

Abstract
Representatives of the C-homoaporphine class of alkaloids have shown interesting biological activities. To date the synthesis of these molecules has never been attempted via a direct arylation strategy. We report herein the first Pd-mediated intramolecular direct arylation in the synthesis of C-homoaporphines via the use of microwaves. Use of tricyclohexylphosphine tetrafluoroborate as ligand gave good percentage conversions and suppressed competing debromination with the substrates evaluated. This arylation strategy should be broadly useful in the synthesis of C-homoaporphine alkaloids as demonstrated herein in the synthesis of (±)-homonantenine.
Keywords: Homoaporphine, Aporphine, Alkaloid, Microwave-assisted, Direct Arylation
1. Introduction
C-Homoaporphine alkaloids (1) are structurally-related to aporphine alkaloids (2, Figure 1); as compared to aporphines these compounds have an additional carbon atom in the C ring. The analogous B-homoaporphines are relatively rare in the literature and have not been reported to occur in nature. Members of the C-homoaporphine series occur naturally in plants of the Liliaceae family, specifically the Merendera, Colchicum, Kreysigia, Androcymbium, Bulbocodium, Iphigenia and Gloriosa genera.1 With regards to biological activity, some C-homoaporphines have been reported to exhibit acetylcholinesterase and butylcholinesterase inhibitory activities and smooth muscle relaxing effects.2
Figure 1.

Approaches to the Synthesis of Homoaporphines.
A number of approaches to the synthesis of C-homoaporphines have been reported. The majority of these methods involve metal-mediated biaryl cyclization of a phenolic or phenol ether tetrahydroisoquinoline precursor as a key step in construction of the homoaporphine core (Figure 1).3 This approach is generally undesirable since toxic metal reagents are used and yields are sometimes quite low. A more recent approach uses a photostimulated intramolecular ortho-arylation of a phenol with an activated aryl ring to construct the biaryl bond.4 Acid treatment of p-quinol acetates, (prepared from phenethylisoquinolines via lead tetraacetate oxidation) has also been employed to prepare C-homoaporphines.5 A fourth approach to the preparation of these molecules, is via ring-expansion of dehydroaporphines.6
Owing to the interesting biological activities exhibited by these compounds and the limited access from natural sources, the development of rapid and efficient methods to prepare libraries of these compounds is desirable.
Microwave promoted reactions have become increasingly popular in the literature since the use of microwaves can enhance reaction rates and yields of several reactions.7a–c The generation of large compound libraries for medicinal chemistry efforts has benefited from advances made in microwave-assisted chemistry.7d The use of microwaves is also a valuable tool for reaction optimization studies.7a–c
The synthesis of C-homoaporphines by direct arylation has never been reported in the literature. In fact, direct arylation for the synthesis of seven-membered rings via coupling of two arene partners is particularly rare.8 We recently reported a microwave-assisted direct arylation protocol for the rapid synthesis of aporphines.9 Perceived challenges notwithstanding and based on our earlier work, we envisaged that microwave-assisted direct arylation might also be applicable to the preparation of C-homoaporphines from tetrahydroisoquinoline precursors. Our efforts in this regard are reported herein.
2. Results and discussion
The phenethyl tetrahydroisoquinoline 10 was prepared as a substrate for studying the microwave-assisted direct arylation reaction (Scheme 1). This substrate was also chosen since it would allow for the synthesis of the ring C-homolog (3) of the aporphine alkaloid nantenine (4) as part of our continuing structure-activity relationship studies on aporphine derivatives at human 5-HT2A receptors.9,10 Thus phenethylamine 5 was coupled to the bromoacid 6 to give amide 8. Bischler-Napieralski cyclization of 8 furnished an intermediate imine which was subjected to NaBH4 reduction to afford amine 9. N- protection of 9 as an ethyl carbamate in the subsequent step gave the arylation substrate 10.
Scheme 1.

Synthesis of arylation substrate 10
To initiate our study, we then attempted the microwave- assisted direct arylation on 10 using similar conditions as we recently reported for the synthesis of 4 and other aporphines (Table 1).8
Table 1.
Initial conditions - microwave-assisted direct arylation on 10.
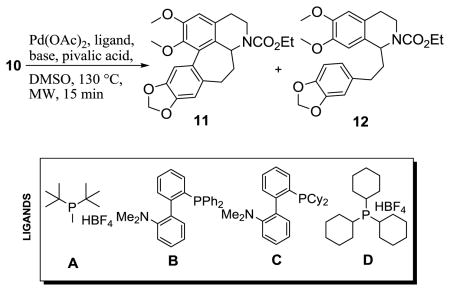 | ||||
|---|---|---|---|---|
| Entry | Ligand | Base | Ratio 11:12a | Yield of 11 (%)b |
| 1 | A | K2CO3 | 1:5 | 16 |
| 2 | B | K2CO3 | 0:1 | 0 |
| 3 | Cc | K2CO3 | 0:1 | 0 |
| 4 | D | Cs2CO3 | 1:1 | 13 |
| 5 | D | K2CO3 | 1:1.5 | 36 |
Based on isolated yields of 11 and 12.
Isolated yield.
Ligand:Pd ratio used was 1:1
These conditions (Table 1, entry 1) furnished the expected cyclized product 11 as well as debrominated product 12 in a ratio of 1:5 giving 11 in only 16% isolated yield. Two biphenyl phosphine ligands B and C were also screened based on the reported utility of these ligands in the synthesis of the structurally similar seven-membered ring containing allocolchicinoids.8 Unfortunately, only debrominated product 12 (Table 1, entries 2 and 3) was obtained with these ligands. However, we were pleased to find that ligand D gave us a slight improvement in the product ratio as compared to ligand A, when either K2CO3 or Cs2CO3 was used as base (Table 1, entries 4 and 5). Spurred by the preceding, we decided to continue our examination of this reaction with other substrates. Our prior success with benzyl ether substrates in the synthesis of nantenine analogs, prompted us to consider similar substrates for this study.8 Thus, compound 13 (Table 2) was prepared from readily available starting materials using a similar route as delineated above for compound 10. We decided to screen a number of ligands for the direct arylation of 13. With ligand A, we obtained the desired cyclized product 14 and the debrominated by-product 15 in addition to starting material (Table 2, entry 1). Although we could chromatographically separate compound 13 from a mixture of 14/15, the mixture of compounds 14 and 15 could not be separated with a variety of chromatographic procedures attempted.
Table 2.
Microwave-assisted Direct Arylation Optimization Study with Substrate 13.
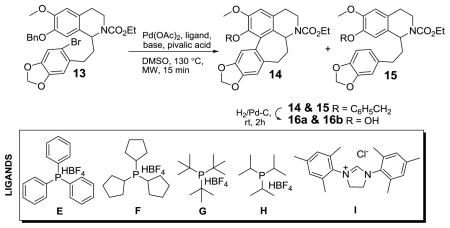 | ||||
|---|---|---|---|---|
| Entry | Ligand | Base | Ligand: Pda | Ratio 14:15:13b |
| 1 | A | K2CO3 | 4:2 | 1:1:1 |
| 2 | E | K2CO3 | 4:2 | 1:4:6 |
| 3 | F | K2CO3 | 4:2 | 1:1:8 |
| 4 | G | K2CO3 | 4:2 | 2:1:2 |
| 5 | H | K2CO3 | 4:2 | 1:3:0 |
| 6 | I | K2CO3 | 4:2 | 3:1:16 |
| 7 | D | K2CO3 | 4:2 | 4:1:0 |
| 8 | D | K2CO3 | 4:1 | 6:1:0 |
| 9 | D | K2CO3 | 8:4 | 1:1:1 |
| 10 | D | Cs2CO3 | 4:2 | 1:4:7 |
| 11 | D | Ag2CO3 | 4:2 | 1:6:0 |
| 12 | D | K2CO3 | 1:1 | 2:1:3 |
ratio relative to starting material
determined by HPLC.
