

Abstract
In the title compound, C17H20N2O2S, the tetrahydropyridine ring adopts an envelope conformation with the N atom at the flap position; the phenyl ring makes a dihedral angle of 81.06 (10)° with the thiophene ring. The amino group links with the carbonyl O atom via intramolecular N—H⋯O hydrogen bonding, forming a six-membered ring. In the crystal, intermolecular N—H⋯O hydrogen bonds link the molecules into infinite chains running along the b axis.
Related literature
For the biological activity of thiophene and its derivatives, see: Kidwai & Mishra (2003 ▶); Amr et al. (2006 ▶); Sherif (1996 ▶).
Experimental
Crystal data
C17H20N2O2S
M r = 316.41
Monoclinic,
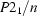
a = 12.197 (3) Å
b = 9.936 (3) Å
c = 13.775 (4) Å
β = 103.430 (4)°
V = 1623.8 (8) Å3
Z = 4
Mo Kα radiation
μ = 0.21 mm−1
T = 293 K
0.25 × 0.19 × 0.14 mm
Data collection
Bruker SMART APEX CCD diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 1999 ▶) T min = 0.953, T max = 0.977
8867 measured reflections
2875 independent reflections
2122 reflections with I > 2σ(I)
R int = 0.030
Refinement
R[F 2 > 2σ(F 2)] = 0.042
wR(F 2) = 0.107
S = 1.04
2875 reflections
205 parameters
3 restraints
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.20 e Å−3
Δρmin = −0.17 e Å−3
Data collection: SMART (Bruker, 1999 ▶); cell refinement: SAINT (Bruker, 1999 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXTL (Sheldrick, 2008) ▶; program(s) used to refine structure: SHELXTL ▶; molecular graphics: SHELXTL ▶; software used to prepare material for publication: SHELXTL ▶.
Supplementary Material
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536810051986/xu5117sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810051986/xu5117Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N2—H1N⋯O1i | 0.81 (2) | 2.17 (2) | 2.972 (2) | 171 (2) |
| N2—H2N⋯O1 | 0.81 (1) | 2.17 (2) | 2.777 (2) | 132 (2) |
Symmetry code: (i) 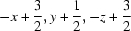 .
.
Acknowledgments
We thank the Natural Science Foundation of Shanxi Province, China (No. 2010011018) for supporting this work.
supplementary crystallographic information
Comment
As part of an investigation of the thiophene and it's derivatives systems due to their diverse biological activities (Kidwai et al., 2003; Amr et al., 2006; Sherif et al., 1996), we present here the crystal structure of the title compound, (I).
In the crystal structure of title compound (Fig.1), all bond lengths and bond angles have standard dimensions.
The fragments (C8 to C12) of piperidine nearly planar (mean deviation from plane within 0.0632 (1) Å) while the the six-membered piperidine ring exhibits half-chair conformation. The amino group are hydrogen bonded to the carbonyl O atom of another molecule (Table 1), forming a one-dimensional supramolecular structure (Fig. 2). In addition, there are intramolecular N—H···O hydrogen-bonding interactions in the crystal.
Experimental
To the solution containing the ethyl 2-cyanoacetate (10 mmol, 1.06 ml), 1-benzylpiperidin-4-one (10 mmol, 1.80 ml) and powdered sulfur (12 mmol, 0.38 g) in DMF (6 ml), was under stirring triethylamine (1.20 ml) dropwise added. When the reaction was finished (TLC monitoring) the mixture was filtered with charcoal and poured into crushed ice. The formed crystals were filtered off and washed with water. The products were crystallized from ethanol.
Refinement
All H atoms bound to C atoms were positioned geometrically and refined as riding, with C—H = 0.93 Å (CH), C—H = 0.97 Å (CH2) and Uiso(H) = 1.2Ueq(C), C—H = 0.96 Å (CH3) and Uiso(H) = 1.5Ueq(C). The H atoms bound to N atoms were located in a difference Fourier map and refined with Uiso(H) = 1.2Ueq(N). The N—H distances were restrained.
Figures
Fig. 1.

Molecular structure of the title compound. Displacement ellipsoids are drawn at the 30% probabilitylevel.
Fig. 2.

View of the one-dimensional supra-molecular chain of the title compound formed by hydrogen bonding (dashed lines). H atoms of C omitted for clarity.
