

Abstract
Permeabilization of the mitochondrial membranes is a crucial step in apoptosis and necrosis. This phenomenon allows the release of mitochondrial death factors which trigger or facilitate different signaling cascades ultimately causing the execution of the cell. The mitochondrial permeability transition pore (mPTP) has long been known as one of the main regulators of mitochondria during cell death. mPTP opening can lead to matrix swelling, subsequent rupture of the outer membrane and a nonspecific release of intermembrane space proteins into the cytosol. While mPTP was purportedly associated with early apoptosis, recent observations suggest that mitochondrial permeabilization mediated by mPTP is generally more closely linked to events of late apoptosis and necrosis. Mechanisms of mitochondrial membrane permeabilization during cell death, involving three different mitochondrial channels, have been postulated. These include the mPTP in the inner membrane, and the mitochondrial apoptosis-induced channel (MAC) and voltage dependent anion-selective channel (VDAC) in the outer membrane. New developments on mPTP structure and function, and the involvement of mPTP, MAC, and VDAC in permeabilization of mitochondrial membranes during cell death are explored.
Keywords: mPTP, mitochondrial permeability transition pore; MAC, mitochondrial apoptosis-induced channel; VDAC, voltage dependent anion-selective channel; Bcl-2 family proteins; patch clamp, pharmacology
1. Introduction
In recent years, the role of mitochondria in both apoptotic and necrotic cell death has received considerable attention. An increase in mitochondrial membrane permeability is one of the key events in apoptotic and necrotic death, although the details of the mechanisms involved remain to be elucidated. Unlike the resting potentials of neurons and cardiomyocytes which are largely governed by basal K+ conductance of their plasma membranes, the value of the mitochondrial resting potential is controlled by the very high resistance of the inner membrane, i.e., low permeability to all ions. While the inner membrane contains several channels, their opening is tightly regulated in order to prevent dissipation of the membrane potential and proton gradient that is the electrochemical energy reservoir for ATP-synthesis and transport. These channels include the putative K+ATP channel and mitochondrial Centum-picoSiemen (mCS), which are cation- and anion-selective channels that monitor metabolic levels and aid in volume regulation, respectively [1–3]. There are also inner membrane channels within the protein import complexes called TIM22 and TIM23 [4, 5]. The inner membrane also expresses multiple calcium channel activities that range from the mitochondrial ryanodine receptor (mRyR) of cardiomyocytes to the more generic calcium uniporter [6, 7]. Uncontrolled opening of any one of these channels may unleash havoc resulting in cell death. For example, exogenous targeting peptides putatively open the pore of the TIM23 complex and induce a rapid and high amplitude swelling of mitochondria [8].
The mitochondrial permeability transition pore, or mPTP, is however the most notorious of all the inner membrane channels. mPTP has not only been linked to a rupture of mitochondrial outer membrane during cell death but also to a myriad of pathologies (Figure 1) [9–11]. More recently, outer membrane channels such as the Mitochondrial Apoptosis-induced Channel (MAC) and the Voltage Dependent Anion Channel (VDAC) were also shown to be directly or indirectly involved in mitochondrial permeabilization during apoptosis and/or necrosis (Figure 1) [12–15]. This review summarizes our current understanding of the role of the mPTP during mitochondrial membrane permeabilization and how this channel may collaborate with MAC and VDAC in order to regulate cell death.
Figure 1. Mitochondrial ion channels in apoptosis and necrosis.

Left, Apoptotic stimuli induce relocation of Bax from the cytosol into the mitochondrial outer membrane (MOM). Bax, Bak, and possibly other unidentified protein(s) oligomerize and form MAC to release cytochrome c. VDAC oligomerization upon apoptotic stimuli was also reported to be involved in cytochrome c release. Right, Necrotic stimuli lead to exacerbated calcium uptake and reactive oxygen species generation by mitochondria. High levels of calcium and reactive oxygen species (ROS) induce a cyclophilin-D (Cyp D)-sensitive opening of mPTP that leads to swelling of the matrix and release of calcium. Swelling disrupts the outer membrane while released calcium activates proteases, phosphatases and nucleases that lead to necrotic degradation. Adapted from [112]. TIM23/22, translocase of the inner membrane complexes 23/22; mRyR, mitochondrial ryanodine receptor; TOM, translocase of the outer membrane; MCU, mitochondrial calcium uniporter; MIM, mitochondrial inner membrane.
2. Separating Fact from Fiction on the Structure and Function of mPTP
The mPTP was originally observed in swelling experiments on isolated mitochondria reported in landmark studies by Hunter and Haworth in 1979 [16–18]. The mPTP has a large caliper pore with low ion-selectivity. Opening of the mPTP increases the permeability of the inner membrane for molecules up to 1.5 kDa that leads to organelle swelling and mitochondrial depolarization. The mPTP can be activated in cells and metabolizing isolated mitochondria by a myriad of effectors but, most notably, calcium plus phosphate and reactive oxygen species (See an extensive review in ref. [19] and a more recent review in ref. [20]). Reversible closure of mPTP occurs upon removal of calcium with EGTA or by the addition of ADP, magnesium, or cyclosporine A (CsA). The principal trigger for mPTP opening is matrix calcium in the presence of phosphate, but the activating concentrations of both are thought to rely on several other factors. For example, higher calcium levels are needed for mPTP opening in the presence of any one of several mPTP inhibitors including high negative membrane potential, low matrix pH, accumulation of adenine nucleotides like ADP, and other divalent cations like magnesium and strontium. Conversely, the calcium levels needed for mPTP activation are lower if adenine nucleotides are depleted or a mitochondrial uncoupler is added to depolarize the membrane potential after the uptake of calcium. Opening of the mPTP in response to oxidative stress has been linked to ischemia-reperfusion injury in heart [9] and more recently to neurodegenerative diseases, like Alzheimer s and multiple sclerosis [10, 11]. Hence, mPTP has become an important pharmacological target for both cardio- and neuroprotection.
The mPTP can also temporarily open or “flicker” leading to transient membrane potential variations in response to certain intracellular signals unrelated to cell death. Then, mPTP closure leads to a “resealing” of the inner membrane and a restoration of the ability to synthesize ATP [21]. This mode of action of the mPTP underlies a mitochondrial version of calcium-induced calcium release between the cytosol and the mitochondrial matrix [22–24]. That is, transient opening of mPTP could provide the pathway for mitochondrial Ca2+ extrusion under relatively normal conditions. The rapid removal of calcium from the cytosol and its subsequent release from mitochondria through mPTP flickering could prevent calcium-inactivation of channels essential to refilling calcium stores as well as allow for signaling in these microdomains. Thus, mPTP flickering could impact processes as diverse as muscle contraction and saliva secretion [25, 26].
Furthermore, brief opening of mPTP causes a transient mitochondrial depolarization and a short burst of ROS production, and reveals a potential role for mPTP in ROS signaling [27]. ROS generated by one mitochondrion might then interact with neighboring mitochondria, perhaps once again transiently opening mPTP. The ensuing depolarization could produce additional ROS. Mitochondria-generated ROS has putative roles in a myriad of cell signaling as evidenced by the redox sensitivity of chaperones, kinases and even gene expression [28]. Hence, mPTP can, at least indirectly, impact many cellular processes that are not linked to cell death. Nevertheless, flickering of the mPTP can result in sufficient production of ROS leading to sustained mPTP opening and eventually cell death [29].