At this juncture, we decided to investigate other bulky alkyltetrafluoroborate phosphine ligands. In this examination, ligands E–F and ligand H gave poor results – low conversion of starting material and/or sub-optimal ratios of 14:15 (Table 2, entries 2, 3 and 5). Ligand G gave a slightly better 14:15 product ratio of 2:1 (Table 2, entry 4). Marginal improvement in this product ratio (3:1) was observed with the N-heterocyclic carbene ligand I, but the conversion was too low to consider for further investigation (Table 2, entry 6). However, we were excited to find that ligand D gave a 4:1 ratio of 14:15 (Table 2, entry 7) with complete consumption of starting material. This ratio improved to 6:1 when a 4:1 ratio of ligand: Pd was employed (Table 2, entry 8). The 14/15 mixture upon hydrogenolysis furnished a mixture of 16a and 16b which was purified by column chromatography to furnish the cyclized phenol 16a in 71% yield over two steps from 13. Other bases were ineffective in improving the product ratio or yields of 14 (Table 2, entries 10 and 11). The cyclization of similar substrates in direct arylation reactions is known to be susceptible to changes in the ligand: Pd ratio.8a Therefore we decided to investigate the effect of the ligand: Pd ratio. This brief study indicated that a 1:1 ratio or 8:4 ratio of ligand: Pd was not optimal in improving the product ratio (Table 2, entries 9 and 12).
In further examinations of the substrate scope of the microwave-assisted arylation protocol, we prepared arylation substrates 17a – 17c using similar methods as above, and subjected these substrates to our optimal conditions (Table 3). As in the case of 13, mixtures of desired cyclized products (18) and the corresponding debrominated products (19) were obtained; no starting material (17) was detected in these reactions.
Table 3.
Direct Arylation with Other Substrates
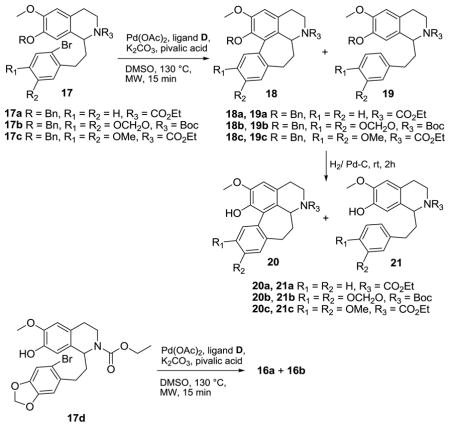 | |||
|---|---|---|---|
| Entry | Substrate | Ratio (18:19/16a:16b)a | Yield of 20/16a (%) |
| 1 | 17a | 6:1 | 72b |
| 2 | 17b | 7:1 | 85b |
| 3 | 17c | 5:1 | 70b |
| 4 | 17d | 5:1 | 69c |
no starting material detected
isolated yield over two steps from 17a–c
isolated yield over single step from 17d
Debenzylation of the 18/19 product mixtures followed by chromatographic purification of the resultant phenols gave the desired cyclized products in good yields. Cyclization of the phenol 17d could also be effected in overall yield comparable to that of the phenol ether substrates (Table 3, entry 4).
Following the development of successful microwave-promoted arylation conditions, we then proceeded to prepare (±)-homonantenine (3). At the outset, this seemed easiest via LAH reduction of compound 11; however, we obtained only a 19% yield of 3 in this transformation. An improved route to 3 was executed wherein compound 20b was sequentially O-methylated (22), Boc-deprotected and N-methylated (Scheme 2).
Scheme 2.

Synthesis of Homonantenine (3)
3. Conclusions
This is the first report on the use of a microwave-assisted direct-arylation strategy for the synthesis of C-homoaporphine alkaloids. Of several ligands screened, ligand D, (tricyclohexylphosphine tetrafluoroborate) proved to be effective for cyclization of a variety of substrates. N-ethyl carbamate and N-Boc protecting groups are tolerated with the conditions used. Based on the substrates investigated, it is apparent that unsubstituted or oxygenated functionalities in the incipient homoaporphine ring D and phenol ether or phenolic moieties in ring A, are also well tolerated. As compared to our previously reported aporphine-targeted arylation, the present method requires slightly longer reaction times (5 min vs 15 min). This is not surprising given the greater entropic cost in cyclizing a seven-membered vs a six-membered ring. This arylation methodology should find general applicability in the synthesis of naturally occurring and biologically active C-homoaporphine alkaloids as well as their analogs. Furthermore, these conditions form a framework for optimizing the challenging seven-membered ring construction in other chemotypes.
4. Experimental
4.1 General experimental procedures
All glass apparatus were oven dried prior to use. A CEM Discover microwave reactor was used to carry out all direct arylation reactions. HRESIMS spectra were obtained using an Agilent 6520 Q-TOF instrument. 1H NMR and 13C NMR spectra were recorded using Bruker DPX-500 spectrometer (operating at 500 MHz for 1H; 125 MHz respectively for 13C) using CDCl3 as solvent. Tetramethylsilane (δ 0.00 ppm) served as an internal standard in 1H NMR and CDCl3 (δ 77.0 ppm) in 13C NMR as solvent unless stated otherwise. Chemical shift (δ 0.00 ppm) values are reported in parts per million and coupling constants in Hertz (Hz). Splitting patterns are described as singlet (s), doublet (d), triplet (t) and multiplet (m). Melting points were obtained on a Mel-Temp capillary electrothermal melting point apparatus. Reactions were monitored by TLC with Whatman Flexible TLC silica gel G/UV 254 precoated plates (0.25 mm). TLC plates were visualized by UV (254 nm) and by staining with vanillin spraying reagent (2 gm vanillin in 1 L of 10% H2SO4) followed by heating. Flash column chromatography was performed with silica gel 60 (EMD Chemicals, 230–400 mesh, 0.04–0.063 μm particle size). All chemicals and reagents were obtained from Sigma-Aldrich and Fischer Scientific (USA) and were used without further purification.
