

Abstract
A growing body of literature highlights the cross-talk between tumor cells and the surrounding peri-tumoral stroma as a key modulator of the processes of hepatocarcinogenesis, epithelial mesenchymal transition (EMT), tumor invasion and metastasis. The tumor microenvironment can be broadly classified into cellular and non-cellular components. The major cellular components include hepatic stellate cells, fibroblasts, immune, and endothelial cells. These cell types produce the non-cellular components of the tumor stroma, including extracellular matrix (ECM) proteins, proteolytic enzymes, growth factors and inflammatory cytokines. The non-cellular component of the tumor stroma modulates hepatocellular carcinoma (HCC) biology by effects on cancer signaling pathways in tumor cells and on tumor invasion and metastasis. Global gene expression profiling of HCC has revealed that the tumor microenvironment is an important component in the biologic and prognostic classification of HCC. There are substantial efforts underway to develop novel drugs targeting tumor–stromal interactions.
In this review, we discuss the current knowledge about the role of the tumor microenvironment in pathogenesis of HCC, the role of the tumor microenvironment in the classification of HCC and efforts to develop treatments targeting the tumor microenvironment.
Keywords: Hepatocellular carcinoma, tumor microenvironment, hepatic stellate cells, cancer-associated fibroblast, Kupffer cell, T cell, TGF-β1, matrix metalloproteinase, tissue inhibitor of metalloproteinase, inflammatory cytokine, gene signature, treatment
1. Introduction
Hepatocellular carcinoma (HCC) is the seventh most common malignancy and the third leading cause of cancer-related death worldwide [1]. Despite the recent advances in diagnosis and treatment of HCC, it remains a highly lethal disease. The main cause of death in HCC patients is tumor progression with metastasis. However, the underlying mechanisms of tumor initiation, progression and metastasis are still not fully understood [2].
The majority of HCC patients have an underlying chronic liver disease; and liver cirrhosis is the main risk factor for the development of HCC[3, 4]. Chronic liver injury is associated with dysregulated growth of hepatocytes and results in the formation of regenerative nodules, dysplastic nodules, and HCC. Nitta et al. demonstrated that cirrhotic liver-derived hepatocytes (CLDH) have a cellular signaling phenotype that indicates a change from a MAPK-independent cell survival pathway to a MAPK-dependent cell survival pathway. The CLDHs have increased vimentin and type 1 collagen expression, which are markers of mesenchymal cells, and morphologic features consistent with the epithelial-mesenchymal transition (EMT), a biologic process in which epithelial cells loose their phenotypic characteristics and acquire features typical of mesenchymal cells[5-7]. EMT is essential during embryonic development, tissue repair in the adult organism and cancer progression, and it is thought to be critical as a connection point between inflammation and the progression of degenerative fibrotic diseases and cancer[8].
Recent literature has highlighted the cross-talk between tumor cells and their surrounding microenvironments as well as a fundamental role of the tumor microenvironment in the pathogenesis of HCC. The tumor microenvironment plays a critical role in modulating the process of liver fibrosis, hepatocarcinogenesis, EMT, tumor invasion and metastasis. The tumor microenvironment largely consists of 1) cells such as hepatic stellate cells, fibroblasts, immune cells - including regulatory and cytotoxic T cells and tumor-associated macrophages, and endothelial cells, 2) growth factors including transforming growth factor β1 (TGF-β1) and platelet derived growth factor (PDGF), 3) proteolytic enzymes such as matrix metalloproteinases (MMPs) and tissue inhibitor of metalloproteinases (TIMPs), 4) extracellular matrix (ECM) proteins, 5) and inflammatory cytokines. In this review, we discuss the current understanding of each component of the tumor microenvironment and their roles in the pathogenesis of HCC. In addition, we examine current treatments targeting the tumor microenvironment as well as directions for future research.
2. Cells in the tumor microenvironment
2-1 Hepatic Stellate Cells (HSCs)
HSCs, which were once known as lipocytes, Ito cells, or peri-sinusoidal cells, are the major cell type responsible for collagen synthesis in the liver[9]. Hepatic stellate cells (HSC) are activated in response to liver damage and trans-differentiate into myofibroblast-like cells when liver injury is repeated, leading to the development of hepatic fibrosis [10, 11]. HSCs undergo phenotypic transformation from quiescent, non-proliferating cells to proliferating, extracellular matrix (ECM) producing cells during the process of liver injury, which involves two steps. The initial phase is represented by the up-regulation of cytoskeletal protein expression including a-SMA, and the perpetuation phase is represented by the release of a multitude of cytokines, chemokines and growths factors [12-14]. Activated HSCs produce the extensive accumulation of ECM during liver fibrosis [11, 15]. Activated HSCs also infiltrate the stroma of liver tumors and localize around tumor sinusoids, fibrous septa and capsules [16].
In addition to their role in development of liver fibrosis, activated HSCs promote HCC cell proliferation. Amann.T et al. demonstrated that the conditioned media collected from HSCs induce proliferation and migration of HCC cells cultured in monolayers and, moreover, they showed that in a 3-dimensional spheroid co-culture system, HSCs promote HCC growth and diminish the extent of central necrosis through the activation of NF kappa B and extracellular-regulated kinase (ERK) pathways [4]. Consistent with these findings, simultaneous in vivo implantation of HSCs and HCC cells into nude mice promoted tumor growth and invasiveness, and inhibited necrosis. PDGF, TGF-β1, MMP-9, JNK, insulin-like growth factor binding protein 5, cathepsins B and D, hepatitis B virus X protein, and HCV nonstructural proteins are all potent inducers of stellate cell activation, proliferation and collagen production, and therefore enhance liver fibrosis and hepatocarcinogenesis [5, 11, 17-23]. In contrast, adiponectin suppresses hepatic stellate cell activation and angiogenesis [24] (Figure 1).
Figure 1. Regulators of stellate cell activation and their roles in liver fibrosis and carcinogenesis.

Stellate cells and CAF activated by several factors induce proliferation, invasion and metastasis in hepatocellular carcinoma. TGF-β plays a critical role in liver fibrosis and tumorigenesis through the epithelial-mesenchymal transition.
2-2 Cancer-Associated Fibroblasts (CAFs)
Cancer-associated fibroblasts (CAFs) are the most prominent cell type within the tumor stroma of many cancers (most notably breast and pancreatic carcinoma) and play a critical role in tumor-stromal interactions [25-27]. They are activated by TGF-β and are responsible for the synthesis, deposition and remodeling of excessive ECM, such as various types of collagen. CAFs modulate the biological activities of HCC. Mazzocca et al. showed that HCC cell growth, intravasation and metastatic spread are dependent upon the presence of CAFs and HCC cells reciprocally stimulate proliferation of CAFs, suggesting a key role for CAFs in tumor-stromal interaction [28]. CAFs from different tumor types express several growth factors, including hepatocyte growth factor (HGF), members of the epidermal growth factor (EGF), fibroblast growth factor (FGF) and Wnt families, and cytokines, such as stromal-derived factor (SDF)-1α and IL-6 [27, 29].
2-3 Lymphocytes and Kupffer Cells
The immune response in the tumor and tumor microenvironment is an important regulator of progression in many cancers, including HCC. Fu et al. showed that CD4(+)CD25(+) regulatory T cells were more predominant than CD8+ T cells in HCC tissues compared with adjacent benign tissue. They also demonstrated that CD4(+)CD25(+) regulatory T cells impair cytotoxic CD8+ T cell proliferation, activation, degranulation, and production of granzyme A, granzyme B, and perforin. In line with these findings, several studies found that low intratumoral CD8+ T cell and high regulatory T cell numbers are associated with a worse prognosis in HCC patients [30, 31]. In addition, dysfunctional regulation of the immune response in the tumor microenvironment by excessive regulatory T cell activity, insufficient B7 costimulation, inhibition by specific ligands such as programmed death ligand-1, or TGF-β mediated impairment of CD8+ T cell anticancer functions are well known mechanisms by which cancers evade the immune response [32]. Similarly, increased densities of NK cells are associated with HCC cell apoptosis and decreased tumor cell proliferation[33].
Although Kupffer cells were initially thought to be involved in antitumor immunity, there is substantial clinical and experimental evidence that suggests that these tumor-associated macrophages (TAMs) enhance tumor progression by impairing cytotoxic CD8+ T cell immune responses[34]. Programmed death 1 (PD 1) is highly expressed in exhausted CD8+ T cells. Its interaction with programmed death ligand-1 (PD-L1) was shown to impair cytotoxic CD8+ T cell function in human HCC. Increased expression of PD-L1 in Kupffer cells is thought to mediate a PD1 and PD-L1 interaction that prevents the cytotoxic activities of CD8+ T cell against tumors. In fact, blocking the interaction between PD-L1 on Kupffer cells and PD1 on CD8+ T cells restores cytotoxic CD8+ T cell function[35]. Kupffer cells also produce IL-6 that stimulates the initiation and development of HCC from hepatocellular damage and compensatory proliferation[36]. Kupffer cells, as well as stellate cells, when activated by inflammatory cytokines (IL-1, TNF alpha, PDGF), produce excessive osteopontin that plays a pivotal role in various cell signaling pathways that promote inflammation, tumor progression and metastasis[37]. In Kupffer cells, NF-kappa B, the master regulator of inflammatory and immune responses, is an important pathway for the integration of signals from the tumor microenvironment that promote carcinogenesis.