Crystal data
| C17H20N2O2S | F(000) = 672 |
| Mr = 316.41 | Dx = 1.294 Mg m−3 |
| Monoclinic, P21/n | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2yn | Cell parameters from 2875 reflections |
| a = 12.197 (3) Å | θ = 2.2–25.1° |
| b = 9.936 (3) Å | µ = 0.21 mm−1 |
| c = 13.775 (4) Å | T = 293 K |
| β = 103.430 (4)° | Block, yellow |
| V = 1623.8 (8) Å3 | 0.25 × 0.19 × 0.14 mm |
| Z = 4 |
Data collection
| Bruker SMART APEX CCD diffractometer | 2875 independent reflections |
| Radiation source: fine-focus sealed tube | 2122 reflections with I > 2σ(I) |
| graphite | Rint = 0.030 |
| ω scans | θmax = 25.1°, θmin = 2.0° |
| Absorption correction: multi-scan (SADABS; Bruker, 1999) | h = −14→14 |
| Tmin = 0.953, Tmax = 0.977 | k = −10→11 |
| 8867 measured reflections | l = −16→14 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.042 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.107 | H atoms treated by a mixture of independent and constrained refinement |
| S = 1.04 | w = 1/[σ2(Fo2) + (0.0533P)2 + 0.2035P] where P = (Fo2 + 2Fc2)/3 |
| 2875 reflections | (Δ/σ)max < 0.001 |
| 205 parameters | Δρmax = 0.20 e Å−3 |
| 3 restraints | Δρmin = −0.17 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| C1 | 0.15299 (16) | 0.6448 (2) | 0.47716 (17) | 0.0480 (5) | |
| H1 | 0.1593 | 0.6354 | 0.4115 | 0.058* | |
| C2 | 0.14619 (18) | 0.7720 (2) | 0.51559 (19) | 0.0583 (6) | |
| H2 | 0.1477 | 0.8472 | 0.4757 | 0.070* | |
| C3 | 0.13735 (19) | 0.7877 (2) | 0.6113 (2) | 0.0610 (6) | |
| H3 | 0.1325 | 0.8734 | 0.6370 | 0.073* | |
| C4 | 0.1356 (2) | 0.6766 (3) | 0.67014 (19) | 0.0630 (6) | |
| H4 | 0.1300 | 0.6873 | 0.7359 | 0.076* | |
| C5 | 0.14210 (18) | 0.5487 (2) | 0.63200 (17) | 0.0539 (6) | |
| H5 | 0.1408 | 0.4740 | 0.6724 | 0.065* | |
| C6 | 0.15054 (15) | 0.5310 (2) | 0.53451 (16) | 0.0416 (5) | |
| C7 | 0.15136 (17) | 0.3930 (2) | 0.48984 (17) | 0.0503 (6) | |
| H7A | 0.0800 | 0.3494 | 0.4889 | 0.060* | |
| H7B | 0.1577 | 0.4022 | 0.4212 | 0.060* | |
| C8 | 0.23336 (17) | 0.17339 (19) | 0.49528 (17) | 0.0480 (5) | |
| H8A | 0.2476 | 0.1822 | 0.4292 | 0.058* | |
| H8B | 0.1571 | 0.1401 | 0.4878 | 0.058* | |
| C9 | 0.31573 (15) | 0.07232 (19) | 0.55515 (16) | 0.0441 (5) | |
| H9A | 0.2875 | 0.0412 | 0.6115 | 0.053* | |
| H9B | 0.3216 | −0.0049 | 0.5136 | 0.053* | |
| C10 | 0.43028 (15) | 0.13375 (19) | 0.59228 (14) | 0.0372 (5) | |
| C11 | 0.44382 (15) | 0.26693 (19) | 0.58452 (15) | 0.0409 (5) | |
| C12 | 0.35245 (15) | 0.36571 (19) | 0.54262 (17) | 0.0471 (5) | |
| H12A | 0.3633 | 0.4473 | 0.5824 | 0.057* | |
| H12B | 0.3552 | 0.3890 | 0.4748 | 0.057* | |
| C13 | 0.53359 (15) | 0.06514 (18) | 0.64255 (14) | 0.0371 (4) | |
