

Abstract
Removal of apoptotic cells from inflammatory sites by macrophages is an important step in the resolution of inflammation. However, the effect of inflammatory modulators on phagocytic clearance of apoptotic cells remains to be clarified. In this paper, we demonstrate that lipopolysaccharide (LPS), a potent inflammatory agent, inhibits the phagocytosis of apoptotic neutrophils by mouse peritoneal macrophages. This inhibition can be attributed to both LPS-mediated induction of tumour necrosis factor (TNF-α) and suppression of growth arrest-specific gene 6 (Gas6) in macrophages. We found that LPS-induced TNF-α production inhibited phagocytic ability of macrophages in an autocrine manner. In contrast, Gas6 expression in macrophages was blocked by LPS, which also contributes to the inhibition of macrophage phagocytosis by LPS. Our data suggest that phagocytic clearance of apoptotic neutrophils by macrophages can be regulated by local pro- and anti-inflammatory factors in two opposite states.
Keywords: inflammation, lipopolysaccharide, macrophage, phagocytosis
Introduction
Cell apoptosis is a mechanism of cell deletion that allows maintenance of tissue homeostasis both under normal conditions and during pathophysiological processes.1 Removal of apoptotic cells by phagocytes is critical in preventing exposure of surrounding tissues to cytotoxic, immunogenic or inflammatory cellular contents.2 The phagocytic clearance of apoptotic cells is an evolutionarily conserved process. The unique signaling pathways and engulfment mechanisms involved in it are different from those mediated by the immunoglobulin G(IgG)/fragment crystallizable receptor and the C3 opsonization/C3 receptor.3 During normal cell differentiation, the rate of apoptosis is sufficiently slow that neighbouring non-professional phagocytes, such as fibroblasts and epithelial cells, can efficiently engulf apoptotic cells. However, when apoptosis becomes large scale during infections and inflammatory responses, professional phagocytes such as macrophages are attracted to the inflammatory site and facilitate the clearance of massive apoptotic cells.
Inflammation involves the infiltration of circulating immune cells, such as neutrophils and mcrophages, into infected or damaged sites to neutralize and eliminate potentially injurious stimuli. The production of inflammatory cytokines by the infiltrated immune cells is a normal physiological defence response against allo- and autopathogens.4 However, this response must be tightly regulated because exaggeration and prolongation of inflammation may lead to chronic tissue damage, such as that occurring in rheumatoid arthritis, atherosclerosis and chronic obstructive pulmonary disease.5 It has been indicated that defective resolution of inflammation is a major contributory factor for the pathogenesis of chronic inflammation.6,7
Efficient resolution of inflammation requires the shutting down of inflammatory factor production. This, in turn, depends on apoptosis of inflammatory cells and their clearance by phagocytes.8 Previous studies have revealed that inflammatory mediators influence the apoptosis of inflammatory cells.9,10 However, the literature concerning the effect of inflammatory modulators on phagocytic clearance of apoptotic cells is limited and contains discrepancies. For example, TNF-α, a key pro-inflammatory factor that is up-regulated at inflammatory sites, has been reported previously to enhance the uptake of apoptotic cells by immature monocyte-derived macrophages.11 Another study demonstrated that TNF-α inhibits the phagocytosis of apoptotic cells by mature macrophages.12 A recent study indicated that the uptake of apoptotic neutrophils by human monocyte-derived macrophages was negatively regulated by TNF-α, which was opposite to the effect of the anti-inflammatory factor interleukin (IL)-10.13
Growth arrest-specific gene 6 (Gas6) is an anti-inflammatory factor.14,15 Gas6 and its receptors – Tyro3, Axl and Mer (TAM) receptor tyrosine kinases– are broadly expressed in various types of phagocyte. The activation of TAM receptors by Gas6 inhibits inflammation responses and promotes the phagocytosis of apoptotic cells by phagocytes.16 In the present study, we found that LPS specifically inhibited mouse macrophage uptake of apoptotic neutrophils through suppression of Gas6 and induction of TNF-α in an autocrine manner. The findings provide novel insights into the effect of inflammatory modulators on phagocytic clearance of apoptotic cells by macrophages.
Materials and methods
Animals
C57BL/6J mice were purchased from the Laboratory Animal Center of Peking Union Medical College (Beijing, China). Toll-like receptor 4 (TLR4) mutant C57BL/10ScN mice (Cat. 003752) were purchased from Jackson Laboratories (Bar Harbor, ME). The animals were housed under specific pathogen-free conditions with a 12-hr light/dark cycle and had free access to food and water. The mice were maintained and treated in accordance with the guidelines for the care and use of laboratory animals established by the Chinese Council on Animal Care. Mice 8–10 weeks old were used in this study.
Reagents
Ultrapure LPS (Escherichia coli 0111:B4) was obtained from InvivoGen (San Diego, CA), and no detectable TNF was produced in TLR4-null (TLR4−/−) macrophages in response to this LPS. TNF-α and neutralizing antibodies against TNF-α were obtained from PeproTech Inc. (Rocky Hill, NJ). Gas6 and neutralizing antibodies against Gas6 were obtained from R & D Systems (Minneapolis, MN).
