

Abstract
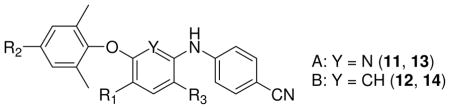
Based on the structures and activities of our previously identified non-nucleoside reverse transcriptase inhibitors (NNRTIs), we designed and synthesized two sets of derivatives, diarylpyridines (A) and diarylanilines (B), and tested their anti-HIV-1 activity against infection by HIV-1 NL4-3 and IIIB in TZM-bl and MT-2 cells, respectively. The results showed that most compounds exhibited potent anti-HIV-1 activity with low nanomolar EC50 values, and some of them, such as 13m, 14c, and 14e, displayed high potency with subnanomolar EC50 values, which were more potent than etravirine (TMC125, 1) in the same assays. Notably, these compounds were also highly effective against infection by multi-RTI-resistant strains, suggesting a high potential to further develop these compounds as a novel class of NNRTIs with improved antiviral efficacy and resistance profile.
Introduction
Acquired immunodeficiency syndrome (AIDS), caused by human immunodeficiency virus (HIV), threatens human health and life, spreading rapidly worldwide and resulting in more than 60 million people infected by HIV and about 25 million patients dying of AIDS (www.unaids.org). Because there is no effective vaccine to prevent HIV infection, development of anti-HIV therapeutics is critical to improve the quality and save the lives of HIV infected individuals. To date, 26 anti-HIV drugs have been approved for the clinical treatment of HIV infection and AIDS (www.fda.gov/oashi/aids/virals.html), including reverse transcriptase inhibitors (RTIs), protease inhibitors (PIs), integrase inhibitors, fusion inhibitor, and entry inhibitor (CCR5 co-receptor antagonist). Highly active antiretroviral therapies (HAART), which use a combination of three to four drugs, can significantly reduce the morbidity and mortality of AIDS. However, as a result of emerging drug-resistant HIV mutants, increasing numbers of HIV-infected patients cannot use or fail to respond to HAART. Therefore, the development of new anti-HIV drugs is urgently required.
HIV-1 reverse transcriptase (RT) is one of the most important viral enzymes and plays a unique role in the HIV-1 life cycle. It has two known drug-target sites, the substrate catalytic site and an allosteric site that is distinct from, but located closely to, the substrate site1,2. Non-nucleoside reverse transcriptase inhibitors (NNRTIs) interact with the allosteric site in a noncompetitive manner to distort the enzyme’s active conformation and thus disrupt the function of the enzyme. The first-generation NNRTI drugs (nevirapine, delavirdine, and efavirenz) exhibit very potent anti-HIV-1 activity and low toxicity. However, rapid drug-resistance emergence, due to single point mutations (especially K103 mutant)3 in the NNRTI binding site, compromises their clinical usefulness. Etravirine (TMC125, 1)4, a diarylpyrimidine (DAPY, Figure 1), was recently approved as a next-generation NNRTI for AIDS therapy. It exhibits high potency against wild-type and a number of mutated viral strains with nanomolar EC50 values and has a higher genetic barrier5 to delay the emergence of drug-resistance. The success of 1 greatly encouraged further research to explore additional novel NNRTIs.6,7,8 The most advanced NNRTI in development is another DAPY derivative rilpivirine (TMC278, 2)9 in Phase III, which showed better potency and pharmacological profiles than 1, such as once-daily administration.10
Figure 1.

Drugs (1 and 2), leads (3 and 4), and new target compounds
By using an isosteric replacement strategy on the central pyrimidine ring of DAPYs, our prior studies discovered two series of active compounds, diarylpyridine (A, DAPD)11 with one pyrimidine nitrogen replaced by carbon and diarylaniline (B, DAAN)12 with both pyrimidine nitrogens replaced by carbon. Exemplary di-para-cyanophenylpyridine compound 3 and di-para-cyanophenylaniline compound 4 (Figure 1) exhibited high potency against HIV-1 wild-type and RT-resistant viral strains. Previous SAR studies also indicated that the para-cyanoaniline moiety (A-ring) is necessary and the amino group on the central ring, either pyridine or benzene (B-ring), ortho to the A-ring is quite crucial for enhancing anti-HIV activity in both the A and B series. On the other hand, the para-substituent on the phenoxy ring (C-ring) was modifiable and could greatly affect the anti-HIV potency. We hypothesized that the amino group on the central ring might form additional H-bonds with the key amino acid K101 on the NNRTI binding site, and thus, compensate for the loss of the H-bond between K101 and the nitrogen on the pyrimidine ring in DAPYs.13 Our previous molecular modeling results provided rational support for our hypothesis.12 In our continuing studies, we have made further structural modifications on the C-ring moiety of A and B series of derivatives. The aims are to investigate how the C-ring moiety will affect anti-HIV activity and develop novel classes of NNRTIs with improved antiviral efficacy. Thus, we report herein the design, synthesis, and anti-HIV activity of two sets of new target A and B compounds (Figure 1).
The crystal structures of complexes HIV-1 RT/2 [protein database (PDB) code: 2zd1] and K103N/Y181C HIV-1 RT/2 (PDB: 3bgr)14 reveal that the linear cyanovinyl in 2 is embedded deep into a cylindrical hydrophobic tunnel, which connects the NNRTI-binding pocket to the nucleic acid-binding cleft, and is favored to interact with the more conservable amino acids W229 and Y183. Thus, the para-cyanovinyl group on the C-ring of 2 is thought to be an important moiety for enhancing activity. On the basis of the information about the binding site and our previous SAR results, we designed new target compounds in the A and B series with diverse para-substituents (R2) on the phenoxy ring (C-ring). The R2 groups included cyanovinyl and other linear substituents, which could likely insert into the cylindrical hydrophobic tunnel of the binding site and reach the conserved amino acid W229. The R1 and R3 substituents on the central pyridine or benzene ring were limited to NH2, NO2, and H.
Chemistry
Totally, 34 target compounds (A and B series, Figure 1) were synthesized via the short routes detailed in Schemes 1–4. Six of them (11a, 11d, 11f, 13a, 13d, and 13f) have been previously described,11 but without the full experimental details presented herein. The starting materials 4-aminobenzonitrile, 2,6-dichloro-3-nitro pyridine (5), 2,4-dichloronitrobenzene (6), and 2,4-dichloro-1,5-dinitrobenzene (7) are inexpensive and commercially available. Coupling of 4-aminobenzonitrile and 5 was performed by heating at 140 °C for 4 h without solvent under nitrogen to provide intermediate 4-(6-chloro-3- nitropyridin-2-ylamino)benzonitrile (8) in 73% yield. The same reaction of dichloronitrobenzene 6 or 7 with 4-aminobenzonitrile took place in DMF in the presence of triethylamine or potassium tert-butoxide (t-BuOK) at room temperature to produce corresponding intermediates N-(4-cyanophenyl)-5-chloro-2-nitroaniline (9) or N-(4-cyanophenyl)-5-chloro-2,4-dinitroaniline (10) in 64% and 89% yields, respectively. Subsequently, intermediate 8 was reacted with a trisubstituted phenol in DMF in the presence of potassium carbonate by traditional heating at 130–140 °C for 6–8 h to afford corresponding 2,6-diaryl-3-nitrophyridines 11a-11c and 11g with yields ranging from 56 to 82%. Alternatively, N1,5-diarylnitroanilines 12a and 12b were prepared from 9 or 10 by coupling with a substituted phenol under microwave irradiation in DMF in the presence of potassium carbonate12. As shown in Scheme 2, a Sonogashira coupling reaction between 11a and 2-methyl-3-butyn-2-ol or ethynylcyclopropane took place in the presence of triethylamine and palladium-copper catalyst to give corresponding 11d and 11e, respectively, with different elongated para-ethynyl substituents on the C-ring. Next, the para-substituent on the C-ring of 11d was removed to convert into 11f by refluxing in dry toluene in the presence of powdered sodium hydroxide in a 69% yield. The nitro-containing compounds 11a-11f were reduced by using sodium hydrosulfite dehydrate15 or nickel(II) chloride hydrate and sodium borohydride to afford corresponding amino-compounds 13a–13f with yields ranging from 43 to 82%. Moreover, the synthesis of other compounds with different elongated R2 substituents is indicated in Scheme 3. The condensation of 11g with nitromethane, malonic acid, or acetone under basic condition afforded 11i, 11j, or 11k, respectively, with a para-linear side chain on the phenoxy ring (Scheme 3). The cyanovinyl compounds 11m, 12c and 12e were prepared by the condensation of diethyl cyanomethyl phosphonate with aldehyde-compounds 11g, 12a or 12b, respectively, in the presence of potassium terbutoxide16 in 53–91% yields. Subsequently, diaryl-nitropyridines 11i-11m and diaryl-mononitroaniline 12c were reduced by sodium hydrosulfite dehydrate to provide corresponding diary-pyridinamines 13i, 13j, and 13m, diaryaniline 14c, and an unexpected reduction product 13k, in which both the nitro group and the para-conjugated double bond on the phenoxy ring were reduced, albeit in low yield. To improve the yield, 13k was then prepared from 11k by using catalytic hydrogenation in THF in 79% yield. Alternatively, selective reduction of diaryl-dinitroanilines 12d and 12e was achieved by using formic acid in the presence of Pd-C (10%) and triethylamine to give corresponding diaryl-4-nitrobenzene-1,2-diamines 14d and 14e respectively, in which the reduced amino group was identified as ortho to the NH-linked aniline.12 Additionally, 12e was reduced completely by using sodium borohydride in the presence of antimony (III) chloride (SbCl3) to provide diamino compound 14f. Scheme 4 shows the syntheses of compounds 15-20 with a 6-cyanonaphthoxy moiety as the C-ring. The coupling of 8 and 10 with 6-cyano-2-naphthol or 1-bromo-6-cyano-2-naphthol by using microwave irradiation followed by the nitro reduction or selective reduction afforded corresponding diarylpyridines 15-16 and diarylanilines 17-20, respectively, in which the para-cyanovinyl group was incorporated into a fused ring system.
Scheme 1.
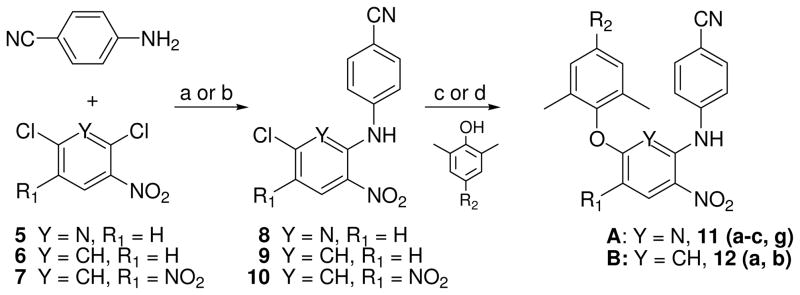
(a) heating 140 °C without solvent, 4 h; (b) t-BuOK/DMF, rt, 24 h.; (c) K2CO3/DMF, 130–140 °C, 6–8 h; (d) K2CO3/DMF, 190 °C, MW, 15 min.