2-4 Endothelial cells and HCC
Endothelial cells in HCC tissues and normal tissues have molecular and functional differences. Tumor-associated endothelial cells have rapid cell turn over, enhanced motility, migration, and high expression of CD105 and TGF-β1. Notably, TGF-β1 plays the role of chemo attractant for CD105 expressing endothelial cells and thus promotes tumor angiogenesis. [38] Recent studies of isolated CD105+ endothelial cells from HCC, showed they had features of increased angiogenesis activity with higher resistance to chemotherapeutic agents and inhibitors of angiogenesis [39]. The expression of PDGF receptor alpha in tumor endothelial cells was reported to be associated with a high risk for metastasis [40].
3. Non-cellular components of the tumor microenvironment
3-1. TGF-β1 and Other Growth Factors
The complex roles of TGF-β1 in HCC have been extensively investigated. TGF-β1 is released in the ECM in a latent form and activated by MMP-2 or MMP-9, which are richly expressed in the tumor microenvironment [41]. When activated, TGF-β1 binds to TGF receptor II, phosphorylates TGF-β RI, and activates down stream signaling through Smad-2 and Smad-3. TGF-β1 is up-regulated in HCC tissues and peri-neoplastic stroma and plays key roles in liver fibrogenesis and hepatocarcinogenesis [42]. TGF-β expression is markedly increased in the cirrhotic liver and is a potent inducer of stellate cell proliferation and collagen production [5, 18]. JNK activity is required for TGF-β1 induced HSC activation and proliferation. Kluwe et al. showed that pan-JNK inhibitors prevent PDGF-, TGF-β- and angiotensin II-induced murine HSC activation and decreased PDGF and TGF-β signaling in human HSC [20]. Connective tissue growth factor (CCN2, also known as CTGF), is a mediator of TGF-β action and plays an important role in HSC-mediated fibrogenesis [43].
Apart from from its role in liver fibrogenesis, TGF-β plays a dual role in HCC pathogenesis. It normally acts as a tumor suppressor in the premalignant state through the inhibition of cell proliferation and activation of apoptosis signals. Anti-proliferative effects are mediated by the mobilization of cyclin-dependent kinase inhibitors and suppression of c-Myc while the proapoptotic mechanisms of TGF-β1 are mediated by down regulation of anti-apoptotic proteins [44]. The tumor suppressor effect of TGF-β not only involves the hepatocyte itself, but also acts through the suppression of tumor stroma mitogens and tumorigenic inflammation [45]. The role of TGF-β1 may shift from tumor suppressor to oncogenic growth factors via several different mechanisms[45, 46]. It has been shown that HBx and HCV can shift hepatocytic TGF-β signaling from the tumor-suppressive pSmad3C pathway to the oncogenic pSmad3L pathway through the activation of c-Jun N-terminal Kinase (JNK) [47, 48] (Figure 2). A recent study suggested that promoter methylation of tristetrapolin (TTP), a negative posttranscriptional regulator of C-Myc, shifts TGF-β1 signaling in HCC tumorigenesis[49]. TGF-β1 was also shown to up-regulate Snail and down-regulate E-cadherin, which is central evidence for the epithelial-mesenchymal transition process [50-52]. Consistent with this molecular evidence, TGF-β1 increases migration, vascular invasion (by modifying the structure of β1 integrin and increasing α3 integrin expression), angiogenesis (by the production of VEGF), tumor-stromal cross-talk, and metastasis (by increasing connective tissue growth factor [28, 53-57]). TGF-β1 also facilitates the EMT process through the activation of the PDGF signaling pathway [58].
Figure 2. Modulation of TGF-β signaling by Hepatitis B or C.
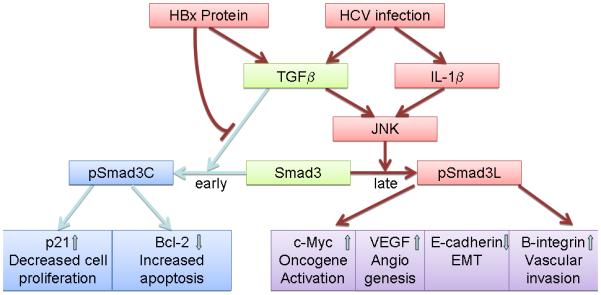
TGF-β normally suppresses cell proliferation and induces cell apoptosis through the activation of the pSmad3C mediated p21 WAF1 pathway and down regulation of anti-apoptotic proteins. HBx proteins and HCV impair pSmad3C/p21 WAF1 tumor suppressive pathways and activate JNK/pSmad3L oncogenic pathways, which in turn activate c-myc and related oncogenic pathways. This also leads to increased angiogenesis via VEGF, vascular invasion through the activation of β1 integrin, and promotion of EMT.
TGF-β1 regulates oncogenic miRNA expression to promote HCC progression. Exposure of hepatocytes to TGF-β1 increases miR-181b expression, which promotes cell growth, survival, migration and invasion of HCC cells [59]. Similarly, TGF-β induces miR-23a, 27a, and 24 (clustered in 1 transcript on chromosome 19), which promotes growth and survival of HCC cells [60].
Other heparin binding growth factors such as PDGF, vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), and hepatocyte growth factor (HGF) play important roles in HCC pathogenesis [17, 61-63]. PDGF plays an important role in the transformation of HSC into myofibroblasts, thus promoting fibrogenesis in the liver and increasing cell proliferation. Campbell et al. showed that over-expression of PDGFC in the liver of the transgenic mouse results in HSC activation, proliferation, tissue fibrosis and subsequent development of hepatocellular carcinoma through the activation of the ERK-1/-2 and PKB/Akt signaling pathways [17]. As HCC is a highly vascular tumor, angiogenesis is a critical step in HCC progression. VEGF is a major growth factor that stimulates angiogenesis in normal and tumor tissues. In the inflammatory condition, the NF-ĸB signaling pathway is activated, which increases VEGF expression[64]. VEGF acts not only on the proliferation of endothelial cells in the vasculature but also on the proliferation of cancer cells expressing VEGF-A receptor through downstream Akt/mTOR signaling[61]. FGFs are growth factors that are involved in tissue regeneration, wound healing, and angiogenesis [65, 66]. Aberrant expression of FGFs has been reported in HCC, and it has been found to promote HCC and endothelial cell proliferation through the activation of downstream Erk and AKT pathways[67].[M1] HGF is a growth factor expressed in hepatic stellate cells or myofibroblasts and is thought to be a mediator of tumor-stromal interactions through which myofibroblasts increase the proliferation and invasion of HCC cells [63].
3-2. Proteolysis enzymes
MMPs are zinc-dependent endopeptidases that were first described in the 1960s. MMPs play roles in physiologic tissue remodeling, development, and regulation during the inflammatory process[68, 69]. There are a total of 23 known human MMPs, and different types of stromal and cancer cells produce various sets of MMPs. The main subtypes of MMPs are; 1) collagenases, MMP-1,-8,-13; 2) gelatinases, MMP-2,-9; 3) matrilysins, MMP-7,-26; 4) membrane type MMPs, MMP-14,-15,-16,-24,-17,-25; and 5) stromelysins, MMP- 3,-10,-11 [70].
MMPs play an important role in the development of liver cirrhosis. Mice with MMP-9 mutations have inhibited fibrogenesis, resulting in decreased portal and periportal accumulation of collagen. MMP-9 mutations suppress trans-differentiation of hepatic stellate cells to the myofibroblast-like phenotype in vitro and in vivo. Moreover, adenoviral application of the mutants MMP-9-H401A and -E402Q led to increased apoptosis of activated hepatic stellate cells, a main modulator of hepatic fibrosis [19].
MMPs lead to tissue remodeling, inflammation, tumor cell growth, migration, invasion and metastasis in many cancers, and they are also major modulators of the tumor microenvironment, playing key roles in HCC tumorigenesis[71]. Tumor invasion is coordinated by increased proteolytic activity of MMPs that degrade the surrounding stroma and allow tumor cell spread. Recent literature has shown that the role of MMPs is not only to degrade ECM but also to modulate cancer signaling pathways. It is well known that MMP-2, -9, and -14 activate TGF-β1, which is a key modulator of epithelial-mesenchymal transition in HCC[72]. TGF-β1 also reciprocally activates MMPs. miR-181b, which is up-regulated by TGF-β1, up-regulates MMP-2 and -9 and promotes migration and invasion of HCC cells[59]. High expression of MMP-9 is associated with activation of the PI3K/PTEN/AKT/Mtor pathways in human HCCs[73, 74]. MMPs also inhibit apoptosis signaling in cancer cells. For example, Fas ligand, which initiates the apoptosis process by binding Fas receptors, cleaved by MMP7 and is then unable to apoptosis[75]. MMP-2, -9, and -14 regulate the bioavailability of VEGF and promote angiogenesis in HCC cells [76, 77]. MMPs are also involved in the modulation of the inflammatory response by regulating inflammatory cytokines and chemokines, which promote cancer progression [69, 78, 79]. MMP9 is highly expressed in HCC and its high expression is associated with capsular infiltration[80]. MMP-9 promotes HCC invasion and metastasis by cleaving the osteopontin precursor into an active form[81].