Isolation of mouse peritoneal macrophages
Peritoneal macrophages were collected from peritoneal fluid as previously described.17 Briefly, mice were anaesthetized with CO2 and then killed by cervical dislocation. The peritoneal cavities were lavaged with 5 ml of cold phosphate-buffered saline (PBS) to collect peritoneal cells. The cells were seeded at 4 × 105 cells/well into a 24-well plate with RPMI-1640 medium (Gibco-BRL, Grand Island, NY) containing 10% fetal calf serum (FCS; Gibco-BRL). After 2 hr in a humidified atmosphere containing 5% CO2 at 37°, non-adherent cells were removed by washing with PBS, and the adherent macrophages (approximately 1 × 105 cells/well) were cultured overnight. The purity and the viability of macrophages were estimated by immunofluorescence staining for F4/80 (a marker of macrophages) and flow cytometery.
Immunofluorescence staining
Macrophages cultured on Lab-Tek chamber slides (Nunc, Naperville, IL) were fixed with pre-cold methanol at −20° for 2 min. The cells were blocked by preincubation with 10% normal goat serum in PBS at room temperature for 30 min, and then incubated with rabbit anti-mouse F4/80 (Abcam, Cambridge, MA) at 37° in a moist chamber for 1 hr. After three washes with PBS, the cells were incubated with the fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit IgG (Zhongshan, Beijing, China) for 30 min. The cells were observed under a fluorescence microscope (IX-71; Olympus, Tokyo, Japan).
Isolation and apoptosis of neutrophils
Mouse neutrophils were isolated from peritoneal fluid as described previously.18 Briefly, the peritoneal cavities were lavaged with 5 ml of cold 1 × PBS to collect peritoneal cells. The peritoneal exudate cells were re-suspended in 1 ml of PBS and mixed with 9 ml of Percoll gradient solution (Sigma, St Louis, MO) at room temperature in a 10-ml ultracentrifuge tube. After centrifugation at 60 000 g for 20 min, the neutrophils were collected. The neutrophils were cultured at 5 × 106 cells/ml in RPMI-1640 medium without serum at 37° in a humidified atmosphere containing 5% CO2 for 24 hr to induce spontaneous apoptosis.19 The purity and apoptosis of neutrophils were assessed by Wright’s Giemsa staining. The rate of apoptosis and secondary necrosis was analysed by flow cytometry after double staining with propidium iodide (Beijing 4A Biotech Co., Ltd, Beijing, China) and FITC-conjugated annexin V (AnxV). Only neutrophils with > 90% apoptosis and < 5% necrosis were labelled with FITC (Sigma), according to the manufacturer’s instructions, and were used as target cells in the phagocytosis assay.
Phagocytosis assays
Macrophages were co-cultured with the following targets: FITC-labelled apoptotic neutrophils at a phagocyte-to-target ratio of 1 : 10; FITC-labelled inactivated yeasts at a ratio of 1 : 30; or 2 μl of FITC-conjugated latex beads (Polysciences Inc., Warrington, PA). At 30 min after co-culture, the cells were extensively washed three times with PBS. The macrophages that had engulfed targets were examined by fluorescence microscopy and flow cytometry. Controls were run by inhibiting actin with 50 μg of cytochalasin B (Sigma). Each condition was tested in duplicate and the experiments were repeated at least three times.
Flow cytometry
Macrophages and neutrophils were washed with cold PBS, and stained with phycoerythrin-conjugated antibodies against F4/80 (BioLegend, San Diego, CA), FITC-conjugated AnxV or propidium iodide following the manufacturer’s instructions. After washes, cells were analysed using a BD FACSSanto flow cytometer (BD Biosciences, San Jose, CA).
Quantitative real-time polymerase chain reaction (Q-PCR)
Total RNA was extracted from macrophages using Trizol reagent (Invitrogen Life Technologies, Carlsbad, CA) following the manufacturer’s instructions. The RNA was reverse-transcribed into cDNA using Moloney murine leukemia virus (MMLV) reverse transcriptase (Promega, Madison, WI). Q-PCRs were performed using the Power SYBR Green PCR Master Mix kit (Applied Biosystems, Warrington, UK) in an ABI PRISM 7300 real-time cycler (Applied Biosystems) according to the supplier’s protocol. The mRNA levels of target genes were normalized to that of β-actin. The primer sequences for TNF-α were: (forward) 5′-CAT CTT CTC AAA ATT CGA GTG ACA A-3′ and (reverse) 5′-TGG GAG TAG ACA AGG TAC AAC CC-3′; those for Gas6 were: (forward) 5′-CGA GTC TTC TCA CAC TGC TGT T-3′ and (reverse) 5′-GCA CTC TTG ATA TCG TGG ATA GAA ATA C-3′; and those for β-actin were: (forward) 5′-GAA ATC GTG CGT GAC ATC AAA G-3′ and (reverse) 5′-TGT AGT TTC ATG GAT GCC ACA G-3′.
Data analysis and statistics
Each experiment was repeated at least three times. Data are presented as mean ± standard error of the mean (SEM). Differences were compared by two-way analysis of variance (ANOVA) and Student’s t-test. The calculations were performed with the statistical software spss version 11.0 (SPSS Inc., Chicago, IL). Statistical significance was defined as P<0·05.