Scheme 4.
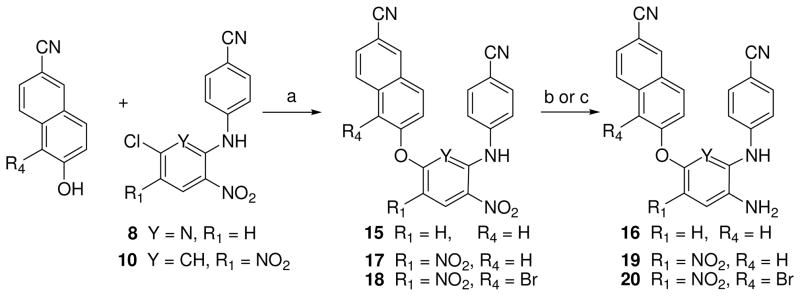
(a) MW, 100–150 °C, 10–20 min; (b) Na2S2O4, NH3·H2O, THF or 1,4-dioxane/H2O (1:1), r.t. 3 h; (c) HCOOH/Pd-C, Et3N/CH3CN, reflux, 1–3 h.
Scheme 2.
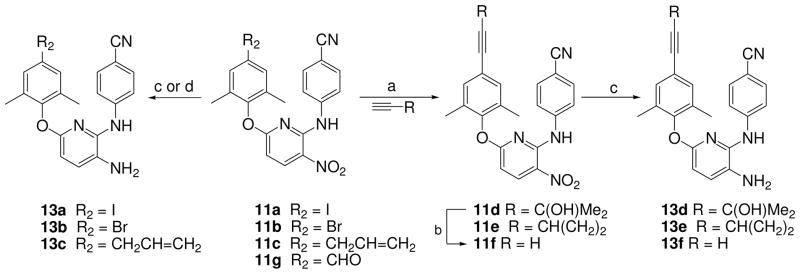
(a) Pd(PPh3)Cl2, CuI, DMF/Et3N, N2 protection; (b) NaOH, toluene, reflux, 16 h; (c) Na2S2O4, NH3·H2O, THF/H2O (v/v 1:1 ), rt; (d) NiCl2·6H2O/NaBH4, THF/MeOH (v/v 1:1 ).
Scheme 3.

(a) CH3NO2, NaOH/THF, rt; (b) CH2(COOH)2, piperidine/Py, 2 h, reflux; (c) (EtO)2P(O)CH2CN, t-BuOK/THF, 0 °C – rt, 48 h; (d) Na2S2O4, NH3·H2O, THF/H2O (1:1), rt, 3 h; (e) 20% HCl/ether in acetone; (f) acetone, NaOH, rt; (g) NaBH4, SbCl3; (h) HCOOH/Pd-C, Et3N/CH3CN, reflux, 1–3 h; (i) H2, Pd-C/THF, 6 h.
Results and discussion
The synthesized diarylpyridines 11, 13, 15, 16 (A series) and diarylanilines 12c-12e, 14, 17-20 (B series) were first tested against infection by wild-type HIV-1 strains NL4-3 and IIIB of TZM-bl and MT-2 cells, respectively, with 1 tested in parallel. HIV-1 was generally quite sensitive to the compounds (low EC50 values), and both assays provided similar patterns of antiviral activity. As shown in Table 1, most compounds, except 11d, 11e, and 11j (EC50 >3 μM), were highly potent in both assays. Notably, most amino-compounds (R3 = NH2), including 13a–13f, 13i-13m, and 14c-14f, were much more potent (at least ten-fold) than the corresponding nitropyridine and nitrobenzene compounds (11 and 12, R3 = NO2). The most potent compounds were 13m, 14c, and 14e with EC50 values ≤ 0.7 nM against HIV-1 NL4-3 infection of TZM-bl cells and <14 nM against HIV-1 IIIB replication in MT-2 cells. These three compounds exhibited great potency than 1 (EC50 = 1.4 and 41 nM against NL4-2 and IIIB, respectively) and comparable potency to 3. These results further support our previous hypothesis that the amino group on the central ring at the ortho-position to the 4-cyanoaniline A-ring is crucial for enhancing anti-HIV activity.
Table 1.
Data of target compounds 11-14 against HIV-1 wild-type strain replication
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| R1 | R2 | R3 | NL4-3 in TZM-bl cell line | IIIB in MT-2 cell line | |||||
| EC50 a (μM) | CC50b (μM) | SI c | EC50 (μM) | CC50 (μM) | SI | ||||
| 11a* | H | I | NO2 | 0.0607 ± 0.024 | >41.15 | >675 | 0.079 ± 0.002 | 22.93 | 283 |
| 13a* | H | I | NH2 | 0.0076 ± 0.0023 | 16.72 | 2200 | 0.054 ± 0.004 | 7.24 | 134 |
| 11b | H | Br | NO2 | 0.0502 ± 0.0068 | >45.66 | >909 | 0.045 ± 0.016 | 65.11 | 1447 |
| 13b | H | Br | NH2 | 0.00729 ± 0.0020 | 15.54 | 2132 | 0.033 ± 0.01 | 9.61 | 291 |
| 11c | H | CH2CH=CH2 | NO2 | NA | 1.34 ± 0.70 | 39..68 | 27.37 | ||
| 13c | H | CH2CH=CH2 | NH2 | 0.00689 ± 0.0014 | 8.46 | 1228 | 0.067 ± 0.003 | 14.02 | 209 |
| 11d* | H | C≡CC(OH)Me2 | NO2 | 41.35 | 45.25 | 1.09 | 8.91 ± 0.00 | 23.28 | 2.61 |
| 13d* | H | C≡CC(OH)Me2 | NH2 | 0.241 ± 0.0147 | 8.73 | 36 | 1.58 ± 0.097 | 3.54 | 2.25 |
| 11e | H | C≡CCH(CH2)2 | NO2 | >47.17 | — | NA | |||
| 13e | H | C≡CCH(CH2)2 | NH2 | 0.0912 ± 0.037 | 7.45 | 82 | 0.41 ± 0.05 | 5.71 | 14 |
| 11f* | H | C≡CH | NO2 | 0.111 ± 0.086 | >52.08 | >469 | 0.247 ± 0.040 | >100 | >405 |
| 13f* | H | C≡CH | NH2 | 0.0067 ± 0.0010 | 9.94 | 1484 | 0. 042 ± 0.004 | 9.28 | 221 |
| 11i | H | CH(OH)CH2NO2 | NO2 | 0.308 ± 0.098 | >44.54 | >145 | 1.71 ± 0.134 | 27.06 | 16 |
| 13i | H | CH(OH)CH2NO2·HCl | NH2 | 0.0101 ± 0.0026 | 7.02 | 695 | 0.068 ± 0.015 | 20.57 | 303 |
| 11j | H | CH=CHCOOH | NO2 | 14.92 | 27.84 | 1.87 | 8.12 ± 0.42 | 34.79 | 4 |
| 13j | H | CH=CHCOOH | NH2 | 0.60 ± 0.043 | 18.0 | 30 | 0.63 ± 0.22 | 15.69 | 25 |
| 11k | H | CH=CHCOMe | NO2 | 0.0244 ± 0.020 | >46.73 | >1915 | 0.608 ± 0.267 | >47.90 | >79 |
| 13k | H | CH2CH2COMe | NH2 | 0.00195 ± 0.00053 | 9.25 | 4744 | 0.055 ± 0.000 | 18.35 | 1835 |
| 11m | H | CH=CHCN | NO2 | 0.0080 ± 0.0020 | >48.66 | >6082 | 0.049 ± 0.01 | >100 | >2041 |
| 13m | H | CH=CHCN | NH2 | 0.00071 ± 0.00058 | 9.75 | 13,732 | <0.014 ± 0.004 | 5.63 | >402 |
| 12c | H | CH=CHCN | NO2 | 0.0104 ± 0.00063 | >48.78 | 4690 | 0.037 ± 0.021 | >100 | >2702 |
| 14c | H | CH=CHCN | NH2 | 0.00055 ± 0.00008 | 25.0 | 45,455 | <0.014 ± 0.002 | 8.83 | >631 |
| 12d | NO2 | CH=CHCOMe | NO2 | 0.146 ± 0.040 | 24.36 | 167 | 0.35 ± 0.17 | >100 | >286 |
| 14d | NO2 | CH=CHCOMe | NH2 | 0.00633 ± 0.0018 | 5.88 | 929 | 0.047 ± 0.023 | 22.63 | 481 |
| 12e | NO2 | CH=CHCN | NO2 | 0.0971 ± 0.0178 | >43.96 | >453 | 0.37 ± 0.04 | 21.69 | 59.35 |
| 14e | NO2 | CH=CHCN | NH2 | 0.00038 ± 0.00007 | >47.06 | >123,842 | <0.014 ± 0.002 | 88.17 | >6298 |
| 14f | NH2 | CH=CHCN | NH2 | 0.0043 ± 0.0005 | 14.43 | 3,356 | <0.014 ± 0.002 | 18.94 | >1353 |
| 3 | H | C≡N | NH2 | 0.00068 ± 0.00003 | 8.98 | 13,206 | 0.015 ± 0.001 | >100 | >6667 |
| 1 | 0.0014 ± 0.0004 | 8.96 | 6400 | 0.041 ± 0.002 | 50.73 | 1237 | |||
Data were reported in Ref 11.
p24 ELISA was used to determine 50% effective concentration (EC50) against HIV-1 strains.
XTT assay was used to determine the CC50 value that causes cytotoxicity to 50% cells.
SI (selective index) = CC50/EC50, NA = not active at the highest concentration tested.
Like the DAPY analog 2, newly synthesized DAPD compounds 13m, 14c, 14e all contain a cyanovinyl group as the R2 substituent at the para-position on the C-ring. This common feature suggests that the para-cyanovinyl moiety may be a favorable structural moiety for anti-HIV activity. This postulate was further confirmed by the high potency of cyanovinyl-compound 11m relative to corresponding compounds with the same R1 and R3 groups but many different R2 substituents (11a-11f, 11i, 11j, and 11k) and even our prior active lead 3. However, incorporation of the cyanovinyl group into a fused ring system, such as the 6-cyanonaphthol moiety in 15-20, resulted in obviously reduced anti-HIV activity (EC50 > 0.1 μM in both assays), much less than most compounds in Table 1. These results suggest that flexibility of the R2 group might also be necessary. Further analysis of different R2 groups indicated that hydrophobic and more linear substituents, such as bromo (Br, 13b), allyl (CH2CH=CH2, 13c), ethynyl (C≡CH, 13f), and conjugated α,β-unsaturated ketone (CH=CHCOMe, 14d), were favorable for anti-HIV activity, irregardless of substituent length. Compound 13k with a more flexible para-substituent (-CH2CH2COCH3) on the C-ring exhibited potent anti-HIV-1 activity comparable to that of 1, but was slightly less active than 13m, 14c, and 14e. In contrast, R2 substituents with increased bulk (11d, 11e, 13d, 13e) or polarity (11g, 11i, 11j, 13i, 13j) led to reduced antiviral potency. Thus, the hydrophobic tunnel on the western wing of the NNRTI binding site might be very narrow and only allow a straight linear, more flexible, hydrophobic substituent to enter and adjust to an appropriate conformation.