4.2 Experimental procedures and characterization data
4.2.1 Synthesis of 3-(6-bromobenzo[d] [1, 3] dioxol-5-yl)-N-(3, 4-dimethoxyphenethyl) propanamide (8)
A solution of 6 (1.375 gm, 5.1 mmol) and 1, 1′-carbonyldiimidazole (0.745 gm, 4.6 mmol, CDI) in anhydrous THF (40 mL) was stirred at 0 °C for 1.5 h and then, at room temperature for 1 h. The mixture was cooled in an ice-bath and stirred for 1h; then 3, 4-dimethoxyphenethylamine (5) (0.762 mL, 4.6 mmol) was added and the solution was stirred at 0 °C for 4h and left stirring overnight at room temperature. The reaction mixture was evaporated under reduced pressure and the residue was dissolved in ethyl acetate and extracted with 1N HCl (25 mL), washed with water (50 mL), satd. NaHCO3 solution (25 mL), then with water (50 mL) and finally with brine (50 mL). The organic layer was dried over anhyd. Na2SO4 and concentrated under reduced pressure. The crude product was crystallized from EtOAc-Diethylether to furnish amide 8 as a white solid (1.538 gm, 77% yield). m.p: 148–150 °C; 1H NMR (500 MHz, CDCl3): δ 6.96 (s, 1H), 6.79 (d, 1H, J = 8.1 Hz), 6.75 (s, 1H), 6.68 (d, 1H, J = 1.8 Hz), 6.65 (dd, 1H, J = 8.1 Hz, 1.8 Hz), 5.93 (s, 2H), 5.37 (bt, 1H), 3.86 (s, 6H), 3.47 (dd, 1H, J = 12.9 Hz, 6.8 Hz), 2.97-2.94 (m, 2H), 2.71 (t, 2H, J = 6.8 Hz), 2.39-2.36 (m, 2H); 13C NMR (125 MHz, CDCl3): δ 171.8 (C), 149.2 (C), 147.9 (C), 147.6 (C), 147.1 (C), 133.2 (C), 131.4 (C), 120.8 (CH), 114.3 (C), 112.8 (CH), 111.9 (CH2), 111.5 (CH), 110.5 (CH), 101.8 (CH2), 56.1 (CH3), 56.0 (CH3), 40.8 (CH2), 36.9 (CH2), 35.4 (CH2), 32.3 (CH2); HRESIMS calculated for C20H22BrNO5 [M+]: 435.0681; Found 435.0684.
4.2.2 Synthesis of 1-(2-(6-bromobenzo[d] [1, 3] dioxol-5-yl) ethyl)-6, 7-dimethoxy-1, 2, 3, 4-tetrahydroisoquinoline (9)
To a magnetically stirred ice-cooled solution of 8 (0.50 g, 1.15 mmol) in dry DCM (25 mL) was added solid phosphorus pentachloride (0.358 g, 1.71 mmol) in several portions over 10 min. The reaction mixture was stirred at 0 °C for 1h and then left to stir at room temperature for 20h. The reaction mixture was then poured onto a saturated solution of satd. NaHCO3 (20 mL) and the contents of the flask was stirred for 1h. The aqueous layer was extracted with dichloromethane (3 × 20 mL). The combined organic layer was washed with sat. NaHCO3 solution (2 × 20 mL), washed with brine (10 mL), dried over anhyd. Na2SO4, and concentrated under reduced pressure. The crude product was purified by flash column chromatography over deactivated silica gel using 0.7% MeOH/DCM as eluant to furnish a crude imine product as a white solid. To a magnetically stirred ice-cooled solution of this crude imine in a mixture of dry MeOH (20 mL) and dry DCM (20 mL), was added powdered NaBH4 (0.437 g, 11.5 mmol) in three portions over 10 min. The reaction mixture was stirred at 0 °C for 2h. The mixture was diluted with water and extracted with dichloromethane (3 × 20 mL). The combined organic layer was washed with brine (10 mL), dried over anhyd. Na2SO4, and concentrated in vacuo. The crude product was purified by flash column chromatography over deactivated silica gel using 0.7% MeOH/DCM as eluant to furnish pure 9 as a white solid (0.45 g, 93% yield from 8). m.p: 82–84 °C; 1H NMR (500 MHz, CDCl3): δ 6.97 (s, 1H), 6.81 (s, 1H), 6.61 (s, 1H), 6.57 (s, 1H), 5.93 (dd, 2H, J = 2.6 Hz, 1.1 Hz), 4.08 (t, 1H, J = 5.5 Hz), 3.85 (s, 3H), 3.84 (s, 3H), 3.36-3.31 (m, 1H), 3.09-3.04 (m, 1H), 2.82 (m, 3H, J = 2.6 Hz), 2.73 (td, 1H, J = 16.1 Hz, 5.1 Hz), 2.0 (dd, 2H, J = 14.3 Hz, 8.0 Hz); 13C NMR (125 MHz, CDCl3): δ 147.7 (C), 147.6 (C), 147.5 (C), 146.8 (C), 134.6 (C), 126.8 (C), 114.3 (C), 112.8 (CH), 111.8 (CH), 110.3 (CH), 109.3 (CH), 101.7 (CH2), 56.2 (CH3), 56.0 (CH3), 55.2 (CH), 41.1 (CH2), 36.4 (CH2), 32.8 (CH2), 28.9 (CH2); HRESIMS calculated for C20H22BrNO4 [M+]: 419.0732; Found 419.0726.
4.2.3 Synthesis of ethyl 1-(2-(6-bromobenzo[d] [1, 3] dioxol-5-yl) ethyl)-6, 7-dimethoxy-3, 4-dihydroisoquinoline-2 (1H)-carboxylate (10)
To a solution of compound 9 (0.46 g, 1.09 mmol) dissolved in dry dichloromethane (25 mL) was added ethyl chloroformate (0.10 mL, 1.09 mmol) and potassium carbonate ( 0.228 mg, 1.63 mmol) at rt. The reaction mixture was stirred at the same temperature for 12h, quenched with satd. NH4Cl solution (10 mL) and extracted with dichloromethane (3 × 20 mL). The organic layer was washed with water (10 mL), dried over anhyd. Na2SO4 and concentrated under reduced pressure. The resultant crude product, on column chromatography over silica gel using EtOAc: Hexanes (20:80) as eluant furnished a rotameric mixture of 10 (0.497 g, 92% yield) as a viscous oil. 1H NMR (500 MHz, CDCl3): δ 6.95 (s, 1H), 6.78-6.72 (m, 1H), 6.58 (s, 2H), 5.91 (s, 2H), 5.91 (s, 2H), 5.16 (d, 2H, J = 5.5 Hz), 4.31-4.10 (m, 3H), 3.84 (s, 3H), 3.36-3.27 (m, 1H), 2.92-2.86 (m, 1H), 2.75 (bs, 2H), 2.65 (d, 1H, J = 15.8 Hz), 2.07-2.01 (m, 1H), 1.97 (bs, 1H), 1.30 (t, 3H, J = 6.9 Hz); 13C NMR (125, MHz, CDCl3): δ 156.0 & 155.8 (2 × C), 147.8 & 147.7 (2 × C), 147.4 & 147.4 (2 × C), 146.7 (C), 134.4 & 134.1 (2 × C), 129.7 & 129.2 (2 × C), 126.2 & 125.8 (2 × C), 114.2 & 114.1 (2 × C), 112.8 & 112.7 (2 × CH), 111.6 & 111.4 (2 × CH), 110.1 & 110.0 (2 × CH), 109.8 (CH), 101.6 (CH2), 61.5 & 61.4 (2 × CH2), 56.0 & 55.9 (2 × CH3), 54.2 (CH), 38.1 & 37.5 (2 × CH2), 37.2 & 37.0 (2 × CH2), 33.4 (CH2), 28.1 & 27.8 (2 × CH2), 14.8 & 14.2 (2 × CH3); HRESIMS calculated for C23H26BrNO6 [M+]: 491.0944; Found 491.0943.