Even though the phenomenon of mitochondrial permeability transition was originally described in the late 1970s, the molecular composition of the mPTP remains a mystery today. The biochemical studies of mitochondrial complexes by Brdiczka s group [30] and the electrophysiological studies of Zoratti s group [31] were among the first to propose that the complexes responsible for mPTP activity spanned both the inner and outer membranes of mitochondria. In fact, they were proposed to be in contact sites, or close junctions, between the two mitochondrial membranes.
These watershed studies also led to the hypothesis that the mPTP contains the outer membrane channel VDAC, which is also often referred to as mitochondrial porin [30, 31]. More recent studies in which the three Vdac isoforms are knocked out have raised doubts about the essential nature of the involvement of outer membrane components in mPTP as these knockouts continue to express mPTP activity [32]. Nevertheless, VDAC continues to be purported as part of the mPTP mechanism in normal cells [33–35]. In other studies, VDAC oligomers were even proposed to be directly responsible for cytochrome c release (Figure 1) [15, 36].
The adenine nucleotide translocator, or ANT, has long been proposed to be an inner membrane component of mPTP. Some of these studies were biochemical, while others were pharmacological [30]. ANT inhibitors like bongkrekic acid and atractyloside lock the translocator in opposite conformations to prevent or induce a mitochondrial permeability transition, respectively [37]. ANT is the most abundant inner membrane protein on a molar basis and is frequently the target of misfolding. In this scenario, ANT undergoes large conformation changes and the atractyloside-stabilized conformation appears to be particularly vulnerable to damage leading to misfolding and mPTP formation (reviewed in [38, 39]). However, the seminal studies of Wallace s group clearly showed the mPTP was in fact still present in Ant knockouts [40]. Hence, Ant does not provide the essential inner membrane permeability pathway for mPTP. Nevertheless, The adenine nucleotide translocator likely plays a regulatory role as the sensitivity of mPTP to certain pharmacological effectors was modified in mitochondria lacking all three isoforms of Ant. Furthermore, knockouts of just Ant1 are more resistant to glutamate-induced excitotoxicity, a well known mPTP-mediated process [41]. Finally, the up-regulation of the uncoupling protein-3 was shown to induce a sensitization of the mPTP to calcium [42, 43], suggesting that inner membrane transporters other than the ANT may also regulate mitochondrial permeability transition.
More recently, the phosphate carrier was proposed to play the central role of inner membrane pore of mPTP [20]. While intriguing, the role of the phosphate carrier in the elusive mPTP awaits molecular studies in which the effects of eliminating the various isoforms of this carrier on the mPTP characteristics are determined. Other putative components have been identified including misfolded proteins [44] and polyphosphates [45]. Finally, other mPTP pore candidates could account for the occurrence of mitochondrial permeabilization in ANT-deficient mitochondria. That is, other anion transporters, such as the aspartate-glutamate exchanger, were also found to form large conductance pores [46–48].
Cyclophilin-D is a known activator of the mPTP as its inhibition by CsA leads to closure of the pore [49, 50]. Cyclophilin-D-depleted mitochondria still undergo a calcium-induced permeability transition but this phenomenon requires higher calcium concentrations and is insensitive to CsA [51]. These observations reinforce the notion that cyclophilin-D is an important regulator of the mPTP but also refute the notion that this protein might be a structural component of the pore. Finally, it was recently postulated that cyclophilin-D favors the open conformation of mPTP by masking a specific site for inorganic phosphate (Pi); the occupancy of this site by Pi leads to a desensitization of the mPTP to calcium [52].
3. mPTP and mechanisms of mitochondrial permeabilization during cell death
Apoptosis is a process of cell suicide that occurs in multi-cellular organisms in response to developmental, homeostatic, or internal damage signals. This programmed cell death involves extrinsic and/or intrinsic apoptotic pathways that converge in a series of biochemical events such as activation of caspases that leads to cell shrinkage and plasma membrane blebbing. On the other hand, necrosis is often caused by external factors, such as infection, toxins, trauma, or ischemia-reperfusion injury that leads to cell swelling and eventually rupture of the plasma membrane. This scenario is in contrast to apoptosis, which is a naturally occurring cause of cellular death [53]. Nevertheless, mitochondria compartmentalize a myriad of death-evoking signaling factors, which wreak havoc upon their release to the cytosol and eventually cause either apoptotic or necrotic cell death.
During early intrinsic apoptosis, permeabilization of the mitochondrial outer membrane releases pro-apoptotic factors, like cytochrome c, from the intermembrane space into the cytosol [12, 54–56]. Importantly, this event is highly regulated through specific interactions between proteins of the Bcl-2 family [12, 54–60]. In this family, effector proteins such as Bax and Bak are essential to the machinery allowing membrane permeabilization. On the other hand, anti-apoptotic members such as Bcl-2 and Bcl-xL, inhibit this process by directly binding to the pro-apoptotic effectors [57]. Finally, BH3-only proteins such as Bad or truncated Bid relay apoptotic stimuli to the mitochondria by interacting with both effectors and anti-apoptotic Bcl2 family members [54, 55, 57]. Therefore, when challenged by an apoptotic stimulus, the combined signaling within the Bcl-2 family dictates the immediate fate of the cell, i.e., whether or not to induce permeabilization of the outer membrane [54]. This phenomenon has been coined Mitochondrial Outer Membrane Permeabilization, or MOMP, and the released cytochrome c leads to an activation of the executioner caspases by proteolysis and eventually to plasma membrane blebbing [55, 61].
Cytochrome c release during MOMP typically occurs through the outer membrane channel MAC. MOMP and MAC formation have been consistently reported in a variety of cell lines during at least two different apoptotic insults, deprivation of interleukin-3 [62] and kinase inhibition [63]. MAC activity is observed before mitochondrial depolarization, is exquisitely regulated by Bcl-2 family proteins, and can initiate release of apoptotic mediators from mitochondria to commit the cell to die [3, 62–65]. Such release can in turn be prevented by overexpressing Bcl-2 or Bcl-xL, or using inhibitors of MAC (iMACs) [62, 66]. Bax oligomers have been identified as part of this channel and knock-out studies have shown that MAC contains neither Vdac1 nor Vdac3 [62, 67]. On the other hand, Vdac2 was shown to regulate the pro-apoptotic protein Bak which is another putative component of MAC [67–69]. Finally, CsA does not prevent MAC-induced cytochrome c release indicating that MAC and mPTP are two separate molecular entities [70]. However, some observations suggest that MAC and mPTP may participate in an amplification loop during cell death and this putative crosstalk is discussed below in section 5 [3, 56].
Opening of the mPTP leads to a transitory permeabilization of the inner membrane also called Mitochondrial Permeability Transition (MPT). Sustained opening of mPTP has catastrophic consequences for these organelles. Mitochondria seem to pop as they rapidly swell; this swelling occurs because the high concentration of proteins in the matrix space exerts a large colloidal osmotic pressure. The cristae unfold as the matrix swells so that the outer membrane ruptures. Depolarized mitochondria become a major liability for the cell as ATP is hydrolyzed in efforts to restore the membrane potential. Furthermore, this unspecific loss of outer membrane integrity also causes a spillage of mitochondrial death factors, like cytochrome c, from the intermembrane space to the cytosol. Finally, the collapse of the membrane potential combined with the leakage of pyridine nucleotides, e.g. NADH, can lead to the generation of reactive oxygen species via the direct transfer of electrons to molecular oxygen [20, 71].
The outer membrane channel VDAC may act indirectly to facilitate mPTP opening. For example, VDAC1 was shown to regulate the transport of calcium, an mPTP activator, across the outer membrane [72]. Intriguingly, partial closure of the channel, but not its opening, increases the calcium permeability of VDAC1 [14, 73]. Interestingly, superoxide but not hydrogen peroxide, induces a mitochondrial permeabilization, which is blocked by VDAC1 antibodies [74]. Therefore, VDAC1 may under certain circumstances act as an mPTP sensitizer even if it may not be a component of the pore [32].