Furthermore, 15 active compounds from the A and B series were tested against two HIV-1 strains resistant to multiple NRTI/NNRTIs, including RTMDR and A17. The data in Table 2 indicate that, in regard to the R3 substituent, the amino-compounds (13 and 14) were again more effective than the nitro-compounds (11 and 12) against the multi-RTI-resistant HIV-1 strains, as shown by comparing the following compound pairs: 11a vs 13a, 11b vs 13b, 11f vs 13f, 11m vs 13m, and 12c vs 14c. Comparison of 14e with 14c and 14f indicated that an additional nitro-group (R1) on the central ring was obviously preferable to NH2 or H at the same position against multi-RTI-resistant HIV-1 strains. Additionally, 14e was also the least cytotoxic of the three compounds (see Table 1). The most potent compounds 13m and 14e against wild-type HIV still exhibited high potency against multi-drug-resistant viral strains HIV-1RTMDR1 and A17 with low EC50 values (0.59 and 0.87 nM; 0.20 and 0.095 μM, respectively) and were more potent than our prior lead 3 (EC50 0.96 nM and 0.39 μM, respectively) and 1 (EC50 1.0 nM and 0.33 μM, respectively). Notably, 14e showed a low resistant fold (RF) of 6.79 against viral strain A17 with double mutant K103N/Y181C. This value is similar to that of 1 (RF = 6) in the same assay and in the literature (RF = 5)17, thus suggesting that 14e would be a promising new lead for further development.
Table 2.
Data against HIV drug-resistant viral strain replication
 | |||||||
|---|---|---|---|---|---|---|---|
| R1 | R2 | R3 | HIV-1 RTMDRa in TZM-bl cell line | A17 b in MT-2 cell line | |||
| EC50 (μM) | RFc | EC50(μM) | RF | ||||
| 11a | H | I | NO2 | 0.0943 ± 0.009 | 1.55 | >10 | >127 |
| 13a | H | I | NH2 | 0.00513 ± 0.00115 | 0.67 | 1.93 ± 0.06 | 36 |
| 11b | H | Br | NO2 | 0.0776 ± 0.0137 | 1.55 | >10 | >222 |
| 13b | H | Br | NH2 | 0.0068 ± 0.0030 | 0.93 | 3.49 ± 0.46 | 106 |
| 11f | H | C≡CH | NO2 | 0.215 ± 0.033 | 1.94 | >10 | >40 |
| 13f | H | C≡CH | NH2 | 0.0090 ± 0.002 | 1.34 | 1.43 ± 0.26 | 34 |
| 11m | H | CH=CHCN | NO2 | 0.0148 ± 0.0024 | 1.85 | >10 | >204 |
| 13m | H | CH=CHCN | NH2 | 0.00059 ± 0.00009 | 0.83 | 0.20 ± 0.01 | 14 |
| 13c | H | CH2CH=CH2 | NH2 | 0.0154 ± 0.0016 | 2.23 | >10 | >149 |
| 13e | H | C≡CCH(CH2)2 | NH2 | 0.121 ± 0.0177 | 1.33 | >10 | >24 |
| 13k | H | CH2CH2COMe | NH2 | 0.0032 ± 0.0008 | 1.64 | 0.73 ± 0.058 | 13 |
| 12c | H | CH=CHCN | NO2 | 0.0170 ± 0.0011 | 1.63 | >10 | >270 |
| 14c | H | CH=CHCN | NH2 | 0.00179 ± 0.0013 | 3.25 | 0.60 ± 0.09 | 43 |
| 14e | NO2 | CH=CHCN | NH2 | 0.00087 ± 0.0001 | 2.29 | 0.095 ± 0.001 | 6.79 |
| 14f | NH2 | CH=CHCN | NH2 | 0.0020 ± 0.0003 | 0.47 | 3.05 ± 0.03 | 128 |
| 3 | H | C≡N | NH2 | 0.00096 ± 0.0003 | 1.41 | 0.39 ± 0.065 | 26 |
| 1 | 0.0010 ± 0.0004 | 0.71 | 0.33 ± 0.02 | 6.00 | |||
Each compound was tested at least in triplicate.
HIV-1 RTMDR (obtained from AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH), which contains mutations in RT amino acid residues L74V, M41L, V106A, and T215Y, is resistant to AZT, ddI, nevirapine, and other non-nucleoside RT inhibitors.
The multi-NRTI-resistant strain A17 from NIH with mutations at amino acids K103N and Y181C in the viral RT domain is highly resistant to NRTIs.19
Resistant fold.
To understand the binding mode of the newly active compounds, molecular modeling was conducted by using the software Discovery Studio 2.5 and crystal structures of the wild-type RT/1 (PDB code: 3mec) complex18 and the K103N/Y181C RT/2 (PDB: 3bgr) complex. As shown in Figure 2, the most active compound (14e) and 1 were superposed into both wild-type and mutant NNRTI binding sites. The compounds showed similar binding orientations, horseshoe conformational shapes, and hydrogen bonds between the secondary amine linker and the main-chain carbonyl of K101 (1.88 Å and 1.85 Å respectively). However, an additional hydrogen bond was observed between the amino group on the central ring of 14e and the K101 carbonyl (2.31 Å) in both sites, which is consistent with our previous results.12 When compared to 1 bound with the wild-type RT structure, the cyanovinyl group of 14e is protruded deeply into a cylindrical open hydrophobic tunnel, which is formed by the side chains of amino acid residues Y188, F227, W229, and L234 and connected to the nucleic acid-binding cleft. Accordingly, the cyanovinyl group might disrupt the viral transcription from RNA to DNA more effectively. As observed in Figure 2B and reported about 2,14 the interactions of the cyanovinyl with the hydrophobic tunnel were conserved despite rearrangements in mutated RT. The cyanovinyl also is likely involved in supplementary interaction with the shifted Y183 (distance 3.53 Å) to compensate the affinity loss of Y181C mutation. Therefore, the extensive interactions of the cyanovinyl group and additional H-bond may explain why 14e, as well as 13m and 14c, is more potent than 1. On the other hand, modeling results indicated that the nitro group was superposed very well with the bromine atom of 1, although no interaction of these groups (NO2 or Br) with the binding site was observed, implying that this position on the central ring would be modifiable for exploring more SAR and new compounds with high potency against both wild-type and mutant viral strains.
Figure 2.
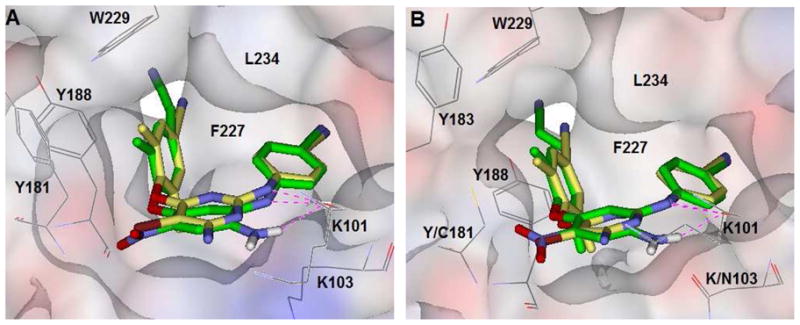
Binding mode of 14e (green) with (A) HIV-1 wild-type RT (PDB: 3mec) and (B) K103N/Y181C mutant RT (PDB: 3bgr) respectively. Drug 1 (yellow) was superposed into the same pocket for comparison. The hydrogen bonds between the inhibitors and K101 are illustrated with dashed lines (pink). The side chains of key amino acids on the binding site surface are indicated with solid lines (gray).
Conclusion
Thirty-five target compounds of A and B series (11-20) were newly synthesized and evaluated for anti-HIV activity. Most of them showed potent anti-HIV activity (EC50 <1 μM), but amino-compounds, 2,6-diarylpyridin-3-amine (13) and 2,4-diarylbenzen-1,2-diamine (14) derivatives, were much more potent against wild-type and multi-drug-resistant viral strains than corresponding nitro-compounds. The most promising compounds 13m and 14e exhibited high potency against wild-type and multi-drug-resistant viral strains with EC50 values at subnanomolar level, and were more potent than 1 in the same assays. The modifications of the C-ring and substituents on the central B-ring provided more structure-activity relationship (SAR) correlations as follows: (1) both 2-pyridinamine and aniline could serve as the central moieties in our compounds; (2) an appropriately positioned amino (R3) group on the central B-ring is crucial for achieving high potency; (3) para-cyanovinyl (R2) on the phenoxy C-ring is a beneficial substituent; (4) a favorable para-R2 substituent on the C-ring should be hydrophobic, flexible, and more linear to insert into the narrow tunnel on the NNRTI binding site; and (5) an additional nitro group (R1) on the central ring orthro to the C-ring could benefit inhibitory activity and reduce cytotoxity. Therefore, 2,6-diarylpyridin-3-amines and 2,4-diarybenzen-1,2-diamines have been discovered as novel NNRTI agents highly potent against wild-type and multi-drug resistant viral strains. The promising data results and readily accessible synthetic methods greatly encourage us to perform further studies for developing and identifying new drug candidates with desirable drug-like properties.
Experimental Section
Chemistry
Melting points were measured with an RY-1 melting apparatus without correction. The proton nuclear magnetic resonance (1H NMR) spectra were measured on a JNM-ECA-400 (400 MHz) spectrometer using tetramethylsilane (TMS) as the internal standard. The solvent used was DMSO-d6, unless otherwise indicated. Mass spectra (MS) were measured on an ABI Perkin-Elmer Sciex API-150 mass spectrometer with electrospray ionization and the relative intensity of each ion peak is presented as percent (%). The purities of target compounds were ≥ 95%, measured by HPLC analyses, which were performed by Agilent 1100 HPLC system with a UV detector, using an Agilent TC-C18 column (250 mm × 4.6 mm, 5 μm) eluting with a mixture of solvents A and B (condition 1: acetonitrile/water with 0.1% formic acid 80:20, flow rate 1.2 mL/min; condition 2: MeOH/water with 0.1% formic acid 80:20, flow rate 1.0 mL/min; UV 254 nm). The microwave reactions were performed on a microwave apparatus from Biotage, Inc. Thin-layer chromatography (TLC) and preparative TLC were performed on silica gel GF254 plates. Silica gel (200–300 mesh) from Qingdao Haiyang Chemical Company was used for column chromatography. Medium-pressure column chromatography was performed using a CombiFlash Companion purification system. All chemicals were obtained from Beijing Chemical Works or Sigma-Aldrich, Inc.