4.2.4 Typical microwave-assisted direct arylation procedure for ethyl 11, 12-dimethoxy-1, 2, 4, 5-tetrahydro-[1, 3] dioxolo [4″, 5″: 4′, 5′] benzo [1′, 2′:6, 7] cyclohepta [1, 2, 3-ij] isoquinoline-3 (3aH)-carboxylate (11)
In a microwave reaction vial, compound 10 (50.0 mg, 0.10 mmol), Pd (OAc)2 (4.57 mg, 0.02 mmol), ligand D (14.9 mg, 0.04 mmol), K2CO3 (42.2 mg, 0.30 mmol), and pivalic acid (3.1 mg, 0.03 mmol) were added and dissolved in Ar-purged anhyd. DMSO (0.5 mL). The mixture was irradiated in the CEM microwave reactor for 15 min at 130 °C with the power level at 200 W. After cooling to room temperature, the reaction mixture was loaded directly onto a deactivated silica gel column and eluted with 10% EtOAc: hexanes to furnish a rotameric mixture of compound 11 (15.2 mg, 36% yield) as a viscous oil. 1H NMR (500 MHz, CDCl3): δ 6.98 & 6.93 (2 × s, 1H), 6.75 & 6.72 (2 × s, 1H), 6.68 & 6.65 (2 × s, 1H), 6.02-5.94 (m, 2H), 4.74 & 4.60 (2 × dd, 1H, J = 10.7 Hz, 6.9 Hz, J = 10.6 Hz, 7.5 Hz), 4.32 & 4.16 (2 × dd, 1H, J = 13.2 Hz, 5.8 Hz, J = 13.5 Hz, 5.5 Hz), 4.15-4.01 (m, 2H), 3.88 (s, 3H), 3.49 (s, 3H), 3.26 & 3.17 (2 × dt, 1H, J = 12.8 Hz, 3.4 Hz, J = 12.9 Hz, 3.2 Hz ), 2.99-2.88 (m, 1H), 2.74-2.69 (m, 1H), 2.46-2.21 (m, 3H), 2.06-2.01 (m, 1H), 1.19 (2 × t, 3H, J = 7.0 Hz); 13C NMR (125 MHz, CDCl3): δ 155.4 & 155.0 (2 × C), 151.9 & 151.8 (2 × C), 147.2 & 147.1 (2 × C), 146.0 & 144.5 (2 × C), 133.1 & 133.0 (2 × C), 132.3 & 132.0 (2 × C), 129.3 & 128.8 (2 × C), 128.2 & 127.9 (2 × C), 127.7 (C), 127.1 & 126.5 (2 × C), 111.8 & 111.6 (2 × CH), 111.1 (CH), 108.5 & 108.2 (2 × CH), 101.1 & 101.0 (2 × CH2), 61.4 (CH2), 60.6 (CH3), 56.0 (CH3), 50.8 & 50.6 (2 × CH), 37.4 & 37.1 (2 × CH2), 36.9 & 36.6 (2 × CH2), 30.5 & 29.9 (2 × CH2), 29.3 & 29.1 (2 × CH2), 14.8 & 14.2 (2 × CH3); HRESIMS calculated for C23H25NO6 [M+]: 411.1682; Found 411.1682.
4.2.5 General procedure for the synthesis of direct arylation substrates 13, 17a, 17b and 17c: (Compound 13 as representative)
To a solution of the secondary amine precursor (2.876 g, 5.8 mmol) dissolved in dry dichloromethane (25 mL) was added ethyl chloroformate (0.630 mL, 7.8 mmol) and potassium carbonate (1.204 mg, 8.7 mmol) at rt. The reaction mixture was stirred at the same temperature for 12h, quenched with satd. NH4Cl solution (50 mL) and extracted with dichloromethane (3 × 50 mL). The organic layer was washed with water (50 mL), dried over anhyd. Na2SO4 and concentrated under reduced pressure. The resultant crude product, on column chromatography over silica gel using EtOAc: Hexanes (20:80) as eluant, furnished a rotameric mixture of 13 (2.571 g, 78% yield) as a white solid.
4.2.5.1 Ethyl 7-(benzyloxy)-1-(2-(6-bromobenzo[d] [1, 3] dioxol-5-yl) ethyl)-6-methoxy-3, 4-dihydroisoquinoline-2 (1H)-carboxylate (13)
m.p: 85–87 °C; 1H NMR (500 MHz, CDCl3): δ 7.41 (d, 2H, J = 6.7 Hz), 7.35-7.32 (m, 2H), 7.27-7.25 (m, 1H), 6.96 (d, 1H, J = 7.7 Hz), 6.72-6.58 (m, 3H), 5.93 (s, 2H), 5.12-5.00 (m, 3H), 4.25-4.06 (m, 3H), 3.85 (s, 3H), 3.29 (m, 1H), 2.90-2.84 (m, 1H), 2.65-2.61 (m, 3H), 1.99-1.91 (m, 1H), 1.82-1.78 (m, 1H), 1.28 (t, 3H, J = 7.1 Hz); 13C NMR (125 MHz, CDCl3): δ 156.1 & 155.9 (2 × C), 148.7 & 148.5 (2 × C), 147.5 (C), 146.7 & 146.7 (2 × C), 146.6 (C), 137.2 (C), 134.5 & 134.2 (2 × C), 129.7 & 129.3 (2 × C), 128.7 (CH), 128.0 (CH), 127.5 (CH), 127.1 & 126.7 (2 × C), 114.4 (C), 113.3 & 113.2 (2 × CH), 112.9 & 112.8 (2 × CH), 112.2 & 112.1 (2 × CH), 110.2 & 109.9 (2 × CH), 101.7 (CH2), 71.5 (CH2), 61.6 & 61.5 (2 × CH), 56.2 (CH3), 54.2 (CH), 38.2 & 37.6 (2 × CH2), 37.2 & 37.1 (2 × CH2), 33.4 (CH2), 28.3 & 28.0 (2 × CH2), 14.9 (CH3); HRESIMS calculated for C29H30BrNO6 [M+]: 567.1257; Found 567.1260.
4.2.5.2 Ethyl 7-(benzyloxy)-1-(2-bromophenethyl)-6-methoxy-3, 4-dihydroisoquinoline-2 (1H)-carboxylate (17a)
m.p: 71–73 °C; 1H NMR (500 MHz, CDCl3): δ 7.52-7.51 (m, 1H), 7.41 (m, 2H), 7.33 (t, 2H, J = 7.1 Hz), 7.24-7.15 (m, 3H), 7.06-7.04 (m, 1H), 6.61-6.59 (m, 2H), 5.15-5.04 (m, 3H), 4.29-4.07 (m, 3H), 3.85 (s, 3H), 3.36-3.24 (m, 1H), 2.93-2.75 (m, 3H), 2.65-2.62 (m, 1H), 2.04-2.00 (m, 1H), 1.87 (m, 1H), 1.28 (t, 3H, J = 7.1 Hz); 13C NMR (125 MHz, CDCl3): δ 156.1 & 156.0 (2 × C), 148.7 & 148.5 (2 × C), 146.6 (C), 141.5 & 141.3 (2 × C), 137.2 (C), 133.0 & 132.9 (2 × CH), 130.6 & 130.2 (2 × CH), 129.7 & 129.3 (2 × C), 128.7 (CH), 128.0 (CH), 127.8 & 127.7 (2 × CH), 127.6 & 127.5 (2 × CH), 127.1 & 126.7 (2 × C), 124.6 & 124.5 (2 × C), 113.3 & 113.2 (2 × CH), 112.2 & 112.1 (2 × CH), 71.58 & 71.52 (2 × CH2), 61.6 & 61.5 (2 × CH2), 56.2 (CH3), 54.3 & 54.2 (2 × CH), 38.2 & 37.6 (2 × CH2), 37.2 & 36.9 (2 × CH2), 33.5 (CH2), 28.3 & 28.1 (2 × CH2), 14.9 (CH3); HRESIMS calculated for C28H30BrNO4 [M+]: 523.1358; Found 523.1365.
