

Abstract
Among the adult organs, liver is unique for its ability to regenerate. A concerted signaling cascade enables optimum initiation of the regeneration process following insults brought about by surgery or a toxicant. Additionally, there exists a cellular redundancy, whereby a transiently amplifying progenitor population appears and expands to ensure regeneration, when differentiated cells of the liver are unable to proliferate in both experimental and clinical scenarios. One such pathway of relevance in these phenomena is Wnt/β-catenin signaling, which is activated relatively early during regeneration mostly through post-translational modifications. Once activated, β-catenin signaling drives the expression of target genes that are critical for cell cycle progression and contribute to initiation of the regeneration process. The role and regulation of Wnt/β-catenin signaling is now documented in rats, mice, zebrafish and patients. More recently, a regenerative advantage of the livers in β-catenin overexpressing mice was reported, as was also the case after exogenous Wnt-1 delivery to the liver paving the way for assessing means to stimulate the pathway for therapeutics in liver failure. β-Catenin is also pertinent in hepatic oval cell activation and differentiation. However, aberrant activation of the Wnt/β-catenin signaling is reported in a significant subset of hepatocellular cancers (HCC). While many mechanisms of such activation have been reported, the most functional means of aberrant and sustained activation is through mutations in the β-catenin gene or in AXIN1/2, which encodes for a scaffolding protein critical for β-catenin degradation. Intriguingly, in experimental models hepatic overexpression of normal or mutant β-catenin is insufficient for tumorigenesis. In fact β-catenin loss promoted chemical carcinogenesis in the liver due to alternate mechanisms. Since most HCC occur in the backdrop of chronic hepatic injury, where hepatic regeneration is necessary for maintenance of liver function, but at the same time serves as the basis of dysplastic changes, this Promethean attribute exhibits a Jekyll and Hyde behavior that makes distinguishing good regeneration from bad regeneration essential for targeting selective molecular pathways as personalized medicine becomes a norm in clinical practice. Could β-catenin signaling be one such pathway that may be redundant in regeneration and indispensible in HCC in a subset of cases?
BACKGROUND
Because of its essential role in regulating developmental decisions as well as adult tissue homeostasis, the Wnt/β-catenin signaling pathway has been the subject of extensive research for the past two decades [1]. Characterization of this evolutionarily well-conserved pathway has shown that Wnt signaling is indispensible in processes as diverse as cell fate, development, proliferation, differentiation, growth, survival, regeneration, and self-renewal [2–6]. For example, Wnt/β-catenin signaling is ongoing in a subset of adult tissues like gut and skin where cell turnover is high.
Specifically, the Wnt pathway has also been shown to play many roles in liver pathobiology, despite the fact that it is mostly quiescent in an adult liver [7]. While it is critical for the highly dynamic environment of developing liver, where it regulates the processes of hepatoblast proliferation, survival and differentiation, the aberrant activation of this pathway has also been established in a subset of liver tumors such as hepatoblastoma and hepatocellular carcinoma (HCC). This pathway can also be reactivated in an adult liver under conditions of experimentally induced controlled growth, such as in liver regeneration after partial hepatectomy. Similarly, this pathway is also involved in instances of abnormal regeneration, as is observed in hepatic progenitor or oval cell–mediated repair following liver injury. Finally, because of its vital role in cell survival and proliferation, this pathway is of essence in cancers of many adult tissues where such processes are revitalized. The present review will discuss the roles of Wnt/β-catenin signaling during normal liver regeneration, progenitor-mediated hepatic repair and discuss aberrant activation of this signaling cascade in HCC. Finally, we will provide a prospective of modulation of this pathway in hepatic regenerative medicine and cancer biology.
WNT/BETA-CATENIN SIGNALING IN THE LIVER
Canonical Wnt Signaling
Wnt genes encode a large family of secreted glycoproteins that act as extracellular signaling molecules. Binding of Wnt proteins initiates a signaling cascade, which results in activation of β-catenin, the central player in the canonical Wnt pathway. However, in most normal unstimulated adult cells, where the Wnt/β-catenin pathway is inactive, this steady-state condition is ensured by the absence of Wnt protein and the degradation of β-catenin. Cytoplasmic β-catenin is bound in a complex with Axin, adenomatous polyposis coli (APC), casein kinase 1 (CK1) and glycogen synthase kinase 3β (GSK3β), which collectively comprise the destruction complex. β-catenin is phosphorylated by casein kinase 1 (CK1) and GSK-3β at specific serine/threonine residues located at the N-terminal region of the protein, which is facilitated by the scaffolding proteins Axin and APC [8]. This sequential phosphorylation targets β-catenin for ubiquitination and ultimate degradation by the proteasome.
Binding of Wnt proteins to the seven-pass transmembrane Frizzled (Fz) receptor on the surface of cells induces association with the low-density lipoprotein receptor related protein (LRP) 5/6. This coreceptor complex triggers activation of the canonical Wnt pathway. Dishevelled (Dvl) is recruited to the Frizzled receptor [9], and the Fz/Dvl complex in turn relocates Axin to LRP5/6 [10]. Axin-bound GSK-3β and CK1 then phosphorylate LRP5/6 [11, 12], which leads to inactivation of GSK-3β [13]. The absence of β-catenin phosphorylation releases it from the Axin/APC/GSK3 complex, resulting in accumulation of cytoplasmic and active β-catenin. Although β-catenin lacks a nuclear localization sequence, it then translocates to the nucleus through an unknown mechanism which may involve interaction with either components of the nuclear pore complex [14] or lymphoid enhancer factor or T-cell factor (LEF/TCF) [15], its nuclear binding partner. Once in the nucleus, β-catenin binds to LEF/TCF, displacing the transcriptional inhibitor Groucho, and in complex with TCF initiates transcription of target genes [4].
Canonical Wnt signaling in adult liver
Despite the expression of several Wnt and Fz genes, β-catenin is observed localizing mostly at the hepatocyte membrane in an adult liver [16]. However, it is present in the cytoplasm in the biliary epithelial and endothelial cells where its role remains unknown. β-Catenin has been implicated in biliary differentiation during development [17–19] and transgenic mice overexpressing β-catenin resolve intrahepatic cholestasis more efficiently than controls [20]. Similarly, the role of Wnt/β-catenin signaling in endothelial cell has been reported recently where Wnt2/Fz4/β-catenin axis seems to be important in sinusoidal endothelial cell proliferation [21]. Wnt/β-catenin signaling has also been identified in hepatic stellate cells, albeit the implications are conflicting. β-Catenin signaling has been implicated in the maintenance of stellate cell quiescence in one study [22]; conversely, β-catenin signaling has also been reported in stellate cell activation [23, 24]. It will be highly relevant to conclusively address the role of Wnt in stellate cell biology to harness its modulation as a means to inhibit hepatic fibrosis.
An important role of β-catenin signaling was identified in imparting zonation in the adult liver. Hepatocytes exhibit molecular heterogeneity based on their location within the hepatic lobule. Interactions between β-catenin, HNF4α and Ras/MAPK/ERK signaling now appear to dictate pericentral versus periportal gene expression and thus these interactions are key to successful hepatic zonation [22, 25–27]. While rarely visible by immunohistochemistry unless more rigorous antigen retrieval methods are applied, pericentral hepatocytes express cytoplasmic and nuclear β-catenin in addition to membranous localization. Here, it is known to regulate expression of genes that encode for enzymes critical in ammonia and xenobiotic metabolism. These include genes encoding for glutamine synthetase (GS), glutamate transporter (Glt1) and cytochrome P450 family members such as CYP2E1 and CYP1A2. Au contraire, the hepatocytes in the periportal zone show higher APC protein expression and as a consequence lower unbound, dephosphorylated β-catenin. These hepatocytes express genes encoding for ammonia metabolism such as arginase 1 and carbamoyl-phosphate synthase.
β-Catenin as a component of Adherens Junctions
In addition to being the key effector molecule in the canonical Wnt signaling pathway, β-catenin also plays a pivotal role maintaining cell-cell adhesion. β-catenin is an essential part of the adherens junctions in epithelial cells, forming a bridge between the actin cytoskeleton and E-cadherin [28–30]. The binding of β-catenin to E-cadherin is controlled by the phosphorylation of β-catenin at a specific tyrosine residue (Y654) [31]. Y654 phosphorylation causes inhibition of the β-catenin/E-cadherin interaction, leading to dissociation of the complex and subsequent degradation of E-cadherin [32]. This results in a disruption of adherens junctions and, in the case of hepatocytes, impairment in the apical trafficking of specific proteins [33]. Loss of cadherin function is an important component of many morphogenetic processes such as development and turnover of adult tissues, and may also contribute to motility and metastasis in the presence of aberrant growth [34].
β-Catenin as a mediator of tyrosine kinase signaling
Another important interaction involving β-catenin in hepatocytes is the association between β-catenin and several receptor tyrosine kinases. The effect of hepatocyte growth factor (HGF), a known mitogen, motogen, and morphogen for the liver [35], is mediated through its receptor, Met, a tyrosine kinase ubiquitously expressed in hepatocytes. Our laboratory has previously shown a subcellular association between Met and β-catenin on the inner surface of the hepatocyte membrane [36, 37]. Binding of HGF to its receptor induces phosphorylated Met to phosphorylate β-catenin at tyrosine residues 654 and 670, which results in its translocation to the nucleus and subsequent upregulation of target genes [36, 38]. In nonconfluent cells cultured in the presence of HGF, active β-catenin regulates cell morphogenesis by promoting migration and cell dedifferentiation [39]. Conversely, in matrigel-induced hepatocyte re-differentiation, Met and β-catenin association increases simultaneous with a decrease in the amount of nuclear and tyrosine-phosphorylated β-catenin [40]. In several human tumor cell lines, β-catenin has also been shown to be tyrosine phosphorylated upon stimulation with HGF [41]. β-Catenin contributes to HGF-induced hepatomegaly in mice through loss of Met/β-catenin association and subsequent activation of β-catenin and increased cyclin-D1 expression [42]. Likewise, cyclin-D1 was shown to be an important mediator of β-catenin and Met induced HCC in genetic models [43]. Further, simultaneous activation of Met and a mutated active form of β-catenin have been found in a subset of human hepatocellular carcinomas [44]. Thus, the Met/β-catenin pathways operate synergistically to induce hepatocyte proliferation in normal and dysregulated growth. β-catenin also appears to crosstalk with the epidermal growth factor receptor (EGFR) signaling pathway, whose ligand, EGF, is another known hepatocyte mitogen. Like Met and E-cadherin, ErbB2, a member of the EGFR family, phosphorylates β-catenin at tyrosine-654 [45], and constitutive tyrosine phosphorylation of β-catenin by ErbB2 has been implicated in the metastasis of cancers [46, 47]. Our laboratory has also identified EGFR to be a transcriptional target of Wnt/β-catenin signaling [48], which may serve to enhance and prolong β-catenin signaling and potentiate its mitogenic effect.
Transcriptional targets of β-Catenin signaling
The number and variety of target genes activated by Wnt/β-catenin signaling are diverse and include target genes such as Met, Jagged, gastrin, MMP7, survivin, and various FGFs [49–56]. β-Catenin also controls the expression of cell-cycle regulators important in proliferation such as cyclin-D1 [57, 58], as well as oncogenes such as c-myc [59]. The majority of Wnt target genes, however, appear to be cell-type specific and are thus regulated both temporally and contextually. In the liver, these genes include glutamine synthetase, cyclin-D1, several cytochrome P450s (2e1, 1a2), EGFR and leukocyte cell-derived chemotaxin 2 (LECT2) as well as our recently discovered target regucalcin or senescent marker protein-30 [48, 60–66]
Interestingly, some of the target genes of the pathway are components of the Wnt signaling pathway itself. For example, β-catenin can activate expression of repressors such as Axin2, Tcf1, and Dkk1 [55, 67, 68], or suppress positive pathway components such as Frizzled and LPR6 [69, 70], indicating the presence of a negative feedback loop to dampen or suppress Wnt signaling. The question that remains unanswered is whether β-catenin activation in the absence of mutations in the β-catenin gene, CTNNB1, or components of degradation complex such as AXIN1/2 will lead to any meaningful target gene expression, when there are several negative feedback opportunities that exist in this signaling cascade. Similarly, as described later, heterogeneity is evident in β-catenin activation due to sites of mutation within the exon-3 of CTNNB1, AXIN1/2 mutations as well as additional modes of β-catenin activation in HCC such as by TGFβ or receptor tyrosine kinases. These disparate mechanisms of β-catenin activation result in variations in target gene expression, which eventually impart a distinct phenotype to the tumor [71–75].
Intriguingly, Wnt/β-catenin signaling also can induce expression of genes such as LEF1, which enhances and prolongs the signal, indicating the presence of a feed-forward mechanism which can be exploited by carcinoma cells [76, 77].
LIVER REGENERATION: NORMAL, REGULATED GROWTH
Model
Adult liver has the unique capacity to regenerate after insult and loss of liver mass, and has thus become a useful model to study organ regeneration and controlled growth. A phenomenon unique to liver regeneration is that rather than utilizing progenitor or stem cells, repopulation of the liver occurs almost exclusively through proliferation of mature cell populations, including hepatocytes, biliary epithelial cells, endothelial cells, and stellate cells [78]. Remarkably, liver regeneration occurs without loss of function; that is, throughout the regenerative process, the liver still performs all the essential functions needed for organism homeostasis. Another property that makes liver regeneration unique is that it can be triggered without causing inflammation or damage to surrounding tissues. The most common method of inducing regeneration experimentally is surgical removal of three of five lobes from the rodent liver, commonly referred to as a 2/3rd or partial hepatectomy (PHx) [79]. The remaining two lobes grow in size until the liver mass is completely restored, which usually occurs after approximately 7 days in the rat [80]. Because these lobes can be resected within minutes, the entire regenerative process can be precisely timed.
Molecular signaling during liver regeneration
Partial hepatectomy triggers a series of cell signaling pathways and cascades that are very tightly regulated. Many of these pathways are the same as those activated in wound healing and innate immunity, although liver resection itself does not generate an immune response. One of the earliest events, which occurs within 30 minutes after PHx, is the induction of “immediate early genes”, including members of the jun, c-fos, and myc families. Transcription of these genes are the result of rapidly activated transcription factors such as Stat3 and NF-κB, which are in turn activated by cytokines such as TNF-α and IL-6 [81]. Genes important for regulation of cell cycle entry are also transcribed either concurrently with cytokine stimulation or immediately following this period in response to growth factors such as HGF and EGF [82, 83]. Interestingly, fetal markers such as alpha-fetoprotein are also upregulated during this time [84], and suggest that regeneration may recapitulate development to some extent.
