

Abstract
BACKGROUND
Defects in the cardiac sodium channel gene, SCN5A, can cause a broad spectrum of inherited arrhythmia syndromes. After genotyping of a proband who presented with syncope, the SCN5A mutant P2006A and the common polymorphism H558R were identified.
OBJECTIVE
The main objective of this study was to determine whether the SCN5A-H558R polymorphism could modify the defective gating kinetics observed in the P2006A mutation and therefore explain why this gain-of-function mutation has been identified in control populations.
METHODS
Mutations were engineered using site directed mutagenesis and heterologously expressed transiently in HEK293 cells. Whole-cell sodium currents were measured at room temperature using the whole-cell patch clamp technique.
RESULTS
In HEK293 cells, P2006A displayed biophysical defects typically associated with long QT syndrome by increasing persistent sodium current, producing a depolarizing shift in voltage dependence of inactivation, and hastening recovery from inactivation. Interestingly, when co-expressed either on the same or different genes, P2006A and H558R displayed currents that behaved like wild-type (WT). We also investigated whether H558R can modulate the gating defects of other SCN5A mutations. Interestingly, the H558R polymorphism also restored the gating defects of the SCN5A mutation V1951L to the WT level.
CONCLUSIONS
Our results suggest that H558R might play an important role in stabilization of channel fast inactivation and may provide a plausible explanation as to why the P2006A gain-of-function mutation has been identified in control populations. Our results also suggest that the SCN5A polymorphism H558R might be a disease modifying gene.
Keywords: Long QT syndrome, Sodium, Mutation, Arrhythmia, Ion channels, Sudden death
INTRODUCTION
Defects in the gene that encodes for the cardiac sodium channel, SCN5A, are now regarded as the cause of a broad spectrum of inherited arrhythmia syndromes including long QT syndrome (LQTS), Brugada syndrome (BrS), and sudden infant death syndrome (SIDS).1–3 To date, mutational analyses have reported close to 200 distinct mutations in SCN5A with at least 84 of them known to be related to LQT3; the majority of these are missense mutations.4,5 Previous functional studies of SCN5A mutations associated with LQT3 indicate that most result in an increase in the persistent or late sodium current (INa). This current leads to a prolongation of the action potential (AP) which manifests as a lengthening of the QT interval on the surface ECG.5 LQT3 is an autosomal dominant disease6,7 that also displays incomplete penetrance, a phenomenon in which some individuals who carry a disease-causing mutation are, nevertheless, asymptomatic.8,9
Interestingly, in clinical studies, a Proline to Alanine mutation at position 2006 (P2006A) of SCN5A has been classified as a rare variant and found in control population, even though it produces in vitro a biophysical phenotype with features typical of LQT3 mutations3,10 This is counterintuitive since the biophysical properties of this particular mutation should produce a prolonged QT interval and lead to arrhythmias, therefore making it hard to reconcile with the fact that it is present in the general population. In the present study, we found a family who exhibited no LQT3 syndrome phenotype even though family members carry SCN5A-P2006A which produces biophysical defects associated with LQT3 as previously described3,10 (Figure 1A). Although SCN5A-P2006A3,10–13 was found in various family members, they displayed no typical sign of LQTS on the resting, supine ECG. Interestingly, two family members were also found to be homozygous for the common sodium channel polymorphism SCN5A-H558R, located in the intracellular domain I–II linker (Figure 1C), and the third affected family member was heterozygous for this polymorphism.
Figure 1.

Pedigree of a family with a SCN5A mutation, location and topology of SCN5A mutations. A, Pedigree shows affected individuals. While the mother (I-1) was homozygous for H558R, the father (I-2) was heterozygous for H558R and P2006A. The proband (II-2) and her brother (II-1) were homozygous for H558R and heterozygous for P2006A. All of them carry the H558R polymorphism on the same allele as P2006A. B, Sequence analysis of SCN5A reveals a change of Histidine to Arginine at position 558 and a change of Proline to Alanine at position 2006. C, Topological diagram of sodium channel showing amino acid residues where polymorphism and mutations occur respectively.
Previous studies demonstrated that the SCN5A-H558R polymorphism can modulate effects of the mutant T512I and M1766L when expressed on the same construct.14,15 Furthermore, it was recently reported that this same SCN5A-H558R polymorphism could restore trafficking of a Brugada Syndrome (BrS) mutation (SCN5A-R282H) on a separate gene thus explaining the incomplete penetrance phenomenon seen in this particular BrS family.16 Therefore, based on the phenotypes that these individuals exhibited, we hypothesized that the SCN5A-H558R polymorphism could modify the defective gating kinetics observed with the P2006A mutation. Here, we show that the common SCN5A-H558R polymorphism, present in 20% of the population, mitigates the gating defects caused by SCN5A-P2006A and by an additional mutation associated with SIDS. These results not only point to a modulatory effect of the SCN5A-H558R polymorphism on the fast inactivation gating defects characteristic of these mutants, but may also provide a plausible explanation as to why SCN5A-P2006A has been identified in control populations. Moreover, our results reinforce previous observations that the SCN5A polymorphism H558R might be a disease modifying gene.