6-Chloro-2-(4′-cyanoaniline)-3-nitropyridine (8)
A mixture of 2,6-dichloro-3-nitropyridine (5, 1.93 g, 10 mmol) and 4-aminobenzonitrile (1.42 g, 12 mmol) was heated at 140 °C without solvent under N2 protection for 4 h. After cooling to room temperature, DMF (ca. 15 mL) was added into the flask to dissolve the mixture. The solution was then poured into ice water and the pH adjusted to 2–3 with aq HCl (5%). The resulting solid was collected and recrystallized from 95% EtOH to give pure 8 (2.01 g, 73% yield), yellow solid, mp 175–178 °C. 1H-NMR δ ppm 10.47 (1H, br s, NH), 8.53 (1H, d, J = 8.4 Hz, ArH-4), 7.86 (2H, d, J = 8.8 Hz, ArH-2′, 6′), 7.70 (2H, d, J = 8.8 Hz, ArH-3′, 5′), 6.96 (1H, d, J = 8.4 Hz, ArH-5). MS m/z (%) 275 (M + 1, 100), 276 (M + 2, 28).
5-Chloro-N1-(4′-cyanophenyl)-2-nitroaniline (9)
To a mixture of 2,4-dichloronitrobenzene (6, 1.92 g, 10 mmols) and 4-aminobenzonitrile (1.30 g, 11 mmols) in DMF (20 mL) was added t-BuOK (2.24 g, 20 mmols) slowly in ice-bath, then stirred at room temperature for 24 h and monitored by TLC. The mixture was poured into ice-water and pH was adjusted to 2–3 with 5% aq HCl. The red solid was collected, washed to neutral, and dried to afford 1.75 g of 9 in 64% yield, mp 221–224 °C. 1H-NMR δ ppm 9.56 (1H, br, NH), 8.20 (1H, d, J = 9.0 Hz, ArH-3), 7.72 (2H, d, J = 9.0 Hz, ArH-2′, 6′), 7.38 (1H, d, J = 2.0 Hz, ArH-6), 7.36 (2H, d, J = 9.0 Hz, ArH-3′, 5′), 6.91 (1H, d, J = 8.4 Hz, ArH-4). MS m/z (%) 272 (M – 1, 100), 274 (M + 1, 28).
5-Chloro-N1-(4′-cyanophenyl)-2,4-dinitroaniline (10)
To a solution of 1,3-dichloro-4,6-dinitrobenzene (7, 237 mg, 1 mmol) and 4-cyanoaniline (130 mg, 1.1 mmol) in DMF (3 mL) was slowly added potassium tert-butoxide (2 equiv) at ice-water bath temperature with stirring and then at room temperature for about 1 h monitored by TLC until the reaction was completed. The mixture was poured into ice-water, pH was adjusted to 6 with 5% aq HCl, and solid was collected and washed with water, The crude product was purified by recrystallization from EtOH to afford 284 mg of 10 in an 89% yield, yellow solid, mp 174 – 176 °C. 1H NMR (CDCl3) δ ppm 7.41 (1H, s, ArH-6), 7.57 (2H, d, J = 8.8 Hz, ArH-2′, 6′), 7.91 ( 2H, d, J = 8.8 Hz, ArH-3′, 5′), 8.90 (1H, s, ArH-3), 10.07 (1H, s, NH). MS m/z (%) 319 (M + 1, 100), 321 (M + 3, 23).
General procedure for the preparation of N2-(4′-cyanophenyl)-3-nitro-6-(4″-substituted-2″,6″-dimethyl)phenoxypyridin-2-amines (11a-11c, 11g)
A mixture of 8 (1 equiv) and a para-substituted 2,6-dimethylphenol (1.2 equiv) in DMF in the presence of anhydrous K2CO3 (excess, 3.5 equiv) was heated at 130–140 °C for 6–8 h. The mixture was poured into ice-water and pH was adjusted to 2–3 with 5% aq HCl. Crude solid product was collected and washed to neutral, followed by silica gel column purification (eluant: CH2Cl2) to give corresponding pure target compounds.
N2-(4′-Cyanophenyl)-6-(2″,6″-dimethyl-4″-iodo)phenoxy-3-nitropyridin-2-amine (11a)
Yield 82%, starting with 275 mg (1.0 mmol) of 8 and 297 mg (1.2 mmol) of 2,6-dimethyl-4-iodophenol to afford 399 mg of 11a, yellow solid, mp 162–165 °C. 1H-NMR δ ppm 10.65 (1H, br, NH), 8.63 (1H, d, J = 8.8 Hz, ArH-4), 7.54 (2H, s, ArH-3″, 5″), 7.38 (2H, d, J = 8.8 Hz, ArH), 7.21 (2H, d, J = 8.8 Hz, ArH), 6.65 (1 H, d, J = 8.8 Hz, ArH-5), 2.06 (6H, s, CH3·2). MS m/z (%) 487 (M + 1, 100). HPLC purity: 98.63%.
6-(4″-Bromo-2″,6″-dimethyl)phenoxy-N2-(4′-cyanophenyl)-3-nitropyridin-2-amine (11b)
Yield 64%, starting with 275 mg (1.0 mmol) of 8 and 241 mg (1.2 mmol) of 2,6-dimethyl-4-bromophenol to afford 280 mg of 11b, yellow solid, mp 193–194 °C. 1H-NMR δ ppm 10.65 (1H, br, NH), 8.63 (1H, d, J = 9.0 Hz, ArH-4), 7.37 (4H, m, ArH), 7.21 (2H, d, J = 8.8 Hz, ArH), 6.65 (1H, d, J = 9.0 Hz, ArH-5), 2.08 (6H, s, CH3·2). MS m/z (%) 439 (M + 1, 100), 441 (M + 3, 90). HPLC purity: 97.24%.
6-(4″-Allyl-2″,6″-dimethyl)phenoxy-N-(4′-cyanophenyl)-3-nitropyridin-2-amine (11c)
Yield 56%, starting with 275 mg (1.0 mmol) of 8 and 194 mg (1.2 mmol) of 4-allyl-2,6-dimethylphenol to afford 225 mg of 11c, yellow solid, mp 193–195 °C. 1H-NMR δ ppm 10.67 (1H, br, NH), 8.61 (1H, d, J = 8.8 Hz, ArH-4), 7.27 (4H, m, ArH), 7.01 (2H, s, ArH), 6.63 (1H, d, J = 8.8 Hz, ArH-5), 6.05 (1H, m, CH=), 5.24 (2H, m, CH2=), 3.44 (2H, d, J = 3.2 Hz, CH2), 2.08 (6H, s, CH3·2). MS m/z (%) 401 (M + 1, 100). HPLC purity: 95.22%.
N-(4′-Cyanophenyl)-6-(2″,6″-dimethyl-4″-formyl)phenoxy-3-nitropyridin-2-amine (11g)
Yield 66%, starting with 275 mg (1.0 mmol) of 8 and 180 mg (1.2 mmol) of 4-hydroxy-3,5-dimethylbenzaldehyde to afford 257 mg of 11g, yellow solid, mp 226–228 °C. 1H-NMR δ ppm 10.68 (1H, br, NH), 10.08 (1H, s, CHO), 8.66 (1H, d, J = 8.8 Hz, ArH-4), 7.75 (2H, s, ArH), 7.20 (4H, s, ArH), 6.69 (1H, d, J = 8.8 Hz, ArH-5), 2.21 (6H, s, CH3·2). MS m/z (%) 389 (M + 1, 100). HPLC purity: 97.90%.
N-(4′-Cyanophenyl)-6-[2″,6″-dimethyl-4″-(3-methylbut-3-ol-1-yn)phenoxy]-3- nitropyridin-2-amine (11d)
To a mixture of 11a (486 mg, 1 mmol), Pd(PPh3)2Cl2 (69 mg, 0.1 mmol), and cuprous iodide (19 mg, 0.1 mmol) in anhydrous DMF (5 mL) in the presence of triethylamine (0.6 mL) was added 2-methylbut-3-yn-2-ol (420 mg, 5 mmol) slowly under N2 protection at room temperature with stirring for 7 h. The mixture was poured into water and extracted with CH2Cl2 three times. After removal of solvent under reduced pressure, residue was purified by flash silica chromatography (gradient eluent: EtOAc/petroleum ether 0–40%) to provide 362 mg of pure 11d, 78% yield, yellow solid, mp 114–116 °C. 1H NMR δ ppm 10.37 (1H, br, NH), 8.67 (1H, d, J = 8.8 Hz, ArH-4), 7.40 and 7.34 (each 2H, d, J = 8.8 Hz, ArH), 7.31 (2H, s, ArH), 6.83 (1 H, d, J = 8.8 Hz, ArH-5), 5.47 (1H, s, OH), 2.00 (6H, s, ArCH3·2), 1.53 (6H, s, CH3·2). MS m/z (%) 465 (M + Na, 25), 425 (M – OH, 100). HPLC purity: 98.25%.
N-(4′-Cyanophenyl)-6-[4″-(2-cyclopropylethynyl)-2″,6″-dimethyl]phenoxy-3-nitro- pyridin-2-amine (11e)
The procedure was the same as for synthesis of 11d, starting with 11a (486 mg, 1 mmol) and ethynylcyclopropane (330 mg, 5 mmol) to provide 290 mg of 11e, in a 68% yield, yellow solid, mp 188–192 °C. 1H-NMR (CDCl3) δ ppm 10.65 (1H, br, NH), 8.61 (1H, d, J = 9.2 Hz, ArH-4), 7.36 (2H, d, J = 8.8 Hz, ArH), 7.22 (2H, s, ArH), 7.19 (2H, d, J = 8.8 Hz, ArH), 6.64 (1H, d, J = 9.2 Hz, ArH-5), 2.05 (6H, s, CH3×2), 1.52 (1H, m, CH), 0.89 (4H, m, CH2 × 2). MS m/z (%) 425 (M + 1, 100). HPLC purity: 96.95%.
N2-(4′-Cyanophenyl)-6-(2″,6″-dimethyl-4″-ethynyl)phenoxy-3-nitropyridin-2-amine (11f)
A solution of 11d (422 mg, 1 mmol) in toluene (20 mL) in the presence of powdered NaOH (16 mg, 0.4 mmol) was refluxed for 24 h under nitrogen protection. After cooling to room temperature, 2 drops of HOAc was dropped into the flask. The organic phase was washed with water and solvent was evaporated. The residue was purified on a flash silica column (gradient eluent: EtOAc/petroleum ether 0–40%) to provide 265 mg of 11f in 69% yield, yellow solid, mp 186–188 °C. 1HNMR (CDCl3) δ ppm 10.66 (1H, br, NH), 8.63 (1H, d, J = 8.8 Hz, ArH-4), 7.35 (2H, d, J = 8.8 Hz, ArH), 7.34 (2H, s, ArH), 7.19 (2H, d, J = 8.8 Hz, ArH), 6.66 (1 H, d, J = 8.8 Hz, ArH-5), 3.17 (1H, s, ≡CH), 2.09 (6H, s, CH3 · 2). MS m/z (%) 385 (M + 1, 100). HPLC purity: 96.63%.