Cellular basis of liver regeneration
The hallmark of liver regeneration is proliferation of adult hepatic cell types. The first peak of DNA synthesis happens in hepatoytes and occurs around 24 hours in the rat and approximately 36 hours in the mice [80]. Hepatocyte DNA synthesis and proliferation proceed in a zonal manner through the hepatic lobules, from periportal to pericentral areas [85]. Since 1/3 of the original hepatocytes remain after PHx, they only need to undergo 1.66 rounds of replication before the original number of hepatocytes is fully restored [86]. In fact, hepatocytes are capable of clonal expansion and have an almost unlimited capacity to proliferate in vivo. Rat liver was able to continue regenerating even after 12 sequential hepatectomies [87], which suggests that unlike other mature cells in the body, hepatocytes are not terminally differentiated and can divide continuously when presented with appropriate stimuli [78]. Proliferating hepatocytes also produce growth factors for other cell types, including stellate cells and endothelial cells, in a paracrine fashion. These cells undergo DNA synthesis 24 hours after hepatocytes, peaking at 48 hours after PHx [88]. A key cell-cycle associated gene critical for initiation of cell proliferation is cyclin-D1, which is expressed as early as 6 hours after PHx [89]. As β-catenin is a key driver of cyclin-D1 expression and cell proliferation, it is logical to assume that β-catenin plays a role in liver regeneration after PHx. Indeed, work by our lab and others have described the function of β-catenin in regeneration and postnatal liver growth, which emphasizes its vital role in liver health and repair.
WNT/β-CATENIN SIGNALING IN LIVER GROWTH AND REGENERATION
Role of β-catenin in postnatal hepatic growth
A hepatic growth spurt is known to occur during first month after birth in mice. The Wnt/β-catenin pathway also plays a key role in contributing to this postnatal liver growth. Our laboratory found an increase in total β-catenin levels in the wild-type mice shortly after birth. In fact an increase in translocation of β-catenin to the nucleus correlates with an increase in cell proliferation evident in livers between 5–20 postnatal days [90]. Further, conditional β-catenin knockout (KO) mice lacking β-catenin in hepatocytes showed a significant decrease in the liver weight/body weight ratio (15–25%) in mice older than two months [65, 66]. This reduction is due to a basal decrease in hepatocyte proliferation as a result of lower expression of cell cycle regulators such as cyclin-D1 in the KO livers.
Growth advantage in livers overexpressing β-catenin
Direct overexpression of β-catenin has also been shown to enhance liver growth and regeneration. Transgenic mice overexpressing liver-specific wild-type β-catenin showed a 15% increase in liver size compared to normal wild-type aged-matched controls, secondary to increased proliferation [48]. Increased cell proliferation leading to hyperplasia and hepatomegaly was also described in a transgenic mouse that expresses an oncogenic form of β-catenin lacking the N-terminus, which is involved in protein stabilization [91]. Another report described enlarged livers shortly after adenoviral infection in a mouse strain carrying a mutant inducible form of β-catenin [92]. It should be noted that none of the mouse models described above developed spontaneous hepatic tumors. Thus, it appears that mutation of β-catenin alone is insufficient to cause tumorigenesis and suggests that only after a “first hit”, such as chemical induction or mutation of another oncogene, does mutant-β-catenin promote tumorigenesis.
Activation of β-catenin signaling after PHx
During liver regeneration, however, the steady-state kinetics of β-catenin changes dramatically. In a rat model, a 2.5-fold increase in β-catenin protein was observed during the early minutes of liver regeneration induced by partial hepatectomy, concomitant with translocation to the nucleus [93]. The mechanism of this increase is a decrease in serine phosphorylation and a subsequent decrease in protein degradation, and not a result of changes in gene expression. Further, this initial increase in β-catenin protein is immediately countered by a downregulation of this pathway and activation of the destruction complex, which limits any sustained elevations in β-catenin protein levels. β-catenin activation after partial hepatectomy was transient, and protein levels returned to normal 48 hours post-PHx. The increased nuclear localization of β-catenin during the early stages of liver regeneration contributes to the increase in cyclin-D1 and c-myc expression observed previously after partial hepatectomy; thus, β-catenin has a positive impact on cellular proliferation [93]. As mentioned above, β-catenin was also identified to be also among the downstream effectors of hepatocyte growth factor, which itself is a significant player in the process of liver regeneration [36, 38, 94].
Impact of β-catenin loss or knockdown on regeneration
Conventional β-catenin knockout results in early embryonic lethality [95]; however, removal of β-catenin gene expression, either through endogenous morpholinos or conditional loss-of-function mutations, has proven to be a valuable tool for the functional analysis of canonical Wnt signaling in many tissues and organs, including liver [96]. The importance of β-catenin to liver regeneration has been elucidated by several studies in which β-catenin is removed or absent from the liver. Ablation of β-catenin transcription by administration of an antisense oligonucleotide simultaneous with partial hepatectomy resulted in a decrease in total and nuclear β-catenin 24 hours after PHx in rats [97]. The antisense-treated group also showed a significant decrease in liver weight/body weight ratio as late as 7 days after PHx, which was a result of decreased hepatocyte proliferation. To further explore the role of β-catenin in liver regeneration, others and we generated conditional knockout (KO) mice containing a hepatocyte-specific disruption of the β-catenin gene. Initial characterization of these mice showed a significant and sustained decrease in liver weight/body weight ratio (15–25%) in mice older than two months [65, 66]. Additionally, when subjected to partial hepatectomy, these mice displayed suboptimal regeneration, the result of a 2-fold decrease in the number of cells in S-phase at the time of peak hepatocyte proliferation in wild-type (40 hours). This decrease in proliferation seen in KO corresponds with deficient expression of cyclins involved in G1 to S transition, including cyclin-A, D, and E [66]. Interestingly, a rebound increase in hepatocyte proliferation was noted in the β-catenin-null mice at day 3 after partial hepatectomy, indicative of a delayed regenerative onset. By day 14, a second, smaller peak in the number of proliferating cells was seen in KO livers, perhaps as a compensatory event to ongoing apoptosis, which was noted at all stages during regeneration. This biphasic trend in proliferation allows for delayed but sufficient regeneration of β-catenin knockout livers after partial hepatectomy [66]. Another laboratory utilized liver-specific β-catenin knockout mice to confirm a delayed onset of DNA synthesis and hepatocyte proliferation after partial hepatectomy, which is likely due to a lack of cyclin-D1 induction [98].
The KO mice were also investigated for addressing the role of β-catenin in toxicant-induced liver regeneration. Sublethal doses of acetaminophen in mice leads to hepatic injury and is immediately followed by enhanced regeneration. We identified an early β-catenin stabilization as a mechanism of hepatocyte proliferation and not injury following acetaminophen exposure. To address the role of β-catenin in acetaminophen-induced regeneration, conditional KO mice were utilized. However, these mice, as discussed earlier, have lower expression of CYP2E1 and CYP1A2 that metabolize the drug to its toxic metabolite and hence are resistant to even lethal doses of acetaminophen [65, 66]. To circumvent this limitation and address the role of β-catenin conclusively in toxicant-induced regeneration, the cytochrome P450s in question were induced in KO mice and regeneration compared between KO and controls at equitoxic doses, which clearly reflected the role of β-catenin in liver regeneration after acetaminophen-induced injury [99].
Effect of β-catenin stimulation on regeneration
While loss of β-catenin reveals defects in liver regeneration, activation of this pathway through gain-of-function mutants, in which β-catenin has been rendered constitutively active, has demonstrated the positive effect of Wnt signaling on liver regeneration. Two studies have demonstrated the effect of β-catenin stabilization indirectly through ablation of the APC gene, a known negative regulator of the pathway. In the first study, Apc-inactivated mice display both a clear increase in liver size and a high incidence of spontaneous hepatocellular cancer [100]. In addition, regeneration was accelerated in Apc+/− zebrafish after 1/3 partial hepatectomy [101]. Inhibition of β-catenin transcription in zebrafish resulted in impaired liver regeneration as expected, confirming the requirement for Wnt/β-catenin signaling after liver resection. These authors further supported this finding by demonstrating a regenerative advantage in APC-mutant mice livers after PHx, as measured by enhanced kinetics of β-catenin expression and proliferation [101]. While β-catenin activation was evident robustly in APC-mutant mice, it is unclear what impact loss of APC has on other signaling pathways, as APC has recently been shown to affect cell processes such as epithelial integrity and DNA replication independent of β-catenin regulation [102, 103].
We have recently utilized transgenic (TG) mice expressing Ser45 mutated β-catenin in hepatocytes to show a growth advantage both in vitro and during liver regeneration through cyclin-D1 regulation. The growth advantage of S45D TG hepatocytes after PHx may be attributed to acceleration of Met/β-catenin dissociation, phosphorylation, and nuclear translocation 40 hours after partial hepatectomy. In addition, hydrodynamic delivery of Wnt-1 gene delivery induced β-catenin activation and hepatocyte proliferation after partial hepatectomy in a mouse model [104]. This report provides evidence for the first time that exogenous manipulation of Wnt/β-catenin activation might be a novel way of inducing regeneration of the liver, when necessary. Clearly, this will be dependent on the availability of modulators of Wnt/β-catenin signaling pathway, which would need additional substantiation in preclinical models. While such modulators are not available currently, agents such as lithium chloride, which are known to inhibit GSK3β, a known negative regulator of β-catenin, might be relevant for further investigation in preclinical scenarios in liver regeneration [105].
β-Catenin activation correlates with hepatocyte proliferation in patients of acute liver failure
In light of the experimental observations, the role of β-catenin is now being explored in clinical scenario. β-Catenin regulates cyclin-D1 expression during regeneration after sublethal doses of acetaminophen in an experimental mouse model [99]. A retrospective study with a limited number of acetaminophen-induced acute liver failure patients was performed to determine β-catenin localization, cell proliferation and patient outcome. β-Catenin activation as evident by its nuclear and cytoplasmic localization correlated with higher spontaneous regeneration in these patients and precluded the need for an orthotopic liver transplant [99]. Conversely, patients that showed low β-catenin expression without any nuclear and cytoplasmic localization usually had lower proliferation and eventually required transplantation. Thus, in addition to having therapeutic potential in treating drug-induced toxicity, activation of the β-catenin may also be a useful biomarker to predict prognosis in cases of acute liver failure.
Wnt/β-catenin signaling and liver regeneration: Future prospects
A positive role of β-catenin in liver regeneration has been identified in rats, mice, zebrafish and humans [66, 93, 97–99, 101]. In addition, a transient regenerative advantage was evident in mutant-β-catenin overexpressing transgenic mouse model after partial hepatectomy, without any long-term consequences of growth dysregulation [104]. At the same time exogenous Wnt1 mediated activation of this pathway demonstrated a distinct regenerative advantage [104]. This work has the potential to enhance the effectiveness of current regenerative technologies in several ways. First, human hepatocytes genetically engineered with stable β-catenin may reduce the number of hepatocytes needed for cell transplant therapy and thus eliminate the need for serial transplants. Second, temporal activation of the Wnt signaling after orthotopic liver transplant in the recipient may promote growth and function of the graft especially in small-for-size grafts. Wnt activation may also be of therapeutic significance in promoting regeneration in hepatic failure. Thus Wnt/β-catenin pathway may truly bridge liver biology, tissue engineering, and hepatic regenerative medicine.
OVAL CELLS: ABNORMAL, REGULATED GROWTH
Overview
Under normal circumstances, stem cells are not required for liver regeneration. However, when hepatocyte proliferation is impaired, as in the case of chronic disease (such as viral hepatitis) or replicative senescence (such as in steatohepatitis), hepatic progenitor cells are activated and function as a reserve stem cell compartment. These progenitors, also called oval cells, have the potential to proliferate, produce multi-lineage progeny, and repopulate the liver when the regenerative capacity of adult hepatocytes is compromised [106–108]. Oval cells, so-called because they have an ovoid nucleus and scanty cytoplasm, are known to reside in the canals of Hering in a quiescent liver [109], although other possible hepatic stem cell niches have also been identified [110]. Oval cells amplify into atypical ductular proliferations (ADP) before differentiating into hepatocytes or bile duct cells [111].
The oval cell population is heterogeneous and contains cells that display a mixed epithelial/mesenchymal phenotype; thus, within a population, oval cells can differ in their differentiation state and lineage commitment [112]. Numerous studies have demonstrated that oval cells play an important role in normal liver biology as well as carcinogenesis [113]. Oval cell proliferation has been shown to precede neoplasia in many rodent hepatocellular carcinomas and has been correlated with the increased risk for development of hepatocellular carcinoma in chronic liver disease [114–116].
Model
Activation of a progenitor cell population can be induced experimentally in rodent models of liver injury, where the adult hepatocytes are rendered incapable of proliferation. Oval cell activation is observed in rats placed on the Solt-Farber protocol, which includes 2-acetylaminoflourene (2-AAF) followed by two-thirds hepatectomy [117]. Bile duct damage has been shown to compromise oval cell activation in this model, providing experimental evidence that oval cells may originate from intraportal or periportal ductules [118, 119]. In mice, feeding a choline-deficient diet supplemented with ethionine or a 3, 5-diethoxycarbonyl-1,4-dihydro-collidine (DDC)-containing diet results in oval cell activation and atypical ductular proliferation (ADP), respectively [120, 121]. Oval cell proliferation has also been observed in other murine models including dipin-induced hepatocarcinogenesis and phenobarbitol/cocaine-induced periportal injury. Intriguingly, the different protocols used for activating the progenitor stem compartment in rodents cause heterogeneity and phenotypically dissimilar populations, resulting in cells that possess unique characteristics and molecular signatures depending upon the treatment used [122]. Oval cells co-express both hepatocellular (Alb, AFP) and biliary (CK-19, OV-6) markers as well as stem cell genes (c-kit, LIF) and thus have the ability to differentiate into either hepatocytes or cholangiocytes [123, 124]. A wide range of markers, such as OV6, A6, Dlk, and EpCAM, have been developed to aid in the identification of putative hepatic progenitor cells.
Stem cells and hepatocellular cancer
Whether a cancer stem cell could be the precursor cell for HCC is controversial. Mature cells of the liver including hepatocytes and biliary epithelial cells are perfectly capable of proliferation as has been shown in numerous studies in models such as partial hepatectomy [80]. In fact there is no evidence of stem cells in the normal process of liver regeneration. However, nothing is ‘normal’ about regeneration in a diseased liver, hence the possibility of subset of HCCs to be contributed by a stem cell or a progenitor is worth entertaining [125]. It is likely that a subset of HCC patients might have a disease contributed by a more primitive cell comparable to those defined as ‘cancer stem cells’ or oval cells in experimental carcinogenesis. The maturation arrest theory implicates arrest of these ‘stem cells’ at different stages of their development to eventually give rise to tumors. Cancer stem cells that follow the stochastic model would be highly significant if these were targets of neoplastic transformation as they have pre-existing characteristics such as self-renewal (reviewed in [113]).