| C14 | 0.62303 (15) | 0.15380 (19) | 0.66948 (15) | 0.0405 (5) | |
| C15 | 0.54785 (16) | −0.07575 (19) | 0.66775 (14) | 0.0400 (5) | |
| C16 | 0.45439 (19) | −0.28970 (19) | 0.65815 (19) | 0.0562 (6) | |
| H16A | 0.5239 | −0.3251 | 0.6456 | 0.067* | |
| H16B | 0.3921 | −0.3307 | 0.6106 | 0.067* | |
| C17 | 0.4472 (2) | −0.3267 (2) | 0.7607 (2) | 0.0746 (8) | |
| H17A | 0.4497 | −0.4229 | 0.7676 | 0.112* | |
| H17B | 0.3777 | −0.2935 | 0.7730 | 0.112* | |
| H17C | 0.5095 | −0.2877 | 0.8080 | 0.112* | |
| O1 | 0.63729 (11) | −0.12830 (13) | 0.70893 (11) | 0.0503 (4) | |
| O2 | 0.45118 (11) | −0.14512 (13) | 0.64303 (12) | 0.0540 (4) | |
| S1 | 0.58197 (4) | 0.31782 (5) | 0.63602 (5) | 0.0502 (2) | |
| N1 | 0.24303 (12) | 0.30618 (15) | 0.54336 (13) | 0.0417 (4) | |
| N2 | 0.73186 (14) | 0.12651 (18) | 0.71338 (16) | 0.0544 (5) | |
| H1N | 0.7738 (18) | 0.1883 (17) | 0.7340 (17) | 0.065* | |
| H2N | 0.7441 (19) | 0.0488 (15) | 0.7300 (17) | 0.065* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| C1 | 0.0413 (11) | 0.0530 (14) | 0.0484 (13) | −0.0033 (10) | 0.0078 (10) | 0.0019 (10) |
| C2 | 0.0566 (14) | 0.0454 (14) | 0.0690 (17) | −0.0080 (11) | 0.0066 (12) | 0.0068 (12) |
| C3 | 0.0595 (15) | 0.0463 (14) | 0.0756 (19) | −0.0028 (11) | 0.0125 (13) | −0.0119 (12) |
| C4 | 0.0689 (16) | 0.0696 (17) | 0.0550 (15) | 0.0006 (13) | 0.0232 (12) | −0.0077 (13) |
| C5 | 0.0573 (14) | 0.0506 (14) | 0.0553 (15) | 0.0017 (11) | 0.0157 (11) | 0.0094 (11) |
| C6 | 0.0290 (10) | 0.0431 (12) | 0.0510 (13) | 0.0035 (8) | 0.0058 (9) | 0.0013 (10) |
| C7 | 0.0397 (11) | 0.0476 (13) | 0.0586 (14) | 0.0062 (10) | 0.0012 (10) | −0.0040 (10) |
| C8 | 0.0384 (11) | 0.0411 (12) | 0.0595 (14) | −0.0027 (9) | 0.0009 (10) | −0.0071 (10) |
| C9 | 0.0378 (11) | 0.0329 (11) | 0.0592 (14) | −0.0022 (8) | 0.0063 (10) | −0.0038 (9) |
| C10 | 0.0346 (10) | 0.0327 (10) | 0.0445 (12) | −0.0012 (8) | 0.0100 (9) | −0.0014 (8) |
| C11 | 0.0332 (10) | 0.0334 (11) | 0.0561 (13) | −0.0011 (8) | 0.0100 (9) | 0.0031 (9) |
| C12 | 0.0373 (11) | 0.0359 (11) | 0.0667 (15) | 0.0017 (9) | 0.0090 (10) | 0.0065 (10) |
| C13 | 0.0356 (10) | 0.0296 (10) | 0.0459 (12) | −0.0007 (8) | 0.0090 (9) | −0.0015 (8) |
| C14 | 0.0355 (10) | 0.0358 (11) | 0.0495 (13) | 0.0015 (8) | 0.0087 (9) | 0.0003 (9) |
| C15 | 0.0387 (11) | 0.0353 (11) | 0.0456 (12) | −0.0004 (9) | 0.0088 (9) | −0.0041 (9) |
| C16 | 0.0553 (14) | 0.0261 (11) | 0.0791 (18) | −0.0048 (9) | −0.0010 (12) | −0.0014 (10) |
| C17 | 0.0791 (18) | 0.0546 (15) | 0.083 (2) | −0.0141 (13) | 0.0051 (15) | 0.0115 (14) |
| O1 | 0.0399 (8) | 0.0375 (8) | 0.0689 (10) | 0.0062 (6) | 0.0032 (7) | 0.0037 (7) |
