

Abstract
We studied the effectiveness of monoclonal anti-CD40 + cytosine–phosphate–guanosine-containing oligodeoxynucleotide 1826 (CpG-ODN) immunotherapy (IT) in mice treated with multidrug chemotherapy (CT) consisting of vincristine, cyclophosphamide and doxorubicin. Combining CT with IT led to synergistic anti-tumour effects in C57BL/6 mice with established B16 melanoma or 9464D neuroblastoma. CT suppressed the functions of T cells and natural killer (NK) cells, but primed naïve peritoneal macrophages (Mφ) to in vitro stimulation with lipopolysaccharide (LPS), resulting in augmented nitric oxide (NO) production. IT, given after CT, did not restore the responsiveness of T cells and NK cells, but further activated Mφ to secrete NO, interferon-γ (IFN-γ) and interleukin (IL)-12p40 and to suppress the proliferation of tumour cells in vitro. These functional changes were accompanied by immunophenotype alterations on Mφ, including the up-regulation of Gr-1. CD11b+F4/80+Mφ comprised the major population of B16 tumour-infiltrating leucocytes. CT + IT treatment up-regulated molecules associated with the M1 effector Mφ phenotype [CD40, CD80, CD86, major histocompatibility complex (MHC) class II, IFN-γ, tumour necrosis factor-α (TNF-α) and IL-12] and down-regulated molecules associated with the M2 inhibitory Mφ phenotype (IL-4Rα, B7-H1, IL-4 and IL-10) on the tumour-associated Mφ compared with untreated controls. Together, the results show that CT and anti-CD40 + CpG-ODN IT synergize in the induction of anti-tumour effects which are associated with the phenotypic repolarization of tumour-associated Mφ.
Keywords: anti-CD40, chemotherapy, CpG-ODN, immunotherapy, monocytes/macrophages, tumour recognition
Introduction
Current cancer treatment frequently involves multiple treatment modalities, including surgery, radiation therapy, chemotherapy (CT) and immunotherapy (IT). The combination of several anti-tumour treatments (e.g. multiple distinct classes of chemotherapy) is more effective than monotherapy because they act on the tumour at multiple sites and via different mechanisms. However, multidrug CT regimens might be expected to interfere with immune-based therapies that depend on the actions of T cells and natural killer (NK) cells that are susceptible to CT-induced immune suppression.1–7 We hypothesized that macrophages (Mφ) might be more resistant to CT than other immune cells, and that IT directed towards the activation of Mφ might therefore be synergistic with CT.
We recently established a preclinical IT protocol in tumour-bearing mice that was effective in suppressing tumour growth, even in the setting of profound immune deficiency, via the activation of Mφ. This regimen combines agonistic monoclonal anti-CD40 and class B cytosine–phosphate–guanosine-containing oligodeoxynucleotide 1826 (CpG-ODN). This IT protocol was effective in controlling the growth of several distinct syngeneic murine tumours.8 Anti-tumour effects were seen against B16 melanoma and NXS2 neuroblastoma (NB), neither of which express receptors for anti-CD40 (CD40) or CpG-ODN (TLR9),8 as well as against CD40/TLR9-expressing B-cell chronic lymphoma.9 In all models tested, the in vivo effect was mediated by Mφ and did not appear to involve any direct cytotoxic/pro-apoptotic effect of anti-CD40 or CpG-ODN. These preclinical results may have clinical relevance as Mφ comprise a major population of tumour-infiltrating leucocytes in many types of cancer.10,11
Multidrug CT protocols, such as VCD (vincristine + cyclophosphamide + doxorubicin), are commonly used clinically for the treatment of solid and haematological malignancies12–14 and are also known for their immunosuppressive effects against lymphoid cells.15 In this study we tested if combining the VCD CT regimen with anti-CD40/CpG-ODN IT would result in augmented anti-tumour effects in immunocompetent C57BL/6 mice. It was found that these CT and IT regimens synergized in inducing anti-tumour effects in vivo despite CT-induced suppression of effector functions of T cells and NK cells. Further analyses demonstrated that CT primed Mφ to subsequent stimulation via CD40 and TLR9 that resulted in augmented Mφ secretory and anti-tumour properties in vitro. Analyses of tumour-associated Mφ (ΤΑΜ) from CT plus IT-treated tumour-bearing mice revealed that in vivo anti-tumour effects were paralleled by the changing of TAM from a pro-tumour (M2) phenotype into Mφ with an effector (M1) phenotype.
Materials and methods
Mice
Six- to ten-week-old C57BL/6 mice (Harlan Sprague Dawley, Madison, WI), were housed, cared for and used in accordance with the Guide for Care and Use of Laboratory Animals (NIH publication 86-23, National Institutes of Health, Bethesda, MD, 1985).
Tumour cell lines
Mouse B16 melanoma and 9464D NB cell lines were grown in complete RPMI-1640 or Dulbecco’s modified Eagle’s minimal essential medium (DMEM), respectively, supplemented with 10% fetal bovine serum (FBS) (Sigma-Aldrich, St Louis, MO), 2 mm l-glutamine, 100 U/ml of penicillin/streptomycin and 0·5 μm 2-mercaptoethanol (2-ME) (Invitrogen Life Technologies, Carlsbad, CA) at 37° in a humidified 5% CO2 atmosphere. The 9464D disialoganglioside-2-positive, N-myc-overexpressing NB cell line was established in the laboratory of Dr Jon Wigginton (NCI), and was derived from spontaneous NB tumours arising in C57BL/6 N-myc transgenic mice developed originally by Dr William A. Weiss (University of California, San Francisco, CA).16
In vivo tumour models and measurement of anti-tumour effects
Subcutaneous B16 melanoma and 9464D NB tumours were established by the injection of 1 × 105 B16 cells/0·05 ml and 2 × 106 9464D cells/0·1 ml phosphatebuffered saline (PBS) in the abdomen. Anti-tumour effects were evaluated by measuring in situ tumour volumes, calculated as follows: smaller diameter (mm) × smaller diameter (mm) × larger diameter (mm) × (π/6), and expressed as mean ± standard error of the mean (SEM) of tumour sizes of all mice of the experimental group.
Intra-adrenal 9464D tumours were established by direct implantation of 1 × 106 tumour cells in 0·03 ml PBS into the left adrenal gland, as previously described.17 Anti-tumour effects were evaluated by measuring the perpendicular sizes of the excised tumour, calculated as follows: smaller diameter (mm) × smaller diameter (mm) × larger diameter (mm) × (π/6), and expressed as mean ± SEM of tumour sizes of all mice of the treatment group. Alternatively, excised peri-renal tumour masses were weighed and the weight of the contralateral normal kidney was subtracted to yield the net tumour weight, which was expressed as mean ± SEM of the net tumour weights of all mice of the experimental group.
CT and IT protocols
CT treatment group
Naïve or tumour-bearing mice were treated with VCD CT (vincristine 0·5 mg/kg, cyclophosphamide 100 mg/kg, doxorubicin 3·3 mg/kg)18 via a single injection in a total volume of 0·25 ml of PBS. Administration was either intraperitoneal or intravenous, and is specified for each experiment. For naïve mice, CT treatment was performed on day 0, followed by 0·25-ml injections of PBS on days 3 and 6 as control treatments for IT. For tumour-bearing mice the treatment was usually started on days 8–10 after tumour implantation, unless specified otherwise for certain experiments.
IT treatment group
Monoclonal anti-CD40 was obtained from ascites of nude mice injected with FGK 45.5 hybridoma cells (a gift from Dr F. Melchers, Basel Institute for Immunology, Basel, Switzerland) and enriched for IgG by ammonium sulphate precipitation. Endotoxin-free CpG1826 (TCCATGACGTTCCTGACGTT; the CpG motifs are underlined) was purchased from Coley Pharmaceuticals Group (Wellesley, MA). Administration was either intraperitoneal. or intravenous, and is specified for each experiment. Naïve mice were treated with agonistic anti-CD40 (0·5 mg/0·25 ml, unless indicated otherwise) on day 3 and CpG-ODN 1826 (0·1 mg/0·25 ml, unless indicated otherwise) on day 6, with these two treatments preceded by injection of 0·25 ml of PBS on day 0 as a control treatment for CT (see above). For tumour-bearing mice the treatment was usually started on days 8–10 after tumour implantation, unless specified otherwise for certain experiments.
CT + IT
In experimental groups where CT and IT were combined, anti-CD40 was given 3 days after CT, and CpG-ODN was given 3 days after treatment with anti-CD40. Control groups received PBS. In some experiments, the CT + IT cycles were repeated, with 3–4 days of rest between cycles.
Splenocyte preparation
Splenocytes were prepared from whole spleens pooled from three C57BL/6 mice by processing the spleens to a single-cell suspension, followed by lysis of erythrocytes by hypotonic shock.
Splenic T-cell proliferation assay
Flat-bottom 96-microwell cell-culture clusters were precoated with 5 μg/ml (0·1 ml/well) of monoclonal anti-CD3 (clone 145-2C11, functional grade purified hamster IgG) or control hamster IgG, both from eBioscience (San Diego, CA), for 24 hr at 4°, and the unbound IgG was removed by washing with ice-cold PBS.
Before plating, the splenocyte preparations from different treatment groups were analyzed by flow cytometry to determine the number of CD3+ cells, and the samples were adjusted to 5 × 105 CD3+ T cells/ml. Fifty-thousand CD3+ cells were cultured in triplicate for 72 hr in complete medium in plates precoated with anti-CD3- or control IgG. To measure cell proliferation, the cells were pulsed with 1 μCi/well of [3H]thymidine ([3H]TdR) for the last 6 hr, and retained radioactivity was counted by β-scintillation of total cells harvested from the wells onto glass fibre filters (Packard Instrument, Meriden, CT), using the Packard Matrix 9600 Direct Beta Counter (Packard Instrument). The results represent the mean ± SEM of β-scintillation counts acquired in 5 min from triplicate samples.