The most relevant evidence that cancer stem cells contribute to the origin of HCC comes from oval cell studies in experimental carcinogenesis in response to various chemicals. Unfortunately, experimental carcinogenesis does not reflect a true patient scenario due to the absence of ongoing fibrosis and cirrhosis, which is the predominant backdrop for HCC in around 80% of clinical scenarios. The most pertinent model that clearly implicates stem or oval cell as a source of HCC is the Solt-Farber model where diethylnitrosamine (DEN) acts as the initiator of transformation and proliferation by causing DNA adduct formation and damage followed by N2-acetylaminofluorene to inhibit proliferation of most hepatocytes and lastly partial hepatectomy which creates the need for cell proliferation [126]. Thus there is selective pressure on transformed oval cells to proliferate, leading to a stem-cell-driven disease. Other chemicals have shown distinct stem cells as precursors of carcinogenesis in various preclinical models; however, their exact correlation to human disease is unknown. Such chemicals include Furan, choline-deficient ethionine supplemented diet, choline-deficient diet with AAF treatment and others.
More than one group has identified CD133, which is a hematopoietic stem cell marker, as the cancer stem cell marker for HCC in patients [127–129]. It was also independently noted that HCC that show fetal hepatoblast, stem cell or CK-19-positive signatures might have a dominant stem cell component and show poor prognosis [114, 130–132]. More recently, EpCAM-positive cells have also been labeled as cancer stem cells in HCC [133]. The relationship between CD133 and EpCAM cells remains unknown. Thus at the present time the overall role and relative significance of oval cells or cancer stem cells as the cells of origin of HCC remains unresolved, and their elucidation will be of significance in predicting disease behavior, combating chemoresistance and devising differentiation therapies.
WNT/β-CATENIN SIGNALING IN OVAL CELL INDUCTION AND EXPANSION
Changes in Wnt/β-catenin signaling during oval cell activation
Wnt/β-catenin signaling has been shown to be at the heart of the phenomena of self-renewal in many stem cells including embryonic and hematopoietic stem cells. Wnt/β-catenin signaling is also implicated in the differentiation process of certain stem cells to specific lineages. Additionally, activation of β-catenin plays a major role in modulating the delicate balance between stemness and differentiation in several adult stem cell niches [134]. However, while Wnt/β-catenin signaling is clearly not a newcomer to the stem cell discipline, its role in hepatic progenitors is more recent and evolving [135].
Several recent studies in rodents have elucidated the role of β-catenin signaling in oval cell induction and proliferation. Wnt signaling was induced in livers harvested from DDC-fed mice, which corresponded to increased oval cell proliferation within atypical ductular proliferations (ADPs). Furthermore, oval cells isolated from these livers showed nuclear localization of β-catenin in response to Wnt3a stimulation in culture [136]. We observed extensive activation of the Wnt/β-catenin pathway in a rat model of oval cell activation 5 and 10 days after 2-AAF+PHx, as evidenced by a significant increase in nuclear β-catenin protein levels in a subset of the oval cells [137] (Figure 1). A vast body of information is available about the role of the stem cell microenvironment in controlling proliferation, differentiation, and/or self-renewal through direct cell–cell interactions and also via various soluble factors, such as Wnts, Hedgehogs, and BMPs. This kind of paracrine signaling was also observed in our model whereby Wnt-1 expressed on the hepatocytes acts on the adjacent Fz-2 expressing oval cells in order to induce the β-catenin activation seen in these oval cells. Activation of β-catenin signaling in response to Wnt ligands was recently observed in a rat hepatic stem-like epithelial cell line, resulting in proliferation of these cells [138]. In another study, significant upregulation of several Wnt genes including Wnt7a, Wnt7b, and Wnt10a accompanied by increased levels of active β-catenin protein was observed in mice fed a DDC diet [139]. Further, activation of the Wnt/β-catenin pathway in vitro was sufficient to induce proliferation of cultured hepatic stem/progenitor cells [139]. Together, these studies demonstrate that Wnt/β-catenin signaling plays a critical role in oval cell biology.
Figure 1. β-Catenin localization in oval cells in rat model of 2-AAF and partial-hepatectomy at 5 and 10 days after surgery.

Total β-catenin is observed in the cytoplasm and nuclei of hepatic oval cells whereas it is evident at the membrane of surrounding hepatocytes. Active-β-catenin (hypophosphorylated at ser37/Thr41) is observed only in oval cells at day 5 and 10 after hepatectomy, which was performed 7 days after insertion of 2-AAF pellet.
Impact of modulation of Wnt/β-catenin signaling on oval cells
As in liver regeneration, the functional importance of Wnt/β-catenin signaling to oval cell activation can be more clearly understood by utilizing loss- and gain-of function mutations of pathway members. β-Catenin KO mice displayed a significantly blunted atypical ductular proliferation in response to DDC diet, further substantiating the requirement of Wnt signaling in the oval cell response. This result was confirmed in a rat model where inhibition of Wnt1 resulted in failure of oval cells to differentiate into hepatocytes and also lead to decreased oval cell proliferation [140]. Interestingly, in both models, long-term injury in the absence of Wnt/β-catenin signaling paradoxically induced a significantly greater ADP, indicating that while β-catenin is necessary for optimum oval cell response, in the presence of constant pressure, oval cells can activate an alternate mechanism in order to continue proliferating. Intriguingly, β-catenin S45D transgenic mice exposed to chronic administration of DDC diet showed an improvement in intrahepatic cholestasis compared to WT, as evidenced by a decrease in ADP and alkaline phosphatase [20]. Thus, overexpression of β-catenin may contribute to liver repair after DDC injury.
Wnt/β-catenin in oval cells in clinical studies
Oval cell activation has been observed in a range of liver injuries, including hepatic fibrosis and cirrhosis in alcoholic liver disease, viral hepatitis and others. Thus, it would be of clinical importance to determine markers for oval cell proliferation and differentiation in liver diseases of varying etiologies. One such study analyzed liver progenitor cells from patients with various diseases, such as acute hepatitis (AH) and primary biliary cirrhosis and identified Wnt signaling as playing a significant role in expanding the progenitor population, especially in patients with AH [141]. These findings have implications in the development of future therapeutic strategies that can be targeted to different subpopulations of progenitor cells.
WNT/β-Catenin signaling in stem cells in HCC
The Wnt/β-catenin pathway has long been observed to be active in a significant subset of HCC patients. More recently, it has also been implicated in cancer stem cells of this tumor type. According to the cancer stem cell theory of tumor origin, cancer might result from the disruption of normal stem cell self-renewal pathways. Due to an important role of Wnt/β-catenin signaling in stem cells, it is a relevant target of transformation. Several recent reports have demonstrated activation of β-catenin in isolated cancer stem cells from patients with HCC. Hepatic stem cells that are c-Kit(−)CD29(+)CD49f(+/low)CD45(−)Ter-119(−), which led to cancer initiation, showed prominent Wnt/β-catenin signaling [142]. Other reports have also demonstrated an important role of the Wnt pathway in tumorigenic cancer stem cells [143]. EpCAM, a hepatic stem cell marker that can be used for HCC prognosis, has been shown to be a target of the Wnt/β-catenin pathway [132, 133]. Inhibition of β-catenin has been shown to kill EpCAM-positive cells. In a more recent report from the same group, numbers of EpCAM-positive cells determined growth and invasiveness of the tumor similar to the HCC that exhibit stem cell signatures [130]. Our recent study identified HCCs harboring mutations in β-catenin gene to be more advanced as they displayed concomitant large size and signs of invasion [72]. In addition a high percentage of these tumors lacked overt cirrhosis and thus might reflect a stem cell driven disease, which is independent of the extent of injury, although this would need additional substantiation. Thus Wnt/β-catenin signaling is now implicated in HCC for its multifactorial role including a role in regulating ‘cancer stem cells’ in a subset of these tumors. Clearly, understanding the precise molecular and cellular basis of altered Wnt/β-catenin signaling may provide new insights into hepatoma biology with significant therapeutic implications [144].
HEPATOCELLULAR CANCER: ABNORMAL, DYSREGULATED GROWTH
Overview
Hepatocellular cancer is the most common liver tumor and a major health burden worldwide. It is the third most fatal cancer worldwide [145]. In the U.S.A, the incidence and associated death rates have steadily increased since the 1980s. According to the American Cancer Society, an estimated 18,910 liver cancer deaths are expected in the 2010. (http://www.cancer.org/acs/groups/content/@epidemiologysurveilance/documents/document/acspc-026238.pdf). Also, both the incidence and mortality rates are higher in men than women. One of the major causes of morbidity and mortality associated with the disease is its poorly understood cellular and molecular basis.
Cellular aberrations in HCC
In around 80% of all HCCs, there is evidence of existing chronic liver disease as a predisposing factor for this form of neoplasia [146]. Virtually any condition that has been linked to the development of fibrosis and cirrhosis has been associated with eventual development of HCC. Viral hepatitis (B and C), alcoholic liver disease, metabolic liver disease including non-alcoholic steatohepatitis, aflatoxin-B1 exposure, primary biliary cirrhosis, primary sclerosing cholangitis, hemochromatosis and hereditary tyrosinemia, are some of the leading risk factors for the development of HCC [147]. While there is some predilection of specific signaling aberrations and genetic and epigenetic mechanisms associated with certain risk factors, a rather common theme exists in terms of specific cellular changes that appear to be at the heart of HCC initiation and progression. Hepatic injury can also be either due to direct cytopathic effect of the inciting agent such as virus or alcohol or due to indirect mechanisms owing to the presence of inflammation, altered redox state of the cell due to free radicals, eventually leading to oncotic cell death and apoptosis. As a consequence of hepatocyte injury, and in response to inflammatory cytokines and oxidative stress, stellate cells and portal fibroblasts undergo activation and myofibroblastic transformation leading to the development of fibrosis and eventually cirrhosis [148]. Under such circumstances, the surviving hepatocytes do what they do best and proliferate as a means to regenerate the injured liver. It is this regenerative response often seen histologically as regenerating nodules in chronic hepatitis of any form that acts as a ‘double-edged sword’. While this ongoing regeneration will be critical for the maintenance of liver functions in such patients, the hepatocytes in these nodules will be now proliferating in the backdrop of continued inflammation and oxidative stress; in the presence of inciting agents capable of directly or indirectly causing genotoxic damage; and in the presence of ongoing fibrosis or cirrhosis. Some of these dividing cells within some of these regenerating nodules will now cumulate certain minimal genetic or epigenetic alterations to mark the inception of neoplastic transformation. It is this cell proliferation within the adverse microenvironment that appears to be driving tumorigenesis within the liver in most of the patients with various forms of chronic liver injuries. However, while cirrhosis is evident in most HCC patients, it is likely that there may be varying thresholds of neoplastic transformations based on cumulated molecular aberrations. Indeed, there have been recent studies where HCC in HCV cases have been observed without the evidence of any significant fibrosis [149]. Also, as pointed out by Dr. Vogelstein, development of solid tumors may depend on the aberrations in three critical signaling pathways ([150]) and the selection of these pathways may further dictate the threshold of neoplastic transformation for any given injury, for any given patient.
Molecular signaling in HCC
Identification of signaling pathways that are utilized by tumor cells to proliferate and survive in HCC will clearly be of high significance for successful targeted therapies. The molecular aberrations may stem from genetic and epigenetic alterations that may be a consequence of genotoxic injury as a result of direct or indirect effects stemming from the inciting agents as described in the previous section. Several epigenetic changes including hypermethylation of p16 and E-cadherin have been identified. Monoallelic chromosomal losses or deletions affecting tumor suppressor genes have been identified frequently at chromosome arms-1p, 4q, 6q, 8p, 9p, 13q, 16p, 16q, 17p and 19p, as have been chromosomal gains at 1q, 7q, 8q and 17q (reviewed in [147]). In HCC, loss of heterozygosity (LOH) has identified following tumor suppressor genes on the following chromosomes - 17p (TP53), 13q (RB1), 16p (AXIN1), 9p (CDKN2A) and 16q (IGF2R) (reviewed in [148]). Additional genetic mechanisms in HCC include mutations in genes such as p53 [151, 152], CTNNB1 [153] and PTEN [154]. As a result of any of the above basic mechanisms or yet unidentified mechanisms, several signaling pathways have been reported to be turned “ON” in HCC. These include the HGF/MET, EGFR/RAS/MAPK, WNT/β-CATENIN, PIK3CA/AKT and IGF signaling cascades [155]. There is ample evidence both pre-clinically and clinically of the efficacy of inhibiting these pathways to inhibit hepatoma cell growth and survival.
Once tumor initiation has occurred, the neoplasia may be self-sustainable and may itself be a source of factors that may result in additional tumor growth in HCC. The hepatoma cells and surrounding microenvironment may demonstrate autocrine and paracrine signaling axes that promote tumor growth. A typical example of such a process is the many angiogenic growth factors such as HGF, EGF, VEGF, PDGF and others that are produced by hepatoma cells enabling angiogenesis and promoting tumor growth [156].
WNT/β-CATENIN SIGNALING IN HEPATOCELLULAR CANCER
β-Catenin genetic models and hepatocarcinogenesis
Is β-catenin sufficient for initiation and progression of HCC? Several β-catenin transgenic mouse models have been generated to test this hypothesis. Intriguingly, none of the mice overexpressing either wild-type or stable-mutants of β-catenin have thus far exhibited spontaneous HCC [48, 91, 92, 104]. These models indicate β-catenin alone to be insufficient in initiating tumorigenesis. However, several studies have now indicated that β-catenin may be collaborating with other signaling pathways to contribute to hepatocarcinogenesis. β-Catenin was shown to cooperate with activated Ha-ras to induce hepatocarcinogenesis [157]. Simultaneous mutations in both the β-catenin and Ha-ras genes caused by adenovirus-cre injection cause hepatocellular carcinoma at an incidence of 100% in this model. Further supporting the idea that dysregulation of β-catenin may promote tumorigenesis in conjunction with another aberration, mice heterozygous for Lkb1 deletion also showed an accelerated progression to HCC when mated with adenovirus-inducible β-catenin mutant mice [158]. Similarly, when chemical carcinogens such as diethylnitrosamine alone, which induce HCC through activation of Ha-ras [159], are injected in serine-45-mutant β-catenin transgenic mouse, which lacks any spontaneous HCC, these animals showed accelerated tumorigenesis [104]. Thus these findings strongly suggest that β-catenin mutation is one of the hits that may be critical to the development of HCC, however additional aberrations are necessary to initiate tumorigenesis.