MMPs are released in inactivated forms due to the interaction between cysteine residue of the pro-domain and the zinc ion of the catalytic site[71]. Twist 1, focal adhesion kinase (FAK), claudin-1, HBV X protein, plasmin, furin, or other MMPs activate MMPs, thus promoting liver fibrosis and HCC progression, invasion and metastasis. [82-87] [88]. The chemopreventive effect of statins against HCC appear to be mediated by deactivation of MMP-2 and -9 due to decreased expression of MMP-14 and TIMP-2[89]. Phase III clinical trials are now ongoing to compare the efficacy of sorafenib alone and sorafenib coupled with pravastatin (NCT01075555).
Active MMPs are regulated by a negative feedback loop to prevent excessive tissue damage and inflammation. MMP activity is regulated at the level of gene transcription, by activation and deactivation of proteolytic enzymes, and by natural inhibitors called TIMPs. TIMPs play complex roles in regulating cell proliferation, apoptosis, MMP activation, and angiogenesis as well as in preventing the excessive degradation of ECM. TIMP3 is a negative regulator of MMPs and is known to inhibit tumor progression, invasion, and metastasis in HCC[90, 91]. High expression of TIMP1 suppresses the proliferative and invasive potential of HCC cell lines [92, 93]. Also of note is ability of TIMP2 to activate as well as inhibit MMPs. At high concentrations, TIMP2 inhibits MMP2 activation while at lower concentrations, it activates MMP2 by triggering MMP2 and MT1-MMP clustering, which is the critical step in MMP2 activation[94, 95]. The enzymatic activities of MMP and TIMP are tightly balanced, and high MMP activity, especially involving MMP-2 and -9, is associated with tumor invasion, metastasis and a poor outcome in HCC [74].
3-3. Extracellular Matrix Proteins
The ECM consists of fibrous proteins and proteoglycans, which have a protein core to which glycosaminoglycans (GAGs) are attached during their synthesis. The main roles of proteoglycans are to maintain the structural framework of the tissue and to store growth factors within the ECM. Heparan sulfate, chondroitin sulfate, and keratan sulfate are the major types of proteoglycans in the ECM. Of these, heparan sulphate proteoglycans (HSPGs) are known to play an important role in the pathogenesis of HCC as key growth factors such as FGF, HGF, PDGF, and VEGF are either stored in HSPGs or utilize HSPGs as co-receptors for binding to their tyrosine kinase receptors [17, 62, 63]. The sulfation of particular saccharide moieties of HSPGs is required for growth factor signaling. Our previous studies have shown that the heparin-degrading endosulfatases, sulfatase 1 (SULF1) and sulfatase 2 (SULF2), play important roles in modulating these heparin-binding growth signaling pathways[96-99]. Although SULF1 and SULF2 are structurally very similar, FGF signaling and its downstream AKT/mitogen-activated protein kinase pathway is activated by SULF2 but abrogated by SULF1[98, 100]. Desulfation of co-receptor type HSPGs by SULF1 inhibits binding of the growth factor to its receptor, abrogating growth factor signaling and producing a tumor suppressing effect [101]. On the other hand, desulfation of HSPGs by SULF2 releases growth factors from the storage subtype of HSPGs and increases binding of growth factors to their receptors, leading to the activation of growth signaling[96]. PI-88, a heparan sulfate mimetic synthesized for targeting heparanases in cancer, has been shown to inhibit SULFs activity [102]. The safety and efficacy of PI-88 as an adjuvant therapy for hepatocellular carcinoma after curative resection was shown recently in a phase II clinical trial [103].
Laminins are cell adhesion proteins in the ECM that form a web-like structure to resist tensile forces in the basal lamina. They consist of three α, β and γ chains, and 15 different heterodimers have been characterized[104]. Of the different subtypes of laminins, laminin-5 is expressed in HCC nodules, and its expression is associated with the metastatic phenotype of HCC[105]. Laminin-5 (Ln-5), together with TGF-β1, was reported to promote EMT [51]. Integrin α3 β1-and α6β4-mediated adhesion, proliferation, migration and invasion of HCC cells are dependent upon Ln-5 [106-109].
Integrins are surface receptor proteins that mediate cell-matrix and cell-cell adhesion. There are more than 20 integrin heterodimers due to alternative splicing and combinations of α and β subunits[110]. β3 integrin was shown to be associated with inhibition of cell growth and promotion of apoptosis[111], and over-expression of β1 integrin inhibits HCC cell proliferation by preventing Skp-2 dependent degradation of p27 via PI3K pathways[112]. Enhanced expression of α3 β1 integrin is associated with increased migration and invasion of HCC cells[109]. Collagens are the most abundant protein in the ECM and provide a structural support for cells. They also promote cell migration and proliferation in HCC. Let-7g, a known tumor suppressor miRNA, down-regulates COL1A2 and inhibits HCC cell migration and growth[113].
3-4 Inflammatory Cytokines
Inflammatory milieu from chronic liver injury contributes to the development of hepatic fibrosis and eventually, HCC. IL-6, TNF-α, and IL-1 are well-established mediators of HCC progression in liver inflammation. IL-6 is a multifunctional inflammatory cytokine produced by Kupffer cells in the liver in response to hepatocyte death that contributes to compensatory hepatocyte proliferation[114]. Serum IL-6 is increased in cirrhosis and high serum IL-6 is associated with increased risk for HCC and a poor prognosis in patients with HCC[115-118]. Estrogen suppresses IL-6 production in Kupffer cells, partly explaining the gender discrepancy in HCC development [119]. A recent study also showed that IL-6 is a link between obesity and HCC as increased expression of IL-6 and TNF in obese mice leads to the activation of the IL-6 signaling pathway via the downstream STAT3 and ERK pathways, thus promoting tumorigenesis in the liver[120].
TNF-α is a multifunctional cytokine produced mainly by Kupffer cells and other immune cells and is an essential cytokine for liver regeneration following liver injury due to the activation of its downstream NF-KB and Akt pathways[121]. Similarly, IL-1 is a pro-inflammatory cytokine that promotes MyD88 adaptor protein-dependent compensatory proliferation of hepatocytes[82]. IL-1 also promotes HSC proliferation, activation, and trans-differentiation into the myofibroblastic phenotype in addition to activating HSCs to produce and activate MMPs, particularly MMP9 [122].
IL-12 is an immune response mediator which induces the production of interferon gamma from NK cells or naïve T cells, promotes helper T cell differentiation, enhances cell-mediated immune responses, and activates cytotoxic lymphocytes[123]. The antitumor effect of IL-12 is thought to be mediated by the activation of tumor specific cytotoxic T lymphocytes and NK cells, and inhibition of angiogenesis. Intra-tumoral injection of IL-12 gene therapy induced lymphocyte infiltration into the tumor and inhibited tumor growth and angiogenesis in a mouse model[34, 124]. The use of IL-12 in clinical practice is limited due to the severe systemic toxicity resulting from high interferon gamma levels in large doses[125] and the minimal efficacy of low doses[126].
4. Tumor microenvironment: Prognostic gene signatures
Since the early 2000s, global gene expression profiling of HCC has provided new insights into the molecular and prognostic classification of HCC. Various subtypes of HCC were defined that have distinctive tumor biologies and altered cell signaling pathways as well as different prognoses. Most importantly, these studies have consistently revealed the significance of the tumor microenvironment in the biological and prognostic classification of HCC.
For TGF-β, consistent with the dual role of TGF-β in HCC pathogenesis, global gene expression profiling of human HCC showed that TGF-β gene signatures can cluster into two homogeneous groups of HCC with early or late TGF-β signatures. The late TGF-β signature is associated with an invasive HCC phenotype and increased risk of tumor recurrence[127]. A recent meta-analysis of gene expression profiling from eight independent HCC cohorts proposed three subclasses of HCC, one of which was characterized by TGF-β-induced Wnt activation and the enrichment of gene sets associated with the EMT process[128]. MMPs and TIMPs have been included in gene signatures linked to poor prognosis. MMP14 was one of the signature genes associated with HCC vascular invasion in humans[129]. Lee et al. integrated gene expression data from rat fetal hepatoblasts and adult hepatocytes with HCC from human and mouse models. HCCs were classified into mature hepatocyte and immature hepatoblast subtypes. MMP1 and TIMP1 were signature genes in the immature hepatoblast subtypes of HCC that was associated with a poor prognosis[130].