Results
LPS inhibits phagocytosis of apoptotic neutrophils by macrophages
Primary mouse peritoneal macrophages and neutrophils were used for phagocytosis assays. Macrophages were identified by immunofluorescence staining for F4/80 (Fig. 1a). The viability and purity of macrophages were quantitatively analysed by flow cytometry after double staining with phycoerythrin (PE)-conjugated antibodies against F4/80 and FITC-conjugated AnxV. The cell populations were not gated for the analysis. The purity of living macrophages was > 95% (Fig. 1b, left; the isotype control is shown in Fig. 1b, right). Mouse peritoneal neutrophils were identified based on characteristic multilobed nuclei after Wright’s Giemsa staining (Fig. 1c, left). The neutrophils with a purity of > 90% were cultured in serum-free medium for 24 hr to attain spontaneous apoptosis. The apoptotic neutrophils were assessed using Wright’s Geimsa staining (Fig. 1c, right), and quantitatively analysed by flow cytometry after double staining with propidium iodide (PI) and FITC-conjugated AnxV. The neutrophils exhibited > 90% AnxV+/PI− (apoptotic) cells with less than 5% AnxV+/PI+ (secondarily necrotic) cells (Fig. 1d, left). Neutrophils without induction of apoptosis were used as a control (Fig. 1d, right).
Figure 1.
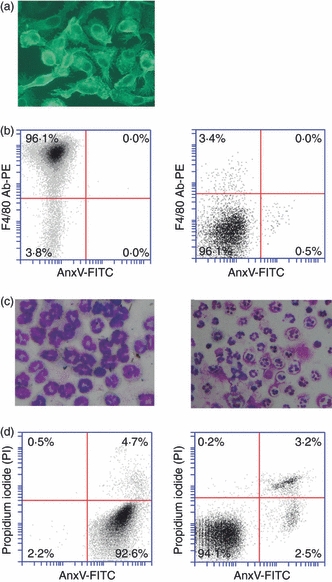
Identification of peritoneal macrophages and neutrophils. (a) The purity of macrophages was determined by immunofluorescence staining using antibodies against F4/80. (b) The viability of macrophages was assessed by flow cytometry after double staining with fluorescein isothiocyanate (FITC)-conjugated annexin V (AnxV-FITC) and phycoerythrin (PE)-conjugated antibodies against F4/80 (F4/80 Ab-PE). The living macrophages were determined to be an F4/80+/AnxV− population (left panel). An isotype control was obtained by labelling cells with FITC-conjugated rat immunoglobulin G2a (IgG2a) (right panel). (c) Wright’s Giemsa staining. Neutrophils were characterized by their multilobed nuclei (left panel), and apoptotic cells by their apoptotic bodies (right panel). (d) Double staining with AnxV-FITC and propidium iodide (PI) was used to determine the proportion of apoptotic (AnxV+/PI−) and secondarily necrotic (AnxV+/PI+) neutrophils after apoptosis induction (left panel). Uninduced neutrophils were used as controls (right panel).
For phagocytosis assays, FITC-labelled apoptotic neutrophils and macrophages tagged with PE-conjugated antibodies against F4/80 were co-cultured. To assess the effect of LPS on macrophage uptake of apoptotic cells, macrophages that had engulfed apoptotic cells were analysed by fluorescence microscopy (Fig. 2a), with confirmation provided by flow cytometry (Fig. 2b). LPS inhibits the phagocytic ability of macrophages in a time-dependent manner (Fig. 2c). A significant decrease in the phagocytic rate was initially observed at 6 hr after treatment with 10 ng/ml LPS. At 8 and 16 hr, the phagocytic rate was decreased two- and threefold, respectively. LPS inhibition of macrophage phagocytosis was also dose-dependent. At 16 hr after treatment, 1 ng/ml LPS significantly inhibited phagocytosis, and remarkable inhibitory effects were observed as the LPS concentration increased (Fig. 2d).
Figure 2.
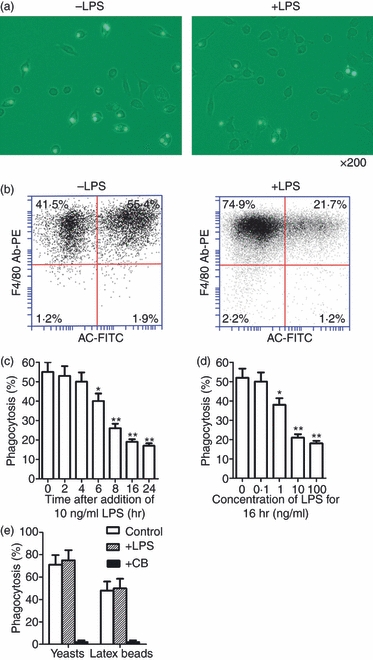
Effects of lipopolysaccharide (LPS) on phagocytosis of apoptotic neutrophils by mouse peritoneal macrophages. (a) Representative images of macrophages that have engulfed fluorescein isothiocyanate (FITC)-labelled apoptotic neutrophils. Magnification × 200. Macrophages were treated with 10 ng/ml LPS for 16 hr and assayed for phagocytic ability. (b) The macrophage phagocytic rate was quantitatively analysed by flow cytometry phagocytic macrophages were double-positive cells for PE (labeled F4/80) and FITC (labeled apoptotic cell, AC). (c, d) Time-dependent (c) and dose-dependent (d) LPS inhibition of phagocytosis by macrophages based on analysis of flow cytometry. (e) Phagocytosis of yeasts and latex beads by macrophages. Macrophages were treated with 10 ng/ml LPS for 16 hr or with 5 μg/ml cytochalasin B (CB) for 1 hr, and then co-cultured with latex beads or FITC-labelled yeasts for 30 min. Data represent the mean ± standard error of the mean for three separate experiments. *P<0·05; **P<0·01.
To determine whether LPS inhibition of phagocytosis was specifically restricted to the engulfment of apoptotic cells, the effect of LPS on the uptake of inactivated yeasts or carboxylate-coated latex beads by macrophages was examined. LPS did not affect macrophage uptake of yeasts or latex beads at 16 hr after treatment (Fig. 2e). In the control, macrophage engulfment of yeasts and latex beads was abolished by inhibiting actin with cytochalasin B.