The exception to this rule is the liver-specific APC deletion mutants, which develop spontaneous HCC secondary to β-catenin activation [100, 160]. However, since APC also inhibits proliferation by binding directly to DNA [102], the relative contribution of β-catenin to HCC in this model will need additional investigation. It is highly likely that β-catenin is one of the hits required in this model and additional genetic dysfunctions due to APC loss may in fact complement development of HCC.
Role of β-Catenin in the models of experimental hepatocarcinogenesis
In the context of a tumor initiator, such as a chemical carcinogen, however, β-catenin mutations can contribute to an oncogenic phenotype in a subset of experimentally induced HCC. An example is the model of DEN-induced carcinogenesis, which provides a selective advantage to Ha-ras mutated hepatocytes [159]. Interestingly, when these mice are fed phenobarbitol (PB) after DEN injection, 80% of the liver tumors that develop show β-catenin mutations whereas Ha-ras mutations are undetectable, suggesting that phenobarbitol positively selects for cells containing β-catenin mutations [161, 162]. Interestingly, in DEN-induced hepatic lesions that progress to HCC, stabilizing mutations in β-catenin were found in over 50% of cases and may represent an important step in the malignant progression to HCC [163]. It should also be discussed, however, that some reports have implicated β-catenin mutations to be a rather common mechanism of mouse hepatocellular neoplasms, including adenomas and carcinomas, in five different chemical treatment groups, suggesting it to be an early event in carcinogenesis [164].
Role of β-Catenin in the models of genetic hepatocarcinogenesis
Genetic models, in which β-catenin is mutated or activated in the presence of another oncoprotein, have also elucidated the role of the Wnt/β-catenin pathway in carcinogenesis. The c-myc/TGF-α transgenic mouse model of liver cancer develops β-catenin activating mutations when treated with phenobarbitol, and these mutations confer a proliferative and invasive advantage to tumors [165]. β-catenin activation is also a dominant event in hepatocarcinogenesis in c-myc/E2F1 transgenic mice and results in accelerated or aggressive carcinogenesis [166]. β-catenin activation was frequent in tumors from c-myc and c-myc/TGFβ transgenic mice with a frequency of 25–33% and represented an early event in carcinogenesis, conferring a selective advantage to tumors of a differentiated phenotype [167]. Similarly, 50% of hepatic tumors in c-myc or H-ras transgenic mice had either point mutations or deletions in β-catenin [153]. Additionally, mouse hepatocytes transformed with Ras showed nuclear accumulation of β-catenin and a resultant dedifferentiation of neoplastic hepatocytes in the presence of TGFβ [168].
β-Catenin loss promotes chemical-induced carcinogenesis: Identification of paradox
Owing to an important role of β-catenin as an oncogene, it was relevant to demonstrate relative protection of the hepatocyte-specific β-catenin KO mice to chemical carcinogenesis. Paradoxically, however, the KO mice showed a dramatic increase in HCC incidence and severity after DEN treatment [169]. This study demonstrates a ‘tumor suppressive’ role of β-catenin in HCC such that its lack promoted hepatocarcinogenesis. A more recent study by Schwarz and colleagues also demonstrated enhanced HCC in β-catenin KO in response to DEN and phenobarbital [170]. In both these reports, lack of β-catenin promoted HCC, which intriguingly was observed in the milieu of increased inflammation, oxidative stress, hepatic fibrosis and regeneration. Further analysis revealed activation of PDGFRα/PIK3CA/AKT signaling in part due to aberrant NF-κB activation, in the β-catenin-deficient livers after DEN-induced genotoxic injury that led to enhanced cell proliferation in the backdrop of chronic liver injury, inflammation and fibrosis, eventually progressing to enhanced carcinogenesis [169]. In fact treatment of these mice with Imatinib led to significantly lower tumor size and number in the KO mice supporting an important role of PDGFRα in this model.
Activation of Wnt signaling in HCC in patients
Role of Wnt/β-catenin signaling in HCC was discovered over a decade ago [153]. Aberrant activation of Wnt/β-catenin signaling has been reported in a wide range of HCC patients. The most well understood and non-controversial mechanism of β-catenin activation in this tumor type is mutations in β-catenin gene or CTNNB1 that have been noted in around 20–40% of all cases [75, 153, 171–174]. Mutations have also been reported in the components of the degradation complex of β-catenin including AXIN1 in around 3–16% [75, 173, 175, 176] and AXIN2 in around 3% of all HCC cases [176]. Additional mechanisms have also been described and include overexpression of FRZ7 [177, 178], inactivation of GSK3β [179], methylation of sFRP1 [180], TGFβ-dependent activation of β-catenin [73], and β-catenin activation by receptor tyrosine kinases especially in fibrolamellar subset of HCCs [72]. Intriguingly, HCC that occur in Hepatitis C virus (HCV) patients showed a high incidence of CTNNB1 mutations in up to 40% of cases [172, 181], while HCV core protein led to increased expression of Wnt-1 in hepatoma cells due to unknown mechanisms [182]. Additional reports in HCC that occur in HBV patients have implicated Hepatitis B virus X protein to induce β-catenin activation in a mutation-independent manner [183, 184]. It is interesting to note that the most functionally relevant mutations that lead to activation of the Wnt pathway are those in CTNNB1, and correlates significantly with nuclear β-catenin by immunohistochemistry (or immunofluoresecence as shown in Figure 2) or GS-positive staining of tumor tissue as a surrogate target of β-catenin in the liver. Intriguingly, there is a significant subset of these tumors that do show increased nuclear or cytoplasmic β-catenin without CTNNB1 or AXIN mutations that may or may not be GS-positive and perhaps do represent the other mechanisms described above. However, their functional equivalence to each other has always been a major question. The eventual result of sustained Wnt/β-catenin signaling is thought to induce enhanced expression of β-catenin-dependent genes that in turn impact overall tumor growth and development [185, 186].
Figure 2. Varying patterns of β-Catenin localization in HCC samples by immunofluorescence.

HCC samples in panels A and B show predominantly membranous β-catenin (red) whereas HCC in panel C shows nuclear translocation of β-catenin (yellow) in subset of tumor cells and retained membranous localization (red) and HCC in panel C shows cytoplasmic (red) and nuclear (yellow) β-catenin in subset of tumor cells with decreased membranous staining. Sytox green was used as a nuclear counterstain.
Phenotype of β-catenin-active HCC
β-Catenin activation in a subset of HCC is undisputable as are the many mechanisms that lead to β-catenin activation [187]. Perhaps due to the heterogeneous mechanisms of β-catenin activation in HCC along with the complex crosstalks with many signaling pathways that are of essence in liver pathobiology, interpretation of the overall phenotype attributable to β-catenin activation has been rather cumbersome. In order to meaningfully decipher several studies that are seemingly conflicting, it will be useful to perhaps define what β-catenin activation really implies. In diagnostic and clinically relevant sense, the presence of non-membranous β-catenin including both cytoplasmic and nuclear β-catenin by immunohistochemistry complemented with the presence of certain targets is perhaps a useful definition. However, what are the true β-catenin targets; is one target sufficiently representative of β-catenin activity; and, do all mutations or modes of β-catenin activation induce similar target genes, are some of the questions that remain unanswered [71, 75]. So far, immunohistchemistry for GS has shown high sensitivity and specificity in detecting β-catenin mutated-tumors, which can be further improved by analysis of additional targets such as G-protein-coupled receptor 49 (GPR49) and glutamate transporter-1 (GLT1) [75]. However, a very careful analysis of differential activation of β-catenin through various reported mutations by sensitive and specific reporter assays will be required to conclusively address their relative efficacy in activating Wnt signaling. It is conceivable that the diverse mutations reported in exon-3 of CTNNB1 alone in HCC may alter the ternary structure of the β-catenin protein differently thereby influencing the interactions with other nuclear components including transcription factors, co-factors and histone acetyltransferases, which may in turn dictate target gene specificity and eventually the tumor phenotype. Indeed β-catenin-active HCC due to mutations in CTNNB1, AXIN1 or additional modes of β-catenin activation, have all been shown to have distinct phenotype in the transcriptomic classification of HCC [73, 75, 188].
Since β-catenin mutations are the predominant mode of activation of the Wnt pathway in HCC and also the most characterized we will discuss the tumor phenotype in this scenario. As it stands now, there are certain recurring themes in regards to the phenotype of the tumors displaying GS positivity as an indicator of β-catenin activation due to CTNNB1 mutations. These tumors have been classically defined to be larger in size, have less or no fibrosis, lack steatosis, are mostly well differentiated and display cholestasis [72, 188, 189]. There do remain contentious tumor attributes in the β-catenin-mutated HCC category as well. β-Catenin mutations have been mostly described as late events in HCC [174, 190, 191] while others report it to be an early event [171, 192]. β-Catenin mutated tumors have been reported to have less vascular invasion [193–195], whereas other reports have identified higher micro- and macro-vascular invasion [72, 174, 196].
What is the overall impact of β-catenin mutations and activation on prognosis in HCC patients? This also remains a debated issue in the field. β-Catenin mutations have been associated with better prognosis and a more differentiated tumor type [188, 195]. However, others have noted high nuclear and cytoplasmic β-catenin in more proliferating and poorly differentiated HCC [73, 190, 192]. Overall prognosis is a complex attribute and in HCC is dependent on not only multiple tumor characteristics including such as vascular invasion, metastasis and nodularity, but also extra-tumoral state of the liver including ongoing fibrosis and cirrhosis, hepatic dysfunction, and extrahepatic involvement. One such example is the lack of overt fibrosis evident in a significant subset of the β-Catenin-mutated HCC, which might be a confounding variable in supporting the observed improved prognosis in the β-catenin-mutated HCC patients. Similarly, additional studies will be critical to address if variable target gene expression due to varying mutations or mode of β-catenin activation in HCC may in fact be responsible for the eventual tumor phenotype as has also been suggested in a few studies [71, 75].
Decreased fibrosis was reported in a few studies and remains an intriguing hallmark of β-catenin mutated tumors [72, 196]. Whether β-catenin mutations are a risk factor for development of HCC independent of cirrhosis or these are merely decreasing the threshold of neoplastic transformation will remain to be further investigated. It is also intriguing to note that in a recent study, a small but significant subset of HCV patients developed HCC without evidence of any fibrosis [149]. Knowing the predilection of HCV in utilizing Wnt pathway as a mechanism of HCC, it might be relevant to pursue the relationship of HCV, fibrosis, Wnt/β-catenin signaling and HCC [172]. Along the same lines, it is relevant to note that the small subset of hepatic adenomas that progress to HCC in patients often exhibit β-catenin gene mutations [197]. This neoplastic transformation of adenomas occurs in a healthy liver without any evidence of fibrosis and further supports the role of β-catenin in fibrosis-independent HCC.
LIVER REGENERATION, β-CATENIN SIGNALING: SORTING THE GOOD FROM THE BAD IN HCC
Liver regeneration: Sorting the good from the bad in HCC?
Liver regeneration is a double-edged sword in chronic liver diseases and while it is critical for maintenance of liver function amidst inflammation, injury (viral, metabolic or toxicant) and cirrhosis, it also is the source of dysplasia when hepatocytes proliferate in an adverse milieu [146]. Once neoplastic transformation occurs, the tumor may still utilize the mitogenic pathways common to regeneration and tumor cells. How feasible will it be to target the signaling pathways that drive the growth of the tumor, but may also be critical for ongoing liver regeneration? For efficacious molecular targeting in HCC especially in chronic liver diseases, it will be critical to identify mitogenic signaling pathways that may be dispensable to hepatic regeneration, but indispensable to HCC growth, to prevent untoward adverse side effects. Based on the above speculation, it will be relevant to, for example, determine if there is any similarity in the molecular mechanisms driving liver regeneration after hepatectomy and carcinogenesis in response to DEN-exposure in β-catenin KO mice [66, 169]. It will also be desirable to identify pathways that when inhibited in β-catenin KO will still allow regeneration but impede tumorigenesis. However, it is also likely that non-transformed hepatocytes in general may have greater molecular redundancy at the outset, thus enabling them to continue to proliferate despite inhibition of a few signaling pathways. Indeed continued regeneration after hepatectomy is observed in many genetic knockout models of known mitogenic pathways (Reviewed in [80]). Conversely, transformed hepatocytes may have only limited redundant molecular mechanisms and thus may succumb to inhibition of specific pathways that these cells may be addicted to for growth and survival. Nonetheless, identification of such escape pathways after suppression of a key pathway for their redundancy in normal versus transformed hepatocytes will also be relevant to successfully treat HCC with undue adverse effects on hepatic function. As recently reported, we discovered PDGFRα signaling to be a major escape pathway following β-catenin suppression and it remains under investigation if sequential β-catenin and PDGFRα inhibition will be dispensable for liver regeneration but essential for tumorigenesis [169].
Wnt/β-catenin signaling: Sorting the good from the bad in HCC?
The origin of hepatocellular cancer (HCC) is mostly in the milieu of chronic injury to the liver with ongoing inflammation and fibrosis [146, 198]. This leads to initiation of regenerating nodules, consisting of proliferating hepatocytes, some of which eventually undergo genetic and/or epigenetic alterations owing to the existing genotoxic microenvironment due to any number of causes, leading eventually to HCC. Intriguingly, a high number of HCCs with β-catenin mutations lacked any significant fibrosis and cirrhosis [72, 196] (Figure 3). Several studies have also demonstrated activation of PIK3CA/AKT signaling in tumors with significant fibrosis and injury, distinct from β-catenin-mutated group [73, 188] (Figure 3). Furthermore, recent studies reporting enhanced chemical-induced carcinogenesis in the β-catenin KO mice through activation of PIK3CA/AKT signaling, reveals a paradoxical role of Wnt signaling in hepatic tumors [169, 170]. All the above observations taken together suggest that β-catenin activation that is observed sometimes in up to 90% of HCC, i.e. in additional 40–60% of HCC patients where no CTNNB1, AXIN1 or AXIN2 mutations are observed, might in fact be exhibiting important role in maintaining hepatocyte function within tumors. Indeed tumors are heterogeneous and composed of tumor and normal cells at varying stages of differentiation and proliferation. Based on its role in hepatocyte proliferation during regeneration and hepatocyte survival during liver growth and development (Reviewed in [63, 187]), β-catenin signaling in the absence of β-catenin mutations in HCC within tumor nodules especially in scenarios of ongoing inflammation, injury and fibrosis, may be playing an important role in maintaining liver function in such patients. Additional studies will be of utmost significance to distinguish the ‘good’ from the ‘bad’ β-catenin signaling in HCC to prevent harmful effects of universal β-catenin suppression as a therapeutic strategy in HCC. Presently, it appears that activation of β-catenin through mutations in CTNNB1 that are often detectable by positive GS immunostaining of the tumor may be the most suitable candidates for β-catenin suppression, when such therapies become a reality [198, 199].