The importance of inflammatory cytokine profiles in the tumor microenvironment has also been recognized in gene expression profiling. Functional enrichment analysis with Gene Ontology categories showed the enrichment of chemotaxis and humoral immune response genes as well as proliferation and development-related functions in the group at high risk of recurrence after surgical resection of HCC [83]. Gene expression signatures from the adjacent benign tissue were reported to predict late recurrence of HCC, this signature was characterized by inflammation-associated pathways and growth factors including NF-κB, TNF-α, and IL-6[131]. IL-6, a major inflammatory cytokine was one of the signature genes in the hepatoblast phenotype signature [130]. In line with this result is the finding that inflammation and immune response-related gene signatures with an increase in Th2 cytokines in adjacent benign tissue can predict venous metastases, recurrence, and prognosis in patients with HCC [132]. Osteopontin, secreted from Kupffer or stellate cells in response to inflammatory cytokines, was also reported to be a leading gene in HCC metastasis signatures[133].
5. Tumor-Stroma interaction: A New Therapeutic Target for HCC
As most systemic chemotherapies fail to improve overall survival in patients with advanced HCC, efforts to develop new drug treatments have shifted from systemic chemotherapy to targeted treatment against the tumor-stromal interaction. The basic rationale for targeting tumor-stromal interaction is to suppress the effect of surrounding tissues or cell types that stimulate hepatocarcinogenesis, tumor progression, invasion, and metastasis while minimizing systemic toxicity by delivering drug effects specifically to tumors and their microenvironment. Each component of the tumor microenvironment shares some functional redundancies. Therefore, targeting one molecular component of the tumor microenvironment dose not necessarily suppress HCC progression. For example, with MMPs, several enzymes display proteolytic activities toward the same ECM proteins[134, 135]. Therefore, current drugs mostly target the tumor-stromal interaction by inhibiting receptors and their downstream signaling pathways, thereby abrogating the cancer-promoting signaling provided by the tumor stroma rather than directly targeting specific components of the tumor stroma.
Sorafenib, an oral multi-kinase inhibitor, is the most successful medication of this kind. It inhibits VEGFR-2/-3 and PDGFR as well as Raf kinase, disrupting tumor-stromal interactions and resulting in decreased cell proliferation and angiogenesis. The efficacy and safety of sorafenib have been demonstrated in Phase III clinical trials, and it is currently the standard of care for patients with advanced stage HCC[131, 136]. Similarly, brivanib, which targets VEGFR2 and FGFR, sunitinib, which targets PDGFR, VEGFR, C-KIT and FLT-3, erlotinib, which targets EGFR, linifanib, which targets VEGFR and PDGFR, ramucirumab, which targets VEGFR2, and PI-88, which targets heparanase as well as sulfatases, are now in Phase III clinical trials for the treatment of HCC (Table 1).
Table 1.
Clinical trials targeting the tumor-stromal interaction for the treatment of HCC
| Treatment | Phase | Target | Trial ID |
|---|---|---|---|
| Brivanib | 3 | VEGFR2, FGFR1 | NCT00858871 |
| Linifanib | 3 | VEGFR, PDGFR | NCT01009593 |
| Sorafenib | 3 | VEGFR, PDGFR, Raf | NCT00492752 |
| Sunitinib | 3 | VEGFR, PDGFR, c-KIT | NCT00699374 |
| Ramucirumab | 3 | VEGFR2 | NCT01140347 |
| Erlotinib | 3 | EGFR | NCT00901901 |
| PI-88 | 3 | Heparanase, SULFs | NCT00568308 |
| Bevacizumab | 2 | VEGF | NCT00162669 |
| Cediranib | 2 | VEGFR, PDGFR, c-KIT | NCT00238394 |
| BIBF-1120 | 2 | VEGFR, PDGFR, FGFR | NCT01004003 |
| E-7080 | 2 | VEGFR, FGFR, PDGFR, c-KIT | NCT00946153 |
| TSU-68 | 2 | VEGFR2, FGFR, PDGFR | NCT00784290 |
| XL-184 | 2 | VEGFR2, MET, RET | NCT00940225 |
| Vandetanib | 2 | VEGFR, EGFR | NCT00508001 |
| Cetuximab | 2 | EGFR | NCT00142428 |
| BIIB-022 | 2 | IGF-1R | NCT00956436 |
| Cixutumumab | 2 | IGF-1R | NCT00639509 |
| CT-011 | 2 | PD-1/2 | NCT00966251 |
| MEDI-575 | 1 | PDGFR | NCT01102400 |
| BAY73-4506 | 1 | VEGFR, PDGFR, FGFR-1, Raf, RET, c-KIT |
NCT01117623 |
| GC33 | 1 | GPC3 | NCT00976170 |
| AVE1642 | 1 | IGF-1R | NCT00791544 |
| Liver NK cell | 1 | Liver NK cell inoculation | NCT01147380 |
Targeted treatment against TGF-β signaling appears to be promising as high expression of TGF-β is a key mediator of liver fibrosis, HCC progression, and the EMT process in addition to being a poor prognostic indicator of HCC. TGF-β receptor 1 kinase inhibitor (LY2109761) deactivates Smad-2, decreasing the migration and (vascular) invasion of HCC cells and up-regulating E cadherin expression in HCC cell membranes, which mediates cell adhesion[28, 53]. More recently, LY2109761 was shown to inhibit tumor specific neoangiogenesis by blocking paracrine cross-talk between HCC and endothelial cells via Smad 2 dependent inhibition of VEGF production with an efficacy that was surprisingly superior to bevacizumab, which specifically targets VEGF [56]. In addition, LY2109761 was also shown to interrupt the cross-talk between HCC cells and cancer-associated fibroblasts through the down-regulation of connective tissue growth factor (CTGF), thus inhibiting tumor progression[57]. Phase I clinical trials targeting TGF-β signaling for the treatment of HCC have not yet been performed.
6. Future prospective and Conclusion
There have been substantial advances in the understanding of the importance of the tumor microenvironment in HCC initiation, progression, invasion, and metastasis over the past few decades. The tumor microenvironment changes dynamically and consequently affects HCC behavior. It is now being recognized as an active component of the tumor rather than merely a passive structural support of tumor growth. In this regard, treatments against the tumor microenvironment and its interaction with HCC cells are under active investigation. Although targeting one specific element of the tumor microenvironment is often ineffective due to the functional redundancies of each component of the tumor microenvironment, targeted treatments (i.e. sorafenib) against tumor-stromal interaction through the inhibition of growth factor receptors have become the standard treatment for advanced stage HCCs in clinical practice. A better understanding of the biological and molecular interactions between each element of the tumor microenvironment and the tumor cells is critical in elucidating the heterogeneous biologic features of HCC and identifying additional effective treatment targets. This insight has the potential to eventually translate into improvements in clinical practice ranging from the prevention and prognostication of HCC to prolonging the survival of patients with advanced stage HCC.
Acknowledgement
We express our appreciation to Christopher Cheung and Sarah Thornburgh for their critical review of the paper.
Grant support: This work was supported by NIH grants CA100882 and CA128633 to LRR.