Does VDAC play a role in cell death outside of modulation of the mPTP? Oligomers of VDAC1 have been proposed to form channel structures to allow the release of cytochrome c through the outer membrane during apoptosis [36]. However, even though VDAC1 oligomers have been observed during apoptosis, their role as components of a functional cytochrome c release channel remains to be determined. VDAC activity is also known to be regulated by anti-apoptotic Bcl-2 family proteins [75]. The NMR solution structure of human VDAC1 identified strands 17 and 18 as a putative binding site for Bcl-xL [76]. VDAC2 complexes with pro-apoptotic Bak and mediates its availability to form the cytochrome c release channel MAC [68, 69, 77]. Interestingly, pro-apoptotic Bax does not change the channel activity of VDAC but the truncated BH3-only protein Bid closes VDAC. This closure could limit metabolite transport and lead mitochondrial dysfunction, and death [78, 79].
The single channel behaviors of MAC and mPTP, as well as VDAC, are quite different as shown in the current traces of Figure 2 and the summary of their channel characteristics provided in Table 1. As expected, blocking MAC with iMACs [66, 80] and mPTP with CsA also impacts onset of cell death. Below, we will further discuss the role of mPTP in apoptosis and necrosis and the possibility that mPTP and MAC may act synergistically during apoptosis.
Figure 2. The channel activities of VDAC, mPTP and MAC.
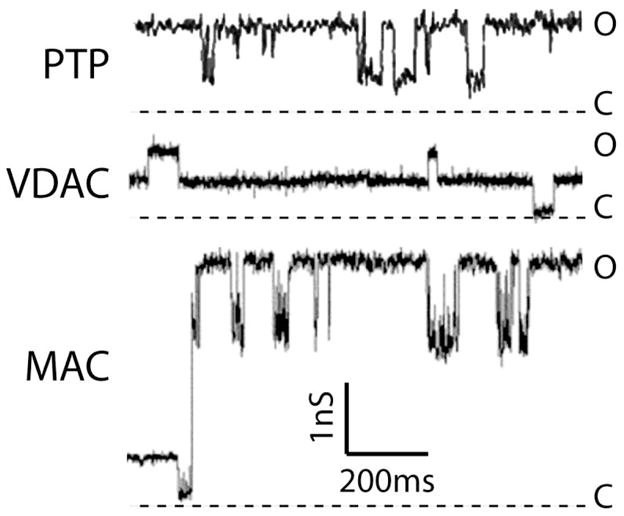
Current traces were recorded from patches excised from either reconstituted outer (MAC and VDAC) or native inner membranes (mPTP) under symmetrical 150 mM KCl. Current traces are represented in the same scale for comparison. Current levels of open (O) channels are shown with downward transitions to closed (C) states in all the traces. Mouse MAC and VDAC traces were adapted from ref. [62], while mouse mPTP was from ref. [56].
Table 1.
Comparison of the channel activities of mPTP, MAC and VDAC*
| mPTP | MAC | VDAC | |
|---|---|---|---|
| Peak conductance (nS) | 1.1 ± 0.1 | 1–5 | 0.7 ± 0.1 |
| Transition size (nS) | 0.3–1.0 | 0.3–2.0 | 0.36 ± 0.04 |
| Ion selectivity | Sl. Cation | Sl. Cation | Anion |
| PK/PCl | 7 | 4.7 ± 1.3 | 0.7 ± 0.1 |
| Voltage dependent | Yes | No | Yes |
| Pore diameter (nm) | 2.8 ± 0.1 | 2.7–6.0 | 2.2 ± 0.05 |
| Physiological Modulators | Ca2+, Pi, ADP [16–18] | TBid, Cyt. c [65, 67] | NADH [14, 109] |
| Pharmacological agents | CsA [49] | iMACs, Bcis [66, 80, 110] | Koenig s Polyanion, G3139, DIDS [36, 111] |
The description of the effects on mPTP, MAC and VDAC of the physiological modulators and pharmacological agents cited is given in sections 2 and 3. Sl, slightly; tBid; truncated Bid; Cyt. c, cytochrome c; CsA, cyclosporine A; iMACs, inhibitors of MAC; Bcis, Bax channel inhibitors; DIDS, 4,4′-diisothiocyanatostilbene-2,29-disulfonic acid.
4. mPTP opening: suicidal, criminal, or both?
In the mid-1990s, the mPTP attracted the attention of investigators in the cell death field, because it was reported that at least some forms of apoptosis could be inhibited by CsA, a well known inhibitor of the pore [81, 82]. Also, the suppression of the calcium-activation of the mPTP by overexpression of Bcl-2 supports a role for this channel in apoptosis [83, 84]. However, the swelling of the matrix as proposed in the standard model of mPTP activation is not observed in all scenarios of apoptosis [85]. Moreover, the permeabilization of the inner membrane is sometimes seen only in late apoptosis, after the release of cytochrome c and caspase activation [86]. Neither extrinsic nor intrinsic pathways of apoptosis were altered in cells deficient in the mPTP activator cyclophilin-D [51, 87, 88]. These findings suggest that mPTP opening does not initiate apoptosis and that this pore is instead central to necrosis [89–91]. These observations suggest that a more selective mechanism of permeabilization such as MAC formation may typically be operating at least during early apoptosis [29, 62, 65, 92, 93].
Even if the pore is not a principal player in apoptotic cell suicide, mPTP is certainly criminal in other ways. The involvement of the mPTP in necrosis and ischemia/reperfusion injury of the heart is a particularly well documented case [20]. The lack of oxygen supply to tissues does not immediately lead to cell death during ischemia. However, the subsequent reperfusion triggers an oxidative stress that causes the demise of the cells. More particularly, it seems that ischemia initiates changes in the cells, such as the increase of free ADP, Pi, and of cytosolic calcium which ultimately leads to superoxide production during reperfusion [9]. Such a scenario would lead to a vicious cycle. The subsequent mPTP opening would further dysregulate calcium homeostasis and inhibit the respiratory chain, resulting in further reactive oxygen species production and mPTP opening. Numerous experimental evidence supports the involvement of the mPTP in ischemia/reperfusion. For example, CsA is the classical inhibitor of the mPTP and this agent can reduce the occurrence of necrosis in cardiomyocytes subjected to anoxia-reperfusion. CsA was also effective in protecting these same cells against reperfusion-induced mitochondrial membrane potential collapse [94]. Another example of mPTP involvement during necrosis can be found in stroke models of primary brain cells [95].
Examination of cyclophilin-D-deficient mice has provided even more compelling evidence that the mPTP plays a crucial role in necrosis [87, 88, 96]. The pore is still present in mitochondria and cells obtained from these mice. However, cyclophilin-D ablation increases the amount of calcium required for mPTP opening and abolishes the sensitivity to CsA [97]. This phenotype has also been associated with the resistance of cyclophilin-D knock out cells to necrotic stimuli such as A23187-induced calcium overload, and to the decrease of heart and brain injury following ischemia/reperfusion [87, 88, 96, 97]. However, cells isolated from these same animals still died in response to treatments with classical apoptotic inducers such as staurosporine or etoposide [88].
Taken together, these data indicate that mPTP opening is chiefly involved in necrosis rather than in triggering cytochrome c release during early apoptosis. Nevertheless, MAC and mPTP opening may act alone or in combination, depending on cell type and death stimulus, to amplify the death signals. In this context, the swelling precipitated by mPTP opening would cause a remodeling of the cristae, which could facilitate a more complete release of cytochrome c and other pro-apoptotic factors from the mitochondria [29, 98].