Figure 3.
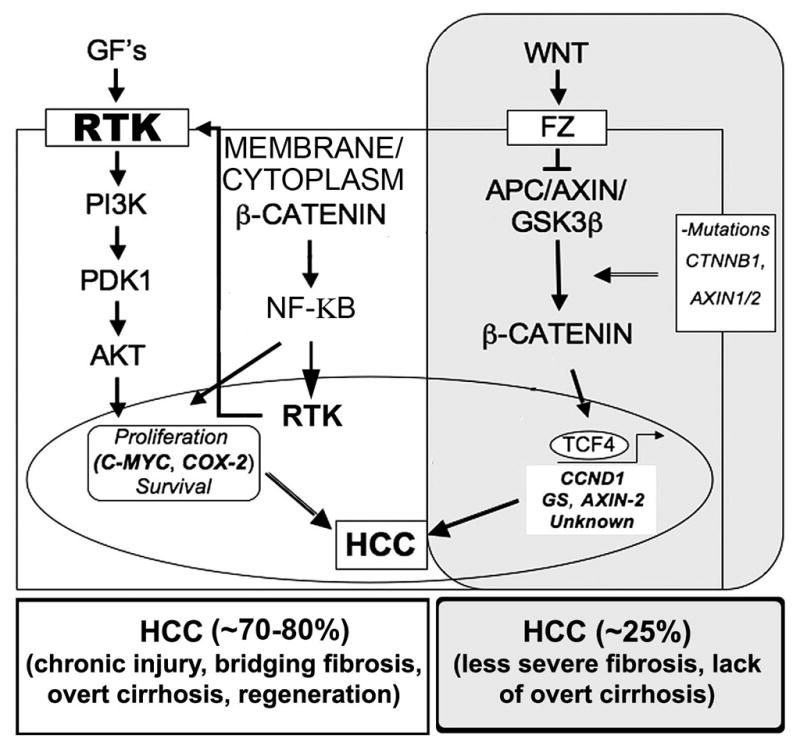
Broad categories of HCC based on β-catenin activation due to mutations versus multifactorial activation of receptor tyrosine kinases and PI3 kinase and AKT signaling.
Acknowledgments
Grant Support: This study was funded by NIH grants 1R01DK62277 and 1R01CA124414 to SPSM and by Rango’s Fund for the Enhancement of Pathology Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wodarz A, Nusse R. Mechanisms of Wnt signaling in development. Annual review of cell and developmental biology. 1998;14:59–88. doi: 10.1146/annurev.cellbio.14.1.59. [DOI] [PubMed] [Google Scholar]
- 2.Cadigan KM, Nusse R. Wnt signaling: a common theme in animal development. Genes Dev. 1997;11:3286–305. doi: 10.1101/gad.11.24.3286. [DOI] [PubMed] [Google Scholar]
- 3.Nusse R. Wnt signaling in disease and in development. Cell Res. 2005;15:28–32. doi: 10.1038/sj.cr.7290260. [DOI] [PubMed] [Google Scholar]
- 4.Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annual review of cell and developmental biology. 2004;20:781–810. doi: 10.1146/annurev.cellbio.20.010403.113126. [DOI] [PubMed] [Google Scholar]
- 5.Peifer M, Polakis P. Wnt signaling in oncogenesis and embryogenesis--a look outside the nucleus. Science (New York, NY) 2000;287:1606–9. doi: 10.1126/science.287.5458.1606. [DOI] [PubMed] [Google Scholar]
- 6.Willert K, Brown JD, Danenberg E, Duncan AW, Weissman IL, Reya T, et al. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature. 2003;423:448–52. doi: 10.1038/nature01611. [DOI] [PubMed] [Google Scholar]
- 7.Thompson MD, Monga SP. WNT/beta-catenin signaling in liver health and disease. Hepatology. 2007;45:1298–305. doi: 10.1002/hep.21651. [DOI] [PubMed] [Google Scholar]
- 8.Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–80. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 9.Wong HC, Bourdelas A, Krauss A, Lee HJ, Shao Y, Wu D, et al. Direct binding of the PDZ domain of Dishevelled to a conserved internal sequence in the C-terminal region of Frizzled. Mol Cell. 2003;12:1251–60. doi: 10.1016/s1097-2765(03)00427-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cliffe A, Hamada F, Bienz M. A role of Dishevelled in relocating Axin to the plasma membrane during wingless signaling. Curr Biol. 2003;13:960–6. doi: 10.1016/s0960-9822(03)00370-1. [DOI] [PubMed] [Google Scholar]
- 11.Zeng X, Huang H, Tamai K, Zhang X, Harada Y, Yokota C, et al. Initiation of Wnt signaling: control of Wnt coreceptor Lrp6 phosphorylation/activation via frizzled, dishevelled and axin functions. Development (Cambridge, England) 2008;135:367–75. doi: 10.1242/dev.013540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zeng X, Tamai K, Doble B, Li S, Huang H, Habas R, et al. A dual-kinase mechanism for Wnt co-receptor phosphorylation and activation. Nature. 2005;438:873–7. doi: 10.1038/nature04185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cadigan KM, Liu YI. Wnt signaling: complexity at the surface. J Cell Sci. 2006;119:395–402. doi: 10.1242/jcs.02826. [DOI] [PubMed] [Google Scholar]
- 14.Yokoya F, Imamoto N, Tachibana T, Yoneda Y. beta-catenin can be transported into the nucleus in a Ran-unassisted manner. Mol Biol Cell. 1999;10:1119–31. doi: 10.1091/mbc.10.4.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huber O, Korn R, McLaughlin J, Ohsugi M, Herrmann BG, Kemler R. Nuclear localization of beta-catenin by interaction with transcription factor LEF-1. Mech Dev. 1996;59:3–10. doi: 10.1016/0925-4773(96)00597-7. [DOI] [PubMed] [Google Scholar]
- 16.Zeng G, Awan F, Otruba W, Muller P, Apte U, Tan X, et al. Wnt’er in liver: expression of Wnt and frizzled genes in mouse. Hepatology. 2007;45:195–204. doi: 10.1002/hep.21473. [DOI] [PubMed] [Google Scholar]
- 17.Decaens T, Godard C, de Reynies A, Rickman DS, Tronche F, Couty JP, et al. Stabilization of beta-catenin affects mouse embryonic liver growth and hepatoblast fate. Hepatology. 2008;47:247–58. doi: 10.1002/hep.21952. [DOI] [PubMed] [Google Scholar]
- 18.Hussain SZ, Sneddon T, Tan X, Micsenyi A, Michalopoulos GK, Monga SP. Wnt impacts growth and differentiation in ex vivo liver development. Exp Cell Res. 2004;292:157–69. doi: 10.1016/j.yexcr.2003.08.020. [DOI] [PubMed] [Google Scholar]
- 19.Monga SP, Monga HK, Tan X, Mule K, Pediaditakis P, Michalopoulos GK. Beta-catenin antisense studies in embryonic liver cultures: role in proliferation, apoptosis, and lineage specification. Gastroenterology. 2003;124:202–16. doi: 10.1053/gast.2003.50000. [DOI] [PubMed] [Google Scholar]
- 20.Thompson MD, Awuah P, Singh S, Monga SP. Disparate cellular basis of improved liver repair in beta-catenin-overexpressing mice after long-term exposure to 3,5-diethoxycarbonyl-1,4-dihydrocollidine. Am J Pathol. 2010;177:1812–22. doi: 10.2353/ajpath.2010.100173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klein D, Demory A, Peyre F, Kroll J, Augustin HG, Helfrich W, et al. Wnt2 acts as a cell type-specific, autocrine growth factor in rat hepatic sinusoidal endothelial cells cross-stimulating the VEGF pathway. Hepatology. 2008;47:1018–31. doi: 10.1002/hep.22084. [DOI] [PubMed] [Google Scholar]
- 22.Kordes C, Sawitza I, Haussinger D. Canonical Wnt signaling maintains the quiescent stage of hepatic stellate cells. Biochem Biophys Res Commun. 2008;367:116–23. doi: 10.1016/j.bbrc.2007.12.085. [DOI] [PubMed] [Google Scholar]
- 23.Cheng JH, She H, Han YP, Wang J, Xiong S, Asahina K, et al. Wnt antagonism inhibits hepatic stellate cell activation and liver fibrosis. Am J Physiol Gastrointest Liver Physiol. 2008;294:G39–49. doi: 10.1152/ajpgi.00263.2007. [DOI] [PubMed] [Google Scholar]
- 24.Jiang F, Parsons CJ, Stefanovic B. Gene expression profile of quiescent and activated rat hepatic stellate cells implicates Wnt signaling pathway in activation. Journal of hepatology. 2006;45:401–9. doi: 10.1016/j.jhep.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 25.Benhamouche S, Decaens T, Godard C, Chambrey R, Rickman DS, Moinard C, et al. Apc tumor suppressor gene is the “zonation-keeper” of mouse liver. Dev Cell. 2006;10:759–70. doi: 10.1016/j.devcel.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 26.Braeuning A, Menzel M, Kleinschnitz EM, Harada N, Tamai Y, Kohle C, et al. Serum components and activated Ha-ras antagonize expression of perivenous marker genes stimulated by beta-catenin signaling in mouse hepatocytes. FEBS J. 2007;274:4766–77. doi: 10.1111/j.1742-4658.2007.06002.x. [DOI] [PubMed] [Google Scholar]
- 27.Colletti M, Cicchini C, Conigliaro A, Santangelo L, Alonzi T, Pasquini E, et al. Convergence of Wnt signaling on the HNF4alpha-driven transcription in controlling liver zonation. Gastroenterology. 2009;137:660–72. doi: 10.1053/j.gastro.2009.05.038. [DOI] [PubMed] [Google Scholar]
- 28.Lilien J, Balsamo J. The regulation of cadherin-mediated adhesion by tyrosine phosphorylation/dephosphorylation of beta-catenin. Curr Opin Cell Biol. 2005;17:459–65. doi: 10.1016/j.ceb.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 29.Orsulic S, Huber O, Aberle H, Arnold S, Kemler R. E-cadherin binding prevents beta-catenin nuclear localization and beta-catenin/LEF-1-mediated transactivation. J Cell Sci. 1999;112 ( Pt 8):1237–45. doi: 10.1242/jcs.112.8.1237. [DOI] [PubMed] [Google Scholar]
- 30.Aberle H, Butz S, Stappert J, Weissig H, Kemler R, Hoschuetzky H. Assembly of the cadherin-catenin complex in vitro with recombinant proteins. J Cell Sci. 1994;107 ( Pt 12):3655–63. doi: 10.1242/jcs.107.12.3655. [DOI] [PubMed] [Google Scholar]
- 31.Piedra J, Martinez D, Castano J, Miravet S, Dunach M, de Herreros AG. Regulation of beta-catenin structure and activity by tyrosine phosphorylation. J Biol Chem. 2001;276:20436–43. doi: 10.1074/jbc.M100194200. [DOI] [PubMed] [Google Scholar]
- 32.Roura S, Miravet S, Piedra J, Garcia de Herreros A, Dunach M. Regulation of E-cadherin/Catenin association by tyrosine phosphorylation. J Biol Chem. 1999;274:36734–40. doi: 10.1074/jbc.274.51.36734. [DOI] [PubMed] [Google Scholar]
- 33.Theard D, Steiner M, Kalicharan D, Hoekstra D, van Ijzendoorn SC. Cell polarity development and protein trafficking in hepatocytes lacking E-cadherin/beta-catenin-based adherens junctions. Mol Biol Cell. 2007;18:2313–21. doi: 10.1091/mbc.E06-11-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gumbiner BM. Regulation of cadherin-mediated adhesion in morphogenesis. Nature reviews. 2005;6:622–34. doi: 10.1038/nrm1699. [DOI] [PubMed] [Google Scholar]
- 35.Zarnegar R. Regulation of HGF and HGFR gene expression. Exs. 1995;74:33–49. doi: 10.1007/978-3-0348-9070-0_3. [DOI] [PubMed] [Google Scholar]
- 36.Monga SP, Mars WM, Pediaditakis P, Bell A, Mule K, Bowen WC, et al. Hepatocyte growth factor induces Wnt-independent nuclear translocation of beta-catenin after Met-beta-catenin dissociation in hepatocytes. Cancer research. 2002;62:2064–71. [PubMed] [Google Scholar]
- 37.Papkoff J, Aikawa M. WNT-1 and HGF regulate GSK3 beta activity and beta-catenin signaling in mammary epithelial cells. Biochem Biophys Res Commun. 1998;247:851–8. doi: 10.1006/bbrc.1998.8888. [DOI] [PubMed] [Google Scholar]
- 38.Zeng G, Apte U, Micsenyi A, Bell A, Monga SP. Tyrosine residues 654 and 670 in beta-catenin are crucial in regulation of Met-beta-catenin interactions. Exp Cell Res. 2006;312:3620–30. doi: 10.1016/j.yexcr.2006.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ishibe S, Haydu JE, Togawa A, Marlier A, Cantley LG. Cell confluence regulates hepatocyte growth factor-stimulated cell morphogenesis in a beta-catenin-dependent manner. Mol Cell Biol. 2006;26:9232–43. doi: 10.1128/MCB.01312-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Monga SP, Micsenyi A, Germinaro M, Apte U, Bell A. beta-Catenin regulation during matrigel-induced rat hepatocyte differentiation. Cell Tissue Res. 2006;323:71–9. doi: 10.1007/s00441-005-0045-8. [DOI] [PubMed] [Google Scholar]
- 41.Shibamoto S, Hayakawa M, Takeuchi K, Hori T, Oku N, Miyazawa K, et al. Tyrosine phosphorylation of beta-catenin and plakoglobin enhanced by hepatocyte growth factor and epidermal growth factor in human carcinoma cells. Cell adhesion and communication. 1994;1:295–305. doi: 10.3109/15419069409097261. [DOI] [PubMed] [Google Scholar]
- 42.Apte U, Zeng G, Muller P, Tan X, Micsenyi A, Cieply B, et al. Activation of Wnt/beta-catenin pathway during hepatocyte growth factor-induced hepatomegaly in mice. Hepatology. 2006;44:992–1002. doi: 10.1002/hep.21317. [DOI] [PubMed] [Google Scholar]
- 43.Patil MA, Lee SA, Macias E, Lam ET, Xu C, Jones KD, et al. Role of cyclin D1 as a mediator of c-Met-and beta-catenin-induced hepatocarcinogenesis. Cancer research. 2009;69:253–61. doi: 10.1158/0008-5472.CAN-08-2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tward AD, Jones KD, Yant S, Cheung ST, Fan ST, Chen X, et al. Distinct pathways of genomic progression to benign and malignant tumors of the liver. Proc Natl Acad Sci U S A. 2007;104:14771–6. doi: 10.1073/pnas.0706578104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shibata T, Ochiai A, Kanai Y, Akimoto S, Gotoh M, Yasui N, et al. Dominant negative inhibition of the association between beta-catenin and c-erbB-2 by N-terminally deleted beta-catenin suppresses the invasion and metastasis of cancer cells. Oncogene. 1996;13:883–9. [PubMed] [Google Scholar]
- 46.Bonvini P, An WG, Rosolen A, Nguyen P, Trepel J, Garcia de Herreros A, et al. Geldanamycin abrogates ErbB2 association with proteasome-resistant beta-catenin in melanoma cells, increases beta-catenin-E-cadherin association, and decreases beta-catenin-sensitive transcription. Cancer research. 2001;61:1671–7. [PubMed] [Google Scholar]
- 47.Wang K, Ma Q, Ren Y, He J, Zhang Y, Zhang Y, et al. Geldanamycin destabilizes HER2 tyrosine kinase and suppresses Wnt/beta-catenin signaling in HER2 overexpressing human breast cancer cells. Oncology reports. 2007;17:89–96. [PubMed] [Google Scholar]
- 48.Tan X, Apte U, Micsenyi A, Kotsagrelos E, Luo JH, Ranganathan S, et al. Epidermal growth factor receptor: a novel target of the Wnt/beta-catenin pathway in liver. Gastroenterology. 2005;129:285–302. doi: 10.1053/j.gastro.2005.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boon EM, van der Neut R, van de Wetering M, Clevers H, Pals ST. Wnt signaling regulates expression of the receptor tyrosine kinase met in colorectal cancer. Cancer research. 2002;62:5126–8. [PubMed] [Google Scholar]