4.2.5.3 tert-butyl 7-(benzyloxy)-1-(2-(6-bromobenzo[d] [1, 3] dioxol-5-yl) ethyl)-6-methoxy-3, 4-dihydroisoquinoline-2(1H)-carboxylate (17b)
m.p: 74–76 °C; 1H NMR (500 MHz, CDCl3): δ 7.41 (bs, 2H), 7.31 (t, 2H, J = 7.2 Hz), 7.25-7.23 (m, 1H), 6.94 (d, 1H, J = 8.6 Hz), 6.62-6.61 (m, 3H), 5.87 (s, 2H), 5.11-5.06 (m, 3H), 4.27-4.03 (2 × d, 1H, J = 9.8 Hz, J = 10.4 Hz), 3.82 (s, 3H), 3.28-3.15 (m, 1H), 2.88-2.82 (m, 1H), 2.68-2.59 (m, 3H), 1.97-1.89 (m, 1H), 1.84 (m, 1H), 1.49 (s, 9H); 13C NMR (125 MHz, CDCl3): δ 155.0 & 154.7 (2 × C), 148.4 & 148.2 (2 × C), 147.3 (C), 146.5 & 146.4 (2 × C), 137.1 (C), 134.4 & 134.1 (2 × C), 129.7 & 129.2 (2 × C), 128.5 (CH), 127.8 (CH), 127.4 & 127.3 (2 × CH), 127.1 & 126.7 (2 × C), 114.2 & 114.1 (2 × C), 113.1 (CH), 112.6 & 112.6 (2 × CH), 112.1 & 111.9 (2 × CH), 110.0 & 109.5 (2 × CH), 101.5 (CH2), 79.9 & 79.5 (2 × C), 71.3 (CH2), 55.9 (CH3), 54.3 & 53.3 (2 × CH), 38.3 & 36.9 (2 × CH2), 37.2 & 36.8 (2 × CH2), 33.3 & 31.5 (2 × CH2), 28.5 (CH3), 28.2 & 28.0 (2 × CH2); HRESIMS calculated for C31H34BrNO6 [M+]: 595.1570; Found 595.1569.
4.2.5.4 Ethyl-7-(benzyloxy)-1-(2-bromo-4, 5-dimethoxyphenethyl)-6-methoxy-3, 4-dihydroisoquinoline-2 (1H)-carboxylate (17c)
m.p: 76–78 °C; 1H NMR (500 MHz, CDCl3): δ 7.41-7.40 (m, 2H), 7.33 (t, 2H, J = 7.0 Hz), 7.26-7.25 (m, 1H), 6.98 (s, 1H), 6.79-6.58 (m, 3H), 5.14-5.05 (m, 3H), 4.30-4.06 (m, 3H), 3.84 (s, 6H), 3.83 (s, 3H), 3.37-3.24 (m, 1H), 2.94-2.85 (m, 1H), 2.69-2.62 (m, 3H), 2.04-2.00 (m, 1H), 1.93-1.85 (m, 1H), 1.29 (t, 3H, J = 7.0 Hz); 13C NMR (125 MHz, CDCl3): δ 156.1 & 155.9 (2 × C), 148.7 & 148.5 (2 × C), 148.0 & 147.9 (2 × C), 146.6 (C), 137.2 (C), 133.4 & 133.2 (2 × C), 129.7 & 129.3 (2 × C), 128.6 (CH), 128.0 (CH), 127.5 (CH), 127.1 & 126.7 (2 × C), 115.7 & 115.6 (2 × CH), 114.1 & 113.9 (2 × C), 113.4 & 113.2 (2 × CH), 112.9 & 112.2 (2 × CH), 112.1 & 112.1 (2 × CH), 71.5 & 71.5 (2 × CH2), 61.6 & 61.5 (2 × CH2), 56.3 (CH3), 56.2 (CH3), 56.1 (CH3), 54.4 & 54.1 (2 × CH), 38.2 & 37.6 (2 × CH2), 37.4 & 36.9 (2 × CH2), 33.3 & 33.2 (2 × CH2), 28.2 & 28.0 (2 × CH2), 14.9 (CH3); HRESIMS calculated for C30H34BrNO6 [M+]: 583.1570; Found 583.1571.
4.2.5.5 Ethyl 1-(2-(6-bromobenzo[d][1,3]dioxol-5-yl)ethyl)-7-hydroxy-6-methoxy-3,4-dihydroisoquinoline-2(1H)-carboxylate (17d)
m.p: 128–130 °C; 1H NMR (500 MHz, CDCl3): δ 6.96-6.56 (m, 4H), 5.92 (s, 2H), 5.55 (s, 1H), 5.17-5.05 (m, 1H), 4.36-4.18 (m, 3H), 3.84 (bs, 3H), 3.33-3.25 (m, 1H), 2.89-2.87 (m, 1H), 2.72-2.60 (m, 3H), 2.53-2.46 & 2.29-2.24 (2 × m, 1H), 2.02-1.96 (m, 2H), 1.30-1.287 (m, 3H); 13C NMR (125 MHz, CDCl3): δ 155.9 (C), 147.5 & 147.5 (2 × C), 147.4 & 147.3 (2 × C), 146.6 (C), 146.6 & 145.6 (2 × C), 145.5 & 145.5 (2 × C), 145.3 (C), 143.9 (C), 135.9 & 135.6 (2 × C), 134.4 & 134.1 (2 × C), 130.4 & 130.0 (2 × C), 125.5 & 125.2 (2 × CH), 121.0 (CH), 114.2 & 114.1 (2 × C), 113.0 & 112.9 (2 × CH), 112.7 & 112.7 (2 × CH), 112.6 (C), 110.8 & 110.7 (2 × CH), 110.1 & 110.0 (2 × CH), 109.7 (CH), 108.8 & 108.2 (2 × CH), 101.6 & 100.6 (2 × CH2), 68.5 (CH2), 61.4 & 61.4 (2 × CH), 55.9 (CH3), 54.0 & 53.9 (2 × CH), 38.8 & 38.1 (2 × CH2), 37.5 & 37.1 (2 × CH2), 33.3 & 32.4 (2 × CH2), 28.2 & 27.9 (2 × CH2), 14.7 (CH3); HRESIMS calculated for C22H24BrNO6 [M+]: 477.0787; Found 477.0792.