Splenic NK cell-medicated cytotoxicity
Splenocytes from mice of different treatment groups were adjusted to 1 × 105 Ly49b+ NK cells/ml and cultured in medium supplemented with 100 U/ml of recombinant human interleukin (IL)-2. At 72 hr of culture, the cells were harvested and readjusted to 5 × 105 viable total splenocytes/0·1 ml, and thereafter tested for lysis of 5 × 103 NK cell-sensitive 51Cr-pulsed YAC-1 cells at different effector-to-target (E/T) ratios (100:1, 50:1, 25:1, 12·5:1) following 4 hr of co-culture in a final volume of 0·2 ml. After 4 hr of incubation, the supernatant was harvested utilizing the Skatron harvesting system (Skatron, McLean, VA) and cytotoxicity values (%) were calculated at each E/T ratio, as reported previously.19
In vitro Mφ-mediated inhibition of tumour cell proliferation
Peritoneal cells (PCs) were obtained from mice by peritoneal cavity lavage. Before plating, PCs from different treatment groups were analyzed by flow cytometry to determine the proportion of CD11b+F4/80+ cells, and thereafter seeded in 96-microwell flat-bottom cell-culture clusters at 2·5–3 × 105 CD11b+F4/80+ cells/0·2 ml. Ninety minutes later, non-adherent cells were removed from the culture by repeated pipetting. This protocol yields a relatively pure population of Mφ, based on co-expression of F4/80 and CD11b on 97% of cells.20 The resultant adherent Mφ were thereafter incubated with B16 tumour cells (5 × 105 cells/ml, 0·05 ml/well, unless otherwise indicated) for 48 hr in medium with or without 10 ng/ml of lipopolysaccharide (LPS) from Salmonella enteritidis (Sigma-Aldrich) in a final volume of 0·2 ml.
To measure tumour cell proliferation, the cell cultures were pulsed with 1 μCi/well of [3H]TdR for the last 6 hr, and retained radioactivity was counted by β-scintillation of total cells harvested from the wells onto glass fibre filters (Packard Instrument), using the Packard Matrix 9600 Direct Beta Counter (Packard Instrument). Under these conditions Mφ incorporate negligible amounts of [3H]TdR, enabling [3H]TdR to reflect the level of proliferation of the tumour cells. Inhibition of proliferation was calculated as c = [(a−b)/a]×100, where c is the inhibition index; a is the mean value of [3H]TdR incorporated into tumour cells cultured in triplicate in medium without Mφ, in the absence or presence of LPS; and b is the mean value of [3H]TdR incorporated into tumour cells cultured in triplicate in medium with Mφ, in the absence or presence of LPS.
Nitric oxide detection
Mφ were cultured with tumour cells, as described above, to determine the inhibition, in vitro, of Mφ-mediated tumour cell proliferation. At 48 hr of co-culture, the Mφ-containing tumour cell culture supernatants were collected without disturbing the cell monolayers. Nitrite accumulation in the cell-culture supernatants was determined by using the Griess reagent (Sigma-Aldrich) as described previously.21
Labelling of PCs with carboxyfluoroscein succinimidyl ester
Carboxyfluoroscein succinimidyl ester (CFSE: excitation, 490 nm; emission, 518 nm) was purchased from Invitrogen (Carlsbad, CA). Before labelling, PCs (pooled from 10–20 naïve C57BL/6 mice) were washed twice with PBS at room temperature to eliminate mouse serum residua, resuspended in 10 ml of 37° PBS supplemented with 5 mm of CFSE and incubated for 15 min at 37°. The PCs were washed once with ice-cold PBS containing 10% FCS (to neutralize free, cell-unbound label) and once with serum-free PBS, resuspended at 2–4 × 106 PC/ml and injected intraperitoneally into mice.
Flow cytometry analysis
Phenotypic and quantitative analysis of splenocytes
Splenocyte preparations were resuspended in ice-cold PBS + 2% FCS at 3 × 106 cells/ml, dispensed in flow cytometry testing Falcon tubes at 3 × 105 cells/0·1 ml and stained with 0·1 μg/sample of anti-CD3-conjugated fluorescein isothiocyanate (FITC) (clone 145-2C11), anti-B220–FITC (RA3-6B2), anti-CD11b–allophycocyanin (M1/70), anti-Ly49b–FITC (DX5), anti-F4/80–FITC (BM8) and anti-Gr1-conjugated phycoerythrin (PE) (RB6-8C5) all from eBioscience, for 40 min on ice, followed by cell washing with ice-cold PBS and fixation with 2% paraformaldehyde. Analysis of F4/80 (BM8) and Gr-1 (RB6-8C5) expression on splenic Mφ was performed by gating on CD11b+ cells. Data acquisition was performed on a FACSCalibur flow cytometer with CellQuest software (BD, Franklin Lakes, NJ). Analysis was performed on FlowJo (TreeStar, Inc., Ashland, OR) software. To calculate the absolute number of a splenocyte subset in the total pool of splenocytes, the absolute number of total splenocytes (obtained via counting the cells on a haemocytometer) was multiplied by the relative prevalence of that subset (%) obtained via flow cytometry analysis. The results were expressed as the absolute number (×106) of the population expressing the antigen of interest.
Phenotypic and quantitative analysis of Mφ
PCs were harvested and counterstained with anti-CD11b–allophycocyanin (M1/70), anti-F4/80–FITC (BM8), anti-Gr1–PE (RB6-8C5), anti-Ly6C-conjugated peridinin chlorophyll protein (PerCP)-Cy5.5 (HK1.4), all from eBioscience, and anti-Ly6G–PE (1A8), from BD Bioscience, on ice for 40 min, followed by repeated cell washing with ice-cold PBS + 2% FCS and fixation in 2% paraformaldehyde. Data acquisition was performed on a FACSCalibur flow cytometer with CellQuest (BD) software. Analyses were performed on FlowJo (Tree Star, Inc., Ashland, OR) software by gating on CD11b+F4/80+ cells.
In separate experiments, collected PCs were seeded in six-well plates at 1 × 106 PC/ml, 5 ml, with subsequent removal of non-adherent cells, and were thereafter cultured for 12 hr in complete medium supplemented with 10 ng/ml of LPS and 1 μm monensin. At 12 hr of culture, the adhesion-purified Mφ were harvested with 0·53 mm EDTA, resuspended in PBS + 2% FCS, then stained with anti-CD11b–allophycocyanin and F4/80–FITC for 40 min on ice. After staining with anti-CD11b/F4/80–allophycocyanin, the cells were washed in ice-cold PBS, fixed and permeabilized as previously described,8 and stained with anti-interferon-γ (IFN-γ)–PE (XMG1.2) and anti-IL-12–PE (C17.8). Flow cytometry analysis was performed as described above. The results are presented as mean fluorescence intensity (MFI) ratios, calculated as the value of MFI of staining with a specific monoclonal antibody (mAb) divided by the value of MFI of staining with isotype-matched control IgG; the MFI ratio equals 1 when an antigen is not expressed. This approach allows for comparison of the same parameter but in different experimental conditions. Quantitative analysis of CD11b+F4/80+PCs was performed as described above for the analysis of CD11b+ splenic Mφ.
Analysis of tumour-associated Mφ
Harvested subcutaneous tumours were mechanically disaggregated and single-cell suspensions were obtained by centrifugation through a Ficoll–Paque density gradient at 450 g using a GH-3.8 rotor for 30 min. The tumour-cell preparations were resuspended in ice-cold PBS + 2% FCS at 5 × 106 total cells/ml, dispensed in flow cytometry testing Falcon tubes at 5 × 105 cells/0·1 ml and stained with 0·1 μg/sample of anti-CD45–allophycocyanin (30-F1), anti-CD11b–FITC or –allophycocyanin (M1/70), anti-F4/80–FITC (BM8), anti-Gr1–PE (RB6-8C5), anti-CD40–PE (1C10), anti-MHC class II–PE (M5/114.15.2), anti-CD80–PE (16-10A1), anti-CD86–PE (GL1) and anti-Ly6C–PerCP-PECy5.5 (HK1.4), all from eBioscience, and anti-Ly6G–PE (1A8), anti-IL-4R-alpha–PE (mIL4R-M1), anti-B7-H1–PE (MIH5), from BD Bioscience, for 40 min on ice, followed by repeated cell washing with ice-cold PBS + 2% FCS and fixation in 2% paraformaldehyde. Analysis was performed, as described above, by gating on CD45+CD11b+ tumour-infiltrating leucocytes or CD11b+F4/80+ Mφ.
Alternatively, tumour-derived single-cell preparations were plated in six macro-well cell-culture clusters at 5 × 106 total cells/ml, in 5 ml, in complete RPMI-1640 supplemented with 1 mm monensin. At 12 hr the cells were harvested with 5% EDTA, resuspended in PBS + 2% FCS, then stained with anti-CD11b–allophycocyanin and anti-F4/80–FITC for 40 min on ice. Subsequently, the cells were washed in ice-cold PBS, fixed and permeabilized as previously described,8 and stained with anti-IL-4–PE (11B11), anti-IL-10–PE (JES5-16E3), anti-IFN-γ–PE (XMG1.2), anti-tumour necrosis factor-α (TNF-α)–PE (MP6-XT22), or anti-IL-12–PE (C17.8). Flow cytometry analysis was performed, as described above, by gating on CD11b+F4/80+ Mφ. The results are presented as MFI ratios.