| O2 | 0.0413 (8) | 0.0291 (8) | 0.0841 (11) | −0.0039 (6) | −0.0006 (7) | 0.0050 (7) |
| S1 | 0.0357 (3) | 0.0324 (3) | 0.0794 (4) | −0.0055 (2) | 0.0068 (3) | 0.0047 (3) |
| N1 | 0.0319 (8) | 0.0328 (9) | 0.0576 (11) | 0.0020 (7) | 0.0048 (8) | −0.0008 (8) |
| N2 | 0.0363 (10) | 0.0392 (10) | 0.0810 (15) | −0.0036 (8) | −0.0002 (9) | 0.0014 (10) |
Geometric parameters (Å, °)
| C1—C2 | 1.380 (3) | C10—C11 | 1.341 (3) |
| C1—C6 | 1.383 (3) | C10—C13 | 1.458 (3) |
| C1—H1 | 0.9300 | C11—C12 | 1.497 (3) |
| C2—C3 | 1.357 (3) | C11—S1 | 1.7445 (19) |
| C2—H2 | 0.9300 | C12—N1 | 1.462 (2) |
| C3—C4 | 1.372 (3) | C12—H12A | 0.9700 |
| C3—H3 | 0.9300 | C12—H12B | 0.9700 |
| C4—C5 | 1.384 (3) | C13—C14 | 1.384 (3) |
| C4—H4 | 0.9300 | C13—C15 | 1.443 (3) |
| C5—C6 | 1.382 (3) | C14—N2 | 1.352 (2) |
| C5—H5 | 0.9300 | C14—S1 | 1.736 (2) |
| C6—C7 | 1.505 (3) | C15—O1 | 1.224 (2) |
| C7—N1 | 1.468 (2) | C15—O2 | 1.340 (2) |
| C7—H7A | 0.9700 | C16—O2 | 1.451 (2) |
| C7—H7B | 0.9700 | C16—C17 | 1.482 (3) |
| C8—N1 | 1.469 (2) | C16—H16A | 0.9700 |
| C8—C9 | 1.521 (3) | C16—H16B | 0.9700 |
| C8—H8A | 0.9700 | C17—H17A | 0.9600 |
| C8—H8B | 0.9700 | C17—H17B | 0.9600 |
| C9—C10 | 1.501 (3) | C17—H17C | 0.9600 |
| C9—H9A | 0.9700 | N2—H1N | 0.807 (15) |
| C9—H9B | 0.9700 | N2—H2N | 0.809 (14) |
| C2—C1—C6 | 121.2 (2) | C10—C11—C12 | 125.70 (17) |
| C2—C1—H1 | 119.4 | C10—C11—S1 | 112.26 (14) |
| C6—C1—H1 | 119.4 | C12—C11—S1 | 121.95 (14) |
| C3—C2—C1 | 120.2 (2) | N1—C12—C11 | 109.34 (16) |
| C3—C2—H2 | 119.9 | N1—C12—H12A | 109.8 |
| C1—C2—H2 | 119.9 | C11—C12—H12A | 109.8 |
| C2—C3—C4 | 119.8 (2) | N1—C12—H12B | 109.8 |
| C2—C3—H3 | 120.1 | C11—C12—H12B | 109.8 |
| C4—C3—H3 | 120.1 | H12A—C12—H12B | 108.3 |
| C3—C4—C5 | 120.3 (2) | C14—C13—C15 | 120.63 (17) |
| C3—C4—H4 | 119.9 | C14—C13—C10 | 111.72 (17) |
| C5—C4—H4 | 119.9 | C15—C13—C10 | 127.61 (17) |
| C6—C5—C4 | 120.6 (2) | N2—C14—C13 | 128.54 (18) |
| C6—C5—H5 | 119.7 | N2—C14—S1 | 119.99 (15) |
| C4—C5—H5 | 119.7 | C13—C14—S1 | 111.45 (14) |
| C5—C6—C1 | 117.87 (19) | O1—C15—O2 | 122.38 (18) |
| C5—C6—C7 | 121.5 (2) | O1—C15—C13 | 124.76 (18) |
| C1—C6—C7 | 120.6 (2) | O2—C15—C13 | 112.84 (16) |
| N1—C7—C6 | 114.01 (16) | O2—C16—C17 | 112.14 (19) |
| N1—C7—H7A | 108.8 | O2—C16—H16A | 109.2 |
| C6—C7—H7A | 108.8 | C17—C16—H16A | 109.2 |
| N1—C7—H7B | 108.8 | O2—C16—H16B | 109.2 |
| C6—C7—H7B | 108.8 | C17—C16—H16B | 109.2 |
| H7A—C7—H7B | 107.6 | H16A—C16—H16B | 107.9 |
| N1—C8—C9 | 112.02 (16) | C16—C17—H17A | 109.5 |
| N1—C8—H8A | 109.2 | C16—C17—H17B | 109.5 |
| C9—C8—H8A | 109.2 | H17A—C17—H17B | 109.5 |
| N1—C8—H8B | 109.2 | C16—C17—H17C | 109.5 |
| C9—C8—H8B | 109.2 | H17A—C17—H17C | 109.5 |