N-(4′-Cyanophenyl)-6-[2″,6″-dimethyl-4″-(2-nitroethan-l-o1)]phenoxy-3-nitropyridin-2- amine (11i)
To a solution of 11g (388 mg, 1 mmol) and CH3NO2 (2 mL, excess) in THF (20 mL) was added dropwise 33% aq NaOH (1 mL) at ice-water bath and stirred for additional 1 h, then at room temperature for 12 h. The mixture was poured into ice-water, pH was adjusted with 5% aq HCl to neutral, and the mixture extracted with CH2Cl2. After removal of organic solvent in vacuo, residue was purified on a silica gel column (eluent: CH2Cl2/MeOH = 60/1) to afford 306 mg of 11i in a 68% yield, yellow solid, mp 230–234 °C. 1HNMR δ ppm 10.43 (1H, br, NH), 8.67 (1H, d, J = 8.8 Hz, ArH-4), 7.49 and 7.40 (each 2H, d, J = 8.4 Hz, ArH on A-ring), 7.33 (2H, s, ArH on C-ring), 6.81 (2H, d, J = 8.8 Hz, ArH-5), 6.32 (1H, d, J = 5.6 Hz, OH), 5.33 and 4.94 (each 1H, m, CH2NO2), 4.66 (1H, m, CHOH), 2.06 (6H, s, CH3 · 2). MS m/z (%) 450 (M + 1, 100), 472 (M + Na, 95). HPLC purity: 97.39%.
(E)-6-(4″-(2-Carboxyvinyl)-2″,6″-dimethyl)phenoxy-N-(4′-cyanophenyl)-3-nitropyridin-2-amine (11j)
A mixture of 11g (388 mg, 1 mmol) in pyridine (5 mL) and malonic acid (416 mg, 4 mmol) was refluxed in the presence of piperidine (0.15 mL) for 2 h. Then the mixture was poured into inc-water and pH was adjusted to 2–3 with 5% aq HCl. After standing overnight, the solid was collected and washed with cold water to afford 377 mg of 11j in 88% yield, yellow solid, mp 258–260 °C. 1HNMR δ ppm 12.43 (1H, br, COOH), 10.38 (1H, br, NH), 8.68 (1H, d, J = 8.8 Hz, ArH-4), 7.65 (1H, d, J = 16.0 Hz, =CHCOOH), 7.62 (2H, s, ArH on C-ring), 7.37 and 7.32 (each 2H, d, J = 8.8 Hz, ArH on A-ring), 6.70 (1H, d, J = 8.8 Hz, ArH-5), 6.60 (1H, d, J = 16.0 Hz, ArCH=), 2.07 (6H, s, CH3 · 2); MS m/z (%) 431 (M + 1, 30), 453 (M + Na, 70), 413 (M − OH, 100). HPLC purity: 98.73%.
(E)-N2-(4′-Cyanophenyl)-6-(4″-(α,β-unsaturated-but-3-)one-2″,6″-dimethyl)phenoxy-3-nitropyridin-2-amine (11k)
To a solution of 11g (388 mg, 1 mmol) in acetone (20 mL) and MeOH (5 mL) was added 10% aq NaOH (2 mL) slowly at ice-water bath temperature. Then the mixture was stirred at room temperature for 1 h, poured into ice-water and pH adjusted to 4–5 with 5% aq HCl. The solid was filtered, washed with water, and purified on a silica gel column (gradient eluent: EtOAc/petroleum ether 0–50%) to obtain 155 mg of 11k in 36% yield, yellow solid, mp 194–197 °C. 1H NMR δ ppm (CDCl3) 10.66 (1H, br, NH), 8.64 (1H, d, J = 9.2 Hz, ArH-4), 7.56 (1H, d, J = 16.0 Hz, ArCH=), 7.39 (2H, s, ArH on C-ring), 7.22 (4H, s, ArH on A-ring) 6.78 (2H, 1H, d, J = 16.0 Hz, =CHCO), 6.66 (1H, d, J = 9.2 Hz, ArH-5), 2.46 (3H, s, COCH3), 2.14 (6H, s, CH3 · 2). MS m/z (%) 429 (M + 1, 100), 387 (M − 43, 50). HPLC purity: 97.99%.
(E)-N-(4′-Cyanophenyl)-6-(4″-cyanovinyl-2″,6″-dimethyl)phenoxy-3-nitropyridin-2-amine (11m)
To a solution of diethyl cyanomethyl phosphonate (266 mg, 1.5 mmol) in THF (15 mL) was added t-BuOK (168 mg, 1.5 mmol) at ice-water bath temperature with stirring for 30 min. Compound 11g (388 mg, 1 mmol) in THF (15 mL) was added dropwise into the above mixture at room temperature and then stirring continued for 2 days. The reaction was then quenched with water and extracted with EtOAc. After removal of solvent under reduced pressure, the residue was purified by silica gel column chromatography (eluent: CH2Cl2/MeOH = 30/1) to afford 333 mg of 11m in an 81% yield, yellow solid, mp 231–233 °C. 1HNMR δ ppm 10.37 (1H, br, NH), 8.68 (1H, d, J = 8.8 Hz, ArH-4), 7.72 (1H, d, J = 16.8 Hz, ArCH=C), 7.56 (2H, s, ArH on C-ring), 7.34 (4H, s, ArH on A-ring), 6.84 (1H, d, J = 8.8 Hz, ArH-5), 6.53 (1H, d, J = 16.8 Hz, =CHCN), 2.06 (6H, s, CH3·2). MS m/z (%) 412 (M + 1, 100), 434 (M + Na, 30). HPLC purity: 98.08%.
(E)-N2-(4′-Cyanophenyl)-4-(4″-cyanovinyl-2″,6″-dimethyl)phenoxy-1-nitroaniline (12c)
The same method as the preparation of 11m, starting with 387 mg (1 mmol) of 12a and 266 mg (1.5 mmol) of (EtO)2P(O)CH2CN to afford 372 mg of 12c, yield 91%, yellow solid, mp 178–181 °C. 1HNMR δ ppm 9.55 (1H, br, NH), 8.18 (1H, d, J = 9.2 Hz, ArH-6), 7.73 (1H, d, J = 8.8 Hz, ArH-3′,5′), 7.60 (1H, d, J = 16.8 Hz, ArCH=), 7.51 (2H, s, ArH-3″, 5″), 7.32 (2H, d, J = 8.8 Hz, ArH-2′, 6′), 6.62 (1H, d, J = 2.8 Hz, ArH-3), 6.51 (1H, dd, J = 9.2 and 2.8 Hz, ArH-4), 6.44 (1H, d, J = 16.8 Hz, =CHCN), 2.08 (6H, s, CH3 · 2). MS m/z (%) 411 (M + 1, 100), 433 (M + Na, 85). HPLC purity: 95.40%
(E)-N1-(4′-Cyanophenyl)-5-[2″,6″-dimethyl-4″-(3-oxobut-1-enyl)]phenoxy-2,4-dinitro-aniline (12d)
The same method as the preparation of 11k, starting with 432 mg of 12b to afford 152 mg of 12d, yield 32%, brown solid, mp 213–215 °C. 1H NMR δ ppm (CDCl3) 9.94 (1H, br, NH), 9.18 (1H, s, ArH-3), 7.53 (2H, d, J = 8.4 Hz, ArH-3′,5′), 7.43 (1H, d, J = 16.4 Hz, ArCH=), 7.31 (2H, s, ArH-3″, 5″), 7.11 (2H, d, J = 8.4 Hz, ArH-2′,6′), 6.68 (1H, d, J = 16.4 Hz, =CHCN), 6.25 (1H, s, ArH-6), 2.42 (3H, s, COCH3), 2.16 (6H, s, CH3 · 2). MS m/z (%) 473 (M + 1, 100), 495 (M + Na, 80). HPLC purity: 99.47%.
(E)-N1-(4′-Cyanophenyl)-5-(4″-cyanovinyl-2″,6″-dimethyl)phenoxy-2,4-dinitroaniline (12e)
The same method as the preparation of 11m, starting with 432 mg (1 mmol) of 12b and 266 mg (1.5 mmol) of (EtO)2P(O)CH2CN to afford 240 mg of 12e, yield 53%, yellow solid, mp 242–244 °C. 1HNMR (CDCl3) δ ppm 9.97 (1H, br, NH), 9.19 (1H, d, J = 9.2 Hz, ArH-3), 7.55 (1H, d, J = 8.8 Hz, ArH-3′,5′), 7.32 (1H, d, J = 16.4 Hz, ArCH=), 7.21 (2H, s, ArH-3″, 5″), 7.12 (2H, d, J = 8.8 Hz, ArH-2′, 6′), 6.26 (1H, d, s, ArH-6), 5.86 (1H, d, J = 16.4 Hz, =CHCN), 2.18 (6H, s, CH3 · 2). MS m/z (%) 456 (M + 1, 100). HPLC purity: 97.15%.
General reduction methods for nitro group(s) on the central ring (pyridine or benzene) to prepare 13a-13f, 13i, 13j, 13m, and 14c
Method A: To a solution of a diaryl-nitropyridine (1 mmol) in THF and water (40 mL, v/v 1:1) was added ammonia (0.5 mL) and Na2S2O4 (90%, 10 mmol) successively. The mixture was stirred at room temperature for 2 h monitored by TLC (CH2Cl2/MeOH: 15/1) until reaction was completed. The mixture was then poured into ice-water and extracted with EtOAc. After removal of solvent in vacuo, the residue was purified on a silica gel column (eluent: CH2Cl2/MeOH = 30/1) to obtain corresponding diarylpyridinamines. Method B: To a solution of a diaryl-nitropyridine or diaryl-nitrobenzene (1 eq) in THF and MeOH (30 mL, v/v 1:1) was added NiCl2·6H2O (0.3 eq) and NaBH4 (4 eq.) successively at ice-water bath temperature. Then the mixture was stirred in the ice-water bath for another 30 min until the starting material disappeared, as monitored by TLC. The mixture was poured into water, pH adjusted to 4–5, warmed to 40–50 °C for 10 min, and then extracted with EtOAc. After removal of solvent in vacuo, crude product was purified on a silica gel column (eluent: CH2Cl2/MeOH = 30/1) to provide corresponding products.
N2-(4′-Cyanophenyl)-6-(2″,6″-dimethyl-4″-iodo)phenoxypyridin-2,3-diamine (13a)
Method A, yield 49%, starting with 486 mg (1.0 mmol) of 11a to afford 225 mg of 13a, brown solid, mp 186–188 °C. 1HNMR (CDCl3) δ ppm 7.50 (2H, s, NH2), 7.33 (2H, d, J = 8.8 Hz, ArH), 7.22 (2H, d, J = 8.8 Hz, ArH), 6.98 (1H, d, J = 8.4 Hz, ArH-4), 6.44 (1H, d, J = 8.4 Hz, ArH-5), 2.09 (6H, s, CH3·2). MS m/z (%) 457 (M + 1, 100). HPLC purity: 95.72%.