Statistical analysis
A two-tailed Student’s t-test was used to determine significant differences between experimental and relevant control values within one experiment. Statistical analysis of inhibition of the tumour cell proliferation under different experimental conditions was performed on original [3H]TdR-incorporation values, which were also used to calculate the percentage of inhibition.
Results
VCD-CT and anti-CD40 + CpG-ODN IT synergize in inducing anti-tumour effects in vivo
We have previously shown that anti-CD40 and CpG-ODN synergize in activating Mφ and suppressing the growth of tumours in various models.8,9 As Mφ are relatively resistant to certain CT regimens,22 we hypothesized that the anti-tumour effects induced by CT will be augmented by anti-CD40 and CpG-ODN IT. To determine the anti-tumour effect of CT alone, mice bearing palpable subcutaneous B16 tumours were given a single cycle of VCD-CT on days 3, 8 or 11 after the tumour had been implanted, and the tumour growth (Fig. 1a) and survival (Fig. 1b) were monitored. Significant suppression of tumour growth was seen when treatment was started on days 3 or 8. However, complete tumour resolution was not achieved, and survival of the mice was extended by less than twofold.
Figure 1.
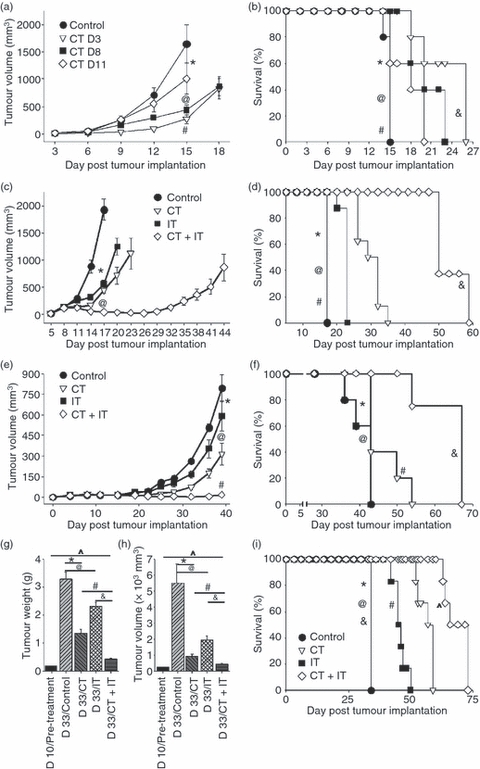
Chemotherapy (CT) and immunotherapy (IT) synergize in inducing anti-tumour effects in vivo. (a,b) C57BL/6 mice (n = 5 per group) bearing subcutaneous B16 melanoma tumours were treated with CT, via a single intraperitoneal injection, on different days after tumour implantation. Tumour volumes, expressed as mean ± standard error of the mean (SEM), (a) and survival (b) were monitored. (a) *P > 0·05 (control versus CT day 11); @P < 0·02 (control group versus CT day 3 or CT day 8); #P > 0·05 (CT day 3 versus CT day 8). (b) *P > 0·05 (control versus CT day 11); @P = 0·031 (control group versus CT day 3); #P = 0·043 (control group versus CT day 8); &P > 0·05 (CT day 3 versus CT day 8). (c,d) C57BL/6 mice (n = 6 per group) bearing subcutaneous B16 melanoma tumours were treated intravenously with CT on days 8 and 17 after tumour implantation and IT (anti-CD40 on days 11 and 20 plus CpG-ODN 1826 on days 14 and 23 after tumour implantation), alone or in combination. Tumour volumes (c) and mouse survival (d) were monitored. (c) *P < 0.001 (control versus CT or IT); @P < 0·001 (CT or IT versus CT + IT). (d) *P > 0·05 (control versus CT); @P = 0·036 (control versus IT); #P < 0·01 (control versus CT + IT); &P < 0·02 (CT + IT versus either CT or IT). (e,f) C57BL/6 mice (n = 5 per group) bearing subcutaneous 9464D neuroblastoma (NB) tumours were treated intraperitoneally with CT on day 15 after tumour implantation, and IT (anti-CD40 on days 18 and 25 plus CpG on days 21 and 28 after tumour implantation), alone or in combination. Tumour volumes (e) and mouse survival (f) were monitored. (e) *P > 0·05 (control versus IT); @P < 0·006 (control versus CT); #P < 0·007 (CT + IT versus CT or IT). (f) *P > 0·05 (control versus IT); @P < 0·05 (control versus CT); #P > 0·05 (CT versus IT); &P < 0·03 (control versus CT + IT); ^P < 0·05 (CT + IT versus CT or IT). (g–i) C57BL/6 mice (n = 9 per group) bearing intra-adrenal 9464D NB tumours were treated with CT on days 10 and 20 after tumour implantation, and with IT (anti-CD40 on days 13 and 23 plus CpG on days 16 and 26 after tumour implantation), alone or in combination. Mice were monitored for tumour growth by left flank palpation every third day. On day 33 after tumour implantation, when animals of the control group showed symptoms as a result of the large tumour size, all mice of the control group and three randomly selected mice from each of the experimental groups were killed, and tumour weight (g) and volume (h) were calculated and compared with those in mice killed on day 10 after tumour implantation, before the treatment started. The rest of the treated animals were also followed for survival (i). (g) *P = 0·0027, @P = 0·0307, #P = 0·002, &P = 0·0001, ^P = 0·0046. (h) *P = 0·0013, @P = 0·0307, #P = 0·015, &P = 0·009, ^P = 0·017. (i) *P = 0·027 (control versus IT); @P = 0·009 (control versus CT); #P = 0·041 (CT versus IT); &P < 0·001 (control versus CT + IT); ^P < 0·025 (CT + IT versus CT or IT). Presented are the results from one experiment (a,b), and representative of three experiments (c,d) or two experiments (e,f and g,i). D3, day 3; D8, day 8; D11, day 11.
Next, we tested whether a combination of CT with IT would result in synergistic anti-tumour effects in mice bearing subcutaneous B16 tumours. CT was given on days 8 and 17 after tumour implantation, and anti-CD40 was given on days 11 and 20 followed by CpG-ODN 3 days later8 (i.e. on days 14 and 23). Animals that received both CT and IT showed the strongest anti-tumour response, resulting in significant inhibition of tumour growth (P < 0·001, CT + IT versus CT or IT alone, Fig. 1c). In addition, the tumours in the CT + IT group showed transient shrinkage of volume, with the tumours in this group being smaller on day 26 than on day 8 (P = 0·0002). This combined treatment also resulted in significant (3·5-fold) prolongation of survival compared with other groups (Fig. 1d).
To confirm the synergy between CT and IT observed in this subcutaneous B16 tumour model, we delivered CT or IT, alone or in combination, to C57BL/6 mice bearing subcutaneous 9464D NB tumours (Fig. 1e,f). The combined CT + IT treatment resulted in very potent suppression of tumour growth (Fig. 1e) as well as significantly prolonged survival of the tumour-bearing mice compared with other groups (Fig. 1f).
The synergy between CT and IT was also seen when used to treat mice bearing intra-adrenal 9464D NB tumours. The CT and IT administered in combination induced the strongest anti-tumour effects based on tumour weight (Fig. 1g), tumour volume (Fig. 1h) and overall mouse survival (Fig. 1i; 33 days for the control group versus 69 ± 1·55 days for the CT + IT treatment group, P < 0·001).
CT + IT treatment results in redistribution of major subsets of spleen leucocytes
CT is known to frequently cause temporary lymphocytopenia, whereas IT may result in the expansion of certain subsets of lymphocytes. We evaluated the total numbers of splenocytes as well as the total numbers of CD3+ T cells, B220+ B cells, CD11b+ Mφ and Ly49b+ NK cells in the spleens of naïve mice after a single cycle of CT and IT, separately or in combination. As shown in Fig. 2, CT caused a statistically significant decrease in the numbers of total splenocytes (Fig. 2a) because of a reduction, at least in part, of the total number of B cells and NK cells (Fig. 2b). In contrast, IT resulted in increased numbers of all four subsets of splenic cells. When IT was administered after CT, it did not significantly change the number of total splenocytes compared with CT alone (Fig. 2a), but led to an increase in the overall number of CD11b+ splenic Mφ (*P = 0·022) while decreasing the numbers of T cells, B cells and NK cells (Fig. 2b).
Figure 2.
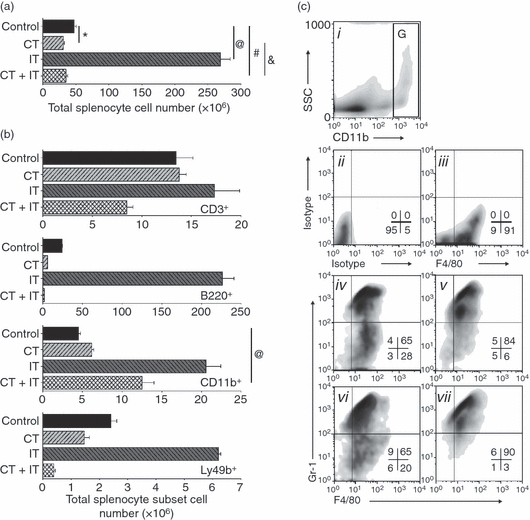
Differential leucodepletion effect of chemotherapy (CT) on splenic T cells, B cells, natural killer (NK) cells and macrophages (Mφ). Naïve non-tumour-bearing C57BL/6 mice (n = 4 per group) were treated (intraperitoneally or intravenously in several different experiments) with CT or immunotherapy (IT), alone or in combination, as described in the Materials and methods, and spleens were harvested on day 7 [i.e. one day after the last treatment; the second injection of phosphate-buffered saline (PBS) for the CT-treatment group, or CpG-ODN for IT and CT + IT treatment groups]. Absolute numbers of total splenocytes (a) or splenic CD3+ T cells, B220+ B cells, CD11b+ Mφ and Ly49+ NK cells (b) were computed based on the total number of cells and cell subset percentages from the flow cytometric analyses. The results are representative of at least three separate experiments. (a) *P = 0·0112, @P = 0·0001, #P = 0·464, &P = 0·3351. (b) (CD11b+) P = 0·022. (c) Splenic cells from mice receiving the treatments indicated below were gated on CD11bhigh cells [region (g) in panel ci for control mice] and analyzed for expression of F4/80 and Gr-1 (ciii–cvii). Panel cii shows the isotype-control staining, in place of the anti-F4/80 and anti-Gr-1. Panel ciii shows staining with anti-F/80 and isotype (for anti-Gr-1) monoclonal antibodies (mAbs). Panels iv–vii show staining with anti-F4/80 and anti-Gr-1 of Mφ from mice treated with vehicles (iv), CT (v), IT (vi) and CT + IT (vii). Numbers in density plots indicate the percentage of events in each quadrant. Data are representative of at least three experiments, with similar results obtained in each.