| H8A—C8—H8B | 107.9 | H17B—C17—H17C | 109.5 |
| C10—C9—C8 | 111.19 (16) | C15—O2—C16 | 118.70 (15) |
| C10—C9—H9A | 109.4 | C14—S1—C11 | 91.59 (9) |
| C8—C9—H9A | 109.4 | C12—N1—C7 | 110.42 (15) |
| C10—C9—H9B | 109.4 | C12—N1—C8 | 109.76 (16) |
| C8—C9—H9B | 109.4 | C7—N1—C8 | 109.22 (15) |
| H9A—C9—H9B | 108.0 | C14—N2—H1N | 118.7 (16) |
| C11—C10—C13 | 112.98 (16) | C14—N2—H2N | 114.6 (16) |
| C11—C10—C9 | 119.74 (17) | H1N—N2—H2N | 124 (2) |
| C13—C10—C9 | 127.21 (17) | ||
| C6—C1—C2—C3 | 0.3 (3) | C9—C10—C13—C15 | −0.3 (3) |
| C1—C2—C3—C4 | 0.2 (4) | C15—C13—C14—N2 | 5.0 (3) |
| C2—C3—C4—C5 | −0.4 (4) | C10—C13—C14—N2 | −177.1 (2) |
| C3—C4—C5—C6 | 0.1 (4) | C15—C13—C14—S1 | −176.97 (15) |
| C4—C5—C6—C1 | 0.4 (3) | C10—C13—C14—S1 | 0.9 (2) |
| C4—C5—C6—C7 | −176.62 (19) | C14—C13—C15—O1 | −3.6 (3) |
| C2—C1—C6—C5 | −0.6 (3) | C10—C13—C15—O1 | 178.95 (19) |
| C2—C1—C6—C7 | 176.49 (19) | C14—C13—C15—O2 | 175.16 (18) |
| C5—C6—C7—N1 | −58.6 (3) | C10—C13—C15—O2 | −2.3 (3) |
| C1—C6—C7—N1 | 124.5 (2) | O1—C15—O2—C16 | −5.2 (3) |
| N1—C8—C9—C10 | −43.7 (2) | C13—C15—O2—C16 | 176.01 (18) |
| C8—C9—C10—C11 | 10.4 (3) | C17—C16—O2—C15 | 86.3 (2) |
| C8—C9—C10—C13 | −172.76 (19) | N2—C14—S1—C11 | 177.73 (18) |
| C13—C10—C11—C12 | −175.99 (19) | C13—C14—S1—C11 | −0.45 (17) |
| C9—C10—C11—C12 | 1.2 (3) | C10—C11—S1—C14 | −0.11 (17) |
| C13—C10—C11—S1 | 0.6 (2) | C12—C11—S1—C14 | 176.65 (18) |
| C9—C10—C11—S1 | 177.86 (15) | C11—C12—N1—C7 | −171.98 (17) |
| C10—C11—C12—N1 | 19.4 (3) | C11—C12—N1—C8 | −51.5 (2) |
| S1—C11—C12—N1 | −156.87 (15) | C6—C7—N1—C12 | −61.0 (2) |
| C11—C10—C13—C14 | −1.0 (3) | C6—C7—N1—C8 | 178.23 (18) |
| C9—C10—C13—C14 | −177.97 (19) | C9—C8—N1—C12 | 66.7 (2) |
| C11—C10—C13—C15 | 176.68 (19) | C9—C8—N1—C7 | −172.09 (18) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N2—H1N···O1i | 0.81 (2) | 2.17 (2) | 2.972 (2) | 171 (2) |
| N2—H2N···O1 | 0.81 (1) | 2.17 (2) | 2.777 (2) | 132 (2) |
Symmetry codes: (i) −x+3/2, y+1/2, −z+3/2.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: XU5117).
References
- Amr, A. G. E., Mohamed, A. M., Mohamed, S. F., Abdel-Hafez, N. A. & Hammam, A. E. F. G. (2006). Bioorg. Med. Chem. 14, 5481–5488. [DOI] [PubMed]
- Bruker (1999). SMART, SAINT and SADABS Bruker AXS Inc., Madison, Wisconsin, USA.
- Kidwai, M. & Mishra, A. D. (2003). Bull Korean Chem. Soc 24, 1038–1040.
- Sheldrick, G. M. (2008). Acta Cryst A64, 112–122. [DOI] [PubMed]
- Sherif, S. M. (1996). Monatsh. Chem. 127, 955–962.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536810051986/xu5117sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810051986/xu5117Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report