- 50.Rodilla V, Villanueva A, Obrador-Hevia A, Robert-Moreno A, Fernandez-Majada V, Grilli A, et al. Jagged1 is the pathological link between Wnt and Notch pathways in colorectal cancer. Proc Natl Acad Sci U S A. 2009;106:6315–20. doi: 10.1073/pnas.0813221106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Koh TJ, Bulitta CJ, Fleming JV, Dockray GJ, Varro A, Wang TC. Gastrin is a target of the beta-catenin/TCF-4 growth-signaling pathway in a model of intestinal polyposis. J Clin Invest. 2000;106:533–9. doi: 10.1172/JCI9476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brabletz T, Jung A, Dag S, Hlubek F, Kirchner T. beta-catenin regulates the expression of the matrix metalloproteinase-7 in human colorectal cancer. Am J Pathol. 1999;155:1033–8. doi: 10.1016/s0002-9440(10)65204-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang T, Otevrel T, Gao Z, Gao Z, Ehrlich SM, Fields JZ, et al. Evidence that APC regulates survivin expression: a possible mechanism contributing to the stem cell origin of colon cancer. Cancer research. 2001;61:8664–7. [PubMed] [Google Scholar]
- 54.Hendrix ND, Wu R, Kuick R, Schwartz DR, Fearon ER, Cho KR. Fibroblast growth factor 9 has oncogenic activity and is a downstream target of Wnt signaling in ovarian endometrioid adenocarcinomas. Cancer research. 2006;66:1354–62. doi: 10.1158/0008-5472.CAN-05-3694. [DOI] [PubMed] [Google Scholar]
- 55.Chamorro MN, Schwartz DR, Vonica A, Brivanlou AH, Cho KR, Varmus HE. FGF-20 and DKK1 are transcriptional targets of beta-catenin and FGF-20 is implicated in cancer and development. Embo J. 2005;24:73–84. doi: 10.1038/sj.emboj.7600460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shimokawa T, Furukawa Y, Sakai M, Li M, Miwa N, Lin YM, et al. Involvement of the FGF18 gene in colorectal carcinogenesis, as a novel downstream target of the beta-catenin/T-cell factor complex. Cancer research. 2003;63:6116–20. [PubMed] [Google Scholar]
- 57.Shtutman M, Zhurinsky J, Simcha I, Albanese C, D’Amico M, Pestell R, et al. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci U S A. 1999;96:5522–7. doi: 10.1073/pnas.96.10.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–6. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 59.He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, et al. Identification of c-MYC as a target of the APC pathway. Science (New York, NY. 1998;281:1509–12. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 60.Cadoret A, Ovejero C, Terris B, Souil E, Levy L, Lamers WH, et al. New targets of beta-catenin signaling in the liver are involved in the glutamine metabolism. Oncogene. 2002;21:8293–301. doi: 10.1038/sj.onc.1206118. [DOI] [PubMed] [Google Scholar]
- 61.Giera S, Braeuning A, Kohle C, Bursch W, Metzger U, Buchmann A, et al. Wnt/beta-catenin signaling activates and determines hepatic zonal expression of glutathione S-transferases in mouse liver. Toxicol Sci. 2010;115:22–33. doi: 10.1093/toxsci/kfq033. [DOI] [PubMed] [Google Scholar]
- 62.Loeppen S, Koehle C, Buchmann A, Schwarz M. A beta-catenin-dependent pathway regulates expression of cytochrome P450 isoforms in mouse liver tumors. Carcinogenesis. 2005;26:239–48. doi: 10.1093/carcin/bgh298. [DOI] [PubMed] [Google Scholar]
- 63.Nejak-Bowen K, Monga SP. Wnt/beta-catenin signaling in hepatic organogenesis. Organogenesis. 2008;4:92–9. doi: 10.4161/org.4.2.5855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ovejero C, Cavard C, Perianin A, Hakvoort T, Vermeulen J, Godard C, et al. Identification of the leukocyte cell-derived chemotaxin 2 as a direct target gene of beta-catenin in the liver. Hepatology. 2004;40:167–76. doi: 10.1002/hep.20286. [DOI] [PubMed] [Google Scholar]
- 65.Sekine S, Lan BY, Bedolli M, Feng S, Hebrok M. Liver-specific loss of beta-catenin blocks glutamine synthesis pathway activity and cytochrome p450 expression in mice. Hepatology. 2006;43:817–25. doi: 10.1002/hep.21131. [DOI] [PubMed] [Google Scholar]
- 66.Tan X, Behari J, Cieply B, Michalopoulos GK, Monga SP. Conditional deletion of beta-catenin reveals its role in liver growth and regeneration. Gastroenterology. 2006;131:1561–72. doi: 10.1053/j.gastro.2006.08.042. [DOI] [PubMed] [Google Scholar]
- 67.Leung JY, Kolligs FT, Wu R, Zhai Y, Kuick R, Hanash S, et al. Activation of AXIN2 expression by beta-catenin-T cell factor. A feedback repressor pathway regulating Wnt signaling. J Biol Chem. 2002;277:21657–65. doi: 10.1074/jbc.M200139200. [DOI] [PubMed] [Google Scholar]
- 68.Roose J, Huls G, van Beest M, Moerer P, van der Horn K, Goldschmeding R, et al. Synergy between tumor suppressor APC and the beta-catenin-Tcf4 target Tcf1. Science (New York, NY) 1999;285:1923–6. doi: 10.1126/science.285.5435.1923. [DOI] [PubMed] [Google Scholar]
- 69.Cadigan KM, Fish MP, Rulifson EJ, Nusse R. Wingless repression of Drosophila frizzled 2 expression shapes the Wingless morphogen gradient in the wing. Cell. 1998;93:767–77. doi: 10.1016/s0092-8674(00)81438-5. [DOI] [PubMed] [Google Scholar]
- 70.Khan Z, Vijayakumar S, de la Torre TV, Rotolo S, Bafico A. Analysis of endogenous LRP6 function reveals a novel feedback mechanism by which Wnt negatively regulates its receptor. Mol Cell Biol. 2007;27:7291–301. doi: 10.1128/MCB.00773-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Austinat M, Dunsch R, Wittekind C, Tannapfel A, Gebhardt R, Gaunitz F. Correlation between beta-catenin mutations and expression of Wnt-signaling target genes in hepatocellular carcinoma. Mol Cancer. 2008;7:21. doi: 10.1186/1476-4598-7-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cieply B, Zeng G, Proverbs-Singh T, Geller DA, Monga SP. Unique phenotype of hepatocellular cancers with exon-3 mutations in beta-catenin gene. Hepatology. 2009;49:821–31. doi: 10.1002/hep.22695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hoshida Y, Nijman SM, Kobayashi M, Chan JA, Brunet JP, Chiang DY, et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer research. 2009;69:7385–92. doi: 10.1158/0008-5472.CAN-09-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hsu HC, Jeng YM, Mao TL, Chu JS, Lai PL, Peng SY. Beta-catenin mutations are associated with a subset of low-stage hepatocellular carcinoma negative for hepatitis B virus and with favorable prognosis. Am J Pathol. 2000;157:763–70. doi: 10.1016/s0002-9440(10)64590-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zucman-Rossi J, Benhamouche S, Godard C, Boyault S, Grimber G, Balabaud C, et al. Differential effects of inactivated Axin1 and activated beta-catenin mutations in human hepatocellular carcinomas. Oncogene. 2007;26:774–80. doi: 10.1038/sj.onc.1209824. [DOI] [PubMed] [Google Scholar]
- 76.Hovanes K, Li TW, Munguia JE, Truong T, Milovanovic T, Lawrence Marsh J, et al. Beta-catenin-sensitive isoforms of lymphoid enhancer factor-1 are selectively expressed in colon cancer. Nat Genet. 2001;28:53–7. doi: 10.1038/ng0501-53. [DOI] [PubMed] [Google Scholar]
- 77.MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Michalopoulos GK, DeFrances MC. Liver regeneration. Science (New York, NY) 1997;276:60–6. doi: 10.1126/science.276.5309.60. [DOI] [PubMed] [Google Scholar]
- 79.Higgins GM, Anderson RM. Experimental pathology of the liver, 1:Restoration of the liver of the white rat following partial surgical removal. Arch Pathol. 1931;12:186–202. [Google Scholar]
- 80.Michalopoulos GK. Liver regeneration. J Cell Physiol. 2007;213:286–300. doi: 10.1002/jcp.21172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Taub R. Liver regeneration 4: transcriptional control of liver regeneration. Faseb J. 1996;10:413–27. [PubMed] [Google Scholar]
- 82.Su AI, Guidotti LG, Pezacki JP, Chisari FV, Schultz PG. Gene expression during the priming phase of liver regeneration after partial hepatectomy in mice. Proc Natl Acad Sci U S A. 2002;99:11181–6. doi: 10.1073/pnas.122359899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.White P, Brestelli JE, Kaestner KH, Greenbaum LE. Identification of transcriptional networks during liver regeneration. J Biol Chem. 2005;280:3715–22. doi: 10.1074/jbc.M410844200. [DOI] [PubMed] [Google Scholar]
- 84.Walker PR, Potter VR. Isozyme studies on adult, regenerating, precancerous and developing liver in relation to findings in hepatomas. Advances in enzyme regulation. 1972;10:339–64. doi: 10.1016/0065-2571(72)90022-2. [DOI] [PubMed] [Google Scholar]
- 85.Rabes HM. Kinetics of hepatocellular proliferation as a function of the microvascular structure and functional state of the liver. Ciba Found Symp. 1977:31–53. doi: 10.1002/9780470720363.ch3. [DOI] [PubMed] [Google Scholar]
- 86.Stocker E, Heine WD. Regeneration of liver parenchyma under normal and pathological conditions. Beitr Pathol. 1971;144:400–8. [PubMed] [Google Scholar]
- 87.Stocker E, Wullstein HK, Brau G. Capacity of regeneration in liver epithelia of juvenile, repeated partially hepatectomized rats. Autoradiographic studies after continous infusion of 3H-thymidine (author’s transl) Virchows Arch B Cell Pathol. 1973;14:93–103. [PubMed] [Google Scholar]
- 88.Grisham JW. A morphologic study of deoxyribonucleic acid synthesis and cell proliferation in regenerating rat liver; autoradiography with thymidine-H3. Cancer research. 1962;22:842–9. [PubMed] [Google Scholar]
- 89.Nelsen CJ, Rickheim DG, Timchenko NA, Stanley MW, Albrecht JH. Transient expression of cyclin D1 is sufficient to promote hepatocyte replication and liver growth in vivo. Cancer research. 2001;61:8564–8. [PubMed] [Google Scholar]
- 90.Apte U, Zeng G, Thompson MD, Muller P, Micsenyi A, Cieply B, et al. beta-Catenin is critical for early postnatal liver growth. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1578–85. doi: 10.1152/ajpgi.00359.2006. [DOI] [PubMed] [Google Scholar]
- 91.Cadoret A, Ovejero C, Saadi-Kheddouci S, Souil E, Fabre M, Romagnolo B, et al. Hepatomegaly in transgenic mice expressing an oncogenic form of beta-catenin. Cancer research. 2001;61:3245–9. [PubMed] [Google Scholar]
- 92.Harada N, Miyoshi H, Murai N, Oshima H, Tamai Y, Oshima M, et al. Lack of tumorigenesis in the mouse liver after adenovirus-mediated expression of a dominant stable mutant of beta-catenin. Cancer research. 2002;62:1971–7. [PubMed] [Google Scholar]
- 93.Monga SP, Pediaditakis P, Mule K, Stolz DB, Michalopoulos GK. Changes in WNT/beta-catenin pathway during regulated growth in rat liver regeneration. Hepatology. 2001;33:1098–109. doi: 10.1053/jhep.2001.23786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pediaditakis P, Lopez-Talavera JC, Petersen B, Monga SP, Michalopoulos GK. The processing and utilization of hepatocyte growth factor/scatter factor following partial hepatectomy in the rat. Hepatology. 2001;34:688–93. doi: 10.1053/jhep.2001.27811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Haegel H, Larue L, Ohsugi M, Fedorov L, Herrenknecht K, Kemler R. Lack of beta-catenin affects mouse development at gastrulation. Development (Cambridge, England) 1995;121:3529–37. doi: 10.1242/dev.121.11.3529. [DOI] [PubMed] [Google Scholar]
- 96.Grigoryan T, Wend P, Klaus A, Birchmeier W. Deciphering the function of canonical Wnt signals in development and disease: conditional loss- and gain-of-function mutations of beta-catenin in mice. Genes Dev. 2008;22:2308–41. doi: 10.1101/gad.1686208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sodhi D, Micsenyi A, Bowen WC, Monga DK, Talavera JC, Monga SP. Morpholino oligonucleotide-triggered beta-catenin knockdown compromises normal liver regeneration. Journal of hepatology. 2005;43:132–41. doi: 10.1016/j.jhep.2005.02.019. [DOI] [PubMed] [Google Scholar]
- 98.Sekine S, Gutierrez PJ, Lan BY, Feng S, Hebrok M. Liver-specific loss of beta-catenin results in delayed hepatocyte proliferation after partial hepatectomy. Hepatology. 2007;45:361–8. doi: 10.1002/hep.21523. [DOI] [PubMed] [Google Scholar]