A number of cells are known to express CD11b (also known as integrin alpha M antigen, macrophage-1 alpha antigen, or complement receptor-3): monocytes, Mφ and microglia express high levels of CD11b, whereas granulocytes, NK cells, CD5+ B-1 cells and certain subsets of dendritic cells express CD11b to a much lower extent. Recent analyses of naïve and tumour-bearing mice have identified populations of CD11b+ cells with varying levels of Gr-1 expression and co-expression of F4/80; their suppressive activity has been used to characterize them as myeloid-derived suppressor cells (MDSCs), but not all cells with these markers are suppressive. We evaluated the CD11bhigh splenocytes (Fig. 2ci, region G) for co-expression of F4/80 (a pan-Mφ marker) as well as Gr-1 (the myeloid differentiation antigen) (Fig. 2cii–vii). It was found that the CD11bbright splenocyte population is represented by two distinct subsets: a major subset of F4/80+GR-1bright cells and a minor subset of F4/80+GR-1dim cells (Fig. 2civ–vii). Interestingly, CT (Fig. 2cv), alone or in combination with IT (Fig. 2cvii), increased the prevalence of F4/80+GR-1bright cells; ∼96% of all CD11bbright splenocytes in mice receiving the combined CT+IT treatment were also Gr-1bright (Fig. 2cvii).
CT treatment results in differential immunosuppression
Having documented quantitative changes in the composition of the splenic cell subset after treatment with CT, alone, or in combination with IT (Fig. 2), we then investigated whether this VCD-CT regimen, with or without IT, could affect the functional characteristics of the major immune effectors involved in anti-tumour responses (Fig. 3a–e). First, we tested splenic CD3+ T cells for their ability to proliferate in response to in vitro stimulation with plate-bound anti-CD3 mAb (Fig. 3a). As shown, T cells from control and IT-treated animals proliferated well after CD3 stimulation. In contrast, splenocytes from the CT-treated group and from the CT + IT treatment group showed minimal proliferative response to the CD3 stimulation. Thus, CT induced profound T-cell suppression, and the IT failed to restore T-cell proliferative function.
Figure 3.
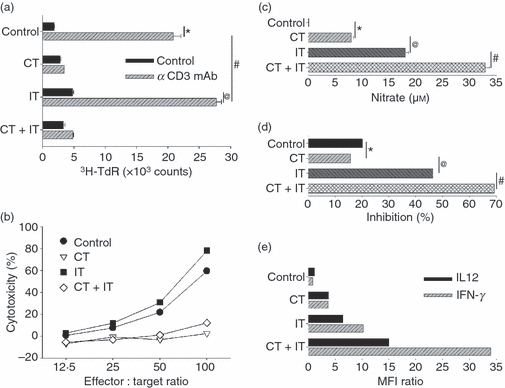
Differential effect of chemotherapy (CT) treatment on the functions of T cells, natural killer (NK) cells and macrophage (Mφ). (a) Splenocytes from naïve (i.e. non-tumour-bearing) C57BL/6 mice (n = 3 per group) treated intraperitoneally with CT and immunotherapy (IT), alone or in combination, as described in the Materials and methods and in the legend for Fig. 2, were adjusted to 5 × 104 CD3+ T cells/0·1 ml and cultured in 96-microwell tissue culture clusters precoated with αCD3 monoclonal antibody (mAb) or control IgG for 72 hr. Proliferation of splenocytes was measured by the incorporation of [3H]thymidine during the last 6 hr of culture. *P = 0·0001, @P = 0·0001, #P = 0·052. (b) Splenocytes from mice (n = 3 per group) treated intraperitoneally with CT and IT, alone or in combination, were adjusted to 1 × 105/ml of Ly49b+ NK cells and cultured in medium supplemented with 100 U/ml of recombinant human IL-2 for 72 hr. After this in vitro stimulation, the cell suspensions were readjusted to 5 × 105 viable total splenocytes/0·1 ml and tested for lysis of 5 × 103 NK cell-sensitive 51Cr-labelled YAC-1 cells/0.1 ml at the indicated effector-to-target ratios following 4 hr of co-culture in a final volume of 0·2 ml. The results are presented as percentage of cytotoxicity. (c–d) Peritoneal cells (PC) from mice treated intraperitoneally with CT and IT, alone or in combination, were adjusted to 2·5 × 105 CD11b+F4/80+Mφ/0·1 ml, seeded in 96-microwell clusters, purified for Mφ by adhesion to plastic, and thereafter tested in vitro for nitric oxide (NO) production (c) and inhibition of B16 tumour cell proliferation following 48 hr of co-culture in vitro with 1 × 103 B16 tumour cells/0·1 ml in medium supplemented with 10 ng/ml of lipopolysaccharide (LPS) (d). (c) *P = 0·01, @P = 0·006, #P = 0·002. (d) *P = 0·7, @P = 0·01, #P = 0·009. (e) PC from CT and/or IT-treated mice (n = 3 per group) were purified for Mφ by adhesion to plastic and cultured for 12 hr in medium supplemented with 1 μm monensin and 10 ng/ml of LPS, followed by flow cytometric assessment of the intracellular expression of IL-12 and IFN-γ. The results are presented as MFI ratios and are representative of at least three separate experiments with similar results obtained in each.
Next, we tested the ability of splenic NK cells to lyse YAC-1 cells in vitro (Fig. 3b). As shown, splenic NK cells from the control and IT-treated groups efficiently lysed YAC-1 cells. In contrast, NK cells from mice that received CT alone or in combination with IT mediated minimal cytotoxicity, even with in vitro IL-2 stimulation for 72 hr, and even at the highest E/T ratio.
To test Mφ function, we used the cells from the peritoneal cavity. Unlike the inhibition of CD3+ T-cell and Ly49+ NK-cell function by CT, peritoneal Mφ exhibited augmented function in vitro in response to LPS following intraperitoneal CT treatment in vivo. This enhanced Mφ function after CT included increased production of nitric oxide (NO) (Fig. 3c) and of IFN-γ and IL-12p40 (Fig. 3e). These effects were mediated by Mφ from the CT group. IT treatment following CT synergistically increased the secretory properties of Mφ (Fig. 3c,e) and their ability to inhibit tumour cell proliferation in vitro (Fig. 3d). Similar functional effects to those shown in Fig. 3 were observed for peritoneal CD11b+ Mφ from mice that received intravenous CT and IT treatments (data not shown) and for mice that received their CT and IT treatments intraperitoneally. Overall, the results presented in Fig. 3 show that this CT regimen suppresses functions of T cells and NK cells but works together with IT in stimulating Mφ functions related to anti-tumour activity.
Effect of CT and IT treatments on Mφ phenotype
We asked investigated whether peritoneal Mφ, unlike the splenic immune effectors (T and NK cells), might be exposed to very high concentrations of either CT drugs or anti-CD40 and CpG-ODN following intraperitoneal injection, which might affect their overall viability or function. To address this, naïve animals were transplanted with syngeneic CFSE-labelled PC and then treated with CT and IT, alone or combination, with the treatments administered either intraperitoneally or intravenously (Fig. 4). Naïve mice were injected intraperitoneally with 2 × 106 syngeneic CFSE-labelled PC obtained from naive donor mice. CT and IT, alone or in combination, were administered 24 hr after PC implantation, using the schedule described in the Materials and methods. Twenty-four hours after the last treatment (i.e. the second PBS treatment for the CT group and CpG-ODN treatment for the IT or CT+IT groups), PC were harvested and tested by flow cytometry for the presence of CFSEbrightCD11bbright Mφ (Fig. 4). These transplanted Mφ (upper right quadrants in Fig. 4) can be easily distinguished from the resident PC Mφ (lower right quadrants in Fig. 4) by their CFSE fluorescence signal. As shown in Fig. 4, CT but not IT, administered intraperitoneally, virtually eliminated the transplanted CFSEbrightCD11bbright Mφ. Resident Mφ (CFSEnegCD11b+) in mice from the group receiving intraperitoneal CT revealed an altered pattern of CD11b expression, becoming CD11bdim in comparison with resident Mφ from the control group (the MFI ratio for CD11b in the CFSEnegCD11b+ cells decreased from 761.3 in the control group to 482.1 in the intraperitoneal CT group in Fig. 4). Whether this represents a change in phenotype of the original naive resident Mφ, or a depletion of the original resident Mφ followed by influx of a new population with an altered phenotype, remains to be clarified. Whereas 97% of CD11b+ Mφ from mice of the control-treatment group were F4/80highGr-1neg, peritoneal Mφ from animals of the CT intraperitoneal-treatment group virtually all became F4/80dimGr-1high (data not shown). In contrast to CT intraperitoneal treatment, CT administered intravenously did not affect the recovery of CFSE+CD11b+ Mφ or the pattern of CD11b expression compared with the control group (Fig. 4).