METHODS
Genotyping
Molecular analyses on the SCN5A gene were performed as previously described.17 The study was approved and performed according to the terms required by our local Ethics Committee, and written informed consent was obtained from all participants.
Cloning of SCN5A mutations and polymorphisms
The SCN5A-H558R, SCN5A-V1951L, SCN5A-H558R-V1951L, SCN5A-P2006A, and SCN5A-H558R-P2006A mutations were created using the Stratagene QuickChange XL Site Directed Mutagenesis Kit on the SCN5A background (PubMed Accession No.NM 198056) expressed in the GFP-IRES vector (BD Biosciences Clonetech, San Joje, Calif).
Expression of SCN5A in heterologous expression system
In this study, to express the cardiac sodium channel, we used transient transfections of SCN5A expressed in GFP-IRES in human embryonic kidney cells (HEK293 cells). The transfections were performed using the Polyfect transfection kit (Qiagen, Valencia, Calif) according to the manufacturer’s protocol.
Cellular Electrophysiological measurements for functional characterization
As previously described, sodium currents from transfected HEK293 cells were recorded at room temperature (22°C to 23°C) 1–2 days after transfection in the whole-cell configuration of the patch-clamp technique.18 To minimize the voltage-clamp errors, series resistance compensation of Axopatch 200A was performed to values >80%. To generate the voltage-clamp command pulses, PCLAMP version 9.02 (Molecular Devices, Sunnyvale, Calif) was used. The intracellular solution contained (in mmol/L, at pH 7.4): NaCl 35, CsF 105, EGTA 10, and Cs-HEPES 10. The external solution contained (mmol/L): Nacl 140, KCl 5, MgCl2 1, CaCl2 2, glucose 10, and HEPES 10 (pH 7.4).
The level of TTX-sensitive persistent current was determined with a 300-ms depolarization to −30 mV as the average current recorded between 200–300 ms and reported as a percentage of peak current after digital subtraction of currents recorded in the presence and absence of 20 µmol/L tetrodotoxin (TTX, Sigma, St Louis, MO).
Data Analysis
For recovery from inactivation, normalized current amplitude was fit to the following equation:
For the voltage dependence of steady-state inactivation normalized current was fit to a Boltzmann distribution:
Statistics
Data are expressed as mean ± S.E.M. Statistical analysis was performed using Student t-test for comparisons of 2 groups. Differences were considered to be significant at a P value <0.05.
RESULTS
Characteristics of the family and genetic findings
After experiencing syncopal episodes, the proband of the family (Figure 1A, II-2) was admitted to the hospital. In the emergency department, prolongation of her QT interval was detected (QTc 500 ms); however, subsequent ECGs during and after the hospitalization failed to show a resting QTc over 460 ms. However, due to these syncopal episodes and the prolonged QT interval, her physician suggested that the patient undergo genetic testing. Sequence analysis of her SCN5A gene revealed a change of Histidine to Arginine at position 558 (H558R) and a change of Proline to Alanine at position 2006 (P2006A). Upon further analysis, it was determined that the proband (Figure 1A, II-2) was homozygous for the H558R polymorphism and heterozygous for the previously reported SCN5A-P2006A mutation (Figure 1B). Since the SCN5A-P2006A mutation which produces a biophysical phenotype with features typical of LQT3 mutations was identified, the genotypic analysis was also performed on other members of her family, all asymptomatic, including her mother, father, and brother. Her mother (Figure 1A, I-1) had a QTc of 450 ms and was homozygous for H558R, her father (Figure 1A, I-2) had a QTc of 410 ms and was heterozygous for H558R and P2006A, and her brother had a QTc of 430 ms and was also found to have the same mutation and was homozygous for the polymorphism (Fig. 1A, II-1).
As a result of the genotyping and manifestations of syncope, the proband was implanted with an ICD. Interestingly though, additional syncopal events that occurred in presence of the ICD demonstrated that they were not of cardiac origin. Yet, this mutation’s biophysical properties are consistent with the typical LQT3 phenotype.3,10–13 In the family under study which carries SCN5A-P2006A, the genotype-phenotype discordance is striking: only one family member has had episodes of syncope which now appear to not even be related to a cardiac event. Since the three individuals carrying SCN5A-P2006A also carry the SCN5A-H558R polymorphism, which has been previously shown to act as a disease modifying gene14–16, a full in vitro biophysical characterization was undertaken to test our hypothesis that the SCN5A-H558R polymorphism can modify the aberrant gating kinetics of the mutated sodium channels P2006A and, as a result, erase any LQT3 phenotype. If proven, this would therefore explain the genotype-phenotype discordance seen in this family.