6-(4″-Bromo-2″,6″-dimethyl)phenoxy-N2-(4′-cyanophenyl)pyridin-2,3-diamine (13b)
Method B, yield 82%, starting with 439 mg (1.0 mmol) of 11b to afford 336 mg of 13b, brown solid, mp 190–193 °C. 1H-NMR δ ppm 8.35 (1H, br s, NH), 7.42 (2H, s, ArH), 7.32 (4H, s, ArH), 7.12 (1H, d, J = 8.4 Hz, ArH-4), 6.44 (1H, d, J = 8.4 Hz, ArH-5), 4.84 (2H, br, NH2), 2.03 (6H, s, CH3 · 2). MS m/z (%) 409 (M + 1, 100), 411 (M + 3, 90). HPLC purity: 97.36%.
6-(4″-Allyl-2″,6″-dimethyl)phenoxy-N2-(4′-cyanophenyl)pyridin-2,3-diamine (13c)
Method A, yield 43%, starting with 400 mg (1.0 mmol) of 11c to afford 160 mg of 13c, brown solid, mp 150–152 °C. 1HNMR δ ppm 8.30 (1H, br, NH ), 7.36 and 7.29 (each 2H, d, J = 8.8 Hz, ArH), 7.11 (1H, d, J = 8.0 Hz, ArH-4), 6.98 (2H, s, ArH), 6.39 (1H, d, J = 8.0 Hz, ArH-5), 6.03 (1H, m, CH=), 5.14 (2H, m, CH2=), 4.76 (2H, br, NH2), 3.36 (2H, d, J = 3.6 Hz, CH2), 2.01 (6H, s, CH3 · 2). MS m/z 371 (M + 1, 100). HPLC purity: 97.40%.
N2-(4′-Cyanophenyl)-6-[2″,6″-dimethyl-4″-(3-methylbut-3-ol-1-yn)phenoxy]pyridin-2,3-di amine (13d)
Method A, yield 48%, starting with 442 mg (1.0 mmol) of 11d to afford 198 mg of 13d, brown solid, mp 155–157 °C. 1HNMR δ ppm 8.33 (1H, br, NH), 8.01 (1H, br, OH), 7.41 (1H, d, J = 8.8 Hz, ArH-4), 7.29 (4H, m, ArH), 7.23 (2H, s, ArH), 6.55 (1 H, d, J = 8.8 Hz, ArH-5), 4.83 (2H, br, NH2), 2.08 (6H, s, Ar-CH3·2), 1.51 (6H, s, CH3·2). MS m/z (%) 413 (M + 1, 100). HPLC purity: 97.16%.
N2-(4′-Cyanophenyl)-6-[4″-(2-cyclopropylethynyl)-2″,6″-dimethylphenoxy]pyridin-2,3-diamine (13e)
Method A, yield 49%, starting with 424 mg (1.0 mmol) of 11e to afford 193 mg of 13e, brown solid, mp 82–85 °C. 1HNMR (CDCl3) δ ppm 7.33 (2H, d, J = 8.4 Hz, ArH-3′, 5′), 7.22 (2H, s, ArH), 7.18 (3H, m, ArH-2′, 6′ and ArH-4), 6.36 (1H, d, J = 8.0 Hz, ArH-5), 2.06 (6H, s, CH3×2), 1.51 (1H, m, CH), 0.89 (4H, m, CH2 × 2). MS m/z (%) 395 (M + 1, 100). HPLC purity: 96.99%.
N2-(4′-Cyanophenyl)-6-(2″,6″-dimethyl-4″-ethynyl)phenoxypyridin-2,3-diamine (13f)
Method A, yield 61%, starting with 384 mg (1.0 mmol) of 11f to afford 217 mg of 13f, brown solid, mp 79–82 °C. 1HNMR δ ppm 8.34 (1H, br, NH), 7.37 (2H, s, ArH on C-ring), 7.03 (4H, m, ArH on A-ring), 6.56 (1H, d, J = 8.4 Hz, ArH-4), 6.44 (1H, d, J = 8.4 Hz, ArH-5), 4.84 (2H, br, NH2), 3.36 (1H, s, CH≡), 2.02 (6H, s, CH3 · 2). MS m/z (%) 355 (M + 1, 100). HPLC purity: 98.03%.
N2-(4′-Cyanophenyl)-6-[2″,6″-dimethyl-4″-(2-nitroethan-l-o1)]phenoxypyridin-2,3-diamine hydrochloride (13i)
Method A, yield 42%, starting with 90 mg (0.2 mmol) of 11i to provide amino-coumpound, which was treated with 20% HCl-ether solution in acetone to afford 38 mg of 13i, white solid, dec. >230 °C. 1HNMR δ ppm 9.28 (1H, br, NH), 7.58 (1H, d, J = 8.4 Hz, ArH-4), 7.43 and 7.38 (each 2H, d, J = 9.2 Hz, ArH on A-ring), 7.27 (2H, s, ArH on C-ring), 6.59 (1H, d, J = 8.4 Hz, ArH-5), 5.29 (1H, dd, J = 3.6 & 9.8 Hz, CH2NO2), 4.90 (1H, dd, J = 3.6 & 12.4 Hz, CH2NO2), 4.62 (1H, dd, J = 9.8 & 12.4 Hz, CH), 3.57 (2H, br. NH2), 2.05 (6H, s, CH3 × 2). MS m/z (%) 420 (M + 1, 100). HPLC purity: 99.44%.
(E)-6-[4″-(2-Acrylic)-2″,6″-dimethyl)phenoxy]-N2-(4′-cyanophenyl)pyridin-2,3-diamine (13j)
Method A, yield 80%, starting with 86 mg (0.2 mmol) of 11j to afford 64 mg of 13j, deep brown solid, mp 245–7 °C. 1HNMR δ ppm 8.41 (1H, s, NH), 7.38 (2H, d, J = 9.2 Hz, ArH), 7.37 (2H, s, ArH-3″, 5″), 7.28 (1H, d, J = 15.6 Hz, ArCH=), 7.24 (2H, d, J = 9.2 Hz, ArH-2′, 6′), 7.11 (1H, d, J = 8.4 Hz, ArH-4), 6.43 (1H, d, J = 15.6 Hz, =CHCN), 6.39 (1H, d, J = 8.4 Hz, ArH-5), 4.83 (2H, br. NH2), 2.05 (6H, s, CH3 × 2). MS m/z (%) 401 (M + 1, 100), 383 (M − OH, 93). HPLC purity: 99.48%.
(E)-N2-(4′-Cyanophenyl)-6-(4″-cyanovinyl-2″,6″-dimethyl)phenoxypyridin-2,3-diamine (13m)
Method A, with a 50% yield, starting with 411 mg (1 mmol) of 11m to afford 192 mg of 13m, brown solid, mp 162–164 °C. 1HNMR δ ppm 8.35 (1H, br, NH), 7.68 (1H, d, J = 16.8 Hz, ArCH=C), 7.50 (2H, s, ArH on C-ring), 7.37 and 7.25 (each 2H, d, J = 8.8 Hz, ArH on A-ring), 7.13 (1H, d, J = 8.0 Hz, ArH-4), 6.45 (1H, d, J = 8.0 Hz, ArH-5), 6.45 (1H, d, J = 16.8 Hz, =CHCN), 4.84 (2H, br. s, NH2), 2.06 (6H, s, CH3 · 2). MS m/z (%) 382 (M + 1, 100). HPLC purity: 98.44%.
(E)-N2-(4′-Cyanophenyl)-4-(4″-cyanovinyl-2″,6″-dimethyl)phenoxybenzene-1,2-diamine (14c)
Method A, starting with 12c (410 mg, 1 mmol) to afford 226 mg of 14c, yield 59%, brown solid, mp 215–218 °C. 1HNMR (CDCl3) δ ppm 7.46 (2H, d, J = 8.8 Hz, ArH-3′,5′), 7.32 (1H, d, J = 16.8 Hz, ArCH=), 7.18 (2H, s, ArH-3″, 5″), 6.74 (1H, d, J = 9.2 Hz, ArH-6), 6.64 (2H, d, J = 8.8 Hz, ArH-2′,6′), 6.58 (1H, d, J = 2.8 Hz, ArH-3), 6.51 (1H, dd, J = 9.2 and 2.8 Hz, ArH-5), 5.80 (1H, d, J = 16.8 Hz, =CHCN), 3.52 (2H, br, NH2), 2.15 (6H, s, CH3 · 2). MS m/z (%) 381 (M + 1, 100). HPLC purity: 97.94%.
6-[(4″-Butan-2-one)-2″,6″-dimethyl]phenoxy-N2-(4′-cyanophenyl)pyridin-2,3-diamine (13k)
A mixture of 11k (50 mg, 0.11 mmol) in THF (20 mL) and excess Pd-C (10%) was hydrogenated at 65 psi for 6 h. After removal of Pd-C and solvent successively, the residue was purified by flash silica gel column chromatography (eluent: CH2Cl2/MeOH = 100:1) to provide 37 mg of pure 13k, yield 79%, gray solid, mp 84–86 °C. 1H NMR δ ppm 8.32 (1H, br, NH), 7.38 and 7.32 (each 2H, d, J = 9.2 Hz, ArH), 7.10 (1H, d, J = 8.0 Hz, ArH-4), 6.99 (2H, s, ArH), 6.37 (1H, d, J = 8.0 Hz, ArH-5), 4.80 (2H, br, NH2), 4.79 (4H, m, CH2CH2), 2.13 (3H, s, COCH3), 2.00 (6H, s, CH3 · 2). MS m/z (%) 401 (M + 1, 100). HPLC purity: 97.35%.
(E)-N1-(4′-Cyanophenyl)-5-[2″,6″-dimethyl-4″-(3-oxobut-1-enyl)]phenoxy-4-nitrobenzene-1,2-diamine (14d)
To a solution of 12d (165 mg, 0.35 mmol) in acetonitrile (3 mL) and triethylamine (3 mL) in the presence of Pd/C (10%, 30 mg) was added formic acid (85%, 2 mL) in acetonitrile (2 mL) keeping the temperature under −15 °C and then stirred for another 1 h. Next it was heated to reflux for 1 h. After filtration of Pd/C, the mixture was poured into ice-water, pH adjusted to neutral, and red solid was collected. The crude product was purified on a silica gel column (eluent: CHCl3/MeOH = 40:1) to yield 81 mg of 14d, yield 53%, red solid, mp 210 – 212 °C; 1H NMR ( CDCl3) δ ppm 9.94 (1H, br, NH), 9.18 (1H, s, ArH-3), 7.31 (2H, s, ArH-3″, 5″), 7.53 (2H, d, J = 8.8 Hz, ArH-3′,5′), 7.44 (1H, d, J = 16.4 Hz, ArCH=), 7.27 (2H, d, J = 8.8 Hz, ArH-2′, 6′), 6.68 (1H, d, J = 16.4 Hz, =CHCO), 6.25 (1H, s, ArH-6), 2.39 (3H, s, COCH3), 2.16 (6H, s, CH3 × 2). MS m/z (%) 443 (M + 1, 100). HPLC purity: 98.21%.