5. Crosstalk between mPTP and MAC links necrosis and apoptosis
A synergistic relationship between apoptosis and necrosis could enhance removal of damaged cells and minimize inflammation, while restoring tissue homeostasis. Crosstalk signaling between mPTP opening resulting in MPT and MAC formation resulting in MOMP could provide such a platform. It is known that MAC formation precedes mPTP opening in many cases, e.g., interleukin-3 withdrawal from FL5.12 cells [3, 62]. However, MAC assembly may also occur after mPTP opening in other situations, e.g., hepatitis in mouse caused by lipopolysaccharide through the tumor necrosis factor receptor pathway [99]. Recently, other modes of interaction between MAC and mPTP have been identified.
As discussed above, mechanisms that block mPTP, like CsA or knocking out cyclophilin-D, reduce necrosis, and, in some cases, also suppress apoptosis [3]. The reversible events leading up to necrotic cell death often include a reduction in available energy sources like ATP and a switch from oxidative phosphorylation to glycolysis as the major source of ATP synthesis. Cells swell as Na+ accumulates in the face of reduced Na+/K+ ATPase activity. Low levels of calcium entry initiate limited mPTP opening as well as activation of proteases and phosphatases. If the irritant is removed, the cells may recover. Persistent irritation or massive harmful stimuli may cause irreversible damage. When cells can no longer cope, plasma membrane integrity is lost and the cells are considered dead by necrosis.
Cellular stresses, which if sustained will result in mPTP opening and necrosis, may also activate intrinsic apoptosis. Clearly, calcium overload opens mPTP, and depolarizes mitochondria, which then jeopardizes buffering of cytosolic calcium transients. The resulting elevated levels of cytosolic calcium may then lead to MAC formation through activation of proteases and phosphatases like calpain and calcineurin (Figure 3). Calpain is a calcium-dependent cysteine protease that can cleave the BH3-only protein Bid. Newly formed truncated Bid can then trigger Bax oligomerization and MAC assembly, leading to MOMP [100]. Similarly, calcineurin is a protein phosphatase that can activate another BH3-only protein Bad. Dephosphorylated Bad interferes with the anti-apoptotic function of Bcl-2 or Bcl-xL, which again facilitates MAC formation [101–103]. Interestingly, dephosphorylated Bad can also directly activate mPTP opening in the absence of truncated Bid, thereby auto-amplifying mPTP-induced mitochondrial permeabilization [103]. Hence, activation of calpain and calcineurin by calcium may amplify mPTP activation, facilitate MAC assembly, and commit cells to die by intrinsic apoptosis. A similar scenario follows excitotoxic injury resulting in necrosis via mPTP opening and then delayed apoptosis through MAC assembly resulting from AMP-kinase activation of the BH3-only protein Bim [104].
Figure 3. Crosstalk between MAC and mPTP amplifies cell death by apoptosis and necrosis.

Elevated cytosolic Ca2+ activates calpain and calcineurin, which facilitate MAC formation through activation of Bid, Bad and Bax. Mitochondrial calcium overload in the matrix induces mPTP opening. Cytochrome c released after either MAC formation or mPTP opening facilitates apoptosome formation and caspase activation. Released cytochrome c causes further dysregulation of calcium homeostasis by interacting with the ER calcium release channel IP3 receptors (IP3R). Anti-apoptotic Bcl-2 suppresses while ROS signaling enhances the formation/activation of MAC, mPTP, and IP3R. Thus, opening of MAC/mPTP, cytochrome c release, and activation of IP3R may form a positive feedback loop to amplify the cell death signal. ER and MITO indicate endoplasmic reticulum and mitochondria, respectively. Adapted from [3].
Positive feedback between MAC formation and mPTP opening may enhance cytochrome c release and progression of cell death processes. In fact, there are several means to release cytochrome c [105]. One report indicates that released cytochrome c binds to and relieves the calcium-dependent inactivation of IP3R in the ER, which causes further Ca2+ release during apoptosis [106]. This additional calcium release may activate mPTP, which would cause matrix swelling. The ensuing cristae remodeling and eventual rupture of the outer membrane caused by mPTP opening would enhance cytochrome c release from the folds of the inner membrane and ultimately caspase activation. These findings suggest that a small release of cytochrome c via MAC may form a positive feedback loop to amplify the cell death signals through further calcium dysregulation and opening of mPTP (Figure 3). This loop utilizes many of the same players recently identified as important in the transfer of calcium from the ER to mitochondria, which is needed to maintain normal bioenergetics for survival and prevent autophagy [107]. One can imagine that inappropriate generation of IP3 could similarly induce opening of mPTP. Another such amplification loop is based on oxidation of cardiolipin by ROS, which is often associated with ischemia-reperfusion injury and early apoptosis in some cell types. Peroxidation of cardiolipin decreases the binding of cytochrome c to the inner membrane, which increases its availability for release to the cytosol [105]. Furthermore, remodeling of the cristae through disruption of OPA1 oligomers by truncated Bid also facilitates cytochrome c release [108]. Finally, caspase degradation of the respiratory chain might also generate ROS. In contrast, cells also contain the means to suppress apoptosis and necrosis, e.g. through proteins like Bcl-2 and HSP70, which are now being explored for novel therapeutics [105].
6. Future Perspectives
The permeability of the mitochondrial membranes is key to decisions regarding survival and death and whether apoptosis or necrosis will take place. Understanding the relationship between these cell death pathways will provide insight into their regulation and may reveal novel therapeutic targets. mPTP opening and MAC formation provide opportunities for crosstalk signaling between apoptosis and necrosis. One compelling problem in this field is lack of information regarding the molecular basis of mPTP and its relationship with other mitochondrial channels like MAC and VDAC. Even though our direct studies of mitochondrial channels are somewhat limited, these channels are, nevertheless, emerging as promising therapeutic targets for aging and diseases including cancer and neurodegenerative diseases, like Parkinson s and Alzheimer s Disease.
Acknowledgments
This work was supported by the National Institutes of Health [Grant GM57249] to KWK. We apologize many important papers were not cited here because of space constraints; many can be found in the cited reviews.