Functional analysis of SCN5A-P2006A with/without the sodium channel polymorphism H558R
The SCN5A-P2006A mutation and the SCN5A-H558R polymorphism were reproduced in vitro on the SCN5A background in order to study the functional consequences of the mutation both with and without the presence of the polymorphism. We expressed SCN5A-WT, SCN5A-P2006A, and SCN5A-H558R-P2006A channels in HEK293 cells for whole-cell voltage-clamp measurements. Figure 2 represents a typical Na+ current (INa) recorded by holding the resting membrane potential at −120 mV and stepping in 10-mV increments from −80 to +60 mV (protocol inset, Figure 2); the whole-cell sodium current amplitudes and reversal potentials were similar between the different groups (Figure 2 and Figure 4A). As expected, P2006A exhibited significantly increased persistent sodium currents (Figure 3A); moreover, P2006A caused a significant +10 mV shift in voltage dependence of steady-state inactivation (Figure 4B and Table 1) and a faster recovery from inactivation (Figure 4C and Table 1) - both suggestive of a typical LQT3 impaired inactivation phenotype. Interestingly, when the mutation was expressed in the presence of the H558R polymorphism, the sodium currents recorded were comparable to those recorded from WT sodium channels (Figure 4 and Table 1), thus suggesting that the polymorphism restored the normal function of this mutated channel.
Figure 2.
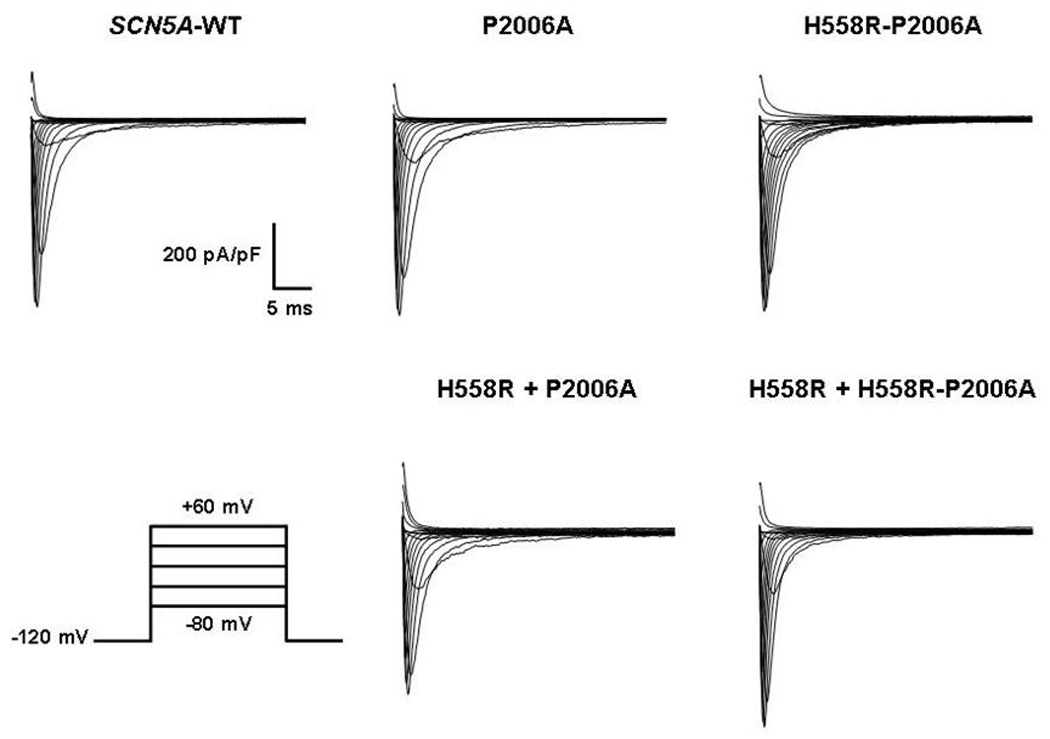
Representative whole-cell sodium current traces from representative experiments of SCN5A-WT, SCN5A-P2006A, SCN5A-H558R-P2006A, SCN5A-H558R + SCN5A-P2006A, and SCN5A-H558R + SCN5A-H558R-P2006A.
Figure 4.

Electrophysiological characterization of SCN5A mutations. A, Voltage dependence of activation for SCN5A-P2006A (n=12), SCN5A-H558R-P2006A (n=20), SCN5A-H558R+SCN5A-P2006A (n=6), and SCN5A-H558R+SCN5A-H558R-P2006A (n=6) in which currents were compared to SCN5A-WT (n=12). B, Steady-state inactivation recorded using the standard protocol as shown in inset and fitted to a Boltzmann distribution. The midpoints of inactivation (V1/2) in mutant channels of SCN5A-P2006A were significantly shifted toward depolarized voltages compared to WT channels. C, Mutant channels of SCN5A-P2006A display faster recovery from inactivation than WT channels.