Exogenous TNF-α inhibits phagocytosis by macrophages
It is known that TNF-α regulates phagocytic clearance of apoptotic cells by macrophages.11,12 We confirmed that exogenous TNF-α inhibited macrophage uptake of apoptotic neutrophils in a dose-dependent manner. Significant inhibition was observed following treatment with 10 ng/ml TNF-α for 4 hr (Fig. 3a). Treatment with 10 ng/ml TNF-α resulted in time-dependent inhibition of phagocytosis. Significant inhibition was observed at 1 hr after addition of TNF-α (Fig. 3b). Notably, the inhibitory effect of TNF-α on macrophage phagocytosis was significantly weaker than that of LPS at 16 hr after treatment (Fig. 3c).
Figure 3.
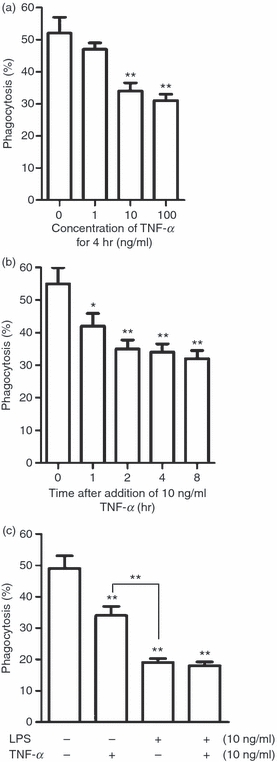
Effect of exogenous tumour necrosis factor (TNF)-α on phagocytosis of apoptotic cells by macrophages. (a, b) Dose-dependent (a) and time-dependent (b) effects of TNF-α on phagocytosis of apoptotic neutrophils by macrophages. Macrophages were assayed for phagocytosis. (c) Comparison between lipopolysaccharide (LPS) and TNF-α inhibition of phagocytosis. Macrophages were treated either with LPS or TNF-α alone or with a combination of both LPS and TNF-α. Note that LPS inhibits phagocytosis more efficiently than TNF-α. Data represent the mean ± standard error of the mean for three separate experiments. *P<0·05; **P<0·01.
LPS-induced TNF-α production inhibits phagocytosis in an autocrine manner
Given that LPS is a powerful inducer of TNF-α production by macrophages, we examined the contribution of LPS-induced TNF-α production to the LPS inhibition of phagocytosis. TNF-α mRNA in macrophages increased rapidly after stimulation with LPS and achieved an 860-fold increase at 2 hr (Fig. 4a). By 16 hr, mRNA levels had declined back to the base level. The TNF-α concentration in the medium peaked at 6 and 8 hr, and then declined dramatically at 16 hr after LPS stimulation (Fig. 4b). The timing of the increase in the TNF-α concentration in the medium corresponded to that of the LPS inhibition of phagocytosis. In particular, the presence of neutralizing antibodies against TNF-α (anti-TNF-α) significantly reduced LPS inhibition of phagocytosis (Fig. 4c). Notably, anti-TNF-α did not completely reverse this inhibition. However, anti-TNF-α fully reversed the exogenous TNF-α-mediated inhibition of phagocytosis (Fig. 4d). In control assays, anti-TNF-α alone did not affect macrophage phagocytosis. These results suggest that the LPS inhibitory effect on the phagocytosis of apoptotic cells by macrophages is partially attributable to LPS-induced TNF-α production, and other mechanisms must be involved in the LPS inhibition of phagocytosis.
Figure 4.
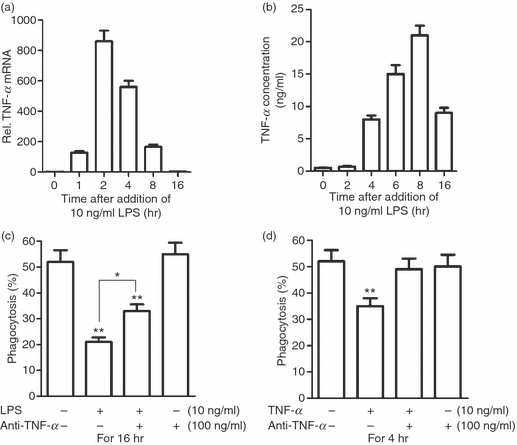
Lipopolysaccharide (LPS)-induced tumour necrosis factor (TNF)-α inhibits phagocytosis in an autocrine manner. (a) Relative TNF-α mRNA level in macrophages after LPS treatment was assessed by quantitative polymerase chain reaction (Q-PCR). (b) The TNF-α concentration in the medium was measured by enzyme-linked immunosorbent assay (ELISA). (c) Effect of neutralizing antibodies against TNF-α (anti-TNF-α) on LPS inhibition of phagocytosis. Note that anti-TNF-α partially rescued LPS inhibition of phagocytosis. (d) Effect of anti-TNF-α on exogenous TNF-α inhibition of phagocytosis. Note that anti-TNF-α reversed TNF-α inhibition of phagocytosis to control levels. Data represent the mean ± standard error of the mean for three experiments. *P<0·05; **P<0·01.
