

Abstract
Group IIA secreted phospholipase A2 (GIIA sPLA2) is a member of the mammalian sPLA2 enzyme family and is associated with various inflammatory conditions. In this study, the synthesis of 2-oxoamides based on α-amino acids and the in vitro evaluation against three secreted sPLA2s (GIIA, GV and GX) are described. The long chain 2-oxoamide GK126 based on the amino acid (S)-leucine displayed inhibition of human and mouse GIIA sPLA2s (IC50 300 nM and 180 nM, respectively). It also inhibited human GV sPLA2 with similar potency, while it did not inhibit human GX sPLA2. The elucidation of the stereoelectronic characteristics that affect the in vitro activity of these compounds was achieved by using a combination of simulated annealing to sample possible conformations before the docking procedure, and molecular docking calculations.
Keywords: GOLD, Molecular docking, 2-Oxoamides, Phospholipase A2, Simulated annealing
1. Introduction
Phospholipase A2 (PLA2) is a superfamily of enzymes which are characterized by their ability to catalyze the hydrolysis of the sn-2 ester bond of glycerophospholipids releasing free fatty acids, including arachidonic acid, and lysophospholipids.1 Arachidonic acid is supplied to the downstream cyclooxygenases COX-1 and COX-2, and to the 5-lipoxygenase for the production of eicosanoids,2 while lysophospholipids, such as lysophosphatidic acid and lysophosphatidylcholine, and their metabolites, such as platelet activating factor, are potent bioactive mediators, acting through their cognate G-protein-coupled receptors.
Cytosolic PLA2 (cPLA2, GIVA cPLA2) plays an important role in inflammation. Various studies on transgenic mice lacking GIVA cPLA2 showed a 90% reduction in the production of prostaglandins and leukotrienes.3, 4 The biochemistry and the role of sPLA2 enzymes have been recently reviewed by Lambeau and Gelb.5 The 10 known mammalian sPLA2s are the groups: IB, IIA, IIC, IID, IIE, IIF, III, V, X and XIIA. Among the members of the mammalian sPLA2 enzymes, the GIIA sPLA2 is an interesting anti-inflammatory drug target because of its potential role in a number of different inflammatory diseases.7
GIIA sPLA2 is a low molecular weight enzyme (14 kDa) with seven disulphide bonds, and was cloned in 1989.6 The crystal structure of the enzyme reveals a highly conserved Ca2+-binding loop and a catalytic dyad consisting of His47/Asp91.7, 8 The substrate hydrolysis proceeds through the activation of a water molecule by the catalytic histidine and subsequent attack of the sn-2 ester carbonyl carbon. Besides this histidine, there is an aspartate residue (Asp48), which together with the other residues of the Ca2+-binding loop (Gly29, Gly31 and His27), act as a ligand cage for the calcium ion. The crystal structure of GIIA sPLA2 enzyme has also defined a conserved active site with a hydrophobic region lined near the N-terminal helix.7, 8
Since PLA2 enzymes have been associated with various inflammatory diseases, the elucidation of their biological roles through the use of small synthetic inhibitors is of high importance. Most recently, Magrioti and Kokotos reviewed the various classes of PLA2 inhibitors discussing their potential role as new anti-inflammatory agents.9 The design of novel GIIA sPLA2 inhibitors may be accomplished using computational methods which are greatly aided by the availability of a number of GIIA sPLA2 crystal structures with or without a ligand bound in the active site. Among the various reported crystal structures of the enzyme deposited in the RCSB protein data bank are the crystal structures with PDB IDs: 1DB4,10 1DB5,10 1J1A11 and 1KVO.12 However, the optimization of the activity of the GIIA sPLA2 inhibitors, requires the better understanding of the receptor-ligand interactions and the development of a strategy to predict the inhibitory activity of new molecules. Techniques based on the field of computational chemistry are very helpful in this case. Previous computational research works on the GIIA sPLA2 inhibitors include molecular docking calculations,13 molecular dynamics simulations (MD)14 and three-dimensional quantitative structure-activity relationship (3D-QSAR) methodologies.15, 16 Among these techniques molecular docking is a powerful technique, which contributes to the understanding of the stereoelectronic factors that affect the inhibitory activity of small molecules against a target receptor.17–19
Recently, a novel class of GIVA cPLA2 inhibitors, the 2-oxoamides (Figure 1), has been developed.20–27 Long chain 2-oxoamides based on δ- and γ-amino acids (m=1 and m=0, respectively) are potent inhibitors of the GIVA cPLA2 enzyme showing interesting in vivo anti-inflammatory and analgesic activity.21, 25 Notably, such 2-oxoamides containing a free carboxyl group do not inhibit the other major intracellular PLA2 enzyme, the Ca+2-independent GVIA iPLA2,24 although a cross-reactivity might be expected because both GIVA cPLA2 and GVIA iPLA2 enzymes are serine hydrolases and share a common catalytic mechanism. However, a few 2-oxoamides displayed some inhibition, although weak, of GV sPLA2.25
Figure 1.