(E)-N1-(4′-Cyanophenyl)-5-(4″-cyanovinyl-2″,6″-dimethyl)phenoxybenzene-4-nitro-1,2-dia mine (14e)
Using the same method as preparation of 14d, starting with 150 mg (0.35 mmol) of 12e to afford 120 mg of 14e as red solid, yield 81%, mp 252–254 °C. 1HNMR (CDCl3) δ ppm 7.63 (1H, s, ArH-3), 7.47 (2H, d, J = 8.8 Hz, ArH-3′,5′), 7.32 (1H, d, J = 16.8 Hz, ArCH=), 7.20 (2H, s, ArH-3″, 5″), 6.76 (2H, d, J = 8.8 Hz, ArH-2′,6′), 6.31 (1H, s, ArH-6), 5.82 (1H, d, J = 16.8 Hz, =CHCN), 3.55 (2H, br. NH), 2.18 (6H, s, CH3 · 2). MS m/z (%) 426 (M + 1, 100), 448 (M + Na, 20). HPLC purity: 97.10%
(E)-N1-(4′-Cyanophenyl)-5-(4″-cyanovinyl-2″,6″-dimethyl)phenoxybenzene-1,2,4-triamine (14f)
To the solution of 12e (100 mg, 0.22 mmol) in EtOH (15 mL) in the presence of SbCl3 (201 mg, 0.33 mmol) was added NaBH4 (168 mg, 4.4 mmol) in portions at ice-bath temperature, and then stirring continued at room temperature for 40 min. The mixture was then poured into ice-water, pH adjusted to 4 with 5% aq HCl, and stirring continued for an additional 1 h. After the pH was adjusted to 9 with Na2CO3, the mixture was extracted with EtOAc three times. After removal of solvent under reduced pressure, the residue was purified on a silica gel column (eluent: CHCl3/CH3OH = 40:1) to afford 56 mg of 14f, yield 64%, red solid, mp 160–162 °C. 1H NMR ( CDCl3) δ ppm 7.37 (2H, d, J = 8.4 Hz, ArH-3′,5′), 7.28 (1H, d, J = 16.4 Hz, ArCH=), 7.16 (2H, s, ArH-3″, 5″), 6.47 (2H, d, J = 8.4 Hz, ArH-2′,6′), 6.30 (1H, s, ArH-3), 5.78 (1H, d, J = 16.4 Hz, =CHCN), 5.32 (1H, s, ArH-6), 4.01 and 3.51 (each 2H, br, NH2), 2.18 (6H, s, CH3 × 2). MS m/z (%) 396 (M + 1, 100). HPLC purity: 100.0%.
6-(6-Cyano-2-naphthoxy)-N2-(4-cyanophenylamino)-3-nitropyridine (15)
A mixture of 8 (54.9 mg, 0.2 mmol), 6-hydroxy-2-naphthonitrile (50.8 mg, 0.3 mmol), and Cs2CO3 (227.5 mg, 0.7 mmol) in DMF (1 mL) was heated at 100 °C for 10 min under microwave irradiation. Then the mixture was poured into water and pH was adjusted to 2–3 with aq HCl (1N). The solid was collected and purified on a silica gel column (gradient eluent: petroleum ether/CH2Cl2 0–60%) to afford 51 mg of 15, yield 63%, yellow solid, mp 264–6 °C. 1HNMR δ ppm 10.27 (1H, s, NH), 8.67 (1H, s, ArH-5″), 8.61 (1H, d, J = 8.8 Hz, ArH-8″), 8.17 (1H, d, J = 8.8 Hz, ArH-4), 8.04 (1H, d, J = 8.8 Hz, ArH-7″), 7.87 (1H, d, J = 2.4Hz, ArH-1″), 7.81 (1H, d, J = 8.8 Hz, ArH-4″), 7.55 (1H, dd, J = 2.4 & 8.8 Hz, ArH-3″), 7.26 (2H, d, J = 8.8 Hz, ArH-3′, 5′), 6.93 (2H, d, J = 8.8 Hz, ArH-2′, 6′), 6.78 (1H, d, J = 8.8 Hz, ArH-5). MS m/z (%) 408 (M + 1, 100). HPLC purity: 99.60%.
6-(6-Cyano-2-naphthoxy)-N2-(4-cyanophenylamino)pyridin-3-amine (16)
To a solution of 15 (51 mg, 0.125 mmol) in 1,4-dioxane (10 mL) was added ammonia (0.5 mL) in water (10 mL) with stirring for 30 min at rt. Then Na2S2O4 (386 mg, excess) was added and stirring continued for an additional 2 h, monitored by TLC until reaction was completed. The mixture was poured into ice-water and extracted with diethyl ether. After removal of solvent in vacuo, the residue was purified by PTLC to obtain 27 mg of 16 in 57% yield, gray solid, mp 204–6 °C. 1HNMR δ ppm 8.66 (1H, s, ArH-5″), 8.50 (1H, s, NH), 8.18 (1H, d, J = 8.8 Hz, ArH-8″), 8.09 (1H, d, J = 8.8 Hz, ArH-7″), 7.82 (1H, dd, J = 1.6 & 8.8 Hz, ArH-3″), 7.66 (1H, d, J = 2.0 Hz, ArH-1″), 7.51 – 7.54 (3H, m, ArH-3′, 5′, 4″), 7.30 (2H, d, J = 8.8 Hz, ArH-2′, 6′), 7.24 (1H, d, J = 8.4 Hz, ArH-4), 6.64 (1H, d, J = 8.4 Hz, ArH-5), 5.11 (2H, s, NH2). MS m/z (%) 378 (M + 1, 100). HPLC purity: 97.53%
5-(6-Cyano-2-naphthoxy)-N1-(4′-cyanophenyl)-2,4-dinitroaniline (17)
Starting 160 mg (0.5 mmol) of 10 and 102 mg (0.75 mmol) of 6-hydroxy-2-naphthonitrile in DMF under microwave irradiation at 150 °C for 20 min to obtain 147 mg of 17 as yellow solid with a 65% yield, mp 230 – 232 °C, 1H NMR (CDCl3) δ ppm 10.00 (1H, s, NH), 9.22 (1H, s, ArH-3), 8.27 (1H, s, ArH-5″), 7.98, 7.82, 7.68 (each 1H, d, J = 8.8 Hz, ArH on C-ring), 7.55 (2 H, d, J = 8.8 Hz, ArH on A-ring), 7.39 (1H, s, ArH-1″), 7.37 (1H, d, J = 8.8 Hz, ArH-3″), 7.25 (2H, d, J = 8.8 Hz, ArH on A-ring), 6.80 (1H, s, ArH-6). MS m/z (%) 450 (M – 1, 100). HPLC purity: 97.28%.
5-(1-Bromo-6-cyano-2-naphthoxy)-N-(4′-cyanophenyl)-2,4-dinitroaniline (18)
The same method as the preparation of 17. Starting with 10 (319 mg, 1.0 mmol) and 5-bromo-6-hydroxy-2-naphthonitrile (248 mg, 1.0 mmol) to afford 390 mg of 18 as yellow solid in a 74% yield, mp 254 – 256 °C. 1H NMR (CDCl3) δ ppm 9.95 (1H, s, NH), 9.24 (1H, s, ArH-3), 8.40 (1H, d, J = 8.8 Hz, ArH on C-ring), 8.29 (1H, s, ArH-5″), 7.95 and 7.86 (each 1H, d, J = 8.8 Hz, ArH on C-ring), 7.44 (2 H, d, J = 8.4 Hz, ArH-3′,5′), 7.34 (1H, d, J = 8.8 Hz, ArH on C-ring), 7.17 (2H, d, J = 8.4 Hz, ArH-2′,6′), 6.45 (1H, s, ArH-6). MS m/z (%) 528 (M – 1, 100), 530 (M+1, 98). HPLC purity: 96.45%.
4-(6-Cyano-2-naphthoxy)-N2-(4′-cyanophenyl)-5-nitrobenzene-1,2-diamine (19)
The method was the same as preparation of 14d. Starting with 17 (188 mg, 0.35 mmol) to obtain 88 mg of 19 as red solid with a 60% yield, mp 196 – 198 °C. 1H NMR (CD3COCD3) δ ppm 10.43 (1H, s, NH), 8.39 (1H, s, ArH-3), 8.06 and 7.94 (each 1H, d, J = 8.8 Hz, ArH on C-ring), 7.72 (1H, s, ArH-5″), 7.64 (1H, dd, J = 2.0 and 8.8 Hz, ArH on C-ring), 7.54 (2 H, d, J = 8.8 Hz, ArH on A-ring), 7.45 (1H, dd, J = 2.0 and 8.8 Hz, ArH on C-ring),7.35 (1H, d, J = 2.0 Hz, ArH on C-ring),7.19 (1H, s, ArH-6), 7.15 (2H, d, J = 8.8 Hz, ArH on A-ring), 5.18 (2H, s, NH2). MS m/z (%) 422 (M + 1, 100). HPLC purity: 98.87%.
4-(1-Bromo-6-cyano-2-naphthoxy)-N1-(4′-cyanophenyl)-5-nitrobenzene-1,2-diamine (20)
The method was the same as preparation of 14d. Starting with 186 mg (0.35 mmol) of 18 to provide 120 mg of 20 as red solid, yield 60%, mp 190 – 192 °C. 1H NMR (CDCl3) δ ppm 8.38 (1H, d, J = 8.8 Hz, ArH on C-ring), 8.19 (1H, s, ArH on C-ring), 7.79 and 7.75 (each 1H, d, J = 8.8 Hz, ArH on C-ring), 7.67 (1H, s, ArH-3), 7.52 (2 H, d, J = 8.8 Hz, ArH on A-ring), 7.09 (1H, d, J = 8.8 Hz, ArH on C-ring), 6.97 (2H, d, J = 8.8 Hz, ArH on A-ring), 6.95 (1H, s, ArH-6). MS m/z (%) 500 (M + 1, 100), 502 (M + 3, 99). HPLC purity: 100.0%.
Assay for measuring the inhibitory activity of compounds on HIV-1 IIIB replication in MT-2 cells
Inhibitory activity of compounds against infection by HIV-1 IIIB and its variant A17, which is resistant to multiple RTIs, was determined as previously described.20 Briefly, MT-2 cells (104 well) were infected with an HIV-1 strain (100 TCID50) in 200 μL RPMI 1640 medium containing 10% FBS in the presence or absence of a test compound at graded concentrations overnight. Then, the culture supernatants were removed and fresh media containing no test compounds were added. On the fourth day post-infection, 100 μL of culture supernatants were collected from each well, mixed with equal volumes of 5% Triton X-100 and assayed for p24 antigen, which was quantitated by ELISA and the percentage of inhibition of p24 production was calculated as previously described.20 The effective concentrations for 50% inhibition (EC50) were calculated using a computer program CalcuSyn.21
HIV-1 infection assay using TZM-bl as reporter cells
Inhibition of HIV-1 infection was measured as reduction in luciferase gene expression after a single round of virus infection of TZM-bl cells as described previously.22 Briefly, 200 TCID50 of virus (NL4-3) was used to infect TZM-bl cells in the presence of various concentrations of compounds. Two days after infection, the culture medium was removed from each well and 100 μl of Bright Glo reagent (Promega, Luis Obispo, CA) was added to the cells for measurement of luminescence using a Victor 2 luminometer. The 50% inhibitory concentration (IC50) was defined as the concentration that caused a 50% reduction of luciferase activity (Relative Light Units) compared to virus control wells.
