Protein C Stimulates the Fibrinolytic Activity of Cultured Endothelial Cells and Decreases Antiactivator Activity. Proc Natl Acad Sci U S A. 1985;82:1121–2215. doi: 10.1073/pnas.82.4.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Fouw NJ, van Hinsbergh VW, de Jong YF, Haverkate F, Bertina RM. The interaction of activated protein C and thrombin with the plasminogen activator inhibitor released from human endothelial cells. Thromb Haemost. 1987;57:176–82. [PubMed] [Google Scholar]
- 32.Keijer J, Linders M, Wegman JJ, Ehrlich HJ, Mertens K, Pannekoek H. On the target specificity of plasminogen activator inhibitor 1: the role of heparin, vitronectin, and the reactive site. Blood. 1991;78:1254–61. [PubMed] [Google Scholar]
- 33.Naski MC, Lawrence DA, Mosher DF, Podor TJ, Ginsburg D. Kinetics of inactivation of a-thrombin by plasminogen activator inhibitor-1. J Biol Chem. 1993;268:12367–72. [PubMed] [Google Scholar]
- 34.Ehrlich HJ, Gebbink RK, Preissner KT, Keijer J, Esmon NL, Mertens K, Pannekoek H. Thrombin neutralizes plasminogen activator inhibitor 1 (PAI-1) that is complexed with vitronectin in the endothelial cell matrix. J Cell Biol. 1991;115:1773–81. doi: 10.1083/jcb.115.6.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ehrlich HJ, Keijer J, Preissner KT, Gebbink RK, Pannekoek H. Functional interaction of plasminogen activator inhibitor type 1 (PAI-1) and heparin. Biochem. 1991;30:1021–8. doi: 10.1021/bi00218a020. [DOI] [PubMed] [Google Scholar]
- 36.Keijer J, Linders M, Wegman JJ, Ehrlich HJ, Mertens K, Pannekoek H. On the target specificity of plasminogen activator inhibitor 1: the role of heparin, vitronectin, and the reactive site. Blood. 1991;78:1254–61. [PubMed] [Google Scholar]
- 37.Lawson JH, Butenas S, Ribarik N, Mann KG. Complex-dependent inhibition of factor VIIa by antithrombin III and heparin. J Biol Chem. 1993;268:767–70. [PubMed] [Google Scholar]
- 38.Olsen OH, Persson E. Cofactor-induced and mutational activity enhancement of coagulation factor VIIa. Cell Mol Life Sci. 2008;65:953–63. doi: 10.1007/s00018-007-7480-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dupont DM, Madsen JB, Kristensen T, Bodker JS, Blouse GE, Wind T, Andreasen PA. Biochemical properties of plasminogen activator inhibitor-1. Front Biosci. 2009;14:1337–61. doi: 10.2741/3312. [DOI] [PubMed] [Google Scholar]
- 40.Fa M, Karolin J, Aleshkov S, Strandberg L, Johansson LB, Ny T. Time-resolved polarized fluorescence spectroscopy studies of plasminogen activator inhibitor type 1: conformational changes of the reactive center upon interactions with target proteases, vitronectin and heparin. Biochem. 1995;34:13833–40. doi: 10.1021/bi00042a015. [DOI] [PubMed] [Google Scholar]
- 41.Jesty J, Wun TC, Lorenz A. Kinetics of the inhibition of factor Xa and the tissue factor-factor VIIa complex by the tissue factor pathway inhibitor in the presence and absence of heparin. Biochem. 1994;33:12686–94. doi: 10.1021/bi00208a020. [DOI] [PubMed] [Google Scholar]
- 42.Baugh RJ, Broze GJ, Jr, Krishnaswamy S. Regulation of extrinsic pathway factor Xa formation by tissue factor pathway inhibitor. J Biol Chem. 1998;273:4378–86. doi: 10.1074/jbc.273.8.4378. [DOI] [PubMed] [Google Scholar]
- 43.Salemink I, Franssen J, Willems GM, Hemker HC, Lindhout T. Inhibition of tissue factor-factor VIIa-catalyzed factor X activation by factor Xa-tissue factor pathway inhibitor. A rotating disc study on the effect of phospholipid membrane composition. J Biol Chem. 1999;274:28225–32. doi: 10.1074/jbc.274.40.28225. [DOI] [PubMed] [Google Scholar]
- 44.van Aken BE, Seiffert D, Thinnes T, Loskutoff DJ. Localization of vitronectin in the normal and atherosclerotic human vessel wall. Histochem Cell Biol. 1997;107:313–20. doi: 10.1007/s004180050116. [DOI] [PubMed] [Google Scholar]
- 45.Stoop AA, Lupu F, Pannekock H. Colocalization of Thrombin, PAI-1, and vitronectin in the artherosclerotic vessel wall. Arterioscler Thromb Vasc Biol. 2000;20:1143–9. doi: 10.1161/01.atv.20.4.1143. [DOI] [PubMed] [Google Scholar]
- 46.Wilcox JN, Noguchi S, Casanova JR, Rasmussen MR. Extrahepatic synthesis of FVII in human atheroma and smooth muscle cells in vitro. Annals New York Academy of Sciences. 2004:433–8. doi: 10.1111/j.1749-6632.2001.tb03980.x. [DOI] [PubMed] [Google Scholar]
- 47.Idell S, Gonzalez K, Bradford H, MacArthur CK, Fein AM, Maunder RJ, Garcia JGN, Griffith DE, Weiland J, Martin TR, Mclarty J, Fair DS, Walsh PN, Colman RW. Procoagulant Activity in Bronchoalveolar Lavage in the adult respiratory distress syndrome - contribution of tissue factor associated with factor VII. Am Rev Respir Dis. 1987;136:1466–74. doi: 10.1164/ajrccm/136.6.1466. [DOI] [PubMed] [Google Scholar]
- 48.Bajaj MS, Pendurthi U, Koenig K, Pueblitz S, Idell S. Tissue factor pathway inhibitor expression by human pleural mesothelial and mesothelioma cells. Eur Respir J. 2000;15:1069–78. doi: 10.1034/j.1399-3003.2000.01515.x. [DOI] [PubMed] [Google Scholar]
- 49.Laake K, Osterud B. Activation of purified plasma factor VII by human plasmin, plasma kallikrein, and activated components of the human intrinsic blood coagulation system. Thromb Res. 1974;5:759–72. doi: 10.1016/0049-3848(74)90119-4. [DOI] [PubMed] [Google Scholar]