Figure 3.

Representative traces of late INa for SCN5A-WT (black, panel A and B), SCN5A-P2006A (light gray, panel A), SCN5A-H558R-P2006A (gray, panel A), SCN5A-H558R+SCN5A-P2006A (gray, panel B), and SCN5A-H558R+SCN5A-H558R-P2006A (light gray, panel B). A, TTX-sensitive persistent sodium currents for SCN5A-WT, SCN5A-P2006A mutation, and SCN5A-P2006A mutation with the sodium channel polymorphism H558R. As expected, the P2006A mutation displays incomplete inactivation. However, in the presence of the H558R polymorphism, the P2006A mutation’s inactivation kinetics are restored to the WT level. B, Superimposition of the normalized sodium currents recorded from SCN5A-WT, SCN5A-H558R+ SCN5A-P2006A, and SCN5A-H558R+SCN5A-H558R-P2006A elicited by clamping at −30 mV. Neither SCN5A-H558R+SCN5A-P2006A nor SCN5A-H558R+SCN5A-H558R-P2006A mutants alter the persistent current compared to WT channels. The right traces represent the magnified regions outlined by the boxes to highlight the persistent sodium currents of the left panels. The dotted lines indicate zero current. Statistically significant differences in late INa comparing mutant channels with SCN5A-WT are indicated (*P<0.05, student t-test).
Table 1. Peak current, Steady-State Inactivation, and Recovery from Inactivation Parameters of Whole-Cell Sodium Current.
Steady-State Inactivation, and Recovery from Inactivation Parameters of Whole-Cell Sodium Current. Functional indexes of sodium channel whole-cell current. The SCN5A-P2006A mutant displays significantly depolarizing shift in voltage dependence of inactivation, and faster recovery from inactivation. SCN5A-H558R can restore the in vitro gating defects caused by SCN5A-P2006A both when co-expressed on the same or on a different SCN5A construct. All other biophysical properties are similar between groups.
| Na Channels hNav1.5 | Peak Current Density (pA/pF) | Steady-State Inactivation (V1/2), mV | Recovery From Inactivation (τrec), ms |
|---|---|---|---|
| SCN5A-WT (n=12) | −598.6 ± 26.9 | −91.2 ± 0.8 | 8.9 ± 0.4 |
| SCN5A-P2006A (n=12) | −591.6 ± 30.1 | −80.6 ± 1.8* | 4.4 ± 0.3* |
| SCN5A-H558R-P2006A (n=20) | −615.5 ± 29.4 | −88.1 ± 1.0 | 8.8 ± 0.4 |
| SCN5A-H558R + SCN5A-P2006A (n=6) | −584.2 ± 81.4 | −87.6 ± 1.9 | 5.9 ± 0.4 |
| SCN5A-H558R + SCN5A-H558R-P2006A (n=6) | −621.2 ± 97.3 | −88.9 ± 2.9 | 6.0 ± 0.6 |
Functional indexes of sodium channel whole-cell current.
P<0.05 compared with wild type.
Given that the H558R polymorphism restored the biophysical defects caused by the P2006A mutation when expressed on the same gene, we also considered whether residual kinetic changes due to the interaction between the SCN5A-H558R and SCN5A-H558R-P2006A could also explain the absent phenotype observed in individuals who are homozygous for H558R. Co-expression of SCN5A-H558R with either SCN5A-P2006A or SCN5A-H558R-P2006A, was performed in HEK293 cells and cells were subsequently voltage clamped after 24-hour incubation. Surprisingly, we found that SCN5A-H558R can mitigate the in vitro gating defects caused by SCN5A-P2006A in both instances of being co-expressed on the same and on different SCN5A construct (Figure 3B, 4 and Table 1). These observations are likely an explanation for the absence of a LQT3 phenotype in the family members carrying the H558R polymorphism in addition to the P2006A mutation.