- 99.Apte U, Singh S, Zeng G, Cieply B, Virji MA, Wu T, et al. Beta-catenin activation promotes liver regeneration after acetaminophen-induced injury. Am J Pathol. 2009;175:1056–65. doi: 10.2353/ajpath.2009.080976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Colnot S, Decaens T, Niwa-Kawakita M, Godard C, Hamard G, Kahn A, et al. Liver-targeted disruption of Apc in mice activates beta-catenin signaling and leads to hepatocellular carcinomas. Proc Natl Acad Sci U S A. 2004;101:17216–21. doi: 10.1073/pnas.0404761101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Goessling W, North TE, Lord AM, Ceol C, Lee S, Weidinger G, et al. APC mutant zebrafish uncover a changing temporal requirement for wnt signaling in liver development. Dev Biol. 2008;320:161–74. doi: 10.1016/j.ydbio.2008.05.526. [DOI] [PubMed] [Google Scholar]
- 102.Qian J, Sarnaik AA, Bonney TM, Keirsey J, Combs KA, Steigerwald K, et al. The APC tumor suppressor inhibits DNA replication by directly binding to DNA via its carboxyl terminus. Gastroenterology. 2008;135:152–62. doi: 10.1053/j.gastro.2008.03.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Prosperi JR, Becher KR, Willson TA, Collins MH, Witte DP, Goss KH. The APC tumor suppressor is required for epithelial integrity in the mouse mammary gland. J Cell Physiol. 2009;220:319–31. doi: 10.1002/jcp.21766. [DOI] [PubMed] [Google Scholar]
- 104.Nejak-Bowen KN, Thompson MD, Singh S, Bowen WC, Jr, Dar MJ, Khillan J, et al. Accelerated liver regeneration and hepatocarcinogenesis in mice overexpressing serine-45 mutant beta-catenin. Hepatology. 2010;51:1603–13. doi: 10.1002/hep.23538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Manisastry SM, Han M, Linask KK. Early temporal-specific responses and differential sensitivity to lithium and Wnt-3A exposure during heart development. Dev Dyn. 2006;235:2160–74. doi: 10.1002/dvdy.20878. [DOI] [PubMed] [Google Scholar]
- 106.Matthews VB, Yeoh GC. Liver stem cells. IUBMB Life. 2005;57:549–53. doi: 10.1080/15216540500215606. [DOI] [PubMed] [Google Scholar]
- 107.Oertel M, Shafritz DA. Stem cells, cell transplantation and liver repopulation. Biochim Biophys Acta. 2008;1782:61–74. doi: 10.1016/j.bbadis.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Oh SH, Hatch HM, Petersen BE. Hepatic oval ‘stem’ cell in liver regeneration. Semin Cell Dev Biol. 2002;13:405–9. doi: 10.1016/s1084952102001271. [DOI] [PubMed] [Google Scholar]
- 109.Farber E. Similarities in the sequence of early histological changes induced in the liver of the rat by ethionine, 2-acetylamino-fluorene, and 3′-methyl-4-dimethylaminoazobenzene. Cancer research. 1956;16:142–8. [PubMed] [Google Scholar]
- 110.Kuwahara R, Kofman AV, Landis CS, Swenson ES, Barendswaard E, Theise ND. The hepatic stem cell niche: identification by label-retaining cell assay. Hepatology. 2008;47:1994–2002. doi: 10.1002/hep.22218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sell S. Comparison of liver progenitor cells in human atypical ductular reactions with those seen in experimental models of liver injury. Hepatology. 1998;27:317–31. doi: 10.1002/hep.510270202. [DOI] [PubMed] [Google Scholar]
- 112.Yovchev MI, Grozdanov PN, Zhou H, Racherla H, Guha C, Dabeva MD. Identification of adult hepatic progenitor cells capable of repopulating injured rat liver. Hepatology. 2008;47:636–47. doi: 10.1002/hep.22047. [DOI] [PubMed] [Google Scholar]
- 113.Sell S, Leffert HL. Liver cancer stem cells. J Clin Oncol. 2008;26:2800–5. doi: 10.1200/JCO.2007.15.5945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Roskams T. Liver stem cells and their implication in hepatocellular and cholangiocarcinoma. Oncogene. 2006;25:3818–22. doi: 10.1038/sj.onc.1209558. [DOI] [PubMed] [Google Scholar]
- 115.de Lima VM, Oliveira CP, Alves VA, Chammas MC, Oliveira EP, Stefano JT, et al. A rodent model of NASH with cirrhosis, oval cell proliferation and hepatocellular carcinoma. Journal of hepatology. 2008;49:1055–61. doi: 10.1016/j.jhep.2008.07.024. [DOI] [PubMed] [Google Scholar]
- 116.Bennoun M, Rissel M, Engelhardt N, Guillouzo A, Briand P, Weber-Benarous A. Oval cell proliferation in early stages of hepatocarcinogenesis in simian virus 40 large T transgenic mice. Am J Pathol. 1993;143:1326–36. [PMC free article] [PubMed] [Google Scholar]
- 117.Evarts RP, Nagy P, Marsden E, Thorgeirsson SS. A precursor-product relationship exists between oval cells and hepatocytes in rat liver. Carcinogenesis. 1987;8:1737–40. doi: 10.1093/carcin/8.11.1737. [DOI] [PubMed] [Google Scholar]
- 118.Paku S, Schnur J, Nagy P, Thorgeirsson SS. Origin and structural evolution of the early proliferating oval cells in rat liver. Am J Pathol. 2001;158:1313–23. doi: 10.1016/S0002-9440(10)64082-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Petersen BE, Zajac VF, Michalopoulos GK. Bile ductular damage induced by methylene dianiline inhibits oval cell activation. Am J Pathol. 1997;151:905–9. [PMC free article] [PubMed] [Google Scholar]
- 120.Akhurst B, Croager EJ, Farley-Roche CA, Ong JK, Dumble ML, Knight B, et al. A modified choline-deficient, ethionine-supplemented diet protocol effectively induces oval cells in mouse liver. Hepatology. 2001;34:519–22. doi: 10.1053/jhep.2001.26751. [DOI] [PubMed] [Google Scholar]
- 121.Preisegger KH, Factor VM, Fuchsbichler A, Stumptner C, Denk H, Thorgeirsson SS. Atypical ductular proliferation and its inhibition by transforming growth factor beta1 in the 3,5-diethoxycarbonyl-1,4-dihydrocollidine mouse model for chronic alcoholic liver disease. Lab Invest. 1999;79:103–9. [PubMed] [Google Scholar]
- 122.Jelnes P, Santoni-Rugiu E, Rasmussen M, Friis SL, Nielsen JH, Tygstrup N, et al. Remarkable heterogeneity displayed by oval cells in rat and mouse models of stem cell-mediated liver regeneration. Hepatology. 2007;45:1462–70. doi: 10.1002/hep.21569. [DOI] [PubMed] [Google Scholar]
- 123.Fausto N. Liver regeneration and repair: hepatocytes, progenitor cells, and stem cells. Hepatology. 2004;39:1477–87. doi: 10.1002/hep.20214. [DOI] [PubMed] [Google Scholar]
- 124.Lazaro CA, Rhim JA, Yamada Y, Fausto N. Generation of hepatocytes from oval cell precursors in culture. Cancer research. 1998;58:5514–22. [PubMed] [Google Scholar]
- 125.Alison MR, Islam S, Lim S. Stem cells in liver regeneration, fibrosis and cancer: the good, the bad and the ugly. J Pathol. 2009;217:282–98. doi: 10.1002/path.2453. [DOI] [PubMed] [Google Scholar]
- 126.Sell S. Cellular origin of hepatocellular carcinomas. Semin Cell Dev Biol. 2002;13:419–24. doi: 10.1016/s1084952102001295. [DOI] [PubMed] [Google Scholar]
- 127.Ma S, Chan KW, Hu L, Lee TK, Wo JY, Ng IO, et al. Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology. 2007;132:2542–56. doi: 10.1053/j.gastro.2007.04.025. [DOI] [PubMed] [Google Scholar]
- 128.Suetsugu A, Nagaki M, Aoki H, Motohashi T, Kunisada T, Moriwaki H. Characterization of CD133+ hepatocellular carcinoma cells as cancer stem/progenitor cells. Biochem Biophys Res Commun. 2006;351:820–4. doi: 10.1016/j.bbrc.2006.10.128. [DOI] [PubMed] [Google Scholar]
- 129.Rountree CB, Senadheera S, Mato JM, Crooks GM, Lu SC. Expansion of liver cancer stem cells during aging in methionine adenosyltransferase 1A-deficient mice. Hepatology. 2008;47:1288–97. doi: 10.1002/hep.22141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lee JS, Heo J, Libbrecht L, Chu IS, Kaposi-Novak P, Calvisi DF, et al. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat Med. 2006;12:410–6. doi: 10.1038/nm1377. [DOI] [PubMed] [Google Scholar]
- 131.Vander Borght S, Komuta M, Libbrecht L, Katoonizadeh A, Aerts R, Dymarkowski S, et al. Expression of multidrug resistance-associated protein 1 in hepatocellular carcinoma is associated with a more aggressive tumour phenotype and may reflect a progenitor cell origin. Liver Int. 2008;28:1370–80. doi: 10.1111/j.1478-3231.2008.01889.x. [DOI] [PubMed] [Google Scholar]
- 132.Yamashita T, Ji J, Budhu A, Forgues M, Yang W, Wang HY, et al. EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology. 2009;136:1012–24. doi: 10.1053/j.gastro.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Yamashita T, Budhu A, Forgues M, Wang XW. Activation of hepatic stem cell marker EpCAM by Wnt-beta-catenin signaling in hepatocellular carcinoma. Cancer research. 2007;67:10831–9. doi: 10.1158/0008-5472.CAN-07-0908. [DOI] [PubMed] [Google Scholar]
- 134.Fodde R, Brabletz T. Wnt/beta-catenin signaling in cancer stemness and malignant behavior. Curr Opin Cell Biol. 2007;19:150–8. doi: 10.1016/j.ceb.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 135.Nusse R, Fuerer C, Ching W, Harnish K, Logan C, Zeng A, et al. Wnt signaling and stem cell control. Cold Spring Harb Symp Quant Biol. 2008;73:59–66. doi: 10.1101/sqb.2008.73.035. [DOI] [PubMed] [Google Scholar]
- 136.Hu M, Kurobe M, Jeong YJ, Fuerer C, Ghole S, Nusse R, et al. Wnt/beta-catenin signaling in murine hepatic transit amplifying progenitor cells. Gastroenterology. 2007;133:1579–91. doi: 10.1053/j.gastro.2007.08.036. [DOI] [PubMed] [Google Scholar]
- 137.Apte U, Thompson MD, Cui S, Liu B, Cieply B, Monga SP. Wnt/beta-catenin signaling mediates oval cell response in rodents. Hepatology. 2008;47:288–95. doi: 10.1002/hep.21973. [DOI] [PubMed] [Google Scholar]
- 138.Zhang Y, Li XM, Zhang FK, Wang BE. Activation of canonical Wnt signaling pathway promotes proliferation and self-renewal of rat hepatic oval cell line WB-F344 in vitro. World J Gastroenterol. 2008;14:6673–80. doi: 10.3748/wjg.14.6673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Itoh T, Kamiya Y, Okabe M, Tanaka M, Miyajima A. Inducible expression of Wnt genes during adult hepatic stem/progenitor cell response. FEBS Lett. 2009;583:777–81. doi: 10.1016/j.febslet.2009.01.022. [DOI] [PubMed] [Google Scholar]
- 140.Williams JM, Oh SH, Jorgensen M, Steiger N, Darwiche H, Shupe T, et al. The role of the Wnt family of secreted proteins in rat oval “stem” cell-based liver regeneration: Wnt1 drives differentiation. Am J Pathol. 2010;176:2732–42. doi: 10.2353/ajpath.2010.080486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Spee B, Carpino G, Schotanus BA, Katoonizadeh A, Vander Borght S, Gaudio E, et al. Characterisation of the liver progenitor cell niche in liver diseases: potential involvement of Wnt and Notch signalling. Gut. 2010;59:247–57. doi: 10.1136/gut.2009.188367. [DOI] [PubMed] [Google Scholar]
- 142.Chiba T, Zheng YW, Kita K, Yokosuka O, Saisho H, Onodera M, et al. Enhanced self-renewal capability in hepatic stem/progenitor cells drives cancer initiation. Gastroenterology. 2007;133:937–50. doi: 10.1053/j.gastro.2007.06.016. [DOI] [PubMed] [Google Scholar]
- 143.Yang W, Yan HX, Chen L, Liu Q, He YQ, Yu LX, et al. Wnt/beta-catenin signaling contributes to activation of normal and tumorigenic liver progenitor cells. Cancer research. 2008;68:4287–95. doi: 10.1158/0008-5472.CAN-07-6691. [DOI] [PubMed] [Google Scholar]
- 144.Kitisin K, Pishvaian MJ, Johnson LB, Mishra L. Liver stem cells and molecular signaling pathways in hepatocellular carcinoma. Gastrointest Cancer Res. 2007;1:S13–21. [PMC free article] [PubMed] [Google Scholar]
- 145.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010 doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 146.Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer. 2006;6:674–87. doi: 10.1038/nrc1934. [DOI] [PubMed] [Google Scholar]
- 147.Villanueva A, Newell P, Chiang DY, Friedman SL, Llovet JM. Genomics and signaling pathways in hepatocellular carcinoma. Semin Liver Dis. 2007;27:55–76. doi: 10.1055/s-2006-960171. [DOI] [PubMed] [Google Scholar]
- 148.Wong CM, Ng IO. Molecular pathogenesis of hepatocellular carcinoma. Liver Int. 2008;28:160–74. doi: 10.1111/j.1478-3231.2007.01637.x. [DOI] [PubMed] [Google Scholar]
- 149.Lok AS, Seeff LB, Morgan TR, di Bisceglie AM, Sterling RK, Curto TM, et al. Incidence of hepatocellular carcinoma and associated risk factors in hepatitis C-related advanced liver disease. Gastroenterology. 2009;136:138–48. doi: 10.1053/j.gastro.2008.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789–99. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 151.Bressac B, Kew M, Wands J, Ozturk M. Selective G to T mutations of p53 gene in hepatocellular carcinoma from southern Africa. Nature. 1991;350:429–31. doi: 10.1038/350429a0. [DOI] [PubMed] [Google Scholar]
- 152.Hsu IC, Metcalf RA, Sun T, Welsh JA, Wang NJ, Harris CC. Mutational hotspot in the p53 gene in human hepatocellular carcinomas. Nature. 1991;350:427–8. doi: 10.1038/350427a0. [DOI] [PubMed] [Google Scholar]