4.2.6 General procedure for the microwave-assisted direct arylation for the synthesis of 16a, 20a, 20b and 20c: (Table 2, entry 8, compound 16a as representative)
In a microwave reaction vial, compound 17 (50.0 mg, 0.088 mmol), Pd(OAc)2 (1.98 mg, 8.8 μmol), ligand D (12.98 mg, 0.035 mmol), K2CO3 (48.74 mg, 0.35 mmol), and pivalic acid (3.60 mg, 0.035 mmol) were added and dissolved in Ar-purged anhyd. DMSO (1.0 mL). The mixture was irradiated in the CEM microwave reactor for 15 min at 130 °C with the power level at 200 W. After cooling to room temperature, the reaction mixture was loaded directly onto a deactivated silica gel column and eluted with 15% EtOAc: hexanes to furnish compounds 14 and 15 in a ratio of 6:1. This mixture on debenzylation with palladium-charcoal furnished the crude cyclized and debrominated mixture of 16a and 16b, which on flash column chromatography over silica gel using EtOAc: DCM (5: 95) as eluant, furnished a rotameric mixture of homoaporphine phenol 16a (25.0 mg, 71% yield from 17) as a white solid.
4.2.6.1 Ethyl 11-hydroxy-12-methoxy-1, 2, 4, 5-tetrahydro-[1, 3] dioxolo [4″, 5″:4′, 5′] benzo [1′, 2′:6, 7] cyclohepta [1, 2, 3-ij] isoquinoline-3(3aH)-carboxylate (16a)
m.p: 140–142 °C; 1H NMR (500 MHz, CDCl3): δ 7.05 & 6.99 (2 × s, 1H), 6.77 & 6.74 (2 × s, 1H), 6.63 & 6.60 (2 × s, 1H), 6.03-5.95 (m, 2H), 5.69 & 5.66 (2 × s, 1H), 4.78 & 4.64 (2 × dd, 1H, J = 10.8 Hz, 7.5 Hz, J = 10.4 Hz, 7.6 Hz), 4.32 & 4.15 (2 × dd, 1H, J = 13.1 Hz, 5.6 Hz, J = 13.0 Hz, 5.6 Hz), 4.11-4.00 (m, 2H), 3.91 (s, 3H), 3.28 & 3.19 (dt, 1H, J = 12.8 Hz, 3.4 Hz), 2.97-2.86 (m, 1H), 2.69 (t, 1H, J = 13.0 Hz), 2.44-2.42 (m, 2H), 2.37-2.21 (m, 1H), 2.04-1.99 (m, 1H), 1.23 & 1.14 (t, 3H, J = 7.0 Hz); 13C NMR (125 MHz, CDCl3): δ 155.4 & 155.0 (2 × C), 147.1 & 147.1 (2 × C), 145.9 & 145.9 (2 × C), 145.8 (C), 140.5 & 140.5 (2 × C), 133.5 & 133.4 (2 × C), 127.2 & 127.1 (2 × C), 126.7 (C), 124.7 & 124.3 (2 × C), 124.1 & 123.8 (2 × C), 110.9 & 110.9 (2 × CH), 110.0 & 109.9 (2 × CH), 108.9 & 108.7 (2 × CH), 101.1 & 101.0 (2 × CH2), 61.4 (CH2), 56.2 (CH3), 50.8 & 50.7 (2 × CH), 37.5 & 37.3 (2 × CH2), 37.0 & 36.8 (2 × CH2), 30.5 & 29.9 (2 × CH2), 29.0 & 28.9 (2 × CH2), 14.7 & 14.7 (2 × CH3); HRESIMS calculated for C22H23NO6 [M+]: 397.1525; Found 397.153.
4.2.6.2 Ethyl 10-hydroxy-11-methoxy-1, 2, 4, 5-tetrahydrobenzo [6, 7] cyclohepta [1, 2, 3-ij] isoquinoline-3 (3aH)-carboxylate (20a)
m.p: 68–70 °C; 1H NMR (500 MHz, CDCl3): δ 7.53 (d, 1H, J = 7.5 Hz), 7.35-7.29 (m, 2H), 6.64 (s, 1H), 5.66 (s, 1H), 4.78 & 4.63 (2 × dd, 1H, J = 10.5 Hz, 7.5 Hz, J = 10.2 Hz, 8.0 Hz), 4.32 & 4.16 (2 × dd, 1H, J = 13.0 Hz, 5.8 Hz, J = 13.1 Hz, 5.5 Hz), 4.09-3.97 (m, 2H), 3.92 (s, 3H), 3.29 & 3.20 (dt, 1H, J = 12.7 Hz, 3.2 Hz), 2.98-2.88 (m, 1H), 2.73-2.67 (m, 1H), 2.57-2.48 (m, 2H), 2.35 & 2.26 (m, 1H), 2.14-2.04 (m, 1H), 1.23 & 1.09 (t, 3H, J = 7.0 Hz); 13C NMR (125 MHz, CDCl3): δ 155.4 (C), 146.0 & 145.9 (2 × C), 140.6 (C), 139.5 & 139.4 (2 × C), 134.4 & 134.3 (2 × C), 130.5 & 130.4 (2 × CH), 131.1 & 130.6 (2 × C), 128.6 & 128.3 (2 × CH), 128.2 & 128.1 (2 × CH), 127.1 & 126.7 (2 × C), 126.2 & 126.1 (2 × C), 124.7 & 124.3 (2 × C), 110.3 & 110.1 (2 × CH2), 61.3 (CH2), 56.2 (CH3), 50.8 & 50.6 (2 × CH), 37.5 & 37.1 (2 × CH2), 37.0 & 36.5 (2 × CH2), 29.9 (CH2), 29.1 & 28.9 (2 × CH2), 14.8 & 14.8 (2 × CH3); HRESIMS calculated for C21H23NO4 [M+]: 353.1627; Found 353.1628.
4.2.6.3 tert-butyl 11-hydroxy-12-methoxy-1, 2, 4, 5-tetrahydro-[1,3] dioxolo [4″, 5″:4′, 5′] benzo [1′, 2′:6, 7] cyclohepta [1, 2, 3-ij] isoquinoline-3 (3aH)-carboxylate (20b)
m.p: 58–60 °C; 1H NMR (500 MHz, CDCl3): δ 7.05 & 6.99 (2 × s, 1H), 6.76 & 6.73 (2 × s, 1H), 6.63 & 6.60 (2 × s, 1H), 5.96 (t, 2H, J = 16.8 Hz), 5.69 & 5.66 (2 × s, 1H), 4.76 & 4.57 (2 × dd, 1H, J = 10.8 Hz, 7.2 Hz, J = 10.4 Hz, 7.6 Hz), 4.26 & 4.10 (2 × dd, 1H, J = 12.8 Hz, 5.8 Hz, J = 13.3 Hz, 5.5 Hz), 3.91 (s, 3H), 3.24 & 3.13 (2 × dt, 1H, J = 12.8 Hz, 3.7 Hz, J = 12.7 Hz, 3.6 Hz), 2.92 & 2.86 (dt, 1H, J = 19.5 Hz, 6.2 Hz, J = 12.5 Hz, 5.9 Hz), 2.72-2.64 (m, 1H), 2.45-2.41 (m, 2H), 2.38-2.21 (m, 1H), 2.02-1.98 (m, 1H), 1.42 (s, 5H), 1.33 (s, 4H); 13C NMR (125 MHz, CDCl3): δ 154.6 & 154.2 (2 × C), 147.1 & 147.0 (2 × C), 145.9 & 145.9 (2 × C), 145.8 & 145.7 (2 × C), 140.4 (C), 133.6 & 133.5 (2 × C), 127.5 & 127.3 (2 × C), 127.2 & 126.9 (2 × C), 124.7 & 124.4 (2 × C), 124.2 & 123.8 (2 × C), 110.9 & 110.8 (2 × CH), 110.0 & 109.9 (2 × CH), 109.0 & 108.4 (2 × CH), 101.0 (CH2), 79.7 (C), 56.2 (CH3), 51.1 & 50.1 (2 × CH), 37.8 & 37.1 (2 × CH2), 36.8 & 36.4 (2 × CH2), 30.6 (CH2), 29.9 & 29.8 (2 × CH2), 29.1 & 29.0 (2 × CH2), 28.6 & 28.5 (2 × CH3); HRESIMS calculated for C24H27NO6 [M+]: 425.1838; Found 425.1869.