Abbreviations
- ECM
extracellular matrix
- FGF
fibroblast growth factor
- HCC
hepatocellular carcinoma
- HGF
hepatocyte growth factor
- HSPG
heparan sulfate proteoglycan
- HS
heparan sulfate
- IL
interleukin
- MMP
matrix metalloproteinase
- MyD88
myeloid differentiation factor 88
- NF-kB
nuclear factor kappa B
- PDGF
platelet- derived growth factor
- STAT3
signal transducer and activator of transcription 3
- Sulf1
sulfatase 1
- Sulf2
sulfatase 2
- TGF
Transforming growth factor
- TNF-α
Tumor necrosis factor-α
- VEGF
Vascular endothelial growth factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure: No Conflict of interest
Reference
- 1.GLOBOCAN 2008 [Google Scholar]
- 2.Tang DJ, Dong SS, Ma NF, Xie D, Chen L, Fu L, et al. Overexpression of eukaryotic initiation factor 5A2 enhances cell motility and promotes tumor metastasis in hepatocellular carcinoma. Hepatology. 2010;51:1255–63. doi: 10.1002/hep.23451. [DOI] [PubMed] [Google Scholar]
- 3.Bruix J, Boix L, Sala M, Llovet JM. Focus on hepatocellular carcinoma. Cancer Cell. 2004;5:215–9. doi: 10.1016/s1535-6108(04)00058-3. [DOI] [PubMed] [Google Scholar]
- 4.Amann T, Bataille F, Spruss T, Muhlbauer M, Gabele E, Scholmerich J, et al. Activated hepatic stellate cells promote tumorigenicity of hepatocellular carcinoma. Cancer Sci. 2009;100:646–53. doi: 10.1111/j.1349-7006.2009.01087.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nitta T, Kim JS, Mohuczy D, Behrns KE. Murine cirrhosis induces hepatocyte epithelial mesenchymal transition and alterations in survival signaling pathways. Hepatology. 2008;48:909–19. doi: 10.1002/hep.22397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–54. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 7.Zeisberg M, Yang C, Martino M, Duncan MB, Rieder F, Tanjore H, et al. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J Biol Chem. 2007;282:23337–47. doi: 10.1074/jbc.M700194200. [DOI] [PubMed] [Google Scholar]
- 8.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–8. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Friedman SL, Roll FJ, Boyles J, Bissell DM. Hepatic lipocytes: the principal collagen-producing cells of normal rat liver. Proc Natl Acad Sci U S A. 1985;82:8681–5. doi: 10.1073/pnas.82.24.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sokolovic A, Sokolovic M, Boers W, Elferink RP, Bosma PJ. Insulin-like growth factor binding protein 5 enhances survival of LX2 human hepatic stellate cells. Fibrogenesis Tissue Repair. 3:3. doi: 10.1186/1755-1536-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bachem MG, Meyer D, Melchior R, Sell KM, Gressner AM. Activation of rat liver perisinusoidal lipocytes by transforming growth factors derived from myofibroblastlike cells. A potential mechanism of self perpetuation in liver fibrogenesis. J Clin Invest. 1992;89:19–27. doi: 10.1172/JCI115561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88:125–72. doi: 10.1152/physrev.00013.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–18. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Friedman SL. Liver fibrosis -- from bench to bedside. J Hepatol. 2003;38(Suppl 1):S38–53. doi: 10.1016/s0168-8278(02)00429-4. [DOI] [PubMed] [Google Scholar]
- 16.Dubuisson L, Lepreux S, Bioulac-Sage P, Balabaud C, Costa AM, Rosenbaum J, et al. Expression and cellular localization of fibrillin-1 in normal and pathological human liver. J Hepatol. 2001;34:514–22. doi: 10.1016/s0168-8278(00)00048-9. [DOI] [PubMed] [Google Scholar]
- 17.Campbell JS, Hughes SD, Gilbertson DG, Palmer TE, Holdren MS, Haran AC, et al. Platelet-derived growth factor C induces liver fibrosis, steatosis, and hepatocellular carcinoma. Proc Natl Acad Sci U S A. 2005;102:3389–94. doi: 10.1073/pnas.0409722102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eghbali-Fatourechi G, Sieck GC, Prakash YS, Maercklein P, Gores GJ, Fitzpatrick LA. Type I procollagen production and cell proliferation is mediated by transforming growth factor-beta in a model of hepatic fibrosis. Endocrinology. 1996;137:1894–903. doi: 10.1210/endo.137.5.8612529. [DOI] [PubMed] [Google Scholar]
- 19.Roderfeld M, Weiskirchen R, Wagner S, Berres ML, Henkel C, Grotzinger J, et al. Inhibition of hepatic fibrogenesis by matrix metalloproteinase-9 mutants in mice. Faseb J. 2006;20:444–54. doi: 10.1096/fj.05-4828com. [DOI] [PubMed] [Google Scholar]
- 20.Kluwe J, Pradere JP, Gwak GY, Mencin A, De Minicis S, Osterreicher CH, et al. Modulation of hepatic fibrosis by c-Jun-N-terminal kinase inhibition. Gastroenterology. 138:347–59. doi: 10.1053/j.gastro.2009.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moles A, Tarrats N, Fernandez-Checa JC, Mari M. Cathepsins B and D drive hepatic stellate cell proliferation and promote their fibrogenic potential. Hepatology. 2009;49:1297–307. doi: 10.1002/hep.22753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martin-Vilchez S, Sanz-Cameno P, Rodriguez-Munoz Y, Majano PL, Molina-Jimenez F, Lopez-Cabrera M, et al. The hepatitis B virus X protein induces paracrine activation of human hepatic stellate cells. Hepatology. 2008;47:1872–83. doi: 10.1002/hep.22265. [DOI] [PubMed] [Google Scholar]
- 23.Schulze-Krebs A, Preimel D, Popov Y, Bartenschlager R, Lohmann V, Pinzani M, et al. Hepatitis C virus-replicating hepatocytes induce fibrogenic activation of hepatic stellate cells. Gastroenterology. 2005;129:246–58. doi: 10.1053/j.gastro.2005.03.089. [DOI] [PubMed] [Google Scholar]
- 24.Kamada Y, Matsumoto H, Tamura S, Fukushima J, Kiso S, Fukui K, et al. Hypoadiponectinemia accelerates hepatic tumor formation in a nonalcoholic steatohepatitis mouse model. J Hepatol. 2007;47:556–64. doi: 10.1016/j.jhep.2007.03.020. [DOI] [PubMed] [Google Scholar]
- 25.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 26.Ostman A, Augsten M. Cancer-associated fibroblasts and tumor growth--bystanders turning into key players. Curr Opin Genet Dev. 2009;19:67–73. doi: 10.1016/j.gde.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 27.Pietras K, Ostman A. Hallmarks of cancer: interactions with the tumor stroma. Exp Cell Res. 316:1324–31. doi: 10.1016/j.yexcr.2010.02.045. [DOI] [PubMed] [Google Scholar]
- 28.Fransvea E, Mazzocca A, Antonaci S, Giannelli G. Targeting transforming growth factor (TGF)-betaRI inhibits activation of beta1 integrin and blocks vascular invasion in hepatocellular carcinoma. Hepatology. 2009;49:839–50. doi: 10.1002/hep.22731. [DOI] [PubMed] [Google Scholar]
- 29.Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature. 2004;432:332–7. doi: 10.1038/nature03096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gao Q, Qiu SJ, Fan J, Zhou J, Wang XY, Xiao YS, et al. Intratumoral balance of regulatory and cytotoxic T cells is associated with prognosis of hepatocellular carcinoma after resection. J Clin Oncol. 2007;25:2586–93. doi: 10.1200/JCO.2006.09.4565. [DOI] [PubMed] [Google Scholar]
- 31.Fu J, Xu D, Liu Z, Shi M, Zhao P, Fu B, et al. Increased regulatory T cells correlate with CD8 T-cell impairment and poor survival in hepatocellular carcinoma patients. Gastroenterology. 2007;132:2328–39. doi: 10.1053/j.gastro.2007.03.102. [DOI] [PubMed] [Google Scholar]
- 32.Gajewski TF, Meng Y, Blank C, Brown I, Kacha A, Kline J, et al. Immune resistance orchestrated by the tumor microenvironment. Immunol Rev. 2006;213:131–45. doi: 10.1111/j.1600-065X.2006.00442.x. [DOI] [PubMed] [Google Scholar]
- 33.Chew V, Tow C, Teo M, Wong HL, Chan J, Gehring A, et al. Inflammatory tumour microenvironment is associated with superior survival in hepatocellular carcinoma patients. J Hepatol. 52:370–9. doi: 10.1016/j.jhep.2009.07.013. [DOI] [PubMed] [Google Scholar]
- 34.Barajas M, Mazzolini G, Genove G, Bilbao R, Narvaiza I, Schmitz V, et al. Gene therapy of orthotopic hepatocellular carcinoma in rats using adenovirus coding for interleukin 12. Hepatology. 2001;33:52–61. doi: 10.1053/jhep.2001.20796. [DOI] [PubMed] [Google Scholar]
- 35.Wu K, Kryczek I, Chen L, Zou W, Welling TH. Kupffer cell suppression of CD8+ T cells in human hepatocellular carcinoma is mediated by B7-H1/programmed death-1 interactions. Cancer Res. 2009;69:8067–75. doi: 10.1158/0008-5472.CAN-09-0901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Benedicto I, Molina-Jimenez F, Barreiro O, Maldonado-Rodriguez A, Prieto J, Moreno-Otero R, et al. Hepatitis C virus envelope components alter localization of hepatocyte tight junction-associated proteins and promote occludin retention in the endoplasmic reticulum. Hepatology. 2008;48:1044–53. doi: 10.1002/hep.22465. [DOI] [PubMed] [Google Scholar]