Down-regulation of Gas6 contributes to LPS inhibition of macrophage phagocytosis
To investigate further the mechanisms underlying LPS-inhibited phagocytosis, we analysed the expression of genes that are known to be involved in the phagocytosis of apoptotic cells in macrophages after treatment with LPS. Notably, Gas6 expression in macrophages could be abolished by LPS. LPS inhibited Gas6 expression in a dose- and time-dependent manner. The Gas6 mRNA level was markedly decreased in macrophages treated with 1 ng/ml LPS for 16 hr, and was abolished by 10 ng/ml LPS (Fig. 5a). A striking down-regulation of Gas6 mRNA was initially observed at 4 hr after treatment with 10 ng/ml LPS, and was abolished at 16 hr (Fig. 5b). An enzyme-linked immunosorbent assay (ELISA) showed that the Gas6 concentration in the medium was significantly decreased at 8 hr after LPS treatment, and declined to a very low level by 16 hr (Fig. 5c).
Figure 5.
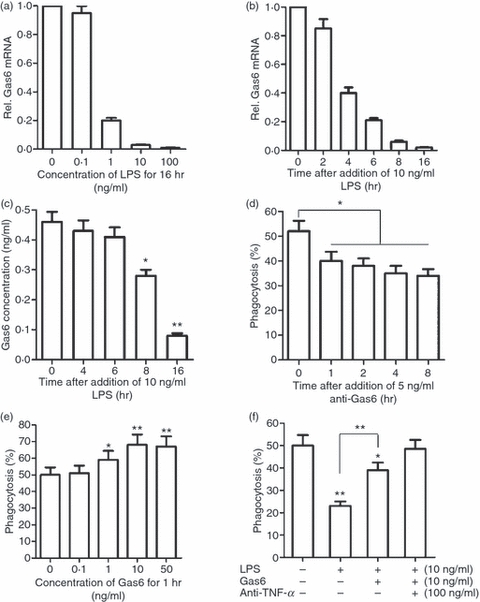
Down-regulation of growth arrest-specific gene 6 (Gas6) expression in macrophages contributes to lipopolysaccharide (LPS) inhibition of phagocytosis. (a) Dose-dependent suppression of Gas6 expression in macrophages by LPS. Gas6 mRNA in macrophages was analysed by quantitative polymerase chain reaction (Q-PCR). (b) Time-dependent suppression of Gas6 mRNA in macrophages by LPS. (c) The Gas6 concentration in the medium was measured by enzyme-linked immunosorbent assay (ELISA). (d) Effect of monoclonal antibodies against Gas6 (anti-Gas6) on macrophage phagocytosis of apoptotic neutrophils. (e) Effect of exogenous Gas6 on macrophage phagocytosis. (f) Effect of Gas6 on LPS inhibition of phagocytosis. Macrophages were treated with LPS alone for 16 hr, or with LPS for 15 hr followed by Gas6 for 1 hr, or with LPS and anti-TNF-α for 15 hr followed by Gas6 for 1 hr. Data represent the mean ± standard error of the mean for three separate experiments. *P<0·05; **P<0·01.
Given that Gas6 specifically promotes phagocytosis of apoptotic cells by macrophages,20 we speculated that LPS inhibition of phagocytosis might be also attributable to the down-regulation of Gas6. We found that neutralizing Gas6 activity with 5 ng/ml anti-Gas6 antibodies, following the manufacturer’s instructions, significantly inhibited macrophage phagocytosis (Fig. 5d), suggesting that Gas6 positively regulated macrophage phagocytosis in an autocrine manner. Exogenous Gas6 increased macrophage phagocytosis in a dose-dependent manner (Fig. 5e). Moreover, exogenous Gas6 significantly reduced the LPS inhibition of phagocytosis (Fig. 5f). In particular, when Gas6 and anti-TNF-α were given to the macrophages simultaneously, they restored LPS-inhibited phagocytosis to a normal level (Fig. 5f).
Down-regulation of Gas6 by LPS is TLR4-independent
Whether TLR4 signalling is necessary for LPS-inhibited Gas6 expression, since it is by activating TLR4 that LPS induces TNF-α production. To address this question, we analysed the effects of LPS on TLR4-deficient (TLR4−/−) macrophages. Gas6 expression in TLR4−/− macrophages was also abolished by LPS, and displayed a similar pattern to that observed in wild-type (WT) macrophages (Fig. 6a). In contrast, LPS-induced TNF-α expression was blocked in TLR4−/− macrophages (Fig. 6b). The concentrations of Gas6 and TNF-α in the medium corresponded to their mRNA levels (Fig. 6c).
Figure 6.