Functional analysis of another SCN5A mutation in presence of the sodium channel polymorphism H558R
The modulatory effect exerted by the SCN5A-H558R polymorphism on the fast inactivation gating defects characteristic of LQT3 mutations may also provide a plausible mechanism for the genotype-phenotype discordances seen in other LQT3 families. Therefore, we tested if H558R can/may also restore defects of other SCN5A mutations. P2006A lies in the C-terminus of SCN5A and consequently, we investigated whether H558R can also modulate the gating defects present in another mutation located in the C-terminus which also affected the inactivation kinetics of the sodium channel. The C-terminal mutation V1951L identified in cases of sudden infant death syndrome10,12 was co-expressed with the sodium channel polymorphism in HEK293 cells. After studying its biophysical properties, we noticed that V1951L also produced a +9 mV shift in steady-state inactivation (SCN5A-WT: −86.6±1.6 mV (n=6), SCN5A-V1951L: −77.8±0.9 mV (n=6), P<0.05) (Figure 5B). However, in the presence of the H558R polymorphism, the channel’s steady-state inactivation properties were not different as compared with those of the WT channels (SCN5A-H558R + SCN5A-V1951L: −82.1±0.9 mV (n=6)). In contrast, the time constants for recovery from inactivation were similar between groups (SCN5A-WT: 3.58±0.2 ms (n=6), SCN5A-V1951L: 4.21±0.5 ms (n=6), and SCN5A-H558R + SCN5A-V1951L: 4.39±0.5 ms (n=6)) (Figure 5C). However, unlike SCN5A-P2006A, neither SCN5A-V1951L nor SCN5A-H558R + SCN5A-V1951L mutants altered the persistent sodium current as compared with those of the WT channels (Figure 5D). Once again, similar to that of the previous studied mutation, co-expression of the H558R polymorphism with V1951L restored the gating defects to the WT level, suggesting that H558R may play an important role in the stabilization of the channel’s fast inactivation.
Figure 5.

Electrophysiological characterization of SCN5A mutations. A, Voltage dependence of activation for SCN5A-V1951L (n=6), SCN5A-H558R+SCN5A-V1951L (n=6) in which currents were compared to SCN5A-WT (n=6). B, The midpoints of steady-state inactivation (V1/2) in mutant channels of SCN5A-V1951L were significantly shifted toward depolarizing voltages compared to WT channels. C, Neither SCN5A-V1951L nor SCN5A-H558R+SCN5A-V1951L mutants modify the time course of recovery from inactivation. D, Superimposition of the normalized sodium currents recorded from SCN5A-WT (black), SCN5A-V1951L (gray), and SCN5A-H558R + SCN5A-V1951L (light gray). The transient sodium currents obtained during depolarization to −30 mV from a holding potential of −120 mV were normalized to illustrate similarity in fast inactivation. Neither SCN5A-V1951L nor SCN5A-H558R + SCN5A-V1951L mutants alter the late sodium current compared to WT channels. The dotted lines indicate zero current.
DISCUSSION
Mutations in the cardiac voltage-gated sodium channel alpha-subunit gene (SCN5A) have been implicated in multiple cardiac diseases, including LQTS, BrS, and SIDS.1–3 Here, we report the case of a 17-year-old girl who presented with episodes of syncope but only rare ECG prolongation. After sequence analysis, the previously reported SCN5A-P2006A mutation was found in this proband. Further sequencing of her family members revealed that her father and brother also carried the same SCN5A-P2006A variant.3,11–13 The functional studies suggested that this mutation could have a potential effect on the prolongation of the QT interval.3,10 Interestingly, although the SCN5A-P2006A gain-of-function mutant was found in three of our studied family members, none demonstrated consistent QT prolongation and only the proband had syncopal episodes. Importantly, even for the proband, it now appears that the syncopal episodes were not of cardiac origin. Since this family also carried the SCN5A-H558R polymorphism, this led us to hypothesize that this polymorphism could modify the gating kinetics of the mutated sodium channels and, as a result, also provide an explanation as to why this SCN5A-P2006A gain-of-function mutation has been identified in control populations.
In the present study, we found that channels containing the P2006A mutation exhibited significantly increased persistent sodium currents. These same mutated channels also displayed noteworthy depolarizing shifts in voltage dependence of steady-state inactivation and faster recovery from inactivation. The alteration in inactivation kinetics of sodium channels containing this SCN5A-P2006A mutation should in theory produce a prolongation of the QTc-interval in individuals who carry this mutation. Interestingly though, when we co-expressed the H558R polymorphism with the P2006A mutation on either the same or different genes, the gating kinetics were restored to the WT level. This observation may contribute to the absence of a typical LQT3 phenotype in those individuals that carry both the mutant and the polymorphism. However, the cause of syncope observed in the proband remains currently unknown.