- 37.Ramaiah SK, Rittling S. Pathophysiological role of osteopontin in hepatic inflammation, toxicity, and cancer. Toxicol Sci. 2008;103:4–13. doi: 10.1093/toxsci/kfm246. [DOI] [PubMed] [Google Scholar]
- 38.Benetti A, Berenzi A, Gambarotti M, Garrafa E, Gelati M, Dessy E, et al. Transforming growth factor-beta1 and CD105 promote the migration of hepatocellular carcinoma-derived endothelium. Cancer Res. 2008;68:8626–34. doi: 10.1158/0008-5472.CAN-08-1218. [DOI] [PubMed] [Google Scholar]
- 39.Xiong YQ, Sun HC, Zhang W, Zhu XD, Zhuang PY, Zhang JB, et al. Human hepatocellular carcinoma tumor-derived endothelial cells manifest increased angiogenesis capability and drug resistance compared with normal endothelial cells. Clin Cancer Res. 2009;15:4838–46. doi: 10.1158/1078-0432.CCR-08-2780. [DOI] [PubMed] [Google Scholar]
- 40.Zhang T, Sun HC, Xu Y, Zhang KZ, Wang L, Qin LX, et al. Overexpression of platelet-derived growth factor receptor alpha in endothelial cells of hepatocellular carcinoma associated with high metastatic potential. Clin Cancer Res. 2005;11:8557–63. doi: 10.1158/1078-0432.CCR-05-0944. [DOI] [PubMed] [Google Scholar]
- 41.Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000;14:163–76. [PMC free article] [PubMed] [Google Scholar]
- 42.Bedossa P, Peltier E, Terris B, Franco D, Poynard T. Transforming growth factor-beta 1 (TGF-beta 1) and TGF-beta 1 receptors in normal, cirrhotic, and neoplastic human livers. Hepatology. 1995;21:760–6. [PubMed] [Google Scholar]
- 43.Gao R, Brigstock DR. Connective tissue growth factor (CCN2) induces adhesion of rat activated hepatic stellate cells by binding of its C-terminal domain to integrin alpha(v)beta(3) and heparan sulfate proteoglycan. J Biol Chem. 2004;279:8848–55. doi: 10.1074/jbc.M313204200. [DOI] [PubMed] [Google Scholar]
- 44.Gomis RR, Alarcon C, Nadal C, Van Poznak C, Massague J. C/EBPbeta at the core of the TGFbeta cytostatic response and its evasion in metastatic breast cancer cells. Cancer Cell. 2006;10:203–14. doi: 10.1016/j.ccr.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 45.Massague J. TGFbeta in Cancer. Cell. 2008;134:215–30. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Siegel PM, Massague J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat Rev Cancer. 2003;3:807–21. doi: 10.1038/nrc1208. [DOI] [PubMed] [Google Scholar]
- 47.Murata M, Matsuzaki K, Yoshida K, Sekimoto G, Tahashi Y, Mori S, et al. Hepatitis B virus X protein shifts human hepatic transforming growth factor (TGF)-beta signaling from tumor suppression to oncogenesis in early chronic hepatitis B. Hepatology. 2009;49:1203–17. doi: 10.1002/hep.22765. [DOI] [PubMed] [Google Scholar]
- 48.Matsuzaki K, Murata M, Yoshida K, Sekimoto G, Uemura Y, Sakaida N, et al. Chronic inflammation associated with hepatitis C virus infection perturbs hepatic transforming growth factor beta signaling, promoting cirrhosis and hepatocellular carcinoma. Hepatology. 2007;46:48–57. doi: 10.1002/hep.21672. [DOI] [PubMed] [Google Scholar]
- 49.Sohn BH, Park IY, Lee JJ, Yang SJ, Jang YJ, Park KC, et al. Functional switching of TGF-beta1 signaling in liver cancer via epigenetic modulation of a single CpG site in TTP promoter. Gastroenterology. 138:1898–908. doi: 10.1053/j.gastro.2009.12.044. [DOI] [PubMed] [Google Scholar]
- 50.Spagnoli FM, Cicchini C, Tripodi M, Weiss MC. Inhibition of MMH (Met murine hepatocyte) cell differentiation by TGF(beta) is abrogated by pre-treatment with the heritable differentiation effector FGF1. J Cell Sci. 2000;113(Pt 20):3639–47. doi: 10.1242/jcs.113.20.3639. [DOI] [PubMed] [Google Scholar]
- 51.Giannelli G, Bergamini C, Fransvea E, Sgarra C, Antonaci S. Laminin-5 with transforming growth factor-beta1 induces epithelial to mesenchymal transition in hepatocellular carcinoma. Gastroenterology. 2005;129:1375–83. doi: 10.1053/j.gastro.2005.09.055. [DOI] [PubMed] [Google Scholar]
- 52.Peinado H, Quintanilla M, Cano A. Transforming growth factor beta-1 induces snail transcription factor in epithelial cell lines: mechanisms for epithelial mesenchymal transitions. J Biol Chem. 2003;278:21113–23. doi: 10.1074/jbc.M211304200. [DOI] [PubMed] [Google Scholar]
- 53.Fransvea E, Angelotti U, Antonaci S, Giannelli G. Blocking transforming growth factor-beta up-regulates E-cadherin and reduces migration and invasion of hepatocellular carcinoma cells. Hepatology. 2008;47:1557–66. doi: 10.1002/hep.22201. [DOI] [PubMed] [Google Scholar]
- 54.Giannelli G, Fransvea E, Marinosci F, Bergamini C, Colucci S, Schiraldi O, et al. Transforming growth factor-beta1 triggers hepatocellular carcinoma invasiveness via alpha3beta1 integrin. Am J Pathol. 2002;161:183–93. doi: 10.1016/s0002-9440(10)64170-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Katabami K, Mizuno H, Sano R, Saito Y, Ogura M, Itoh S, et al. Transforming growth factor-beta1 upregulates transcription of alpha3 integrin gene in hepatocellular carcinoma cells via Ets-transcription factor-binding motif in the promoter region. Clin Exp Metastasis. 2005;22:539–48. doi: 10.1007/s10585-005-5260-x. [DOI] [PubMed] [Google Scholar]
- 56.Mazzocca A, Fransvea E, Lavezzari G, Antonaci S, Giannelli G. Inhibition of transforming growth factor beta receptor I kinase blocks hepatocellular carcinoma growth through neo-angiogenesis regulation. Hepatology. 2009;50:1140–51. doi: 10.1002/hep.23118. [DOI] [PubMed] [Google Scholar]
- 57.Mazzocca A, Fransvea E, Dituri F, Lupo L, Antonaci S, Giannelli G. Down-regulation of connective tissue growth factor by inhibition of transforming growth factor beta blocks the tumor-stroma cross-talk and tumor progression in hepatocellular carcinoma. Hepatology. 51:523–34. doi: 10.1002/hep.23285. [DOI] [PubMed] [Google Scholar]
- 58.van Zijl F, Mair M, Csiszar A, Schneller D, Zulehner G, Huber H, et al. Hepatic tumor-stroma crosstalk guides epithelial to mesenchymal transition at the tumor edge. Oncogene. 2009;28:4022–33. doi: 10.1038/onc.2009.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang B, Hsu SH, Majumder S, Kutay H, Huang W, Jacob ST, et al. TGFbeta-mediated upregulation of hepatic miR-181b promotes hepatocarcinogenesis by targeting TIMP3. Oncogene. 29:1787–97. doi: 10.1038/onc.2009.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huang S, He X, Ding J, Liang L, Zhao Y, Zhang Z, et al. Upregulation of miR-23a approximately 27a approximately 24 decreases transforming growth factor-beta-induced tumor-suppressive activities in human hepatocellular carcinoma cells. Int J Cancer. 2008;123:972–8. doi: 10.1002/ijc.23580. [DOI] [PubMed] [Google Scholar]
- 61.Yang JC, Teng CF, Wu HC, Tsai HW, Chuang HC, Tsai TF, et al. Enhanced expression of vascular endothelial growth factor-A in ground glass hepatocytes and its implication in hepatitis B virus hepatocarcinogenesis. Hepatology. 2009;49:1962–71. doi: 10.1002/hep.22889. [DOI] [PubMed] [Google Scholar]
- 62.Ogasawara S, Yano H, Iemura A, Hisaka T, Kojiro M. Expressions of basic fibroblast growth factor and its receptors and their relationship to proliferation of human hepatocellular carcinoma cell lines. Hepatology. 1996;24:198–205. doi: 10.1053/jhep.1996.v24.pm0008707262. [DOI] [PubMed] [Google Scholar]
- 63.Neaud V, Faouzi S, Guirouilh J, Le Bail B, Balabaud C, Bioulac-Sage P, et al. Human hepatic myofibroblasts increase invasiveness of hepatocellular carcinoma cells: evidence for a role of hepatocyte growth factor. Hepatology. 1997;26:1458–66. doi: 10.1053/jhep.1997.v26.pm0009397985. [DOI] [PubMed] [Google Scholar]
- 64.Liu LP, Liang HF, Chen XP, Zhang WG, Yang SL, Xu T, et al. The role of NF-kappaB in Hepatitis b virus X protein-mediated upregulation of VEGF and MMPs. Cancer Invest. 28:443–51. doi: 10.3109/07357900903405959. [DOI] [PubMed] [Google Scholar]
- 65.Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature. 2008;453:314–21. doi: 10.1038/nature07039. [DOI] [PubMed] [Google Scholar]
- 66.Presta M, Dell'Era P, Mitola S, Moroni E, Ronca R, Rusnati M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005;16:159–78. doi: 10.1016/j.cytogfr.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 67.Lai JP, Chien JR, Moser DR, Staub JK, Aderca I, Montoya DP, et al. hSulf1 Sulfatase promotes apoptosis of hepatocellular cancer cells by decreasing heparin-binding growth factor signaling. Gastroenterology. 2004;126:231–48. doi: 10.1053/j.gastro.2003.09.043. [DOI] [PubMed] [Google Scholar]