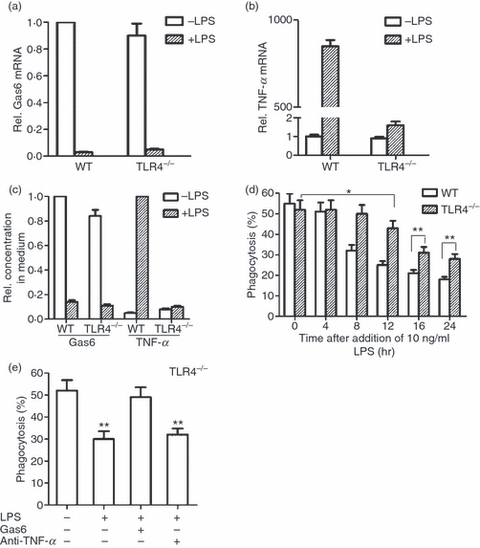
Lipopolysaccharide (LPS) inhibition of phagocytosis by Toll-like receptor 4 (TLR4)−/− macrophages. (a) Wild-type (WT) and TLR4−/− macrophages were treated with 10 ng/ml LPS for 16 hr. Growth arrest-specific gene 6 (Gas6) mRNA was analysed by quantitative polymerase chain reaction (Q-PCR). (b) Macrophages were treated with 10 ng/ml LPS for 2 hr, and tumour necrosis factor (TNF)-α mRNA was analysed by Q-PCR. (c) Relative concentrations of Gas6 and TNF-α in the medium. Macrophages were treated with 10 ng/ml LPS for 16 hr to measure Gas6, or for 8 hr to measure TNF-α concentrations in the medium by enzyme-linked immunosorbent assay (ELISA). (d) Comparison of the LPS inhibition of phagocytosis by WT and TLR4−/− macrophages. Note that LPS inhibition of phagocytosis by TLR4−/− macrophage was significantly lower and delayed compared with that by WT macrophages. (e) Effects of anti-TNF-α and exogenous Gas6 on LPS inhibition of TLR4−/− macrophage phagocytosis. TLR4−/− macrophages were treated with 10 ng/ml LPS alone, or LPS and 100 ng/ml anti-TNF-α for 16 hr, or LPS for 15 hr followed by 10 ng/ml exogenous Gas6 for 1 hr. Note that Gas6, but not anti-TNF-α, reversed LPS inhibition of phagocytosis by TLR4−/− macrophages. Data represent the mean ± standard error of the mean for three separate experiments. *P<0·05; **P<0·01.
Next, we analysed the phagocytosis of apoptotic cells by TLR4−/− macrophages. In the absence of LPS, the phagocytic ability of TLR4−/− macrophages was similar to that of WT controls (Fig. 6d). Although LPS significantly inhibited phagocytosis of apoptotic cells by TLR4−/− macrophages, there was a latency in this inhibitory effect compared with WT macrophages. The LPS inhibition of phagocytosis by TLR4−/− macrophages was initially observed at 12 hr after treatment, and the inhibition became more evident at 16 and 24 hr (Fig. 6d). Moreover, the LPS-inhibited phagocytosis by TLR4−/− macrophages was significantly reduced compared with that by WT controls (Fig. 6d).
Anti-TNF-α did not affect LPS inhibition of phagocytosis by TLR4−/− macrophages (Fig. 6e). In contrast, exogenous Gas6 reversed LPS-inhibited phagocytosis by TLR4−/− macrophages to the control level. These observations suggest that down-regulation of Gas6 production is entirely responsible for LPS inhibition of phagocytosis by TLR4−/− macrophages. Taken together, the results suggest that TNF-α and Gas6 act independently of one another in the regulation of macrophage phagocytosis of apoptotic neutrophils.
Discussion
Apoptosis of inflammatory cells and their phagocytic clearance by phagocytes are critical for the resolution of inflammation.7 LPS triggers inflammatory responses by inducing inflammatory cytokine production and thus influences the rate of inflammatory cell apoptosis.9,21 In this study, we focused on the role of LPS and LPS-induced inflammatory modulators in regulating phagocytosis of apoptotic cells by macrophages. We demonstrated that LPS significantly inhibited phagocytosis of apoptotic neutrophils by mouse peritoneal macrophages via LPS-driven induction of TNF-α and suppression of Gas6 production.
Macrophage phagocytosis prevents apoptotic cells from undergoing secondary necrosis and releasing their histotoxic contents. As different macrophage subpopulations exhibit different phagocytic features, so macrophages at different stages of maturity.11,12,22 A recent study reported that LPS inhibits the ability of human monocyte-derived macrophages to ingest apoptotic neutrophils.13 In agreement with this report, the present study showed that LPS significantly inhibited phagocytosis of apoptotic neutrophils by mouse peritoneal macrophages. However, we found that the LPS inhibition of phagocytosis occurred at an earlier time-point (8 hr) after LPS treatment than that (96 hr) reported in the previous study.13 This discrepancy may be explained by the different macrophage types used in the two studies.
We have provided evidence that LPS-mediated induction of TNF-α was partially responsible for LPS inhibition of phagocytosis. TNF-α can be rapidly released by macrophages after stimulation with LPS, and is one of the most abundant inflammatory factors in inflamed sites.23 TNF-α is actively involved in the development of both chronic inflammation and autoimmune disease.24 Consequently, the blockade of TNF-α activity, using a neutralizing antibody or a soluble TNF-α receptor, has been shown to have a therapeutic benefit in the treatment of chronic inflammatory diseases.25 However, the mechanisms underlying the role of TNF-α in the development of chronic inflammation remain to be clarified. In the present study, we have provided convincing evidence that LPS-induced TNF-α inhibits the phagocytosis of apoptotic neutrophils by mouse peritoneal macrophages. This result suggests that excess TNF-α in inflamed tissue may result in inefficient removal of apoptotic cells. This would lead to secondary cell necrosis and damage of the surrounding tissue, which in turn will delay the resolution of inflammation. However, TNF-α does not sufficiently account or LPS inhibition of phagocytosis, because neutralization of TNF-α activity by antibodies did not completely reverse the LPS inhibitory effect. In addition, LPS inhibition of phagocytosis was also observed in TLR4−/− macrophages, which had no capacity to induce TNF-α. These observations suggest that other mechanisms are involved in LPS inhibition of macrophage phagocytosis.