4.2.6.4 Ethyl 10-hydroxy-7, 8, 11-trimethoxy-1, 2, 4, 5-tetrahydrobenzo [6, 7] cyclohepta[1, 2, 3-ij] isoquinoline-3 (3aH)-carboxylate (20c)
m.p: 208–210 °C; 1H NMR (500 MHz, CDCl3): δ 7.11 & 7.06 (2 × s, 1H), 6.80 & 6.77 (2 × s, 1H), 6.64 & 6.61 (2 × s, 1H), 5.68 & 5.65 (2 × s, 1H), 4.79 & 4.63 (2 × dd, 1H, J = 10.7 Hz, 7.3 Hz, J = 10.4 Hz, 7.6 Hz), 4.33 & 4.16 (2 × dd, 1H, J = 13.1 Hz, 5.8 Hz, J = 13.6 Hz, 5.8 Hz), 4.14-4.07 & 4.04-4.00 (m, 2H), 3.94 & 3.91 (s, 3H), 3.92 & 3.92 (s, 3H), 3.88 & 3.85 (s, 3H), 3.28 & 3.19 (dt, 1H, J = 12.9 Hz, 3.4 Hz), 2.98-2.87 (m, 1H), 2.73-2.67 (m, 1H), 2.50-2.46 (m, 1H), 2.41-2.33 & 2.31-2.23 (m, 1H), 2.12-2.09 (m, 1H), 1.23 & 1.11 (t, 3H, J = 7.0 Hz); 13C NMR (125 MHz, CDCl3): δ 163.9 (C), 159.5 (C), 155.2 (C), 148.4 & 148.2 (2 × C), 146.9 & 146.8 (2 × C), 145.8 & 145.7 (2 × C), 140.2 (C), 132.2 & 132.1 (2 × C), 127.1 (C), 126.6 (C), 126.0 & 125.9 (2 × C), 124.7 (C), 124.2 (C), 123.8 (C), 113.7 & 113.6 (CH), 111.6 & 111.3 (CH), 109.8 & 109.6 (2 × CH), 61.2 (CH2), 56.1 & 56.0 (2 × CH3), 56.0 (CH3), 55.9 & 55.8 (2 × CH3), 50.8 & 50.6 (2 × CH), 37.3 & 37.2 (2 × CH2), 36.8 & 36.8 (2 × CH2), 30.1 & 29.7 (2 × CH2), 28.9 & 28.7 (2 × CH2), 14.7 (CH3); HRESIMS calculated for C23H27NO6 [M+]: 413.1838; Found 413.1846.
4.2.7 Preparation of tert-butyl 11, 12-dimethoxy-1, 2, 4, 5-tetrahydro-[1,3] dioxolo [4″, 5″:4′, 5′] benzo [1′, 2′:6, 7] cyclohepta [1,2,3-ij] isoquinoline-3(3aH)-carboxylate (22)
To a solution of 20b (0.025 g, 0.058 mmol) and methyl iodide (0.073 mL, 1.17 mmol) dissolved in dry acetone (20 mL) was added potassium carbonate (0.081 gm, 0.58 mmol) and potassium iodide (0.097 g, 0.58 mmol) and the mixture refluxed for 6h. The acetone was evaporated; water (10 mL) was added to the residue and extracted with dichloromethane (3 × 10 mL). The combined organic layer was dried over anhyd. Na2SO4 and concentrated under reduced pressure. The crude product on flash column chromatography over deactivated silica gel using EtOAc: Hexane (1: 9) as eluant, furnished 22 (0.0215 g, 83% yield) as a white solid (mixture of rotamers). m.p: 74–76 °C; 1H NMR (500 MHz, CDCl3): δ 6.97 & 6.92 (2 × s, 1H), 6.75 & 6.72 (2 × s, 1H), 6.68 & 6.65 (2 × s, 1H), 5.98-5.91 (s, 2H), 4.73 & 4.53 (2 × dd, 1H, J = 11.1 Hz, 7.1 Hz, J = 10.7 Hz, 7.3 Hz), 4.26 & 4.10 (2 × dd, 1H, J = 13.0 Hz, 5.4 Hz, J = 13.1 Hz, 5.8 Hz), 3.88 (s, 3H), 3.49 (s, 3H), 3.23 & 3.12 (2 × dt, 1H, J = 12.9 Hz, 3.6 Hz, J = 12.7 Hz, 3.4 Hz ), 2.96-2.84 (m, 1H), 2.75-2.67 (m, 1H), 2.47-2.43 (m, 2H), 2.41-2.28 (m, 1H), 2.02-1.98 (m, 1H), 1.42 (s, 6H), 1.33 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 154.3 (C), 151.6 & 151.5 (2 × C), 146.9 (C), 145.8 (C), 144.3 (C), 132.8 & 132.1 (2 × C), 128.7 & 127.5 (2 × C), 127.1 & 126.5 (2 × C), 111.4 & 111.0 (2 × CH), 108.3 & 107.7 (2 × CH), 100.8 (CH2), 79.5 (C), 60.4 (CH3), 55.8 (CH3), 50.8 & 49.9 (2 × CH), 37.5 & 36.6 (2 × CH2), 36.3 & 36.1 (2 × CH2), 30.3 & 29.7 (2 × CH2), 29.2 & 29.0 (2 × CH2); HRESIMS calculated for C25H29NO6 [M+]: 439.1995; Found 439.1994.
4.2.8 Preparation of (±)-Homonantenine 11, 12-dimethoxy-3-methyl-1, 2, 3, 3a, 4, 5-hexahydro [1, 3] dioxolo [4″, 5″:4′, 5′] benzo [1′, 2′:6, 7] cyclohepta [1,2,3-ij] isoquinoline (3)
To a solution of 22 (9.0 mg, 0.02 mmol) in dry dichloromethane (10 mL), was added dry zinc bromide (18.46 mg, 0.08 mmol). The reaction mixture was allowed to stir at rt for 45 min. The reaction was quenched with water (10 mL) and extracted with dichloromethane (3 × 10 mL). The combined organic layer was washed with water (10 mL), then with brine (10 mL), dried over anhyd. Na2SO4 and concentrated to give the crude amine. To a solution of this amine in dichloromethane (10 mL) was added 37% formaldehyde solution (0.017 mL, 0.607 mmol) and sodium triacetoxyborohydride (21.8 mg, 0.10 mmol). The reaction mixture was allowed to stir at room temperature for 1h. The reaction was quenched with water (10 mL) and extracted with dichloromethane (3 × 10 mL). The combined organic layer was washed with water (10 mL), then with brine (10 mL), dried over anhyd. Na2SO4 and concentrated to give crude 3, which on column chromatography over deactivated silica gel using methanol/dichloromethane (2:98) as eluant, furnished 3 (4.5 mg, 66% yield) as a viscous oil. 1H NMR (500 MHz, CDCl3): δ 6.98 (s, 1H), 6.73 (s, 1H), 6.66 (s, 1H), 5.98 (dd, 2H, J = 12.1 Hz, 1.1 Hz), 3.88 (s, 3H), 3.48 (s, 3H), 3.27 (dd, 1H, J = 11.1 Hz, 6.8 Hz), 3.15 (dt, 1H, J = 11.4 Hz, 3.8 Hz), 3.06-2.99 (m, 1H), 2.81 (dd, 1H, J = 12.0 Hz, 5.2 Hz), 2.66-2.63 (bs, 1H), 2.46 (dd, 1H, J = 12.9 Hz, 5.6 Hz), 2.40 (s, 3H), 2.32 (dt, 1H, J = 12.9 Hz, 6.7 Hz), 2.06-2.00 (m, 1H); 13C NMR (125 MHz, CDCl3): δ 151.4 (C), 146.7 (C), 145.6 (C), 144.1 (C), 133.1 (C), 132.3 (C), 130.0 (C), 127.9 (C), 111.4 (CH), 110.7 (CH), 108.0 (CH), 100.8 (CH2), 60.3 (CH3), 58.10 (CH), 55.8 (CH3), 45.0 (CH2), 41.7 (CH3), 34.7 (CH2), 30.3 (CH2), 29.7 (CH2); HRESIMS calculated for C21H23NO4 [M+]: 353.1627; Found 353.1627.