- 68.Gross J, Lapiere CM. Collagenolytic activity in amphibian tissues: a tissue culture assay. Proc Natl Acad Sci U S A. 1962;48:1014–22. doi: 10.1073/pnas.48.6.1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Parks WC, Wilson CL, Lopez-Boado YS. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol. 2004;4:617–29. doi: 10.1038/nri1418. [DOI] [PubMed] [Google Scholar]
- 70.Bourboulia D, Stetler-Stevenson WG. Matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs): Positive and negative regulators in tumor cell adhesion. Semin Cancer Biol. doi: 10.1016/j.semcancer.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 141:52–67. doi: 10.1016/j.cell.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yu Q, Stamenkovic I. Localization of matrix metalloproteinase 9 to the cell surface provides a mechanism for CD44-mediated tumor invasion. Genes Dev. 1999;13:35–48. doi: 10.1101/gad.13.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cheng JC, Chou CH, Kuo ML, Hsieh CY. Radiation-enhanced hepatocellular carcinoma cell invasion with MMP-9 expression through PI3K/Akt/NF-kappaB signal transduction pathway. Oncogene. 2006;25:7009–18. doi: 10.1038/sj.onc.1209706. [DOI] [PubMed] [Google Scholar]
- 74.Chen JS, Wang Q, Fu XH, Huang XH, Chen XL, Cao LQ, et al. Involvement of PI3K/PTEN/AKT/mTOR pathway in invasion and metastasis in hepatocellular carcinoma: Association with MMP-9. Hepatol Res. 2009;39:177–86. doi: 10.1111/j.1872-034X.2008.00449.x. [DOI] [PubMed] [Google Scholar]
- 75.Mitsiades N, Yu WH, Poulaki V, Tsokos M, Stamenkovic I. Matrix metalloproteinase-7-mediated cleavage of Fas ligand protects tumor cells from chemotherapeutic drug cytotoxicity. Cancer Res. 2001;61:577–81. [PubMed] [Google Scholar]
- 76.Littlepage LE, Sternlicht MD, Rougier N, Phillips J, Gallo E, Yu Y, et al. Matrix metalloproteinases contribute distinct roles in neuroendocrine prostate carcinogenesis, metastasis, and angiogenesis progression. Cancer Res. 70:2224–34. doi: 10.1158/0008-5472.CAN-09-3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kohga K, Tatsumi T, Takehara T, Tsunematsu H, Shimizu S, Yamamoto M, et al. Expression of CD133 confers malignant potential by regulating metalloproteinases in human hepatocellular carcinoma. J Hepatol. 52:872–9. doi: 10.1016/j.jhep.2009.12.030. [DOI] [PubMed] [Google Scholar]
- 78.Manicone AM, McGuire JK. Matrix metalloproteinases as modulators of inflammation. Semin Cell Dev Biol. 2008;19:34–41. doi: 10.1016/j.semcdb.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lin WW, Karin M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest. 2007;117:1175–83. doi: 10.1172/JCI31537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Arii S, Mise M, Harada T, Furutani M, Ishigami S, Niwano M, et al. Overexpression of matrix metalloproteinase 9 gene in hepatocellular carcinoma with invasive potential. Hepatology. 1996;24:316–22. doi: 10.1053/jhep.1996.v24.pm0008690399. [DOI] [PubMed] [Google Scholar]
- 81.Takafuji V, Forgues M, Unsworth E, Goldsmith P, Wang XW. An osteopontin fragment is essential for tumor cell invasion in hepatocellular carcinoma. Oncogene. 2007;26:6361–71. doi: 10.1038/sj.onc.1210463. [DOI] [PubMed] [Google Scholar]
- 82.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 83.Woo HG, Park ES, Cheon JH, Kim JH, Lee JS, Park BJ, et al. Gene expression-based recurrence prediction of hepatitis B virus-related human hepatocellular carcinoma. Clin Cancer Res. 2008;14:2056–64. doi: 10.1158/1078-0432.CCR-07-1473. [DOI] [PubMed] [Google Scholar]
- 84.Ou DP, Tao YM, Tang FQ, Yang LY. The hepatitis B virus X protein promotes hepatocellular carcinoma metastasis by upregulation of matrix metalloproteinases. Int J Cancer. 2007;120:1208–14. doi: 10.1002/ijc.22452. [DOI] [PubMed] [Google Scholar]
- 85.Yu FL, Liu HJ, Lee JW, Liao MH, Shih WL. Hepatitis B virus X protein promotes cell migration by inducing matrix metalloproteinase-3. J Hepatol. 2005;42:520–7. doi: 10.1016/j.jhep.2004.11.031. [DOI] [PubMed] [Google Scholar]
- 86.Lara-Pezzi E, Gomez-Gaviro MV, Galvez BG, Mira E, Iniguez MA, Fresno M, et al. The hepatitis B virus X protein promotes tumor cell invasion by inducing membrane-type matrix metalloproteinase-1 and cyclooxygenase-2 expression. J Clin Invest. 2002;110:1831–8. doi: 10.1172/JCI200215887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhao XL, Sun T, Che N, Sun D, Zhao N, Dong XY, et al. Promotion of hepatocellular carcinoma metastasis through matrix metalloproteinase activation by epithelial-mesenchymal transition regulator twist1. J Cell Mol Med. doi: 10.1111/j.1582-4934.2010.01052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Taras D, Blanc JF, Rullier A, Dugot-Senant N, Laurendeau I, Vidaud M, et al. Pravastatin reduces lung metastasis of rat hepatocellular carcinoma via a coordinated decrease of MMP expression and activity. J Hepatol. 2007;46:69–76. doi: 10.1016/j.jhep.2006.06.015. [DOI] [PubMed] [Google Scholar]
- 90.Qi JH, Ebrahem Q, Moore N, Murphy G, Claesson-Welsh L, Bond M, et al. A novel function for tissue inhibitor of metalloproteinases-3 (TIMP3): inhibition of angiogenesis by blockage of VEGF binding to VEGF receptor-2. Nat Med. 2003;9:407–15. doi: 10.1038/nm846. [DOI] [PubMed] [Google Scholar]
- 91.Zhang H, Wang YS, Han G, Shi Y. TIMP-3 gene transfection suppresses invasive and metastatic capacity of human hepatocarcinoma cell line HCC-7721. Hepatobiliary Pancreat Dis Int. 2007;6:487–91. [PubMed] [Google Scholar]
- 92.Xia D, Yan LN, Xie JG, Tong Y, Yan ML, Wang XP, et al. Overexpression of TIMP-1 mediated by recombinant adenovirus in hepatocellular carcinoma cells inhibits proliferation and invasion in vitro. Hepatobiliary Pancreat Dis Int. 2006;5:409–15. [PubMed] [Google Scholar]
- 93.Kim JR, Kim CH. Association of a high activity of matrix metalloproteinase-9 to low levels of tissue inhibitors of metalloproteinase-1 and -3 in human hepatitis B-viral hepatoma cells. Int J Biochem Cell Biol. 2004;36:2293–306. doi: 10.1016/j.biocel.2004.04.022. [DOI] [PubMed] [Google Scholar]
- 94.Strongin AY, Collier I, Bannikov G, Marmer BL, Grant GA, Goldberg GI. Mechanism of cell surface activation of 72-kDa type IV collagenase. Isolation of the activated form of the membrane metalloprotease. J Biol Chem. 1995;270:5331–8. doi: 10.1074/jbc.270.10.5331. [DOI] [PubMed] [Google Scholar]
- 95.Kinoshita T, Sato H, Okada A, Ohuchi E, Imai K, Okada Y, et al. TIMP-2 promotes activation of progelatinase A by membrane-type 1 matrix metalloproteinase immobilized on agarose beads. J Biol Chem. 1998;273:16098–103. doi: 10.1074/jbc.273.26.16098. [DOI] [PubMed] [Google Scholar]
- 96.Mejean A, Lebret T. [The metastatic cascade: angiogenesis and new concepts] Prog Urol. 2008;18(Suppl 7):S156–66. doi: 10.1016/S1166-7087(08)74538-X. [DOI] [PubMed] [Google Scholar]
- 97.El-Zayadi AR. Heavy smoking and liver. World J Gastroenterol. 2006;12:6098–101. doi: 10.3748/wjg.v12.i38.6098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wang S, Ai X, Freeman SD, Pownall ME, Lu Q, Kessler DS, et al. QSulf1, a heparan sulfate 6-O-endosulfatase, inhibits fibroblast growth factor signaling in mesoderm induction and angiogenesis. Proc Natl Acad Sci U S A. 2004;101:4833–8. doi: 10.1073/pnas.0401028101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Uchimura K, Morimoto-Tomita M, Bistrup A, Li J, Lyon M, Gallagher J, et al. HSulf-2, an extracellular endoglucosamine-6-sulfatase, selectively mobilizes heparin-bound growth factors and chemokines: effects on VEGF, FGF-1, and SDF-1. BMC Biochem. 2006;7:2. doi: 10.1186/1471-2091-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lai JP, Sandhu DS, Yu C, Han T, Moser CD, Jackson KK, et al. Sulfatase 2 up-regulates glypican 3, promotes fibroblast growth factor signaling, and decreases survival in hepatocellular carcinoma. Hepatology. 2008;47:1211–22. doi: 10.1002/hep.22202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bosch FX, Ribes J, Diaz M, Cleries R. Primary liver cancer: worldwide incidence and trends. Gastroenterology. 2004;127:S5–S16. doi: 10.1053/j.gastro.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 102.Hossain MM, Hosono-Fukao T, Tang R, Sugaya N, van Kuppevelt TH, Jenniskens GJ, et al. Direct detection of HSulf-1 and HSulf-2 activities on extracellular heparan sulfate and their inhibition by PI-88. Glycobiology. 20:175–86. doi: 10.1093/glycob/cwp159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Liu CJ, Lee PH, Lin DY, Wu CC, Jeng LB, Lin PW, et al. Heparanase inhibitor PI-88 as adjuvant therapy for hepatocellular carcinoma after curative resection: a randomized phase II trial for safety and optimal dosage. J Hepatol. 2009;50:958–68. doi: 10.1016/j.jhep.2008.12.023. [DOI] [PubMed] [Google Scholar]