We have demonstrated that Gas6 expression in macrophages was blocked by LPS, and that the down-regulation of Gas6 also contributed to the LPS inhibition of phagocytosis. This result is consistent with a previous observation that Gas6-deficient macrophages exhibit impaired phagocytosis of apoptotic cells.26 Gas6 has been reported to mediate specifically phagocytosis of apoptotic cells by phagocytes.27,28 Accordingly, we demonstrated that LPS inhibition of phagocytosis is restricted to the uptake of apoptotic cells. One key signal for engulfment of apoptotic cells is an externalized phosphatidylserine (PS) on the apoptotic cell surface.29 Gas6 binds, through its gamma- carboxyglutamic (GLA) domains, to PS exposed on cell surfaces.30 As a common ligand, Gas6 activates the TAM receptors through its carboxy-terminal immunoglobulin-like domains. Of these, Mer is critical for initiating phagocytosis signalling.27,31 Notably, Gas6 is a potent inhibitor of the production of pro-inflammatory cytokines, including TNF-α.32 It is reasonable to speculate that Gas6 may also facilitate phagocytosis through suppressing TNF-α. We noted a significant latency of the maximal inhibitory effect of LPS on phagocytosis in comparison to TNF-α. The reduction in the Gas6 level was also delayed in comparison to the induction of TNF-α in the medium after treatment with LPS. Therefore, we speculate that LPS-induced TNF-α is responsible for the LPS inhibition of macrophage phagocytosis in the earlier time after LPS treatment, and that LPS suppression of Gas6 production is responsible for the inhibition of phagocytosis at a later time after the challenge.
LPS induces TNF-α production in macrophages by activating TLR4. However, we showed that Gas6 expression in macrophages was suppressed by LPS in a TLR4-independent manner, as LPS suppression of Gas6 expression and inhibition of phagocytosis also occurred in TLR4−/− macrophages. This finding suggests that TNF-α and Gas6 act independently of one another in regulating the phagocytosis of apoptotic cells by macrophages. Understanding the mechanism underlying the LPS inhibition of Gas6 expression may have clinical implications.
In conclusion, this article demonstrated that LPS inhibits the engulfing of apoptotic neutrophils by mouse peritoneal macrophages through LPS-mediated induction of TNF-α in a TLR4-dependent manner and suppression of Gas6 in a TLR4-independent manner in macrophages. These findings provide new insights into the role of inflammatory modulators in regulating phagocytic removal of apoptotic cells, which may be helpful in developing therapeutic approaches to the resolution of inflammation.
Acknowledgments
This work was supported by the Special Funds for Major State Basic Research Project of China (Grant No. 2007CB947504) and the National Natural Science Foundation of China (Grant No. 30971459).
Disclosures
The authors indicated no potential conflicts of interest.
References
- 1.deCathelineau AM, Henson PM. The final step in programmed cell death: phagocytes carry apoptotic cells to the grave. Essays Biochem. 2003;39:105–17. doi: 10.1042/bse0390105. [DOI] [PubMed] [Google Scholar]
- 2.Greenberg S, Grinstein S. Phagocytosis and innate immunity. Curr Opin Immunol. 2002;14:136–45. doi: 10.1016/s0952-7915(01)00309-0. [DOI] [PubMed] [Google Scholar]
- 3.Henson PM. Engulfment: ingestion and migration with Rac, Rho and TRIO. Curr Biol. 2005;15:R29–30. doi: 10.1016/j.cub.2004.12.017. [DOI] [PubMed] [Google Scholar]
- 4.Weissmann G, Smolen JE, Korchak HM. Release of inflammatory mediators from stimulated neutrophils. N Engl J Med. 1980;303:27–34. doi: 10.1056/NEJM198007033030109. [DOI] [PubMed] [Google Scholar]
- 5.Nathan C. Points of control in inflammation. Nature. 2002;420:846–52. doi: 10.1038/nature01320. [DOI] [PubMed] [Google Scholar]
- 6.Gilroy DW, Lawrence T, Perretti M, Rossi AG. Inflammatory resolution: new opportunities for drug discovery. Nat Rev Drug Discov. 2004;3:401–16. doi: 10.1038/nrd1383. [DOI] [PubMed] [Google Scholar]
- 7.Serhan CN, Brain SD, Buckley CD, et al. Resolution of inflammation: state of the art, definitions and terms. FASEB J. 2007;21:325–32. doi: 10.1096/fj.06-7227rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chung EY, Kim SJ, Ma XJ. Regulation of cytokine production during phagocytosis of apoptotic cells. Cell Res. 2006;16:154–61. doi: 10.1038/sj.cr.7310021. [DOI] [PubMed] [Google Scholar]
- 9.Lee A, Whyte MK, Haslett C. Inhibition of apoptosis and prolongation of neutrophil functional longevity by inflammatory mediators. J Leukoc Biol. 1993;54:283–8. [PubMed] [Google Scholar]
- 10.Rossi AG, Sawatzky DA, Walker A, et al. Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis. Nat Med. 2006;12:1056–64. doi: 10.1038/nm1468. [DOI] [PubMed] [Google Scholar]
- 11.Ren Y, Savill J. Proinflammatory cytokines potentiate thrombospondin-mediated phagocytosis of neutrophils undergoing apoptosis. J Immunol. 1995;154:2366–74. [PubMed] [Google Scholar]