Figure 4.
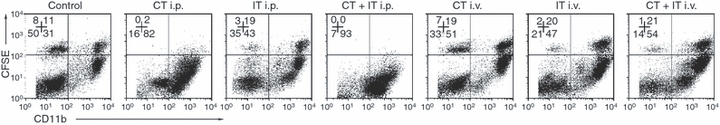
Route of chemotherapy (CT) and immunotherapy (IT) administration affects macrophage (Mφ) survival and phenotype. Twenty-four hours after intraperitoneal implantation of carboxyfluoroscein succinimidyl ester (CFSE)-labelled syngeneic peritoneal cells (PC), C57BL/6 mice (n = 3 per group) received one cycle of CT (day 1 after PC implantation) or IT (anti-CD40 – day 4 post PC implantation followed by CpG-ODN on day 7 post PC implantation), alone or in combination, either intraperitoneally or intravenously On day 8 of the experiment, total (both resident and transplanted) PCs were harvested and tested by flow cytometry for the presence of CFSE+Mφ. To distinguish CFSE+Mφ from other CFSE+PC, as well as from resident CFSE−Mφ, the total PC samples were additionally stained with anti-CD11–allophycocyanin. The numbers indicate the percentage of events in each quadrant. The data shown are representative of three experiments. i.p., intraperitoneal; i.v., intravenous.
The absolute numbers of total PC and PC-derived CD11b+F4/80+ Mφ from mice that received CT and IT, alone or in combination, intraperitoneally or intravenously, are shown Table 1. While the intraperitoneal IT induced an increase in total PC number (*P ≤ 0·05) and the intraperitoneal CT (alone or with IT) induced a decrease in recovered PC (**P ≤ 0·05), the numbers of PC-derived CD11b+F4/80+ Mφ were similar for all four treatment groups and similar for intraperitoneal versus intravenous treatment.
Table 1.
Quantitative changes in splenic leucocyte subsets after treatment with chemotherapy (CT) and/or immunotherapy (IT)
| Intraperitoneal | Intravenous | |||
|---|---|---|---|---|
| Total PC (×106) | CD11b+F4/80+Mφ (×106) | Total PC (×106) | CD11b+F4/80+Mφ (×106) | |
| Control | 4·5 ± 0·32 | 1·2 ± 0·34 | 4·6 ± 0·39 | 1·26 ± 0·35 |
| CT | 2·5 ± 0·41 | 1·3 ± 0·36 | 3·9 ± 0·31 | 1·48 ± 0·54 |
| IT | 8·7 ± 0·81* | 1·2 ± 0·5 | 5·6 ± 0·42 | 1·4 ± 0·36 |
| CT + IT | 1·8 ± 0·18** | 1·3 ± 0·4 | 3·7 ± 0·32 | 1·58 ± 0·46 |
Absolute numbers of total peritoneal cells (PC) and CD11b+PC from mice that received CT or IT, alone or in combination, intraperitoneally or intravenously, are shown. Results are presented as mean ± standard error of the mean (SEM) of three mice per treatment group. *P ≤ 0·05; **P ≤ 0·05 compared with the control group in that column. The data shown are representative of three experiments.
CD11b+ Mφ comprise the major population of tumour-infiltrating leucocytes
The results presented in Fig. 3a–e show that CT (alone or in combination with IT) inhibits NK and T-cell function, but enables Mφ, in naïve non-tumour-bearing mice, to remain active and even primed to further stimulation with IT. In the next series of experiments we investigated whether CT and IT, alone or in combination, influenced the phenotype of TAM.
C57BL/6 mice bearing established subcutaneous B16 tumours, received a single intravenous cycle of CT and IT, alone or in combination, starting on day 8 after tumour implantation. Twenty-four hours after the last treatment (i.e. on day 15 after tumour implantation), the tumours were harvested, processed to a single-cell suspension and tested by flow cytometry for TAM in a pool of tumour-infiltrating leucocytes (TIL) by counterstaining for CD45 (pan-leucocyte marker) and CD11b (Fig. 5). The results indicate that CD11b+TAM are the major leucocyte population among CD45+TIL, and that CD11b+Mφ comprised 89–95% of the total CD45+TIL in all treatment groups. The results also suggest that CT + IT treatment results in a relative increase in the percentage of CD11b+TAM in the tumour-derived single cell suspension compared with control cells, probably via reduction of the total number of B16 tumour cells in the preparation.
Figure 5.
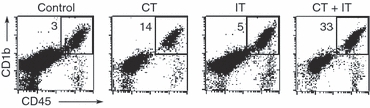
CD11b+ macrophage (Mφ) comprise the major population of CD45+ tumour-infiltrating leucocytes (TIL) in B16 tumours. C57BL/6 mice [n = 3 for control, chemotherapy (CT) and immunotherapy (IT) treatment groups, n = 6 for the CT + IT treatment group] were injected subcutaneously with B16 cells (day 0). Mice were treated with one cycle of CT (day 8) or IT (anti-CD40 on day 11, CpG on day 14), alone or in combination. On day 15 after tumour implantation, the tumours were harvested, processed to a single-cell suspension, and viable cells were studied by flow cytometry for the prevalence of CD45+ (pan-leucocyte marker)/CD11b+ (pan-Mφ marker) cells. The numbers in the right upper quadrants represent the percentage of CD45+CD11b+Mφ (gate) in the pool of total (tumour cells + non-tumour stromal cells + TIL) viable cells. Of the total CD45+ TILs, CD11b+ cells comprised 89% in the control group, 95% in the CT-treatment group, 91% in the IT-treatment group and 94% in the CT + IT-treatment group.
In the next experiment we analyzed the phenotype of TAM within B16 tumours from mice treated with CT, alone or in combination with IT, or with vehicles alone. The results suggest that TAM are present in two subsets – CD11b+F4/80+Gr-1bright and CD11b+F4/80+Gr-1dim– and that CT, administered alone or in combination with IT, and to a lesser degree IT alone, enriches CD11b+TAM for the F4/80+Gr-1bright subset compared with control treatment (data not shown).
Treatment of mice with CT and IT shifts the TAM phenotype from M2 to M1
The above studies confirmed results previously obtained by others10,11,23 of a high prevalence of TAM in growing tumours. We also demonstrated that most of these CD11b+F4/80+ TAM may have a bimodal expression pattern of Gr-1 (data not shown). The F4/80+Gr-1bright phenotypic profile has been associated with MDSCs.24–26 Although in vivo treatment with anti-CD40 plus CpG-ODN did not result in notable changes of CD11b expression on the TAM (Fig. 5), we tested whether it would affect the expression of other surface antigens and cytokines that are characteristic for Mφ in general and for MDSC/TAM in particular.
C57BL/6 mice, bearing established subcutaneous B16 tumours, were treated with two cycles of CT and IT, alone or in combination, beginning on day 8 after tumour implantation. Twenty-four hours after the last treatment, tumours were harvested, processed to a single-cell suspension and CD11b+F4/80+ TAM were gated by flow cytometry and tested for surface expression of CD40, CD80, CD86, MHC class II antigen, CD124 (IL-4Rα) and CD274 (B7-H1) and for intracellular expression of IL-10, IL-4, IFN-γ, TNF-α and IL-12p40 (Fig. 6). As shown, CD11b+F4/80+ TAM from control mice treated with vehicle alone expressed the M2 phenotype, in that they expressed high levels of surface IL-4Rα and B7-H1, and produced significant amounts of IL-10 and IL-4, but did not secrete any IFN-γ, TNF-α or IL-12. Chemotherapy resulted in the down-regulation of B7-H1 and in the up-regulation of IFN-γ. Immunotherapy down-regulated the expression of B7-H1, IL-10 and IL-4, and up-regulated the expression of CD40, CD80, CD86, MHC class II, IFN-γ, TNF-α and IL-12. When these two therapies were combined, CD11b+F4/80+TAM expressed the highest levels of CD40, CD80, CD86, MHC class II, IFN-γ, TNF-α and IL-12, and the lowest levels of IL-4Rα, B7-H1 and IL10, as well as a low level of IL-4. Thus, following the combination of CT and IT, the phenotype of Mφ within a tumour changed from a suppressive M2 pattern to an effector M1 pattern. It is possible that this change in phenotype, from M2 to M1, for TAM following combined CT + IT treatment may play a role in the observed synergistic anti-tumour effect (Fig. 1).
Figure 6.
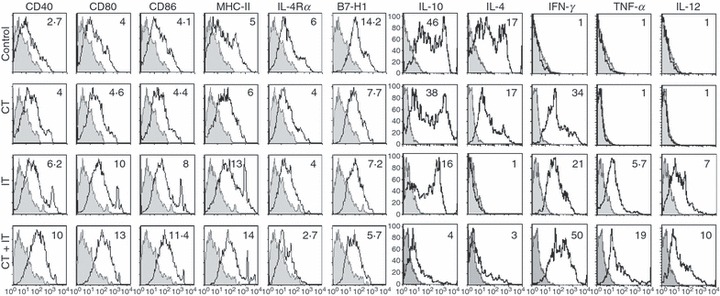
Chemotherapy (CT) and immunotherapy (IT) alter the phenotype and cytokine profile of tumour-associated macrophages (TAM). C57BL/6 mice (n = 3 for control, CT- and IT-treatment groups; n = 6 for the CT + IT-treatment group) bearing established subcutaneous B16 tumours were treated with two cycles of CT or IT, alone or in combination, as described in Fig. 1(c,d). One day after the last treatment [the second phosphate-buffered saline (PBS) injection for the CT-treatment group, or CpG-ODN for the IT- and the CT + IT-treatment groups], the mice were killed and the tumours were harvested and processed to a single-cell suspension. Tumour-associated cells were gated on CD11b+F4/80+ macrophages (Mφ) and analyzed by flow cytometry for expression of various surface antigens and intracellular cytokines (open histograms) as compared with isotype controls (shaded histograms). The mean fluorescence intensity (MFI) ratios (numbers in histograms) were computed as described in the Materials and methods. Data are representative of two separate experiments, with similar results obtained on each occasion.