Abbreviation list
- mPTP
mitochondrial permeability transition pore
- MAC
mitochondrial apoptosis-induced channel
- VDAC
voltage dependent anion-selective channel
- CsA
cyclosporine A
- ROS
reactive oxygen species
- MPT
mitochondrial permeability transition
- MOMP
mitochondrial outer membrane permeabilization
- iMACs
inhibitors of MAC
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- 1.Inoue I, Nagase H, Kishi K, Higuti T. ATP-sensitive K+ channel in the mitochondrial inner membrane. Nature. 1991;352:244–7. doi: 10.1038/352244a0. [DOI] [PubMed] [Google Scholar]
- 2.Sorgato MC, Keller BU, Stuhmer W. Patch-clamping of the inner mitochondrial membrane reveals a voltage-dependent ion channel. Nature. 1987;330:498–500. doi: 10.1038/330498a0. [DOI] [PubMed] [Google Scholar]
- 3.Ryu SY, Peixoto PM, Teijido O, Dejean LM, Kinnally KW. Role of mitochondrial ion channels in cell death. Biofactors. 2010 doi: 10.1002/biof.101. [DOI] [PubMed] [Google Scholar]
- 4.Lohret TA, Jensen RE, Kinnally KW. Tim23, a protein import component of the mitochondrial inner membrane, is required for normal activity of the multiple conductance channel, MCC. J Cell Biol. 1997;137:377–86. doi: 10.1083/jcb.137.2.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peixoto PM, Grana F, Roy TJ, Dunn CD, Flores M, Jensen RE, Campo ML. Awaking TIM22, a dynamic ligand-gated channel for protein insertion in the mitochondrial inner membrane. J Biol Chem. 2007;282:18694–701. doi: 10.1074/jbc.M700775200. [DOI] [PubMed] [Google Scholar]
- 6.Beutner G, Sharma VK, Lin L, Ryu SY, Dirksen RT, Sheu SS. Type 1 ryanodine receptor in cardiac mitochondria: transducer of excitation-metabolism coupling. Biochim Biophys Acta. 2005;1717:1–10. doi: 10.1016/j.bbamem.2005.09.016. [DOI] [PubMed] [Google Scholar]
- 7.Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 2004;427:360–4. doi: 10.1038/nature02246. [DOI] [PubMed] [Google Scholar]
- 8.Kushnareva YE, Campo ML, Kinnally KW, Sokolove PM. Signal presequences increase mitochondrial permeability and open the multiple conductance channel. Arch Biochem Biophys. 1999;366:107–15. doi: 10.1006/abbi.1999.1190. [DOI] [PubMed] [Google Scholar]
- 9.Crompton M, Costi A, Hayat L. Evidence for the presence of a reversible Ca2+-dependent pore activated by oxidative stress in heart mitochondria. Biochem J. 1987;245:915–8. doi: 10.1042/bj2450915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singh P, Suman S, Chandna S, Das TK. Possible role of amyloid-beta, adenine nucleotide translocase and cyclophilin-D interaction in mitochondrial dysfunction of Alzheimer’s disease. Bioinformation. 2009;3:440–5. doi: 10.6026/97320630003440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Su KG, Banker G, Bourdette D, Forte M. Axonal degeneration in multiple sclerosis: the mitochondrial hypothesis. Curr Neurol Neurosci Rep. 2009;9:411–7. doi: 10.1007/s11910-009-0060-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dejean LM, Martinez-Caballero S, Kinnally KW. Is MAC the knife that cuts cytochrome c from mitochondria during apoptosis? Cell Death Differ. 2006;13:1387–95. doi: 10.1038/sj.cdd.4401949. [DOI] [PubMed] [Google Scholar]
- 13.Dejean LM, Martinez-Caballero S, Manon S, Kinnally KW. Regulation of the mitochondrial apoptosis-induced channel, MAC, by BCL-2 family proteins. Biochim Biophys Acta. 2006;1762:191–201. doi: 10.1016/j.bbadis.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 14.Rostovtseva TK, Tan W, Colombini M. On the role of VDAC in apoptosis: fact and fiction. J Bioenerg Biomembr. 2005;37:129–42. doi: 10.1007/s10863-005-6566-8. [DOI] [PubMed] [Google Scholar]
- 15.Shoshan-Barmatz V, De Pinto V, Zweckstetter M, Raviv Z, Keinan N, Arbel N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol Aspects Med. 2010;31:227–85. doi: 10.1016/j.mam.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 16.Haworth RA, Hunter DR. The Ca2+-induced membrane transition in mitochondria. II. Nature of the Ca2+ trigger site. Arch Biochem Biophys. 1979;195:460–7. doi: 10.1016/0003-9861(79)90372-2. [DOI] [PubMed] [Google Scholar]
- 17.Hunter DR, Haworth RA. The Ca2+-induced membrane transition in mitochondria. I. The protective mechanisms. Arch Biochem Biophys. 1979;195:453–9. doi: 10.1016/0003-9861(79)90371-0. [DOI] [PubMed] [Google Scholar]
- 18.Hunter DR, Haworth RA. The Ca2+-induced membrane transition in mitochondria. III. Transitional Ca2+ release. Arch Biochem Biophys. 1979;195:468–77. doi: 10.1016/0003-9861(79)90373-4. [DOI] [PubMed] [Google Scholar]
- 19.Gunter T, Pfeiffer D. Mechanisms by which mitochondria transport calcium. Am J Physiol. 1990;258:C755–86. doi: 10.1152/ajpcell.1990.258.5.C755. [DOI] [PubMed] [Google Scholar]
- 20.Halestrap AP. What is the mitochondrial permeability transition pore? J Mol Cell Cardiol. 2009;46:821–31. doi: 10.1016/j.yjmcc.2009.02.021. [DOI] [PubMed] [Google Scholar]
- 21.Pastorino JG, Tafani M, Rothman RJ, Marcinkeviciute A, Hoek JB, Farber JL. Functional consequences of the sustained or transient activation by Bax of the mitochondrial permeability transition pore. J Biol Chem. 1999;274:31734–9. doi: 10.1074/jbc.274.44.31734. [DOI] [PubMed] [Google Scholar]
- 22.Ichas F, Jouaville LS, Mazat JP. Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell. 1997;89:1145–53. doi: 10.1016/s0092-8674(00)80301-3. [DOI] [PubMed] [Google Scholar]
- 23.Jouaville LS, Ichas F, Mazat JP. Modulation of cell calcium signals by mitochondria. Mol Cell Biochem. 1998;184:371–6. [PubMed] [Google Scholar]
- 24.Gunter TE, Sheu SS. Characteristics and possible functions of mitochondrial Ca(2+) transport mechanisms. Biochim Biophys Acta. 2009;1787:1291–308. doi: 10.1016/j.bbabio.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Altschuld RA, Hohl CM, Castillo LC, Garleb AA, Starling RC, Brierley GP. Cyclosporin inhibits mitochondrial calcium efflux in isolated adult rat ventricular cardiomyocytes. Am J Physiol. 1992;262:H1699–704. doi: 10.1152/ajpheart.1992.262.6.H1699. [DOI] [PubMed] [Google Scholar]
- 26.Ryu SY, Peixoto PM, Won JH, Yule DI, Kinnally KW. Extracellular ATP and P2Y2 receptors mediate intercellular Ca(2+) waves induced by mechanical stimulation in submandibular gland cells: Role of mitochondrial regulation of store operated Ca(2+) entry. Cell Calcium. 2010;47:65–76. doi: 10.1016/j.ceca.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang W, Fang H, Groom L, Cheng A, Zhang W, Liu J, Wang X, Li K, Han P, Zheng M, Yin J, Wang W, Mattson MP, Kao JP, Lakatta EG, Sheu SS, Ouyang K, Chen J, Dirksen RT, Cheng H. Superoxide flashes in single mitochondria. Cell. 2008;134:279–90. doi: 10.1016/j.cell.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Veal EA, Day AM, Morgan BA. Hydrogen peroxide sensing and signaling. Mol Cell. 2007;26:1–14. doi: 10.1016/j.molcel.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 29.De Giorgi F, Lartigue L, Bauer MK, Schubert A, Grimm S, Hanson GT, Remington SJ, Youle RJ, Ichas F. The permeability transition pore signals apoptosis by directing Bax translocation and multimerization. Faseb J. 2002;16:607–9. doi: 10.1096/fj.01-0269fje. [DOI] [PubMed] [Google Scholar]