- 153.de La Coste A, Romagnolo B, Billuart P, Renard CA, Buendia MA, Soubrane O, et al. Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc Natl Acad Sci U S A. 1998;95:8847–51. doi: 10.1073/pnas.95.15.8847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Yao YJ, Ping XL, Zhang H, Chen FF, Lee PK, Ahsan H, et al. PTEN/MMAC1 mutations in hepatocellular carcinomas. Oncogene. 1999;18:3181–5. doi: 10.1038/sj.onc.1202659. [DOI] [PubMed] [Google Scholar]
- 155.Zender L, Villanueva A, Tovar V, Sia D, Chiang DY, Llovet JM. Cancer gene discovery in hepatocellular carcinoma. Journal of hepatology. 2010;52:921–9. doi: 10.1016/j.jhep.2009.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Finn RS, Zhu AX. Targeting angiogenesis in hepatocellular carcinoma: focus on VEGF and bevacizumab. Expert Rev Anticancer Ther. 2009;9:503–9. doi: 10.1586/era.09.6. [DOI] [PubMed] [Google Scholar]
- 157.Harada N, Oshima H, Katoh M, Tamai Y, Oshima M, Taketo MM. Hepatocarcinogenesis in mice with beta-catenin and Ha-ras gene mutations. Cancer research. 2004;64:48–54. doi: 10.1158/0008-5472.can-03-2123. [DOI] [PubMed] [Google Scholar]
- 158.Miyoshi H, Deguchi A, Nakau M, Kojima Y, Mori A, Oshima M, et al. Hepatocellular carcinoma development induced by conditional beta-catenin activation in Lkb1+/− mice. Cancer Sci. 2009;100:2046–53. doi: 10.1111/j.1349-7006.2009.01284.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Bauer-Hofmann R, Klimek F, Buchmann A, Muller O, Bannasch P, Schwarz M. Role of mutations at codon 61 of the c-Ha-ras gene during diethylnitrosamine-induced hepatocarcinogenesis in C3H/He mice. Mol Carcinog. 1992;6:60–7. doi: 10.1002/mc.2940060110. [DOI] [PubMed] [Google Scholar]
- 160.Haramis AP, Hurlstone A, van der Velden Y, Begthel H, van den Born M, Offerhaus GJ, et al. Adenomatous polyposis coli-deficient zebrafish are susceptible to digestive tract neoplasia. EMBO Rep. 2006;7:444–9. doi: 10.1038/sj.embor.7400638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Aydinlik H, Nguyen TD, Moennikes O, Buchmann A, Schwarz M. Selective pressure during tumor promotion by phenobarbital leads to clonal outgrowth of beta-catenin-mutated mouse liver tumors. Oncogene. 2001;20:7812–6. doi: 10.1038/sj.onc.1204982. [DOI] [PubMed] [Google Scholar]
- 162.Schwarz M, Wanke I, Wulbrand U, Moennikes O, Buchmann A. Role of connexin32 and beta-catenin in tumor promotion in mouse liver. Toxicol Pathol. 2003;31:99–102. doi: 10.1080/01926230390173932. [DOI] [PubMed] [Google Scholar]
- 163.Ogawa K, Yamada Y, Kishibe K, Ishizaki K, Tokusashi Y. Beta-catenin mutations are frequent in hepatocellular carcinomas but absent in adenomas induced by diethylnitrosamine in B6C3F1 mice. Cancer research. 1999;59:1830–3. [PubMed] [Google Scholar]
- 164.Devereux TR, Anna CH, Foley JF, White CM, Sills RC, Barrett JC. Mutation of beta-catenin is an early event in chemically induced mouse hepatocellular carcinogenesis. Oncogene. 1999;18:4726–33. doi: 10.1038/sj.onc.1202858. [DOI] [PubMed] [Google Scholar]
- 165.Calvisi DF, Ladu S, Factor VM, Thorgeirsson SS. Activation of beta-catenin provides proliferative and invasive advantages in c-myc/TGF-alpha hepatocarcinogenesis promoted by phenobarbital. Carcinogenesis. 2004;25:901–8. doi: 10.1093/carcin/bgh083. [DOI] [PubMed] [Google Scholar]
- 166.Calvisi DF, Conner EA, Ladu S, Lemmer ER, Factor VM, Thorgeirsson SS. Activation of the canonical Wnt/beta-catenin pathway confers growth advantages in c-Myc/E2F1 transgenic mouse model of liver cancer. Journal of hepatology. 2005;42:842–9. doi: 10.1016/j.jhep.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 167.Calvisi DF, Factor VM, Loi R, Thorgeirsson SS. Activation of beta-catenin during hepatocarcinogenesis in transgenic mouse models: relationship to phenotype and tumor grade. Cancer research. 2001;61:2085–91. [PubMed] [Google Scholar]
- 168.Zulehner G, Mikula M, Schneller D, van Zijl F, Huber H, Sieghart W, et al. Nuclear beta-catenin induces an early liver progenitor phenotype in hepatocellular carcinoma and promotes tumor recurrence. Am J Pathol. 2010;176:472–81. doi: 10.2353/ajpath.2010.090300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Zhang XF, Tan X, Zeng G, Misse A, Singh S, Kim Y, et al. Conditional beta-catenin loss in mice promotes chemical hepatocarcinogenesis: role of oxidative stress and platelet-derived growth factor receptor alpha/phosphoinositide 3-kinase signaling. Hepatology. 2010;52:954–65. doi: 10.1002/hep.23747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Rignall B, Braeuning A, Buchmann A, Schwarz M. Tumor Formation in Liver of Conditional {beta}-Catenin-Deficient Mice Exposed to a Diethylnitrosamine/Phenobarbital Tumor Promotion Regimen. Carcinogenesis. 2010 doi: 10.1093/carcin/bgq226. [DOI] [PubMed] [Google Scholar]
- 171.Fujie H, Moriya K, Shintani Y, Tsutsumi T, Takayama T, Makuuchi M, et al. Frequent beta-catenin aberration in human hepatocellular carcinoma. Hepatol Res. 2001;20:39–51. doi: 10.1016/s1386-6346(00)00116-9. [DOI] [PubMed] [Google Scholar]
- 172.Huang H, Fujii H, Sankila A, Mahler-Araujo BM, Matsuda M, Cathomas G, et al. Beta-catenin mutations are frequent in human hepatocellular carcinomas associated with hepatitis C virus infection. Am J Pathol. 1999;155:1795–801. doi: 10.1016/s0002-9440(10)65496-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kim YD, Park CH, Kim HS, Choi SK, Rew JS, Kim DY, et al. Genetic alterations of Wnt signaling pathway-associated genes in hepatocellular carcinoma. J Gastroenterol Hepatol. 2008;23:110–8. doi: 10.1111/j.1440-1746.2007.05250.x. [DOI] [PubMed] [Google Scholar]
- 174.Kondo Y, Kanai Y, Sakamoto M, Genda T, Mizokami M, Ueda R, et al. Beta-catenin accumulation and mutation of exon 3 of the beta-catenin gene in hepatocellular carcinoma. Jpn J Cancer Res. 1999;90:1301–9. doi: 10.1111/j.1349-7006.1999.tb00712.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Satoh S, Daigo Y, Furukawa Y, Kato T, Miwa N, Nishiwaki T, et al. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat Genet. 2000;24:245–50. doi: 10.1038/73448. [DOI] [PubMed] [Google Scholar]
- 176.Taniguchi K, Roberts LR, Aderca IN, Dong X, Qian C, Murphy LM, et al. Mutational spectrum of beta-catenin, AXIN1, and AXIN2 in hepatocellular carcinomas and hepatoblastomas. Oncogene. 2002;21:4863–71. doi: 10.1038/sj.onc.1205591. [DOI] [PubMed] [Google Scholar]
- 177.Merle P, de la Monte S, Kim M, Herrmann M, Tanaka S, Von Dem Bussche A, et al. Functional consequences of frizzled-7 receptor overexpression in human hepatocellular carcinoma. Gastroenterology. 2004;127:1110–22. doi: 10.1053/j.gastro.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 178.Nambotin SB, Lefrancois L, Sainsily X, Berthillon P, Kim M, Wands JR, et al. Pharmacological inhibition of Frizzled-7 displays anti-tumor properties in hepatocellular carcinoma. Journal of hepatology. 2010 doi: 10.1016/j.jhep.2010.06.033. [DOI] [PubMed] [Google Scholar]
- 179.Ban KC, Singh H, Krishnan R, Seow HF. GSK-3beta phosphorylation and alteration of beta-catenin in hepatocellular carcinoma. Cancer Lett. 2003;199:201–8. doi: 10.1016/s0304-3835(03)00421-x. [DOI] [PubMed] [Google Scholar]
- 180.Shih YL, Shyu RY, Hsieh CB, Lai HC, Liu KY, Chu TY, et al. Promoter methylation of the secreted frizzled-related protein 1 gene SFRP1 is frequent in hepatocellular carcinoma. Cancer. 2006;107:579–90. doi: 10.1002/cncr.22023. [DOI] [PubMed] [Google Scholar]
- 181.Wong CM, Fan ST, Ng IO. beta-Catenin mutation and overexpression in hepatocellular carcinoma: clinicopathologic and prognostic significance. Cancer. 2001;92:136–45. doi: 10.1002/1097-0142(20010701)92:1<136::aid-cncr1301>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 182.Fukutomi T, Zhou Y, Kawai S, Eguchi H, Wands JR, Li J. Hepatitis C virus core protein stimulates hepatocyte growth: correlation with upregulation of wnt-1 expression. Hepatology. 2005;41:1096–105. doi: 10.1002/hep.20668. [DOI] [PubMed] [Google Scholar]
- 183.Cha MY, Kim CM, Park YM, Ryu WS. Hepatitis B virus X protein is essential for the activation of Wnt/beta-catenin signaling in hepatoma cells. Hepatology. 2004;39:1683–93. doi: 10.1002/hep.20245. [DOI] [PubMed] [Google Scholar]
- 184.Lian Z, Liu J, Li L, Li X, Clayton M, Wu MC, et al. Enhanced cell survival of Hep3B cells by the hepatitis B x antigen effector, URG11, is associated with upregulation of beta-catenin. Hepatology. 2006;43:415–24. doi: 10.1002/hep.21053. [DOI] [PubMed] [Google Scholar]
- 185.Polakis P. Wnt signaling and cancer. Genes Dev. 2000;14:1837–51. [PubMed] [Google Scholar]
- 186.Paul S, Dey A. Wnt signaling and cancer development: therapeutic implication. Neoplasma. 2008;55:165–76. [PubMed] [Google Scholar]
- 187.Monga SP. Role of Wnt/beta-catenin signaling in liver metabolism and cancer. Int J Biochem Cell Biol. 2009 doi: 10.1016/j.biocel.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Boyault S, Rickman DS, de Reynies A, Balabaud C, Rebouissou S, Jeannot E, et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology. 2007;45:42–52. doi: 10.1002/hep.21467. [DOI] [PubMed] [Google Scholar]
- 189.Audard V, Grimber G, Elie C, Radenen B, Audebourg A, Letourneur F, et al. Cholestasis is a marker for hepatocellular carcinomas displaying beta-catenin mutations. J Pathol. 2007;212:345–52. doi: 10.1002/path.2169. [DOI] [PubMed] [Google Scholar]
- 190.Nhieu JT, Renard CA, Wei Y, Cherqui D, Zafrani ES, Buendia MA. Nuclear accumulation of mutated beta-catenin in hepatocellular carcinoma is associated with increased cell proliferation. Am J Pathol. 1999;155:703–10. doi: 10.1016/s0002-9440(10)65168-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Park JY, Park WS, Nam SW, Kim SY, Lee SH, Yoo NJ, et al. Mutations of beta-catenin and AXIN I genes are a late event in human hepatocellular carcinogenesis. Liver Int. 2005;25:70–6. doi: 10.1111/j.1478-3231.2004.0995.x. [DOI] [PubMed] [Google Scholar]
- 192.Suzuki T, Yano H, Nakashima Y, Nakashima O, Kojiro M. Beta-catenin expression in hepatocellular carcinoma: a possible participation of beta-catenin in the dedifferentiation process. J Gastroenterol Hepatol. 2002;17:994–1000. doi: 10.1046/j.1440-1746.2002.02774.x. [DOI] [PubMed] [Google Scholar]
- 193.Mao TL, Chu JS, Jeng YM, Lai PL, Hsu HC. Expression of mutant nuclear beta-catenin correlates with non-invasive hepatocellular carcinoma, absence of portal vein spread, and good prognosis. J Pathol. 2001;193:95–101. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH720>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 194.Yuan RH, Jeng YM, Chen HL, Hsieh FJ, Yang CY, Lee PH, et al. Opposite roles of human pancreatitis-associated protein and REG1A expression in hepatocellular carcinoma: association of pancreatitis-associated protein expression with low-stage hepatocellular carcinoma, beta-catenin mutation, and favorable prognosis. Clin Cancer Res. 2005;11:2568–75. doi: 10.1158/1078-0432.CCR-04-2039. [DOI] [PubMed] [Google Scholar]
- 195.Yuan RH, Jeng YM, Hu RH, Lai PL, Lee PH, Cheng CC, et al. Role of p53 and beta-catenin Mutations in Conjunction with CK19 Expression on Early Tumor Recurrence and Prognosis of Hepatocellular Carcinoma. J Gastrointest Surg. 2010 doi: 10.1007/s11605-010-1373-x. [DOI] [PubMed] [Google Scholar]
- 196.Dal Bello B, Rosa L, Campanini N, Tinelli C, Torello Viera F, D’Ambrosio G, et al. Glutamine synthetase immunostaining correlates with pathologic features of hepatocellular carcinoma and better survival after radiofrequency thermal ablation. Clin Cancer Res. 2010;16:2157–66. doi: 10.1158/1078-0432.CCR-09-1978. [DOI] [PubMed] [Google Scholar]
- 197.Zucman-Rossi J, Jeannot E, Nhieu JT, Scoazec JY, Guettier C, Rebouissou S, et al. Genotype-phenotype correlation in hepatocellular adenoma: new classification and relationship with HCC. Hepatology. 2006;43:515–24. doi: 10.1002/hep.21068. [DOI] [PubMed] [Google Scholar]
- 198.Llovet JM, Bruix J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology. 2008;48:1312–27. doi: 10.1002/hep.22506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Yang JD, Nakamura I, Roberts LR. The tumor microenvironment in hepatocellular carcinoma: Current status and therapeutic targets. Semin Cancer Biol. 2010 doi: 10.1016/j.semcancer.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]