Discussion
CT and IT have been considered as potentially antagonistic forms of cancer treatment because of the lymphopenia, immunosuppression and myelosuppression that can be induced by many cytotoxic drugs. This paradigm has been challenged by preclinical and clinical observations that have demonstrated therapeutic synergy between certain CT and IT regimens.27–29 Several proposed mechanisms for CT + IT synergy include increased tumour antigen cross-presentation,30 partial activation of antigen-presenting cells31 and their priming to CD40-ligation,32 tumour debulking,33 partial sensitization of tumour cells to lysis by cytotoxic T cells,34 reduction in function of negative regulatory cells,35 increased access of immune cells to tumours after CT36 and others.37,38 In this study we describe, to our knowledge for the first time, a different mechanism of synergy between multidrug CT and IT that is based on TAM activation and their partial repolarization from Mφ with an M2 tumour-promoting phenotype into Mφ with an M1 anti-tumour effector phenotype.
The VCD-CT regimen is used frequently in combination with other drugs for the treatment of patients with solid and haematological malignancies. Considerable myelosuppression, neutropenia, lymphopenia and thrombocytopenia are among the most frequent side effects of this protocol. Consistent with the results of clinical reports, in our experiments a single cycle of CT resulted in some anti-tumour effects and lymphopenia with overall redistribution of major subsets of immune cells in the spleen. This CT regimen also compromised the ability of splenic T cells and NK cells to respond to stimulation via CD3 or IL-2 receptors, respectively, at least within the first 7–8 days after a single cycle of CT treatment. Taking into consideration that Mφ are more resistant than lymphocytes to the lethal effects of certain CT,22,39 we decided to combine CT with IT with the hope of stimulating Mφ to induce an anti-tumour effect that would augment the anti-tumour activity of the CT. In our recent preclinical studies of IT we found that treatment with anti-CD40 and CpG-ODN induced strong activation of Mφ that were capable of killing tumour cells in vitro as well as mediating anti-tumour effects in vivo in the absence of T cells and/or NK cells.8,9 In this study, functional in vitro tests revealed that a cycle of treatment with anti-CD40 and CpG-ODN was not able to restore the CT-induced suppressed responses of T cells or NK cells. In contrast to T cells and NK cells, peritoneal Mφ from CT-treated mice became primed to both anti-CD40 and CpG-ODN stimulation in vivo and to LPS stimulation in vitro, as measured by secretion of NO, IL-12 and IFN-γ. This Mφ activation was associated with the statistically significant increase in the number of CD11b+ splenic Mφ in CT + IT-treated mice. In vivo, combined administration of CT with IT resulted in synergistic anti-tumour effects against the B16 and 9464D tumours, especially when animals received two versus just one cycle of CT + IT. In a separate study, using an intraperitoneal B16 model, we found that the anti-tumour effect of cyclophosphamide + anti-CD40 + CpG-ODN IT was reduced by Mφ depletion with clodronate-containing liposomes, identifying Mφ as in vivo anti-tumour effector cells (data not shown).
In experiments where CT and/or IT treatments were administered intraperitoneally, we investigated whether peritoneal Mφ, unlike the splenic immune effectors, might be exposed to very high concentrations of either CT drugs or anti-CD40 and CpG-ODN following intraperitoneal injection, which might affect their overall viability or function. To address this, naïve animals were transplanted with syngeneic CFSE-labelled PC and then treated with CT and IT, alone or in combination, with the treatments administered either intraperitoneally or intravenously. PC samples from the group treated intraperitoneally with CT contained no CFSE-labelled donor cells [neither CD11b+ PC (Mφ) nor CD11b− PC]. This suggested that local (intraperitoneal) CT administration was toxic for the mature CFSE+ donor Mφ, resulting in their apoptosis. Interestingly, 1 day (data not shown) and even 8 days (Table 1 and Fig. 4) after a single cycle of intraperitoneal treatment, PC samples from the mice treated intraperitoneally with CT still retained relatively normal numbers of endogenous cells, the majority of which were CD11bdimF4/80dim GR-1bright. This suggests that the CT administered intraperitoneally may be directly toxic to the mature CD11b+ Mφ (both from the donor and from the host), and that by the time the PCs are harvested, a new population of host bone marrow-derived CD11bdim Mφ responsive to IT has repopulated the peritoneum. In contrast to intraperitoneal CT, intraperitoneal administration of anti-CD40 plus CpG-ODN did not affect the viability of CFSE+ PC, which were readily identifiable in the pool of total PC. Indeed, in our previous studies, when naïve adherent Mφ were exposed in vitro to anti-CD40 plus LPS or CpG-ODN, a sustained Mφ activation of both secretory and cytotoxic function was documented.8,40,41 Either treatment (CT or IT), used alone or in combination but administered intravenously, did not affect donor Mφ viability in the peritoneum, as transplanted CFSE+ Mφ were easily detectable in samples of total PC.
Following CT, IT or CT + IT treatment, we noted the expansion of a group of PC that express intermediate amounts of CD11b (Fig. 4), which are otherwise infrequent in the pool of PC cells from control mice. These CD11bdim PC were also F4/80dim Gr-1bright (compared with Mφ from control-treated mice), thereby expressing an ‘immature’ phenotype. Some CD11bbrightF4/80bright cells in samples of PC from IT groups treated intraperitoneally, CT groups treated intravenously, IT groups treated intravenously and CT + IT groups treated intravenously, also expressed Gr-1 (data not shown). Whereas CD11bdimF4/80dimGr-1bright PC cells could be immature myeloid cells that have recently migrated into the peritoneal cavity (and probably, into other compartments) from the bone marrow, the CD11bbrightF4/80brightGr-1bright cells could be mature, resident peritoneal Mφ that were induced by the treatments to express Gr-1. Indeed, IT, when administered intraperitoneally, caused slight down-regulation of F4/80 and considerable up-regulation of Gr-1 on virtually all naïve Mφ that possessed a classical ‘mature’ phenotype (CD11bbrightF4/80brightGr-1neg) before implantation, and which retained that phenotype in animals that received control treatment (data not shown). Whereas down-regulation of F4/80 on mature Mφ is known to be associated with the state of activation42–44 and involves IFN-γ, TNF-α and other cytokines, the phenomenon of induced expression of Gr-1 on mature Mφ merits additional analysis.
It has previously been suggested that the RB6-8C5 clone of anti-Gr-1 may cross-react with two distinct members of the Ly6 family of proteins, namely Ly6C and Ly6G (Gr-1).45,46 Thus, we retested the IT treatment-induced Gr-1 expression of previously Gr-1-negative CD11b+F4/80+ peritoneal Mφ by using a Ly6G-specific mAb, clone 1A8, and a Ly6C-specific mAb, clone HK1.4, and found that the majority (∼85%) of CD11b+F4/80+ Mφ expressed Ly6G (Gr-1), whereas up-regulated expression of Ly6C was found only on 15–25% of IT-stimulated CD11b+F4/80+ Mφ. Importantly, Ly6G and Ly6C were co-expressed on anti-CD40 + CpG-ODN-stimulated PC Mφ. Very little Ly6G and Ly6C were expressed on CD11b+F4/80+ Mφ from the control group (I.N. Buhtoiarov, unpublished observations). As for the mechanisms accounting for Gr-1 up-regulation, it was reported that Ly6C+Gr-1+CD11b+Mφ can be generated in vitro by culturing the lineage phenotype-negative, c-kit-positive haematopoietic progenitor cells in the presence of granulocyte–macrophage colony-stimulating factor (GM-CSF), stem-cell factor (SCF) and IFN-γ.47 Thus, it might be possible that CT and/or IT induced some of these factors, which alone or in combination could alter Ly6G/Ly6C expression on naïve Mφin vivo. This last concept might be consistent with our inability to induce Gr-1 expression on adhesion-purified CD11b+F4/80+Gr-1neg naïve Mφ by stimulating them with anti-CD40 plus CpG-ODN in vitro (data not shown), which may reflect a necessity for exogenous (i.e. not Mφ-derived) factors to facilitate the process of this phenotypic change.
When analyzing TAM, we found that, at least in subcutaneous B16 tumours, CD11b+ Mφ comprise the overwhelming majority of total CD45+TIL, and that both CT and IT treatments enrich tumours for these CD11b+ cells. CD11b+ TAM in the tumour were present in two different subsets: F4/80+Gr-1bright and F4/80+Gr-1dim (data not shown). The pattern of F4/80 and Gr-1 co-expression by CD11b+ TAM strikingly resembled the phenotypic distribution of naïve splenic CD11b+Mφ, with a slight difference in the prevalence of these subsets. In both compartments (tumour and spleen), the therapies increased the percentage of the CD11b+F4/80+Gr-1bright subset and decreased the percentage of the CD11b+F4/80+Gr-1dim subset, without significant alteration of CD11b expression. Whether this effect was caused by the selective depletion of CD11b+F4/80+Gr-1dim Mφ, or the up-regulation of Gr-1 on this subset, remains unclear. Given the observation that the number of CD11b+F4/80+ Mφ increased in the spleens of CT+IT treated mice, it seems likely that Gr-1 is up-regulated on previously CD11b+F4/80+Gr-1dim Mφ by the CT + IT treatment.