- 30.Beutner G, Ruck A, Riede B, Brdiczka D. Complexes between porin, hexokinase, mitochondrial creatine kinase and adenylate translocator display properties of the permeability transition pore. Implication for regulation of permeability transition by the kinases. Biochim Biophys Acta. 1998;1368:7–18. doi: 10.1016/s0005-2736(97)00175-2. [DOI] [PubMed] [Google Scholar]
- 31.Szabo I, De Pinto V, Zoratti M. The mitochondrial permeability transition pore may comprise VDAC molecules. II. The electrophysiological properties of VDAC are compatible with those of the mitochondrial megachannel. FEBS Lett. 1993;330:206–10. doi: 10.1016/0014-5793(93)80274-x. [DOI] [PubMed] [Google Scholar]
- 32.Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol. 2007;9:550–5. doi: 10.1038/ncb1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krestinina OV, Grachev DE, Odinokova IV, Reiser G, Evtodienko YV, Azarashvili TS. Effect of peripheral benzodiazepine receptor (PBR/TSPO) ligands on opening of Ca2+-induced pore and phosphorylation of 3.5-kDa polypeptide in rat brain mitochondria. Biochemistry (Mosc) 2009;74:421–9. doi: 10.1134/s0006297909040105. [DOI] [PubMed] [Google Scholar]
- 34.Kusano T, Tateda C, Berberich T, Takahashi Y. Voltage-dependent anion channels: their roles in plant defense and cell death. Plant Cell Rep. 2009;28:1301–8. doi: 10.1007/s00299-009-0741-z. [DOI] [PubMed] [Google Scholar]
- 35.Tomasello F, Messina A, Lartigue L, Schembri L, Medina C, Reina S, Thoraval D, Crouzet M, Ichas F, De Pinto V, De Giorgi F. Outer membrane VDAC1 controls permeability transition of the inner mitochondrial membrane in cellulo during stress-induced apoptosis. Cell Res. 2009;19:1363–76. doi: 10.1038/cr.2009.98. [DOI] [PubMed] [Google Scholar]
- 36.Shoshan-Barmatz V, Keinan N, Abu-Hamad S, Tyomkin D, Aram L. Apoptosis is regulated by the VDAC1 N-terminal region and by VDAC oligomerization: release of cytochrome c, AIF and Smac/Diablo. Biochim Biophys Acta. 2010;1797:1281–1291. doi: 10.1016/j.bbabio.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 37.Zamzami N, Kroemer G. The mitochondrion in apoptosis: how Pandora’s box opens. Nat Rev Mol Cell Biol. 2001;2:67–71. doi: 10.1038/35048073. [DOI] [PubMed] [Google Scholar]
- 38.Leung AW, Varanyuwatana P, Halestrap AP. The mitochondrial phosphate carrier interacts with cyclophilin D and may play a key role in the permeability transition. J Biol Chem. 2008;283:26312–23. doi: 10.1074/jbc.M805235200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kumarswamy R, Chandna S. Putative partners in Bax mediated cytochrome-c release: ANT, CypD, VDAC or none of them? Mitochondrion. 2009;9:1–8. doi: 10.1016/j.mito.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 40.Kokoszka J, Waymire K, Levy S, Sligh J, Cai J, Jones D, MacGregor G, Wallace D. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature. 2004;427:461–5. doi: 10.1038/nature02229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee J, Schriner SE, Wallace DC. Adenine nucleotide translocator 1 deficiency increases resistance of mouse brain and neurons to excitotoxic insults. Biochim Biophys Acta. 2009;1787:364–70. doi: 10.1016/j.bbabio.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Camara Y, Mampel T, Armengol J, Villarroya F, Dejean L. UCP3 expression in liver modulates gene expression and oxidative metabolism in response to fatty acids, and sensitizes mitochondria to permeability transition. Cell Physiol Biochem. 2009;24:243–52. doi: 10.1159/000233249. [DOI] [PubMed] [Google Scholar]
- 43.Dejean L, Camara Y, Sibille B, Solanes G, Villarroya F. Uncoupling protein-3 sensitizes cells to mitochondrial-dependent stimulus of apoptosis. J Cell Physiol. 2004;201:294–304. doi: 10.1002/jcp.20048. [DOI] [PubMed] [Google Scholar]
- 44.He L, Lemasters JJ. Regulated and unregulated mitochondrial permeability transition pores: a new paradigm of pore structure and function? FEBS Lett. 2002;512:1–7. doi: 10.1016/s0014-5793(01)03314-2. [DOI] [PubMed] [Google Scholar]
- 45.Abramov AY, Fraley C, Diao CT, Winkfein R, Colicos MA, Duchen MR, French RJ, Pavlov E. Targeted polyphosphatase expression alters mitochondrial metabolism and inhibits calcium-dependent cell death. Proc Natl Acad Sci U S A. 2007;104:18091–6. doi: 10.1073/pnas.0708959104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dierks T, Salentin A, Heberger C, Kramer R. The mitochondrial aspartate/glutamate and ADP/ATP carrier switch from obligate counterexchange to unidirectional transport after modification by SH-reagents. Biochim Biophys Acta. 1990;1028:268–80. doi: 10.1016/0005-2736(90)90176-o. [DOI] [PubMed] [Google Scholar]
- 47.Dierks T, Salentin A, Kramer R. Pore-like and carrier-like properties of the mitochondrial aspartate/glutamate carrier after modification by SH-reagents: evidence for a performed channel as a structural requirement of carrier-mediated transport. Biochim Biophys Acta. 1990;1028:281–8. doi: 10.1016/0005-2736(90)90177-p. [DOI] [PubMed] [Google Scholar]
- 48.Schroers A, Kramer R, Wohlrab H. The reversible antiport-uniport conversion of the phosphate carrier from yeast mitochondria depends on the presence of a single cysteine. J Biol Chem. 1997;272:10558–64. doi: 10.1074/jbc.272.16.10558. [DOI] [PubMed] [Google Scholar]
- 49.Broekemeier K, Pfeiffer D. Cyclosporin A-sensitive and insensitive mechanisms produce the permeability transition in mitochondria. Biochem Biophys Res Commun. 1989;163:561–6. doi: 10.1016/0006-291x(89)92174-8. [DOI] [PubMed] [Google Scholar]
- 50.Broekemeier KM, Carpenter-Deyo L, Reed DJ, Pfeiffer DR. Cyclosporin A protects hepatocytes subjected to high Ca2+ and oxidative stress. FEBS Lett. 1992;304:192–4. doi: 10.1016/0014-5793(92)80616-o. [DOI] [PubMed] [Google Scholar]
- 51.Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J Biol Chem. 2005;280:18558–61. doi: 10.1074/jbc.C500089200. [DOI] [PubMed] [Google Scholar]