General structure of 2-oxoamide inhibitors of GIVA cPLA2.
The aim of this work was to identify 2-oxoamides able to inhibit sPLA2s. In the present report, the synthesis of new 2-oxoamides based on α-amino acids and the in vitro evaluation against three human sPLA2s (GIIA, GV and GX sPLA2) are described. To understand the binding mode of 2-oxoamides to GIIA sPLA2, molecular docking calculations were performed.
2. Results and discussion
2.1 Synthesis of inhibitors
The synthesis of the new 2-oxoamides is depicted in Schemes 1 and 2. The methyl ester of (S)-leucine (1a) was coupled with 2-hydroxyhexadecanoic acid using 1-(3-dimethylaminopropyl)-3-ethyl carbodiimide (WSCI) as a condensing agent in the presence of 1-hydroxybenzotriazole (HOBt) (Scheme 1). After saponification of compound 2a, oxidation of the 2-hydroxyamide 3a was carried out by the NaOCl/AcNH-TEMPO method21 leading to the target compound GK126. Starting from the ethyl ester of (R)-leucine (1b), compound GK145, the enantiomer of GK126 was prepared. Compound GK144 was synthesized starting from the methyl ester of (S)-leucine by coupling with hexadecanoic acid and subsequent saponification.
Scheme 1.

Reagents and conditions: (a) CH3(CH2)13CHOHCOOH, Et3N, WSCI, HOBt, CH2Cl2; (b) 1 N NaOH, dioxane/H2O 9:1; (c) NaOCl, AcNH-TEMPO, NaBr, NaHCO3, EtOAc/PhCH3/H2O 3:3:0.5, 0 °C.
Scheme 2.

Reagents and conditions: (a) CH3(CH2)13CHOHCOOH, Et3N, WSCI, HOBt, CH2Cl2; (b) Dess-Martin reagent, CH2Cl2; (c) 50% TFA/CH2Cl2.
Compounds GK111, GK112, GK122 and GK141 were prepared by coupling 2-hydroxyhexadecanoic acid with tert-butyl glycinate (4a), β-alaninate (4b), δ-aminovalerate (4c) and (S)-phenylalaninate (4d), respectively (Scheme 2). Oxidation of 2-hydroxyamides 5a–d was carried out by the Dess-Martin method,28 followed by treatment of compounds 6a–d with trifluoroacetic acid, to afford the target compounds. The synthesis of AX115 is described elsewhere.26
2.2 In vitro inhibition of GIIA sPLA2, GV sPLA2 and GX sPLA2
The activity of compounds GK111, GK112, GK122, GK126, GK141, GK144, GK145, and AX115 was studied against three different human enzymes using a continuous fluorimetric assay described previously.29 The results for GIIA sPLA2, GV sPLA2 and GX sPLA2 are summarized in Table 1. 2-Oxoamides GK111, GK112 and GK122, based on glycine, β-alanine and δ-aminovaleric acid, respectively, inhibited GIIA sPLA2 in the micromolar range (IC50 4.20-11.67 µM). At 16.6 µM concentration, none of them showed any inhibition of GV sPLA2 and GX sPLA2. It seems that for the inhibition of GIIA sPLA2, the optimum distance between the 2-oxoamide group and the carboxyl group corresponds to one carbon atom. Increasing the length leads to less potent inhibitors. Comparing the results for GK111 and AX115, it was obvious that a free carboxyl group was necessary for the inhibition of GIIA sPLA2. The introduction of a side chain corresponding to leucine increased the inhibitory activity for GIIA sPLA2 by an order of magnitude. GK126 inhibited GIIA sPLA2 with an IC50 value of 0.30 µM. This 2-oxoamide also inhibited GV sPLA2 at the same level (IC50 0.44 µM), while it did not show any inhibition of GX sPLA2. The inhibitory activity of GK126 was also measured against the corresponding mouse enzymes. Similar IC50 values for mouse GIIA sPLA2 and GV sPLA2 were found (0.18 µM and 2.60 µM, respectively). Compound GK145, based on (R)-leucine, displayed six times lower potency for GIIA sPLA2 and four times lower potency for GV sPLA2, indicating that the (S)-configuration of the α-amino acid makes more favorable contacts with the enzyme active site. Replacement of either the 2-oxoamide functionally of GK126 by an amide functionality (GK144) or the side chain by a chain corresponding to phenylalanine (GK141) led to inactive compounds. The most potent inhibitor against sPLA2 (GK126) did not present any significant inhibition against cPLA2 (not higher than 12% inhibition at 1µµ). Thus, we identified a 2-oxoamide based on the natural α-amino acid leucine, which displayed submicromolar inhibition of GIIA sPLA2.