- 12.McPhillips K, Janssen WJ, Ghosh M, et al. TNF-alpha inhibits macrophage clearance of apoptotic cells via cytosolic phospholipase A2 and oxidant-dependent mechanisms. J Immunol. 2007;178:8117–26. doi: 10.4049/jimmunol.178.12.8117. [DOI] [PubMed] [Google Scholar]
- 13.Michlewska S, Dransfield I, Megson IL, Rossi AG. Macrophage phagocytosis of apoptotic neutrophils is critically regulated by the opposing actions of pro-inflammatory and anti-inflammatory agents: key role for TNF-alpha. FASEB J. 2009;23:844–54. doi: 10.1096/fj.08-121228. [DOI] [PubMed] [Google Scholar]
- 14.Hafizi S, Dahlback B. Gas6 and protein S. Vitamin K-dependent ligands for the Axl receptor tyrosine kinase subfamily. FEBS J. 2006;273:5231–44. doi: 10.1111/j.1742-4658.2006.05529.x. [DOI] [PubMed] [Google Scholar]
- 15.Bellido Martin L, de Frutos PG. Vitamin K-dependent actions of Gas6. Vitam Horm. 2008;78:185–209. doi: 10.1016/S0083-6729(07)00009-X. [DOI] [PubMed] [Google Scholar]
- 16.Lemke G, Rothlin CV. Immunobiology of the TAM receptors. Nat Rev Immunol. 2008;8:327–36. doi: 10.1038/nri2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McPhillips KA, Erwig LP. Assessment of apoptotic cell phagocytosis by macrophages. Methods Mol Biol. 2009;559:247–56. doi: 10.1007/978-1-60327-017-5_17. [DOI] [PubMed] [Google Scholar]
- 18.Licht R, Jacobs CW, Tax WJ, Berden JH. An assay for the quantitative measurement of in vitro phagocytosis of early apoptotic thymocytes by murine resident peritoneal macrophages. J Immunol Methods. 1999;223:237–48. doi: 10.1016/s0022-1759(98)00212-9. [DOI] [PubMed] [Google Scholar]
- 19.Savill JS, Wyllie AH, Henson JE, Walport MJ, Henson PM, Haslett C. Macrophage phagocytosis of aging neutrophils in inflammation. Programmed cell death in the neutrophil leads to its recognition by macrophages. J Clin Invest. 1989;83:865–75. doi: 10.1172/JCI113970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ishimoto Y, Ohashi K, Mizuno K, Nakano T. Promotion of the uptake of PS liposomes and apoptotic cells by a product of growth arrest-specific gene, gas6. J Biochem. 2000;127:411–7. doi: 10.1093/oxfordjournals.jbchem.a022622. [DOI] [PubMed] [Google Scholar]
- 21.Murray J, Barbara JA, Dunkley SA, et al. Regulation of neutrophil apoptosis by tumor necrosis factor-alpha: requirement for TNFR55 and TNFR75 for induction of apoptosis in vitro. Blood. 1997;90:2772–83. [PubMed] [Google Scholar]
- 22.Shao WH, Zhen Y, Eisenberg RA, Cohen PL. The Mer receptor tyrosine kinase is expressed on discrete macrophage subpopulations and mainly uses Gas6 as its ligand for uptake of apoptotic cells. Clin Immunol. 2009;133:138–44. doi: 10.1016/j.clim.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kodama S, Davis M, Faustman DL. The therapeutic potential of tumor necrosis factor for autoimmune disease: a mechanistically based hypothesis. Cell Mol Life Sci. 2005;62:1850–62. doi: 10.1007/s00018-005-5022-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clark IA. How TNF was recognized as a key mechanism of disease. Cytokine Growth Factor Rev. 2007;18:335–43. doi: 10.1016/j.cytogfr.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 25.Feldmann M, Maini RN. Lasker Clinical Medical Research Award. TNF defined as a therapeutic target for rheumatoid arthritis and other autoimmune diseases. Nat Med. 2003;9:1245–50. doi: 10.1038/nm939. [DOI] [PubMed] [Google Scholar]
- 26.Angelillo Scherrer A, Burnier L, Lambrechts D, et al. Role of Gas6 in erythropoiesis and anemia in mice. J Clin Invest. 2008;118:583–96. doi: 10.1172/JCI30375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scott RS, McMahon EJ, Pop SM, Reap EA, Caricchio R, Cohen PL, Earp HS, Matsushima GK. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature. 2001;411:207–11. doi: 10.1038/35075603. [DOI] [PubMed] [Google Scholar]
- 28.Seitz HM, Camenisch TD, Lemke G, Earp HS, Matsushima GK. Macrophages and dendritic cells use different Axl/Mertk/Tyro3 receptors in clearance of apoptotic cells. J Immunol. 2007;178:5635–42. doi: 10.4049/jimmunol.178.9.5635. [DOI] [PubMed] [Google Scholar]
- 29.Fadok VA, Bratton DL, Rose DM, Pearson A, Ezekewitz RA, Henson PM. A receptor for phosphatidylserine-specific clearance of apoptotic cells. Nature. 2000;405:85–90. doi: 10.1038/35011084. [DOI] [PubMed] [Google Scholar]
- 30.Nakano T, Ishimoto Y, Kishino J, et al. Cell adhesion to phosphatidylserine mediated by a product of growth arrest-specific gene 6. J Biol Chem. 1997;272:29411–4. doi: 10.1074/jbc.272.47.29411. [DOI] [PubMed] [Google Scholar]
- 31.Wu Y, Singh S, Georgescu MM, Birge RB. A role for Mer tyrosine kinase in alphavbeta5 integrin-mediated phagocytosis of apoptotic cells. J Cell Sci. 2005;118:539–53. doi: 10.1242/jcs.01632. [DOI] [PubMed] [Google Scholar]
- 32.Rothlin CV, Ghosh S, Zuniga EI, Oldstone MB, Lemke G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell. 2007;131:1124–36. doi: 10.1016/j.cell.2007.10.034. [DOI] [PubMed] [Google Scholar]