- 52.Basso E, Petronilli V, Forte MA, Bernardi P. Phosphate is essential for inhibition of the mitochondrial permeability transition pore by cyclosporin A and by cyclophilin D ablation. J Biol Chem. 2008;283:26307–11. doi: 10.1074/jbc.C800132200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Galluzzi L, Aaronson SA, Abrams J, Alnemri ES, Andrews DW, Baehrecke EH, Bazan NG, Blagosklonny MV, Blomgren K, Borner C, Bredesen DE, Brenner C, Castedo M, Cidlowski JA, Ciechanover A, Cohen GM, De Laurenzi V, De Maria R, Deshmukh M, Dynlacht BD, El-Deiry WS, Flavell RA, Fulda S, Garrido C, Golstein P, Gougeon ML, Green DR, Gronemeyer H, Hajnoczky G, Hardwick JM, Hengartner MO, Ichijo H, Jaattela M, Kepp O, Kimchi A, Klionsky DJ, Knight RA, Kornbluth S, Kumar S, Levine B, Lipton SA, Lugli E, Madeo F, Malomi W, Marine JC, Martin SJ, Medema JP, Mehlen P, Melino G, Moll UM, Morselli E, Nagata S, Nicholson DW, Nicotera P, Nunez G, Oren M, Penninger J, Pervaiz S, Peter ME, Piacentini M, Prehn JH, Puthalakath H, Rabinovich GA, Rizzuto R, Rodrigues CM, Rubinsztein DC, Rudel T, Scorrano L, Simon HU, Steller H, Tschopp J, Tsujimoto Y, Vandenabeele P, Vitale I, Vousden KH, Youle RJ, Yuan J, Zhivotovsky B, Kroemer G. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ. 2009;16:1093–107. doi: 10.1038/cdd.2009.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chipuk JE, Green DR. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008;18:157–64. doi: 10.1016/j.tcb.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Danial NN. BCL-2 family proteins: critical checkpoints of apoptotic cell death. Clin Cancer Res. 2007;13:7254–63. doi: 10.1158/1078-0432.CCR-07-1598. [DOI] [PubMed] [Google Scholar]
- 56.Kinnally KW, Antonsson B. A tale of two mitochondrial channels, MAC and PTP, in apoptosis. Apoptosis. 2007;12:857–68. doi: 10.1007/s10495-007-0722-z. [DOI] [PubMed] [Google Scholar]
- 57.Leber B, Lin J, Andrews DW. Embedded together: the life and death consequences of interaction of the Bcl-2 family with membranes. Apoptosis. 2007;12:897–911. doi: 10.1007/s10495-007-0746-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- 59.Er E, Oliver L, Cartron PF, Juin P, Manon S, Vallette FM. Mitochondria as the target of the pro-apoptotic protein. Bax Biochim Biophys Acta. 2006;1757:1301–11. doi: 10.1016/j.bbabio.2006.05.032. [DOI] [PubMed] [Google Scholar]
- 60.Antignani A, Youle RJ. How do Bax and Bak lead to permeabilization of the outer mitochondrial membrane? Curr Opin Cell Biol. 2006;18:685–9. doi: 10.1016/j.ceb.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 61.Fadeel B, Orrenius S. Apoptosis: a basic biological phenomenon with wide-ranging implications in human disease. J Intern Med. 2005;258:479–517. doi: 10.1111/j.1365-2796.2005.01570.x. [DOI] [PubMed] [Google Scholar]
- 62.Pavlov EV, Priault M, Pietkiewicz D, Cheng EH, Antonsson B, Manon S, Korsmeyer SJ, Mannella CA, Kinnally KW. A novel, high conductance channel of mitochondria linked to apoptosis in mammalian cells and Bax expression in yeast. J Cell Biol. 2001;155:725–31. doi: 10.1083/jcb.200107057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dejean LM, Martinez-Caballero S, Guo L, Hughes C, Teijido O, Ducret T, Ichas F, Korsmeyer SJ, Antonsson B, Jonas EA, Kinnally KW. Oligomeric Bax is a component of the putative cytochrome c release channel MAC, mitochondrial apoptosis-induced channel. Mol Biol Cell. 2005;16:2424–32. doi: 10.1091/mbc.E04-12-1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Martinez-Caballero S, Dejean LM, Jonas EA, Kinnally KW. The role of the mitochondrial apoptosis induced channel MAC in cytochrome c release. J Bioenerg Biomembr. 2005;37:155–64. doi: 10.1007/s10863-005-6570-z. [DOI] [PubMed] [Google Scholar]
- 65.Guo L, Pietkiewicz D, Pavlov EV, Grigoriev SM, Kasianowicz JJ, Dejean LM, Korsmeyer SJ, Antonsson B, Kinnally KW. Effects of cytochrome c on the mitochondrial apoptosis-induced channel MAC. Am J Physiol Cell Physiol. 2004;286:C1109–17. doi: 10.1152/ajpcell.00183.2003. [DOI] [PubMed] [Google Scholar]
- 66.Peixoto PM, Ryu SY, Bombrun A, Antonsson B, Kinnally KW. MAC inhibitors suppress mitochondrial apoptosis. Biochem J. 2009;423:381–7. doi: 10.1042/BJ20090664. [DOI] [PubMed] [Google Scholar]
- 67.Martinez-Caballero S, Dejean LM, Kinnally MS, Oh KJ, Mannella CA, Kinnally KW. Assembly of the mitochondrial apoptosis-induced channel, MAC. J Biol Chem. 2009;284:12235–45. doi: 10.1074/jbc.M806610200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cheng EH, Sheiko TV, Fisher JK, Craigen WJ, Korsmeyer SJ. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science. 2003;301:513–7. doi: 10.1126/science.1083995. [DOI] [PubMed] [Google Scholar]
- 69.Roy SS, Ehrlich AM, Craigen WJ, Hajnoczky G. VDAC2 is required for truncated BID-induced mitochondrial apoptosis by recruiting BAK to the mitochondria. EMBO Rep. 2009;10:1341–7. doi: 10.1038/embor.2009.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Martinez-Caballero S, Dejean LM, Kinnally KW. Some amphiphilic cations block the mitochondrial apoptosis-induced channel, MAC. FEBS Lett. 2004;568:35–8. doi: 10.1016/j.febslet.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 71.Bernardi P, Krauskopf A, Basso E, Petronilli V, Blachly-Dyson E, Di Lisa F, Forte MA. The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 2006;273:2077–99. doi: 10.1111/j.1742-4658.2006.05213.x. [DOI] [PubMed] [Google Scholar]
- 72.Hajnoczky G, Csordas G, Yi M. Old players in a new role: mitochondria-associated membranes, VDAC, and ryanodine receptors as contributors to calcium signal propagation from endoplasmic reticulum to the mitochondria. Cell, Calcium. 2002;32:363–77. doi: 10.1016/s0143416002001872. [DOI] [PubMed] [Google Scholar]
- 73.Tan W, Colombini M. VDAC closure increases calcium ion flux. Biochim Biophys Acta. 2007;1768:2510–5. doi: 10.1016/j.bbamem.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Madesh M, Hajnoczky G. VDAC-dependent permeabilization of the outer mitochondrial membrane by superoxide induces rapid and massive cytochrome c release. J Cell Biol. 2001;155:1003–15. doi: 10.1083/jcb.200105057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vander Heiden MG, Li XX, Gottleib E, Hill RB, Thompson CB, Colombini M. Bcl-xL promotes the open configuration of the voltage-dependent anion channel and metabolite passage through the outer mitochondrial membrane. J Biol Chem. 2001;276:19414–9. doi: 10.1074/jbc.M101590200. [DOI] [PubMed] [Google Scholar]
- 76.Hiller S, Garces RG, Malia TJ, Orekhov VY, Colombini M, Wagner G. Solution structure of the integral human membrane protein VDAC-1 in detergent micelles. Science. 2008;321:1206–10. doi: 10.1126/science.1161302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ren D, Kim H, Tu HC, Westergard TD, Fisher JK, Rubens JA, Korsmeyer SJ, Hsieh JJ, Cheng EH. The VDAC2-BAK rheostat controls thymocyte survival. Sci Signal. 2009;2:ra48. doi: 10.1126/scisignal.2000274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rostovtseva TK, Bezrukov SM. VDAC regulation: role of cytosolic proteins and mitochondrial lipids. J Bioenerg Biomembr. 2008;40:163–70. doi: 10.1007/s10863-008-9145-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rostovtseva TK, Antonsson B, Suzuki M, Youle RJ, Colombini M, Bezrukov SM. Bid, but not Bax, regulates VDAC channels. J Biol Chem. 2004;279:13575–83. doi: 10.1074/jbc.M310593200. [DOI] [PubMed] [Google Scholar]