Table 1.
In vitro inhibition of the enzymatic activity of human GIIA, GV and GX sPLA2s by 2-oxoamides.
| Code | Structure | IC50 (µM) | ||
|---|---|---|---|---|
| hGIIA sPLA2 | hGV sPLA2 | hGX sPLA2 | ||
| GK111 |
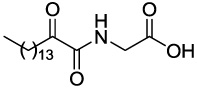
|
4.20 ± 0.20 | 43%a | 23%a |
| GK112 | 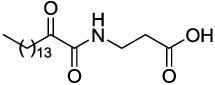 |
5.60 ± 0.25 | 47%a | 18%a |
| GK122 | 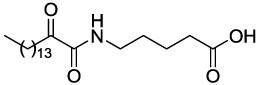 |
11.67 ± 0.50 | 49%a | 7%a |
| AX115 | 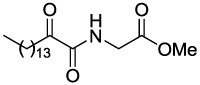 |
3%a | 9%a | 22%a |
| GK126 | 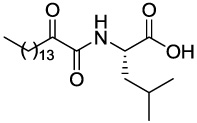 |
0.30 ± 0.06 (0.18 ± 0.04)b | 0.44 ± 0.04 (2.60 ± 0.33)c | 50%a (58%a)d |
| GK145 | 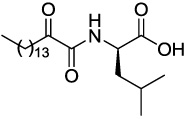 |
1.75 ± 0.07 | 1.50 ± 0.23 | >1.66 |
| GK144 | 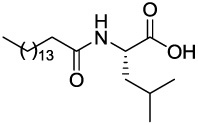 |
>1.66 | >1.66 | >1.66 |
| GK141 | 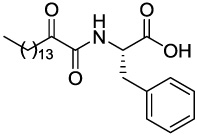 |
>1.66 | >1.66 | >1.66 |
At 16.6 µM concentration.
For mouse GIIA sPLA2.
For mouse GV sPLA2.
For mouse GX sPLA2.
2.3 Molecular docking calculations
The 2-oxoamide inhibitors were initially designed to interact with the hydroxyl group of the active site serine of GIVA cPLA2.20 Recently, the location of the 2-oxoamide inhibitor AX007 within the active site of GIVA cPLA2 was determined by a combination of molecular dynamics and deuterium exchange mass spectrometry.30 However, the binding mode of 2-oxoamides with GIIA sPLA2 is not obvious because GIIA sPLA2 is not a serine hydrolase and it utilizes a different catalytic mechanism than GIVA cPLA2. Thus, to understand how the 2-oxoamides interact with GIIA sPLA2, we decided to perform molecular docking calculations. Four 2-oxoamide inhibitors based on α-amino acids, GK126, GK145, GK111 and GK141, were selected for this study. The molecular docking calculations were performed using the genetic docking algorithm GOLD 4.1.31–33
The active site of GIIA sPLA2 consists of a hydrophilic region and a hydrophobic region. The hydrophilic region, where catalytic activity occurs, is formed by the residues His47 and Asp91, and by the Ca2+-binding loop. The Ca2+-binding loop consists of the residues Gly29, Gly31, His27 and Asp48. The calcium ion is hepta-coordinated with a pentagonal bipyramidal geometry providing two binding sites for the substrate or the inhibitor.34 The hydrophobic region, which binds the fatty acid tails of the substrate, is formed by aliphatic and aromatic residues within or closed to the N-terminal helix, including Leu2, Phe5, Ile9, Ala17, Ala18, Tyr21 and Phe98.