- 104.Aumailley M, Bruckner-Tuderman L, Carter WG, Deutzmann R, Edgar D, Ekblom P, et al. A simplified laminin nomenclature. Matrix Biol. 2005;24:326–32. doi: 10.1016/j.matbio.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 105.Giannelli G, Fransvea E, Bergamini C, Marinosci F, Antonaci S. Laminin-5 chains are expressed differentially in metastatic and nonmetastatic hepatocellular carcinoma. Clin Cancer Res. 2003;9:3684–91. [PubMed] [Google Scholar]
- 106.Carter WG, Kaur P, Gil SG, Gahr PJ, Wayner EA. Distinct functions for integrins alpha 3 beta 1 in focal adhesions and alpha 6 beta 4/bullous pemphigoid antigen in a new stable anchoring contact (SAC) of keratinocytes: relation to hemidesmosomes. J Cell Biol. 1990;111:3141–54. doi: 10.1083/jcb.111.6.3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Giannelli G, Bergamini C, Fransvea E, Marinosci F, Quaranta V, Antonaci S. Human hepatocellular carcinoma (HCC) cells require both alpha3beta1 integrin and matrix metalloproteinases activity for migration and invasion. Lab Invest. 2001;81:613–27. doi: 10.1038/labinvest.3780270. [DOI] [PubMed] [Google Scholar]
- 108.Bergamini C, Sgarra C, Trerotoli P, Lupo L, Azzariti A, Antonaci S, et al. Laminin-5 stimulates hepatocellular carcinoma growth through a different function of alpha6beta4 and alpha3beta1 integrins. Hepatology. 2007;46:1801–9. doi: 10.1002/hep.21936. [DOI] [PubMed] [Google Scholar]
- 109.Mizuno H, Ogura M, Saito Y, Sekine W, Sano R, Gotou T, et al. Changes in adhesive and migratory characteristics of hepatocellular carcinoma (HCC) cells induced by expression of alpha3beta1 integrin. Biochim Biophys Acta. 2008;1780:564–70. doi: 10.1016/j.bbagen.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 110.Hynes RO. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- 111.Wu Y, Zuo J, Ji G, Saiyin H, Liu X, Yin F, et al. Proapoptotic function of integrin beta(3) in human hepatocellular carcinoma cells. Clin Cancer Res. 2009;15:60–9. doi: 10.1158/1078-0432.CCR-08-1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Fu Y, Fang Z, Liang Y, Zhu X, Prins P, Li Z, et al. Overexpression of integrin beta1 inhibits proliferation of hepatocellular carcinoma cell SMMC-7721 through preventing Skp2-dependent degradation of p27 via PI3K pathway. J Cell Biochem. 2007;102:704–18. doi: 10.1002/jcb.21323. [DOI] [PubMed] [Google Scholar]
- 113.Ji J, Zhao L, Budhu A, Forgues M, Jia HL, Qin LX, et al. Let-7g targets collagen type I alpha2 and inhibits cell migration in hepatocellular carcinoma. J Hepatol. 52:690–7. doi: 10.1016/j.jhep.2009.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cressman DE, Greenbaum LE, DeAngelis RA, Ciliberto G, Furth EE, Poli V, et al. Liver failure and defective hepatocyte regeneration in interleukin-6-deficient mice. Science. 1996;274:1379–83. doi: 10.1126/science.274.5291.1379. [DOI] [PubMed] [Google Scholar]
- 115.Tilg H, Wilmer A, Vogel W, Herold M, Nolchen B, Judmaier G, et al. Serum levels of cytokines in chronic liver diseases. Gastroenterology. 1992;103:264–74. doi: 10.1016/0016-5085(92)91122-k. [DOI] [PubMed] [Google Scholar]
- 116.Wong VW, Yu J, Cheng AS, Wong GL, Chan HY, Chu ES, et al. High serum interleukin-6 level predicts future hepatocellular carcinoma development in patients with chronic hepatitis B. Int J Cancer. 2009;124:2766–70. doi: 10.1002/ijc.24281. [DOI] [PubMed] [Google Scholar]
- 117.Nakagawa H, Maeda S, Yoshida H, Tateishi R, Masuzaki R, Ohki T, et al. Serum IL-6 levels and the risk for hepatocarcinogenesis in chronic hepatitis C patients: an analysis based on gender differences. Int J Cancer. 2009;125:2264–9. doi: 10.1002/ijc.24720. [DOI] [PubMed] [Google Scholar]
- 118.Zhu AX, Sahani DV, Duda DG, di Tomaso E, Ancukiewicz M, Catalano OA, et al. Efficacy, safety, and potential biomarkers of sunitinib monotherapy in advanced hepatocellular carcinoma: a phase II study. J Clin Oncol. 2009;27:3027–35. doi: 10.1200/JCO.2008.20.9908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317:121–4. doi: 10.1126/science.1140485. [DOI] [PubMed] [Google Scholar]
- 120.Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 140:197–208. doi: 10.1016/j.cell.2009.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Fausto N, Campbell JS, Riehle KJ. Liver regeneration. Hepatology. 2006;43:S45–53. doi: 10.1002/hep.20969. [DOI] [PubMed] [Google Scholar]
- 122.Han YP, Zhou L, Wang J, Xiong S, Garner WL, French SW, et al. Essential role of matrix metalloproteinases in interleukin-1-induced myofibroblastic activation of hepatic stellate cell in collagen. J Biol Chem. 2004;279:4820–8. doi: 10.1074/jbc.M310999200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3:133–46. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 124.Yamashita YI, Shimada M, Hasegawa H, Minagawa R, Rikimaru T, Hamatsu T, et al. Electroporation-mediated interleukin-12 gene therapy for hepatocellular carcinoma in the mice model. Cancer Res. 2001;61:1005–12. [PubMed] [Google Scholar]
- 125.Leonard JP, Sherman ML, Fisher GL, Buchanan LJ, Larsen G, Atkins MB, et al. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood. 1997;90:2541–8. [PubMed] [Google Scholar]
- 126.Sangro B, Mazzolini G, Ruiz J, Herraiz M, Quiroga J, Herrero I, et al. Phase I trial of intratumoral injection of an adenovirus encoding interleukin-12 for advanced digestive tumors. J Clin Oncol. 2004;22:1389–97. doi: 10.1200/JCO.2004.04.059. [DOI] [PubMed] [Google Scholar]
- 127.Coulouarn C, Factor VM, Thorgeirsson SS. Transforming growth factor-beta gene expression signature in mouse hepatocytes predicts clinical outcome in human cancer. Hepatology. 2008;47:2059–67. doi: 10.1002/hep.22283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hoshida Y, Nijman SM, Kobayashi M, Chan JA, Brunet JP, Chiang DY, et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res. 2009;69:7385–92. doi: 10.1158/0008-5472.CAN-09-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Chen X, Cheung ST, So S, Fan ST, Barry C, Higgins J, et al. Gene expression patterns in human liver cancers. Mol Biol Cell. 2002;13:1929–39. doi: 10.1091/mbc.02-02-0023.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lee JS, Heo J, Libbrecht L, Chu IS, Kaposi-Novak P, Calvisi DF, et al. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat Med. 2006;12:410–6. doi: 10.1038/nm1377. [DOI] [PubMed] [Google Scholar]
- 131.Hoshida Y, Villanueva A, Kobayashi M, Peix J, Chiang DY, Camargo A, et al. Gene expression in fixed tissues and outcome in hepatocellular carcinoma. N Engl J Med. 2008;359:1995–2004. doi: 10.1056/NEJMoa0804525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Budhu A, Forgues M, Ye QH, Jia HL, He P, Zanetti KA, et al. Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell. 2006;10:99–111. doi: 10.1016/j.ccr.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 133.Ye QH, Qin LX, Forgues M, He P, Kim JW, Peng AC, et al. Predicting hepatitis B virus-positive metastatic hepatocellular carcinomas using gene expression profiling and supervised machine learning. Nat Med. 2003;9:416–23. doi: 10.1038/nm843. [DOI] [PubMed] [Google Scholar]
- 134.Giannelli G, Falk-Marzillier J, Schiraldi O, Stetler-Stevenson WG, Quaranta V. Induction of cell migration by matrix metalloprotease-2 cleavage of laminin-5. Science. 1997;277:225–8. doi: 10.1126/science.277.5323.225. [DOI] [PubMed] [Google Scholar]
- 135.Koshikawa N, Giannelli G, Cirulli V, Miyazaki K, Quaranta V. Role of cell surface metalloprotease MT1-MMP in epithelial cell migration over laminin-5. J Cell Biol. 2000;148:615–24. doi: 10.1083/jcb.148.3.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S, Kim JS, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009;10:25–34. doi: 10.1016/S1470-2045(08)70285-7. [DOI] [PubMed] [Google Scholar]