Interestingly, we demonstrated that the SCN5A-H558R polymorphism is able to mitigate the in vitro gating defects caused by the SCN5A gain-of-function mutant either when SCN5A-H558R and SCN5A-P2006A are co-expressed on the same or different SCN5A constructs. Suggestive of an interaction between sodium channel alpha subunits, similar results were obtained with another mutation associated with SIDS also located in the C-terminal region of the sodium channel. Experiments with the mutation V1951L demonstrated that co-expression with the H558R polymorphism on a separate gene also rescued the gating defects associated with this mutation. These interesting findings are contributing to the growing evidence suggesting that sodium channel alpha subunits may interact19–22 despite the fact that, as opposed to those of potassium channels, the sodium channel alpha subunit alone is sufficient to form a functional conducting pore. These new pieces of evidence may have important implications in our understanding of genotype-phenotype correlations in inherited arrhythmias where SCN5A mutations are involved. In fact, our group has shown that this same SCN5A-H558R polymorphism also interacts with a BrS mutation (SCN5A-R282H) within intracellular compartments of the cell before protein trafficking to the membrane. This interaction restores the trafficking defect of the BrS R282H mutations thereby explaining the absence of a BrS phenotype in genotype-positive family members.16
The H558R polymorphism’s ability to mitigate the inactivation gating defects caused by the P2006A and V1951L mutations suggests that H558R might play an important role in the stabilization of these channels’ fast inactivation. It is interesting to note that previous studies demonstrated that LQT3 mutations located in the cytoplasmic linker between domains I and II (A572D, A572F, delAL586–587, G615E, L619F, and R680H) can either increase the persistent Na+ current or shorten recovery times from inactivation.10,23,24 This suggests that the domain I–II linker, in addition to the domain III–IV linker and the C-terminus region25,26–29, might also be involved in stabilizing the channels’ fast inactivation. Consequently, based on our results, it is possible to speculate that the H558R polymorphism found in the domain I–II linker might produce a change in the channel structure. This structural modification may allow for the stabilization of fast inactivation, thus correcting the gating defects seen in the P2006A and V1951L mutations.
As a result of the genotyping and manifestations of syncope, the proband (Figure 1A, II-2) was implanted with an ICD. Interestingly though, additional syncopal events that occurred in presence of the ICD demonstrated that they were not of cardiac origin. This therefore supports our electrophysiological recordings which demonstrate that the SCN5A-H558R polymorphism restores the gating defects of the SCN5A-P2006A variant. Thus, based on our electrophysiological findings and consistent with the clinical profiles of the three family members I-2 and II-1 and II-2 (Figure 1A), an individual who carries both the SCN5A-H558R polymorphism and the SCN5A-P2006A mutation should be asymptomatic. Notably, the proband, similar to her brother and father, has an ECG and QTc interval uncharacteristic of someone carrying a LQT3 mutation. We thus speculate that this could be attributable to the presence of the H558R polymorphism and its modification of the defective P2006A sodium channel that we observed in vitro. Hence, our results suggest that the H558R polymorphism might act as a disease-modifying gene that most likely, plays a vital role in the genotype-phenotype discordance seen in this family.
Importantly, a growing number of clinical studies have found this P2006A gain-of-function mutation in apparently healthy volunteers (whites, and blacks).30–32 This is somewhat counterintuitive since, according to its biophysical properties, this mutation, by itself, should produce a prolonged QT interval and lead to arrhythmias therefore making it hard to reconcile with the fact that it is present in the general population. However based on our findings, in presence of the H558R polymorphism the P2006A gating defects are restored which would explain why this mutation could be present in the general population without any diseased phenotype. This was actually the case for the members of the family studied which all carried the polymorphism in addition to the mutation. This then raises the interesting issue of whether an individual who carries P2006A may also carry the H558R polymorphism. Interestingly, after further inquiries to investigate this hypothesis, we have found that in fact for the reported carriers of P2006A, they were also carriers of the H558R polymorphism.3,13 Based on the fact that this polymorphism is present in about 20% of the population, we would therefore expect the polymorphism to be absent in some of these individuals who carry P2006A; however, none have been identified so far. Thus, it appears as if P2006A may co-segregate with H558R. This in itself is extremely interesting and might provide an explanation as to why P2006A has been identified in control populations.
However, the issue remains that P2006A has also been found in some patients who are victims of life-threatening arrhythmias.3 Interestingly though, these have been SIDS cases in which there is a possibility that the death could have been related to something else unrelated to the ion channel mutation identified by genotyping. Another possibility is that we obviously cannot be certain as to which level the H558R polymorphism can actually mitigate the P2006A defects in vivo. Further studies will be necessary to resolve this. Nevertheless, the fact that the P2006A mutation might co-segregate with H558R could at least provide an explanation as to why it has been found in healthy controls.
CONCLUSIONS AND STUDY LIMITATION
Our data are consistent with the hypothesis that the SCN5A-H558R polymorphism has the ability to modify gating kinetics in SCN5A mutants. These observations provide a plausible mechanism for decreased arrhythmogenic events in patients who not only carry these mutants, but also carry a sodium channel polymorphism. However, further studies are required to elucidate the definite mechanisms of this modification observed in the present study. One of the limitations of the present study lies in the fact that the characterizations of the SCN5A mutations were carried out in a heterologous mammalian expression system, thereby creating conditions that may be different from native cardiomyocytes that contain other components of the sodium channel macromolecular complex. Moreover, the relatively small number of affected individuals in this family precludes us from reaching a definitive conclusion. Nevertheless, our electrophysiological findings can provide an explanation as to why a mutation that produces biophysical defects characteristic of LQT3 might actually be found in the general population. However, further studies in a larger population will be necessary before our findings can be used to support clinical decision making.