4.3 HPLC & microwave conditions
HPLC was conducted with an Agilent 1200 system with the following conditions: flow rate: 1 mL/min; detection wavelength: 254 nm; retention times: Compound 14: 8.33 min, Compound 15: 9.08 min, Compound 13: 11.27 min; Column: Agilent Eclipse XDB-C18; Eluant 80:20 (Water: Methanol)
Microwave reactions were conducted in closed vessel in a CEM Discover microwave using the following conditions: temperature: 130 °C; power: 200W; pressure: 175 Psi; mode: Standard
Supplementary Material
Acknowledgments
This publication was made possible by Grant Number RR03037 and R03DA025910 from the National Center for Research Resources (NCRR) and National Institute of Drug Abuse (NIDA) respectively, components of the National Institutes of Health. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH or its divisions.
Footnotes
1H NMR and 13C NMR spectra of compounds 8, 9, 10, 11, 13, 16a, 17a, 17b, 17c, 17d, 20a, 20b, 20c, 22 and 3. Supplementary data associated with this article can be found in the online version, at
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Tojo E. J Nat Prod. 1989;52:909–921. [Google Scholar]; (b) Alikulov RV, Levina EV, Yusupov MK. Chem Nat Compd. 2003;39:212–214. [Google Scholar]; (c) Husek A, Sutlupinar N, Potesilova H, Dvorackova S, Hanus V, Sedmera P, Malon P, Simanek V. Phytochemistry. 1989;28:3217–3219. [Google Scholar]; (d) Tojo E, Abu Zarga MH, Sabri SS, Freyer AJ, Shamma M. J Nat Prod. 1989;52:1055–1059. [Google Scholar]
- 2.(a) Basova NE, Rozengart EV, Suvorov AA. J Evol Biochem Phys. 2006;42:408–416. [Google Scholar]; (b) Abu-Zarga MH, Abu Ghalyun YY. Int J Pharmacol. 2006;2:451–454. [Google Scholar]; (c) Rozengart EV, Basova NE, Suvorov AA. Comparative and Ontogenic Biochemistry. 2006;42:328–334. [Google Scholar]
- 3.(a) Schwartz MA, Rose BF, Holton RA, Scott SW, Vishnuvajjala B. J Am Chem Soc. 1977;99:2571–2578. [Google Scholar]; (b) Herbert RB, Kattah AE, Murtagh AJ, Sheldrake PW. Tetrahedron Lett. 1995;36:5649–5650. [Google Scholar]; (c) Kupchan SM, Dhingra OP, Kim C, Kameswaran V. J Org Chem. 1976;41:4047–4049. [Google Scholar]; (d) Kupchan SM, Dhingra OP, Kim C. J Org Chem. 1976;41:4049–4050. [Google Scholar]; (e) Kupchan SM, Dhingra OM, Kim C, Kameswaran V. J Org Chem. 1978;43:2521–2529. [Google Scholar]; (f) Taylor EC, Andrade JG, Rall GJH, McKillop A. J Am Chem Soc. 1980;102:6513–6519. [Google Scholar]; (g) Landais Y, Rambault D, Robin JP. Tetrahedron Lett. 1987;28:543–546. [Google Scholar]; (h) Rao TVP. Curr Sci India. 1976;45:453–454. [Google Scholar]; (i) Hara H, Hoshino O, Umezawa B, Iitaka Y. J Chem Soc Perkin Trans 1: Organic and Bio-organic Chemistry. 1979;11:2657–2663. [Google Scholar]; (j) Kametani T, Fukumoto K, Yagi H, Satoh F. Chem Commun. 1967;17:878–879. [Google Scholar]
- 4.Barolo SM, Teng X, Cuny GD, Rossi RA. J Org Chem. 2006;71:8493–8499. doi: 10.1021/jo061478+. [DOI] [PubMed] [Google Scholar]
- 5.(a) Hoshino O, Toshioka T, Ohyama K, Umezawa B. Chem Pharm Bull. 1974;22:1307–1312. [Google Scholar]; (b) Hoshino O, Hara H, Serizawa N, Umezawa B. Chem Pharm Bull. 1975;23:2048–2053. [Google Scholar]; (c) Hara H, Shinoki H, Komatsu T, Hoshino O, Umezawa B. Chem Pharm Bull. 1986;34:1924–1928. [Google Scholar]
- 6.(a) Castro JL, Castedo L, Riguera R. J Org Chem. 1987;52:3579–3584. [Google Scholar]; (b) Castro JL, Castedo L, Riguera R. Tetrahedron Lett. 1985;26:1561–1564. [Google Scholar]
- 7.(a) Kapppe CO. Angew Chem Int Ed. 2004;43:6250–6284. doi: 10.1002/anie.200400655. [DOI] [PubMed] [Google Scholar]; (b) Hayes BL. Aldrichim Acta. 2004;37:66–77. [Google Scholar]; (c) Kappe CO, Dallinger D. Nat Rev Drug Discov. 2005;5:51–63. doi: 10.1038/nrd1926. [DOI] [PubMed] [Google Scholar]; (d) Ersmark K, Larhed M, Wannberg J. Curr Opin Drug Discov Develop. 2004;7:417–427. [PubMed] [Google Scholar]
- 8.(a) Leblanc M, Fagnou K. Org Lett. 2005;7(14):2849–2852. doi: 10.1021/ol0505959. [DOI] [PubMed] [Google Scholar]; (b) Alberico D, Scott ME, Lautens M. Chem Rev. 2007;107:174–238. doi: 10.1021/cr0509760. [DOI] [PubMed] [Google Scholar]
- 9.Chaudhary S, Pecic S, LeGendre O, Harding WW. Tetrahedron Lett. 2009;50:2437–2439. doi: 10.1016/j.tetlet.2009.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chaudhary S, Pecic S, LeGendre O, Navarro HA, Harding WW. Bioorg Med Chem Lett. 2009;19:2530–2532. doi: 10.1016/j.bmcl.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.