- 80.Hetz C, Vitte PA, Bombrun A, Rostovtseva TK, Montessuit S, Hiver A, Schwarz MK, Church DJ, Korsmeyer SJ, Martinou JC, Antonsson B. Bax channel inhibitors prevent mitochondrion-mediated apoptosis and protect neurons in a model of global brain ischemia. J Biol Chem. 2005;280:42960–70. doi: 10.1074/jbc.M505843200. [DOI] [PubMed] [Google Scholar]
- 81.Crompton M. On the involvement of mitochondrial intermembrane junctional complexes in apoptosis. Curr Med Chem. 2003;10:1473–84. doi: 10.2174/0929867033457197. [DOI] [PubMed] [Google Scholar]
- 82.Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–9. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 83.Murphy AN, Bredesen DE, Cortopassi G, Wang E, Fiskum G. Bcl-2 potentiates the maximal calcium uptake capacity of neural cell mitochondria. Proc Natl Acad Sci U S A. 1996;93:9893–8. doi: 10.1073/pnas.93.18.9893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Murphy RC, Diwan JJ, King M, Kinnally KW. Two high conductance channels of the mitochondrial inner membrane are independent of the human mitochondrial genome. FEBS Lett. 1998;425:259–62. doi: 10.1016/s0014-5793(98)00245-2. [DOI] [PubMed] [Google Scholar]
- 85.Desagher S, Martinou JC. Mitochondria as the central control point of apoptosis Trends. Cell Biol. 2000;10:369–77. doi: 10.1016/s0962-8924(00)01803-1. [DOI] [PubMed] [Google Scholar]
- 86.Bossy-Wetzel E, Newmeyer DD, Green DR. Mitochondrial cytochrome c release in apoptosis occurs upstream of DEVD-specific caspase activation and independently of mitochondrial transmembrane depolarization. Embo J. 1998;17:37–49. doi: 10.1093/emboj/17.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–62. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 88.Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–658. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 89.Halestrap AP. Calcium, mitochondria and reperfusion injury: a pore way to die. Biochem Soc Trans. 2006;34:232–7. doi: 10.1042/BST20060232. [DOI] [PubMed] [Google Scholar]
- 90.Halestrap A. Biochemistry: a pore way to die. Nature. 2005;434:578–9. doi: 10.1038/434578a. [DOI] [PubMed] [Google Scholar]
- 91.Halestrap AP. Mitochondrial permeability: dual role for the ADP/ATP translocator? Nature. 2004;430:1. doi: 10.1038/nature02816. following 983. [DOI] [PubMed] [Google Scholar]
- 92.Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 93.Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–30. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J. 1999;341(Pt 2):233–49. [PMC free article] [PubMed] [Google Scholar]
- 95.Jacobson J, Duchen MR. Mitochondrial oxidative stress and cell death in astrocytes--requirement for stored Ca2+ and sustained opening of the permeability transition pore. J Cell Sci. 2002;115:1175–88. doi: 10.1242/jcs.115.6.1175. [DOI] [PubMed] [Google Scholar]
- 96.Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci U S A. 2005;102:12005–10. doi: 10.1073/pnas.0505294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rasola A, Sciacovelli M, Pantic B, Bernardi P. Signal transduction to the permeability transition pore FEBS. Lett. 2010;584:1989–96. doi: 10.1016/j.febslet.2010.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Scorrano L, Korsmeyer SJ. Mechanisms of cytochrome c release by proapoptotic BCL-2 family members. Biochem Biophys Res Commun. 2003;304:437–44. doi: 10.1016/s0006-291x(03)00615-6. [DOI] [PubMed] [Google Scholar]
- 99.Guihard G, Bellot G, Moreau C, Pradal G, Ferry N, Thomy R, Fichet P, Meflah K, Vallette FM. The mitochondrial apoptosis-induced channel (MAC) corresponds to a late apoptotic event. J Biol Chem. 2004;279:46542–50. doi: 10.1074/jbc.M405153200. [DOI] [PubMed] [Google Scholar]
- 100.Chen M, Won DJ, Krajewski S, Gottlieb RA. Calpain and mitochondria in ischemia/reperfusion injury. J Biol Chem. 2002;277:29181–6. doi: 10.1074/jbc.M204951200. [DOI] [PubMed] [Google Scholar]
- 101.Springer JE, Azbill RD, Nottingham SA, Kennedy SE. Calcineurin-mediated BAD dephosphorylation activates the caspase-3 apoptotic cascade in traumatic spinal cord injury. J Neurosci. 2000;20:7246–51. doi: 10.1523/JNEUROSCI.20-19-07246.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wang HG, Pathan N, Ethell IM, Krajewski S, Yamaguchi Y, Shibasaki F, McKeon F, Bobo T, Franke TF, Reed JC. Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science. 1999;284:339–43. doi: 10.1126/science.284.5412.339. [DOI] [PubMed] [Google Scholar]
- 103.Roy SS, Madesh M, Davies E, Antonsson B, Danial N, Hajnoczky G. Bad targets the permeability transition pore independent of Bax or Bak to switch between Ca2+-dependent cell survival and death. Mol Cell. 2009;33:377–88. doi: 10.1016/j.molcel.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Concannon CG, Tuffy LP, Weisova P, Bonner HP, Davila D, Bonner C, Devocelle MC, Strasser A, Ward MW, Prehn JH. AMP kinase-mediated activation of the BH3-only protein Bim couples energy depletion to stress-induced apoptosis. J Cell Biol. 2010;189:83–94. doi: 10.1083/jcb.200909166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Garrido C, Galluzzi L, Brunet M, Puig PE, Didelot C, Kroemer G. Mechanisms of cytochrome c release from mitochondria. Cell Death Differ. 2006;13:1423–33. doi: 10.1038/sj.cdd.4401950. [DOI] [PubMed] [Google Scholar]
- 106.Boehning D, Patterson RL, Sedaghat L, Glebova NO, Kurosaki T, Snyder SH. Cytochrome c binds to inositol (1,4,5) trisphosphate receptors, amplifying calcium-dependent apoptosis. Nat Cell Biol. 2003;5:1051–61. doi: 10.1038/ncb1063. [DOI] [PubMed] [Google Scholar]
- 107.Cardenas C, Miller RA, Smith I, Bui T, Molgo J, Muller M, Vais H, Cheung KH, Yang J, Parker I, Thompson CB, Birnbaum MJ, Hallows KR, Foskett JK. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell. 2010;142:270–83. doi: 10.1016/j.cell.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, Bartoli D, Polishuck RS, Danial NN, De Strooper B, Scorrano L. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006;126:177–89. doi: 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 109.Rostovtseva T, Colombini M. ATP flux is controlled by a voltage-gated channel from the mitochondrial outer membrane. J Biol Chem. 1996;271:28006–8. doi: 10.1074/jbc.271.45.28006. [DOI] [PubMed] [Google Scholar]
- 110.Bombrun A, Gerber P, Casi G, Terradillos O, Antonsson B, Halazy S. 3,6-dibromocarbazole piperazine derivatives of 2-propanol as first inhibitors of cytochrome c release via Bax channel modulation. J Med Chem. 2003;46:4365–8. doi: 10.1021/jm034107j. [DOI] [PubMed] [Google Scholar]
- 111.Lai JC, Tan W, Benimetskaya L, Miller P, Colombini M, Stein CA. A pharmacologic target of G3139 in melanoma cells may be the mitochondrial VDAC. Proc Natl Acad Sci U S A. 2006;103:7494–9. doi: 10.1073/pnas.0602217103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Peixoto PM, Ryu SY, Kinnally KW. Mitochondrial ion channels as therapeutic targets. FEBS Lett. 2010;584:2142–52. doi: 10.1016/j.febslet.2010.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]