Acknowledgement of Funding
This study was supported by an American Heart Association Scientist Development Grant (0635295N) and R01HL094450 (ID) and an AHA Pre-Doctoral Fellowship from the Great Rivers Affiliate (KS).
GLOSSARY OF ABBREVIATION
- SIDS
Sudden infant death syndrome
- LQT3
Long QT syndrome type 3
- LQTS
Long QT syndrome
- BrS
Brugada Syndrome
- WT
Wild-type
- ECG
Electrocardiogram
- INa
Sodium current
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest: none
REFERENCES
- 1.Chen Q, Kirsch GE, Zhang D, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998;392:293–296. doi: 10.1038/32675. [DOI] [PubMed] [Google Scholar]
- 2.Wang Q, Shen J, Splawski I, et al. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80:805–811. doi: 10.1016/0092-8674(95)90359-3. [DOI] [PubMed] [Google Scholar]
- 3.Arnestad M, Crotti L, Rognum TO, et al. Prevalence of long-QT syndrome gene variants in sudden infant death syndrome. Circulation. 2007;115:361–367. doi: 10.1161/CIRCULATIONAHA.106.658021. [DOI] [PubMed] [Google Scholar]
- 4.Tfelt-Hansen J, Winkel BG, Grunnet M, et al. Inherited Cardiac Diseases Caused by Mutations in the Nav1.5 Sodium Channel. J Cardiovasc Electrophysiol. 2009 doi: 10.1111/j.1540-8167.2009.01633.x. [DOI] [PubMed] [Google Scholar]
- 5.Zimmer T, Surber R. SCN5A channelopathies--an update on mutations and mechanisms. Prog Biophys Mol Biol. 2008;98:120–136. doi: 10.1016/j.pbiomolbio.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 6.Li H, Fuentes-Garcia J, Towbin JA. Current concepts in long QT syndrome. Pediatr Cardiol. 2000;21:542–550. doi: 10.1007/s002460010132. [DOI] [PubMed] [Google Scholar]
- 7.Bennett PB, Yazawa K, Makita N, et al. Molecular mechanism for an inherited cardiac arrhythmia. Nature. 1995;376:683–685. doi: 10.1038/376683a0. [DOI] [PubMed] [Google Scholar]
- 8.Beaufort-Krol GC, van den Berg MP, Wilde AA, et al. Developmental aspects of long QT syndrome type 3 and Brugada syndrome on the basis of a single SCN5A mutation in childhood. J Am Coll Cardiol. 2005;46:331–337. doi: 10.1016/j.jacc.2005.03.066. [DOI] [PubMed] [Google Scholar]
- 9.Schulze-Bahr E, Eckardt L, Breithardt G, et al. Sodium channel gene (SCN5A) mutations in 44 index patients with Brugada syndrome: different incidences in familial and sporadic disease. Hum Mutat. 2003;21:651–652. doi: 10.1002/humu.9144. [DOI] [PubMed] [Google Scholar]
- 10.Wang DW, Desai RR, Crotti L, et al. Cardiac sodium channel dysfunction in sudden infant death syndrome. Circulation. 2007;115:368–376. doi: 10.1161/CIRCULATIONAHA.106.646513. [DOI] [PubMed] [Google Scholar]
- 11.Ruan Y, Liu N, Priori SG. Sodium channel mutations and arrhythmias. Nat Rev Cardiol. 2009;6:337–348. doi: 10.1038/nrcardio.2009.44. [DOI] [PubMed] [Google Scholar]
- 12.Shim SH, Ito M, Maher T, et al. Gene sequencing in neonates and infants with the long QT syndrome. Genet Test. 2005;9:281–284. doi: 10.1089/gte.2005.9.281. [DOI] [PubMed] [Google Scholar]
- 13.Adabag AS, Peterson G, Apple FS, et al. Etiology of sudden death in the community: results of anatomical, metabolic, and genetic evaluation. Am Heart J. 2010;159:33–39. doi: 10.1016/j.ahj.2009.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Viswanathan PC, Benson DW, Balser JR. A common SCN5A polymorphism modulates the biophysical effects of an SCN5A mutation. J Clin Invest. 2003;111:341–346. doi: 10.1172/JCI16879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ye B, Valdivia CR, Ackerman MJ, et al. A common human SCN5A polymorphism modifies expression of an arrhythmia causing mutation. Physiol Genomics. 2003;12:187–193. doi: 10.1152/physiolgenomics.00117.2002. [DOI] [PubMed] [Google Scholar]
- 16.Poelzing S, Forleo C, Samodell M, et al. SCN5A polymorphism restores trafficking of a Brugada syndrome mutation on a separate gene. Circulation. 2006;114:368–376. doi: 10.1161/CIRCULATIONAHA.105.601294. [DOI] [PubMed] [Google Scholar]