CD11b+F4/80+ TAM from control-treated tumour-bearing mice expressed IL-4Rα, B7-H1 and high amounts of IL-10 and IL-4. Whereas tumour-associated CD11b+F4/80+Mφ were represented by two distinct subsets, only MHC class II, IL-10 and IL-4 expression showed a bimodal pattern of distribution, with the rest of the antigens examined (Fig 6) being expressed more uniformly. CT combined with IT caused a marked decrease in expression of suppressor Mφ-associated surface antigens (IL-4Rα and B7-H1) and cytokines (IL-4 and IL-10), and in parallel augmented the expression of CD40, CD80, CD86, MHC class II, IFN-γ, TNF-α and IL-12. Hence, after two consecutive cycles of CT + IT, CD11b+ Mφ were repolarized (at least partially) from the M2 into the M1 functional phenotype. These results also suggest that changes in expression of ‘signature’ phenotype antigens, such as CD11b, F4/80 and Gr-1, may have limited predictive value, as patterns of alterations of their expression did not prognostically correlate with changes of expression of other tested surface antigens or intracellular cytokines.
Whereas several mechanisms of synergy between CT and IT have been proposed, the mechanism of synergy between VCD-CT and IT with anti-CD40 and CpG-ODN remains uncharacterized and probably involves several components. CT-induced leucopenia is associated with the production of endogenous granulocyte colony-stimulating factor (G-CSF), GM-CSF and several other proinflammatory cytokines, including IL-12;36,48,49 these might facilitate the mobilization and migration of immature myeloid cells from the bone marrow, and alter the phenotype of the resident mature Mφ in different compartments and their priming to the subsequent CD40 and TLR9 stimulation via endogenous/autocrine IFN-γ production.8,41 Even Mφ with an ‘immature’ CD11dimF4/80dimGr-1bright phenotype that appeared to have recently migrated into the peritoneum following intraperitoneal CT treatment were found to be able to respond to CD40/TLR9 stimulation in vivo and to LPS in vitro.
Previously published reports suggest that CT may synergize with IT via the selective depletion of Gr-1+ suppressor cells.50 Whereas this mechanism may be involved in augmenting T-cell-mediated anti-tumour effects, it seems less important in facilitating T-cell-independent mechanisms of tumour destruction, such as those induced by anti-CD40 plus CpG-ODN IT.8 Indeed, we did not observe any augmented anti-tumour effects of anti-CD40 plus CpG-ODN IT in B16 melanoma-bearing mice that received concomitant treatment with anti-Gr-1 (RB6-8C5 clone) mAb to deplete Gr-1+ cells.8 It is likely that the cytotoxic mechanisms utilized by myeloid suppressor Gr-1+ cells to suppress T-cell immunity,51,52 such as NO and IFN-γ, can also be involved in tumour cell destruction, especially when tumours are weakly immunogenic and cytotoxic T-cell-mediated mechanisms are not elicited. In agreement with that, we previously showed that the anti-tumour effects of anti-CD40-activated Mφ do not require T cells and involve IFN-γ41 and NO.21 The results of this study further demonstrate that clinically relevant, multidrug CT, although immunosuppressive for T cells and NK cells, can synergize with Mφ-activating IT via the repolarization of TAM and the induction of Mφ-mediated anti-tumour effects.
Acknowledgments
This work was supported by The National Institutes of Health Grants CA87025 and CA032685 (to PMS), grants from the Midwest Athletes Against Childhood Cancer Fund (to PMS, ALR and INB), and the grant from the UW Cure Kids Cancer Coalition (INB and PMS). The authors thank Drs Jacquelyn A. Hank and Jacek Gan of The University of Wisconsin for helpful discussions. INB is a UICC fellow.
Disclosures
I.B. designed/conducted experiments, analyzed data and wrote the manuscript; P.S. and A.R. directed the project, interpreted results and helped to write and revise the manuscript; J.W. developed and provided a crucial mouse model; T.B., E.Y. and D.M. assisted with experiments.
References
- 1.Borella L, Green AA, Webster RG. Immunologic rebound after cessation of long-term chemotherapy in acute leukemia. Blood. 1972;40:42–51. [PubMed] [Google Scholar]
- 2.Lanfranchi A, Andolina M, Tettoni K, Porta F, Locatelli F, De Manzini A, Candotti F, Albertini A. Functional depletion of T cells by vincristine and methylprednisolone as an in vitro model for the prevention of graft versus host disease. Haematologica. 1992;77:11–5. [PubMed] [Google Scholar]
- 3.Multhoff G, Botzler C, Allenbacher A, Issels R. Effects of ifosfamide on immunocompetent effector cells. Cancer Immunol Immunother. 1996;42:251–4. doi: 10.1007/s002620050278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferraro C, Quemeneur L, Prigent AF, Taverne C, Revillard JP, Bonnefoy-Berard N. Anthracyclines trigger apoptosis of both G0-G1 and cycling peripheral blood lymphocytes and induce massive deletion of mature T and B cells. Cancer Res. 2000;60:1901–7. [PubMed] [Google Scholar]
- 5.Kimhi O, Drucker L, Neumann A, et al. Fluorouracil induces apoptosis and surface molecule modulation of peripheral blood leukocytes. Clin Lab Haematol. 2004;26:327–33. doi: 10.1111/j.1365-2257.2004.00629.x. [DOI] [PubMed] [Google Scholar]
- 6.Mozaffari F, Lindemalm C, Choudhury A, et al. NK-cell and T-cell functions in patients with breast cancer: effects of surgery and adjuvant chemo- and radiotherapy. Br J Cancer. 2007;97:105–11. doi: 10.1038/sj.bjc.6603840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuppner MC, Bleifuss E, Noessner E, Mocikat R, Hesler C, Mayerhofer C, Issels RD. Differential effects of ifosfamide on dendritic cell-mediated stimulation of T cell interleukin-2 production, natural killer cell cytotoxicity and interferon-gamma production. Clin Exp Immunol. 2008;153:429–38. doi: 10.1111/j.1365-2249.2008.03708.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buhtoiarov IN, Lum HD, Berke G, Sondel PM, Rakhmilevich AL. Synergistic activation of macrophages via CD40 and TLR9 results in T cell independent antitumor effects. J Immunol. 2006;176:309–18. doi: 10.4049/jimmunol.176.1.309. [DOI] [PubMed] [Google Scholar]
- 9.Wu QL, Buhtoiarov IN, Sondel PM, Rakhmilevich AL, Ranheim EA. Tumoricidal effects of activated macrophages in a mouse model of chronic lymphocytic leukemia. J Immunol. 2009;182:6771–8. doi: 10.4049/jimmunol.0801847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barrera CN, Mazzolli AB, Bustuoabad OD, Andreetta AM, Pasqualini CD. Macrophages and tumor growth. II. Cell kinetics at the site of allogeneic tumor growth. Cancer Invest. 1985;3:7–13. doi: 10.3109/07357908509040603. [DOI] [PubMed] [Google Scholar]
- 11.Allavena P, Sica A, Garlanda C, Mantovani A. The Yin-Yang of tumor-associated macrophages in neoplastic progression and immune surveillance. Immunol Rev. 2008;222:155–61. doi: 10.1111/j.1600-065X.2008.00607.x. [DOI] [PubMed] [Google Scholar]
- 12.De Campos ES, Menasce LP, Radford J, Harris M, Thatcher N. Metastatic carcinoma of uncertain primary site: a retrospective review of 57 patients treated with vincristine, doxorubicin, cyclophosphamide (VAC) or VAC alternating with cisplatin and etoposide (VAC/PE) Cancer. 1994;73:470–5. doi: 10.1002/1097-0142(19940115)73:2<470::aid-cncr2820730236>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 13.Arndt CA, Nascimento AG, Schroeder G, et al. Treatment of intermediate risk rhabdomyosarcoma and undifferentiated sarcoma with alternating cycles of vincristine/doxorubicin/cyclophosphamide and etoposide/ifosfamide. Eur J Cancer. 1998;34:1224–9. doi: 10.1016/s0959-8049(98)00080-x. [DOI] [PubMed] [Google Scholar]
- 14.Kushner BH, Meyers PA. How effective is dose-intensive/myeloablative therapy against Ewing’s sarcoma/primitive neuroectodermal tumor metastatic to bone or bone marrow? The Memorial Sloan-Kettering experience and a literature review. J Clin Oncol. 2001;19:870–80. doi: 10.1200/JCO.2001.19.3.870. [DOI] [PubMed] [Google Scholar]
- 15.Solomayer EF, Feuerer M, Bai L, et al. Influence of adjuvant hormone therapy and chemotherapy on the immune system analyzed in the bone marrow of patients with breast cancer. Clin Cancer Res. 2003;9:174–80. [PubMed] [Google Scholar]
- 16.Norris MD, Burkhart CA, Marshall GM, Weiss WA, Haber M. Expression of N-myc and MRP genes and their relationship to N-myc gene dosage and tumor formation in a murine neuroblastoma model. Med Pediatr Oncol. 2000;35:585–9. doi: 10.1002/1096-911x(20001201)35:6<585::aid-mpo20>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 17.Salcedo R, Stauffer JK, Lincoln E, et al. IL-27 mediates complete regression of orthotopic primary and metastatic murine neuroblastoma tumors: role for CD8+ T cells. J Immunol. 2004;173:7170–82. doi: 10.4049/jimmunol.173.12.7170. [DOI] [PubMed] [Google Scholar]