Assessment of in vitro cytotoxicity in MT-2 cells
The in vitro cytotoxicity of compounds on MT-2 cells was measured using a colorimetric XTT assay.23 Briefly, 100 μL of the test compound at graded concentrations were added to equal volumes of cells (5 · 105/mL) in wells of a 96-well plate. After incubation at 37 °C for 4 days, 50 μL of XTT solution (1 mg/mL) containing 0.02 μM of phenazine methosulphate (PMS) was added. After 4 h, the absorbance at 450 nm was measured with an ELISA reader. The CC50 (concentration for 50% cytotoxicity) values were calculated using the CalcuSyn computer program as described above.
Molecular modeling
The docking studies were conducted by using software Discovery Studio 2.5 (DS 2.5) and two crystal structures of wild-type RT/1 complex (PDB: 3mec) and K103N/Y181C mutant RT/2 complex (PDB: 3bgr). Following the “Guide-Docking ligands” in the software tool, the proteins and ligands were prepared, the dockings were performed by the CDOCK module, and results were analyzed in the DS 2.5 Base with relevant modules. The conformation energies of ligands, 1 and 14e, were minimized with the modified parameters algorithm (smart minimizer), max steps (2000), and RMS gradient (0.05). The radius of the binding site sphere was defined as 10 Å. Comparing to crystal structure, the root mean squared deviation (RMSD) of the modeling was 0.29, thus ensuring the docking method was reliable.
Supplementary Material
Acknowledgments
This investigation was supported by grants 30930106, 20472114, and 7052057 from the Natural Science Foundation of China (NSFC) and Beijing municipal government, respectively, awarded to L. Xie, and US NIH grants awarded to S. Jiang (AI46221), C. H. Chen (AI65310), and K. H. Lee (AI33066).
Abbreviations
- AIDS
acquired immunodeficiency syndrome
- CC50
concentration for 50% cytotoxicity
- DAPYs
diarylpyrimidines
- DAPDs
diarylpyridines
- DAANs
diarylanilines
- EC50
effective concentration for 50% inhibition
- HAART
highly active antiretroviral therapies
- HIV
human immunodeficiency virus, NNRTI, non-nucleoside reverse transcriptase inhibitor
- NRTI
nucleoside reverse transcriptase inhibitor
- PDB
protein data base
- RF
resistant fold
- RT
reverse transcriptase
- RTI
reverse transcriptase inhibitor
- SAR
structure-activity relationship
- SI
selective index (ratio of CC50/EC50)
Footnotes
Supporting Information Available: HPLC conditions and summary of HPLC purity data for final target compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Esnouf R, Ren J, Ross C, Jones Y, Stammers D, Stuart D. Mechanism of inhibition of reverse transcriptase by nonnucleoside inhibitors. Nat Struct Biol. 1995;2:303–308. doi: 10.1038/nsb0495-303. [DOI] [PubMed] [Google Scholar]
- 2.Pauwels R. New non-nucleoside reverse transcriptase inhibitors (NNRTIs) in development for the treatment of HIV infections. Curr Opin Pharmacol. 2004;4:437–446. doi: 10.1016/j.coph.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 3.Sweeney ZK, Klumpp K. Improving non-nucleoside reverse trancriptase inhibitors for first-line treatment of HIV infection: the development pipeline and recent clinical data. Curr Opin Drug Discov Devel. 2008;11(4):458–470. [PubMed] [Google Scholar]
- 4.Minudo JJ, Haubrich R. Entravirine: a second-generation NNRTI for treatment- experienced adults with resistant HIV-1 infection. Futur HIV Ther. 2008;2(6):525–537. doi: 10.2217/17469600.2.6.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sarafianos SG, Marchand B, Das K, Himmel DM, Parniak MA, Hughes SH, Arnold E. Structure and function of HIV-1 reverse transcriptase: molecular mechanisms of polymerization and inhibition. J Mol Biol. 2009;385:693–713. doi: 10.1016/j.jmb.2008.10.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rossotti R, Rusconi S. Efficacy and resistance of recently developed non-nucleoside reverse transcriptase inhibitors for HIV-1. HIV Ther. 2009;3(1):63–77. [Google Scholar]
- 7.Lai MT, Munshi V, Touch S, Tynebor RM, Tucker TJ, McKenna PM, Williams TM, DiStefano DJ, Hazuda DJ, Miller MD. Antiviral activity of MK-4965; a novel non-nucleoside reverse transcriptase inhibitor. Antimicrob Agents Chemother. 2009;53(6):2424–2431. doi: 10.1128/AAC.01559-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tucker TJ, Sisko JT, Tynebor RM, Williams TM, Felock PJ, Flynn JA, Lai MT, Liang YX, Gaughey GM, Liu MQ, Miller M, Moyer G, Munshi V, Perlow-Poehnelt R, Prasad S, Reid JC, Sanchez R, Torrent M, Vacca JP, Wan BL, Yan YW. Discovery of 3-{5-[(6-Amino-1H-pyrazolo [3,4-b]pyridine-3- yl)methoxy]-2- chlorophenoxy}-5-chlorobenzonitrile (MK-4965): a potent, orally bioavailable HIV-1 non-nucleoside reverse transcriptase inhibitor with improved potency against key mutant viruses. J Med Chem. 2008;51:6503–6511. doi: 10.1021/jm800856c. [DOI] [PubMed] [Google Scholar]
- 9.Ripamonti D, Maggiolo F. Rilpivirine, a non-nucleoside reverse transcriptase inhibitor for the treatment of HIV infection. Current Opinion in investigational drugs. 2008;9(8):899–912. [PubMed] [Google Scholar]
- 10.Pecora Fulco P, McNicholl IR. Ereavirine and rilpivirine: nonnucleoside reverse transcriptase inhibitors with activity against human immunodeficiency virus type 1 strains resistant to previous nonnucleoside agents. Pharmacotherapy. 2009;29(3):281–294. doi: 10.1592/phco.29.3.281. [DOI] [PubMed] [Google Scholar]
- 11.Tian XT, Qin BJ, Lu H, Lai W, Jiang S, Lee KH, Chen CH, Xie L. Discovery of diarylpyridine derivatives as novel non-nucleoside HIV-1 reverse transcriptase inhibitors. Bioorg Med Chem Lett. 2009;19:3482–5485. doi: 10.1016/j.bmcl.2009.07.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qin BJ, Jiang XK, Lu H, Tian XT, Barbault F, Huang L, Qian K, Chen CH, Huang R, Jiang S, Lee KH, Xie L. Diarylaniline derivatives as a distinct class of HIV-1 non-nucleoside reverse transcriptase inhibitors. J Med Chem. 2010;53:4906–4916. doi: 10.1021/jm1002952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Corte BL. From 4,5,6,7-tetrahydro-5-methylimidazo[4,5,1-jk](1,4)benzodiazepine-2(1H)-one (TIBO) to etravirine (TMC125): fifteen years of research on non-nucleoside inhibitors of HIV-1 reverse transcriptase. J Med Chem. 2005;48:1689–1696. doi: 10.1021/jm040127p. [DOI] [PubMed] [Google Scholar]
- 14.Das K, Bauman JD, Clark ADJ, Frenkel YV, Lewi PJ, Shatkin AJ, Hughes SH, Arnold E. High-resolution structures of HIV-1 reverse transcriptase/TMC-278 complexes: Strategic flexibility explains potency against resistance mutations. Proc Natl Acad Sci USA. 2008;105(5):1466–1471. doi: 10.1073/pnas.0711209105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Redemann CT, Redemann CE. 5-Amino-2,3-dihydro-1,4-phthalazinedione. Org Synth. 1949;29:8–9. [Google Scholar]
- 16.Mordant C, Schmitt B, Pasquier E, Demestre C, Queguiner L, Masungi C, Peeters A, Smeulders L, Bettens E, Hertogs K, Heeres J, Lewi P, Guillemont J. Synthesis of novel diarylpyrimidine analogues of TMC278 and their antiviral activity against HIV-1 wild-type and mutant strains. E J Med Chem. 2007;42:567–569. doi: 10.1016/j.ejmech.2006.11.014. [DOI] [PubMed] [Google Scholar]
- 17.Andries K, Azijn H, Thielemans T, Ludovici D, Kukla M, Heeres J, Janssen P, De Corte B, Vingerhoets J, Pauwels R, de Bethune MP. TMC125, a novel next- Generation nonnucleoside reverse transcriptase inhibitor active against nonnucleoside reverse transcriptase inhibitor-resistant human immunodeficiency virus type 1. Antimicrob Agents Chemother. 2004;48:4680–4686. doi: 10.1128/AAC.48.12.4680-4686.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lansdon EB, Broendza KM, Hung M, Wang R, Mukund S, Jin D, Birkus G, Kutty N, Liu X. Crystal structures of HIV-1 reverse transcriptase with etravirine (TMC125) and rilpivirine (TMC278): implications for drug design. J Med Chem. 2010;53:4295–4299. doi: 10.1021/jm1002233. [DOI] [PubMed] [Google Scholar]
- 19.Obiol-Pardo C, Rubio-Martinez J. Comparative evaluation of MMPBSA and XSCORE to compute binding free energy in XIA-peptide complexes. J Chem Inf Model. 2007;47:134–142. doi: 10.1021/ci600412z. [DOI] [PubMed] [Google Scholar]
- 20.Jiang S, Lu H, Liu S, Zhao Q, He Y, Debnath AK. N-substituted pyrrole derivatives as novel human immunodeficiency virus type 1 entry inhibitors that interfere with the gp41 six-helix bundle formation and block virus fusion. Antimicrob Agents Chemother. 2004;48:4349–4359. doi: 10.1128/AAC.48.11.4349-4359.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 22.Dang Z, Lai W, Qian K, Lee KH, Chen CH, Huang L. Betulinic Acid Derivatives as Human Immunodeficiency Virus Type 2 (HIV-2) Inhibitors. J Med Chem. 2009;52:7887–7891. doi: 10.1021/jm9004253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao Q, Ma L, Jiang S, Lu H, Liu S, He Y, Strick N, Neamati N, Debnath AK. Identification of N-phenyl-N′-(2,2,6,6-tetramethylpiperidin-4-yl)-oxalamides as a new class of HIV-1 entry inhibitors that prevent gp120 binding to CD4. Virology. 2005;339:213–225. doi: 10.1016/j.virol.2005.06.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.