- 17.Pitzalis MV, Anaclerio M, Iacoviello M, et al. QT-interval prolongation in right precordial leads: an additional electrocardiographic hallmark of Brugada syndrome. J Am Coll Cardiol. 2003;42:1632–1637. doi: 10.1016/j.jacc.2003.07.005. [DOI] [PubMed] [Google Scholar]
- 18.Deschenes I, Baroudi G, Berthet M, et al. Electrophysiological characterization of SCN5A mutations causing long QT (E1784K) and Brugada (R1512W and R1432G) syndromes. Cardiovasc Res. 2000;46:55–65. doi: 10.1016/s0008-6363(00)00006-7. [DOI] [PubMed] [Google Scholar]
- 19.Aldrich RW, Corey DP, Stevens CF. A reinterpretation of mammalian sodium channel gating based on single channel recording. Nature. 1983;306:436–441. doi: 10.1038/306436a0. [DOI] [PubMed] [Google Scholar]
- 20.Iwasa K, Ehrenstein G, Moran N, et al. Evidence for interactions between batrachotoxin-modified channels in hybrid neuroblastoma cells. Biophys J. 1986;50:531–537. doi: 10.1016/S0006-3495(86)83491-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Undrovinas AI, Fleidervish IA, Makielski JC. Inward sodium current at resting potentials in single cardiac myocytes induced by the ischemic metabolite lysophosphatidylcholine. Circ Res. 1992;71:1231–1241. doi: 10.1161/01.res.71.5.1231. [DOI] [PubMed] [Google Scholar]
- 22.Keller DI, Rougier JS, Kucera JP, et al. Brugada syndrome and fever: genetic and molecular characterization of patients carrying SCN5A mutations. Cardiovasc Res. 2005;67:510–519. doi: 10.1016/j.cardiores.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 23.Albert CM, Nam EG, Rimm EB, et al. Cardiac sodium channel gene variants and sudden cardiac death in women. Circulation. 2008;117:16–23. doi: 10.1161/CIRCULATIONAHA.107.736330. [DOI] [PubMed] [Google Scholar]
- 24.Wehrens XH, Rossenbacker T, Jongbloed RJ, et al. A novel mutation L619F in the cardiac Na+ channel SCN5A associated with long-QT syndrome (LQT3): a role for the I–II linker in inactivation gating. Hum Mutat. 2003;21:552. doi: 10.1002/humu.9136. [DOI] [PubMed] [Google Scholar]
- 25.Deschenes I, Trottier E, Chahine M. Implication of the C-terminal region of the alpha-subunit of voltage-gated sodium channels in fast inactivation. J Membr Biol. 2001;183:103–114. doi: 10.1007/s00232-001-0058-5. [DOI] [PubMed] [Google Scholar]
- 26.Cormier JW, Rivolta I, Tateyama M, et al. Secondary structure of the human cardiac Na+ channel C terminus: evidence for a role of helical structures in modulation of channel inactivation. J Biol Chem. 2002;277:9233–9241. doi: 10.1074/jbc.M110204200. [DOI] [PubMed] [Google Scholar]
- 27.Motoike HK, Liu H, Glaaser IW, et al. The Na+ channel inactivation gate is a molecular complex: a novel role of the COOH-terminal domain. J Gen Physiol. 2004;123:155–165. doi: 10.1085/jgp.200308929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Glaaser IW, Bankston JR, Liu H, et al. A carboxyl-terminal hydrophobic interface is critical to sodium channel function. Relevance to inherited disorders. J Biol Chem. 2006;281:24015–24023. doi: 10.1074/jbc.M605473200. [DOI] [PubMed] [Google Scholar]
- 29.Kass RS. Sodium channel inactivation in heart: a novel role of the carboxy-terminal domain. J Cardiovasc Electrophysiol. 2006;17 Suppl 1:S21–S25. doi: 10.1111/j.1540-8167.2006.00381.x. [DOI] [PubMed] [Google Scholar]
- 30.Ackerman MJ, Splawski I, Makielski JC, et al. Spectrum and prevalence of cardiac sodium channel variants among black, white, Asian, and Hispanic individuals: implications for arrhythmogenic susceptibility and Brugada/long QT syndrome genetic testing. Heart Rhythm. 2004;1:600–607. doi: 10.1016/j.hrthm.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 31.Schwartz PJ, Stramba-Badiale M, Crotti L, et al. Prevalence of the congenital long-QT syndrome. Circulation. 2009;120:1761–1767. doi: 10.1161/CIRCULATIONAHA.109.863209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kapplinger JD, Tester DJ, Alders M, et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm. 2010;7:33–46. doi: 10.1016/j.hrthm.2009.09.069. [DOI] [PMC free article] [PubMed] [Google Scholar]