- 18.Mohammad RM, Wall NR, Dutcher JA, Al-Katib AM. The addition of bryostatin 1 to cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) chemotherapy improves response in a CHOP-resistant human diffuse large cell lymphoma xenograft model. Clin Cancer Res. 2000;6:4950–6. [PubMed] [Google Scholar]
- 19.Neal ZC, Yang JC, Rakhmilevich AL, et al. Enhanced activity of hu14.18-IL2 immunocytokine against murine NXS2 neuroblastoma when combined with interleukin 2 therapy. Clin Cancer Res. 2004;10:4839–47. doi: 10.1158/1078-0432.CCR-03-0799. [DOI] [PubMed] [Google Scholar]
- 20.Buhtoiarov IN, Rakhmilevich AL, Lanier LL, Ranheim EA, Sondel PM. Naive mouse macrophages become activated following recognition of L5178Y lymphoma cells via concurrent ligation of CD40, NKG2D, and CD18 molecules. J Immunol. 2009;182:1940–53. doi: 10.4049/jimmunol.0800443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lum HD, Buhtoiarov IN, Schmidt BE, Berke G, Paulnock DM, Sondel PM, Rakhmilevich AL. Tumoristatic effects of anti-CD40 mAb-activated macrophages involve nitric oxide and tumour necrosis factor-alpha. Immunology. 2006;118:261–70. doi: 10.1111/j.1365-2567.2006.02366.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wheeler HR, Geczy CL. Induction of macrophage procoagulant expression by cisplatin, daunorubicin and doxorubicin. Int J Cancer. 1990;46:626–32. doi: 10.1002/ijc.2910460413. [DOI] [PubMed] [Google Scholar]
- 23.Evans R, Lawler EM. Macrophage content and immunogenicity of C57BL/6J and BALB/cByJ methylcholanthrene-induced sarcomas. Int J Cancer. 1980;26:831–5. doi: 10.1002/ijc.2910260618. [DOI] [PubMed] [Google Scholar]
- 24.Kusmartsev S, Gabrilovich DI. STAT1 signaling regulates tumor-associated macrophage-mediated T cell deletion. J Immunol. 2005;174:4880–91. doi: 10.4049/jimmunol.174.8.4880. [DOI] [PubMed] [Google Scholar]
- 25.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–74. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol. 2009;182:4499–506. doi: 10.4049/jimmunol.0802740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buzaid AC, Grimm EA, Ali-Osman F, et al. Mechanism of the anti-tumour effect of biochemotherapy in melanoma: preliminary results. Melanoma Res. 1994;4:327–30. doi: 10.1097/00008390-199410000-00010. [DOI] [PubMed] [Google Scholar]
- 28.Soulié P, Ruffié P, Trandafir L, et al. Combined systemic chemoimmunotherapy in advanced diffuse malignant mesothelioma. Report of a phase I-II study of weekly cisplatin/interferon alfa-2a. J Clin Oncol. 1996;14:878–85. doi: 10.1200/JCO.1996.14.3.878. [DOI] [PubMed] [Google Scholar]
- 29.Grimm EA, Smid CM, Lee JJ, Tseng CH, Eton O, Buzaid AC. Unexpected cytokines in serum of malignant melanoma patients during sequential biochemotherapy. Clin Cancer Res. 2000;6:3895–903. [PubMed] [Google Scholar]
- 30.Salem ML, Díaz-Montero CM, Al-Khami AA, El-Naggar SA, Naga O, Montero AJ, Khafagy A, Cole DJ. Recovery from cyclophosphamide-induced lymphopenia results in expansion of immature dendritic cells which can mediate enhanced prime-boost vaccination antitumor responses in vivo when stimulated with the TLR3 agonist poly(I:C) J Immunol. 2009;182:2030–40. doi: 10.4049/jimmunol.0801829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nowak AK, Lake RA, Marzo AL, Scott B, Heath WR, Collins EJ, Frelinger JA, Robinson BW. Induction of tumor cell apoptosis in vivo increases tumor antigen cross-presentation, cross-priming rather than cross-tolerizing host tumor-specific CD8 T cells. J Immunol. 2003;170:4905–13. doi: 10.4049/jimmunol.170.10.4905. [DOI] [PubMed] [Google Scholar]
- 32.Nowak AK, Robinson BW, Lake RA. Synergy between chemotherapy and immunotherapy in the treatment of established murine solid tumors. Cancer Res. 2003;63:4490–6. [PubMed] [Google Scholar]
- 33.Lundy J, Lovett EJ, Hinchliffe D, Schor M. Chemoimmunotherapy of a murine fibrosarcoma: critical factors for success of combined modality therapy. J Surg Oncol. 1977;9:339–45. doi: 10.1002/jso.2930090404. [DOI] [PubMed] [Google Scholar]
- 34.Yang S, Haluska FG. Treatment of melanoma with 5-fluorouracil or dacarbazine in vitro sensitizes cells to antigen-specific CTL lysis through perforin/granzyme- and Fas-mediated pathways. J Immunol. 2004;172:4599–608. doi: 10.4049/jimmunol.172.7.4599. [DOI] [PubMed] [Google Scholar]
- 35.Polak L, Turk JL. Reversal of immunological tolerance by cyclophosphamide through inhibition of suppressor cell activity. Nature. 1974;249:654–6. doi: 10.1038/249654a0. [DOI] [PubMed] [Google Scholar]
- 36.Bracci L, Moschella F, Sestili P, et al. Cyclophosphamide enhances the antitumor efficacy of adoptively transferred immune cells through the induction of cytokine expression, B-cell and T-cell homeostatic proliferation, and specific tumor infiltration. Clin Cancer Res. 2007;13:644–53. doi: 10.1158/1078-0432.CCR-06-1209. [DOI] [PubMed] [Google Scholar]
- 37.Dudley ME, Wunderlich JR, Robbins PF, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–4. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol. 2003;4:1191–8. doi: 10.1038/ni1009. [DOI] [PubMed] [Google Scholar]
- 39.Ibe S, Qin Z, Schüler T, Preiss S, Blankenstein T. Tumor rejection by disturbing tumor stroma cell interactions. J Exp Med. 2001;194:1549–59. doi: 10.1084/jem.194.11.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buhtoiarov IN, Sondel PM, Eickhoff JC, Rakhmilevich AL. Macrophages are essential for antitumour effects against weakly immunogenic murine tumours induced by class B CpG-oligodeoxynucleotides. Immunology. 2007;120:412–23. doi: 10.1111/j.1365-2567.2006.02517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Buhtoiarov IN, Lum H, Berke G, Paulnock DM, Sondel PM, Rakhmilevich AL. CD40 ligation activates murine macrophages via an IFN-gamma-dependent mechanism resulting in tumor cell destruction in vitro. J Immunol. 2005;174:6013–22. doi: 10.4049/jimmunol.174.10.6013. [DOI] [PubMed] [Google Scholar]
- 42.Ezekowitz R, Alan B, Gordon S. Down-regulation of mannosyl receptor-mediated endocytosis and antigen F4/80 in Bacillus Calmette-Guerin-activated mouse macrophages. J Exp Med. 1982;155:1623–37. doi: 10.1084/jem.155.6.1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Benedict CA, De Trez C, Schneider K, Ha S, Patterson G, Ware CF. Specific Remodeling of Splenic Architecture by Cytomegalovirus. PLoS Pathog. 2006;2:0164–73. doi: 10.1371/journal.ppat.0020016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Menson EN, Wilson RA. Lung-phase immunity to Schistosorna rnansoni: definition of alveolar macrophage phenotypes after vaccination and challenge of mice. Parasite Immunol. 1990;12:353–66. doi: 10.1111/j.1365-3024.1990.tb00974.x. [DOI] [PubMed] [Google Scholar]
- 45.Fleming TJ, Fleming ML, Malek TR. Selective expression of Ly-6G on myeloid lineage cells in mouse bone marrow. RB6-8C5 mAb to granulocyte-differentiation antigen (Gr-1) detects members of the Ly-6 family. J Immunol. 1993;151:2399–408. [PubMed] [Google Scholar]
- 46.Nagendra S, Schlueter AJ. Absence of cross-reactivity between murine Ly-6C and Ly-6G. Cytometry. 2004;58:195–200. doi: 10.1002/cyto.a.20007. [DOI] [PubMed] [Google Scholar]
- 47.Ferret-Bernard S, Saï P, Bach JM. In vitro induction of inhibitory macrophage differentiation by granulocyte-macrophage colony-stimulating factor, stem cell factor and interferon-gamma from lineage phenotypes-negative c-kit-positive murine hematopoietic progenitor cells. Immunol Lett. 2004;91:221–7. doi: 10.1016/j.imlet.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 48.Bruserud O, Foss B, Petersen H. Hematopoietic growth factors in patients receiving intensive chemotherapy for malignant disorders: studies of granulocyte-colony stimulating factor (G-CSF), granulocyte-macrophage colony stimulating factor (GM-CSF), interleukin-3 (IL-3) and Flt-3 ligand (Flt3L) Eur Cytokine Netw. 2001;12:231–8. [PubMed] [Google Scholar]
- 49.Banning U, Mauz-Körholz C, Muhammad Q, et al. Endogenous pro-inflammatory cytokines in children and adolescents during chemotherapy-induced neutropenia. Pediatr Hematol Oncol. 2002;19:561–8. doi: 10.1080/08880010290097413. [DOI] [PubMed] [Google Scholar]
- 50.Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res. 2005;11:6713–21. doi: 10.1158/1078-0432.CCR-05-0883. [DOI] [PubMed] [Google Scholar]
- 51.Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, De Baetselier P, Van Ginderachter JA. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood. 2008;111:4233–44. doi: 10.1182/blood-2007-07-099226. [DOI] [PubMed] [Google Scholar]
- 52.Watanabe S, Deguchi K, Zheng R, Tamai H, Wang LX, Cohen PA, Shu S. Tumor-induced CD11b+Gr-1+ myeloid cells suppress T cell sensitization in tumor-draining lymph nodes. J Immunol. 2008;181:3291–300. doi: 10.4049/jimmunol.181.5.3291. [DOI] [PubMed] [Google Scholar]