Four GIIA sPLA2 inhibitor X-ray structures have been selected in order to test if GOLD 4.1 is able to reproduce experimental crystallographic data (see Table 1 in Supplementary Data).10–12 For each inhibitor, the best score pose has been selected to compare with the crystallographic one. The RMSD values after the superimposition of the crystallographic conformation and the conformation predicted by GOLD are smaller or equal to 1.0 Å, indicating that the two conformations are almost identical. Thus, the main interactions of each inhibitor with the GIIA sPLA2 active site, reported in the literature,10–12 were reproduced by GOLD.
Six inhibitors (Table 2) have been chosen for the molecular docking studies since they possess a wide variety of biological activity. The high flexibility of the molecules imposed the use of a conformational sampling in an attempt to obtain low-energy conformers and to avoid false positives.35 For this purpose the simulated annealing module in the SYBYL 8.0 molecular modeling package36 was used in order to generate 100 annealed structures for each inhibitor, before the docking procedure. The annealed structures have subsequently been docked in the enzyme active site using GOLD. For each inhibitor the best score pose was chosen as the one that represents better the putative bioactive conformation. The docking results are summarized in Table 2. A good correlation between the IC50 values and the GOLDscore Fitness values (r2 = 0.798, N = 5) was observed. Five inhibitors with an experimentally determined IC50 value have been used for the correlation procedure. However, the present study has focused on 2-oxoamides which are based on α-amino acids (GK126, GK145, GK111 and GK141). The most active inhibitor GK126 is scored with the highest GOLDscore Fitness. The inhibitor GK145, which is the (R)-enantiomer of GK126, shows both a higher IC50 value against GIIA sPLA2 and a lower GOLDscore Fitness than the inhibitor GIK126. The GK111 inhibitor displays a higher IC50 value and a lower GOLDscore Fitness than those of the inhibitors GK126 and GK145. The lowest GOLDscore Fitness was obtained for inhibitor GK141, which also showed the lowest inhibition against GIIA sPLA2.
Table 2.
The GOLDscore Fitness for the GIIA sPLA2 2-oxoamide inhibitors based on the α-amino acids.
| Compound | Structure | IC50 (µM) | Gsc. Fit. |
|---|---|---|---|
| GK126 (S)-Leu |
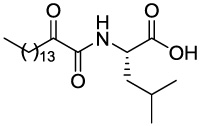
|
0.30 ± 0.06 | 85.91 |
| GK145 (R)-Leu |  |
1.75 ± 0.07 | 84.91 |
| GK111 Gly |  |
4.20 ± 0.20 | 78.89 |
| GK112 | 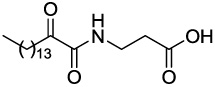 |
5.60 ± 0.25 | 77.33 |
| GK122 | 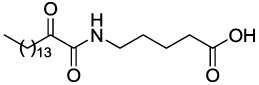 |
11.67 ± 0.50 | 75.69 |
| GK141 (S)-Phe | 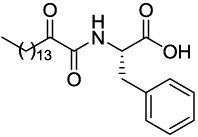 |
>1.60 | 73.09 |
Using the inhibitor-enzyme complexes which were calculated using GOLD, it was possible to understand the main stereoelectronic characteristics that affect the inhibition of these compounds. Figure 2 presents the binding of the inhibitor GK126 in the GIIA sPLA2 active site. The 2-oxoamide group participates in two hydrogen bonds with Gly29. The 2-carbonyl group participates in a hydrogen bond with the N-H of Gly29 (C=O…H-N 2.50 Å, O…N 3.30 Å) and the N-H of the 2-oxoamide group forms a hydrogen bond with the carbonyl group of Gly29 (N-H… O=C 1.65 Å, N…O 2.65). The 2-carbonyl group also interacts with the calcium ion (NHCOCO…Ca2+ 2.40 Å). The carboxylate group of the inhibitor interacts with the calcium ion (COO−…Ca2+ 2.87 Å) and is involved in a hydrogen bond with Lys62 through a water molecule placed near the hydrophilic region of the active site (O…H-OH 1.94 Å, O…O 2.90 Å and H2O…H-N 1.89 Å, O…N 2.89 Å). The side chain of (S)-leucine is in a suitable orientation to interact with Leu2 of the active site in aliphatic/aliphatic interactions. The long aliphatic 2-oxoacyl chain is accommodated in the hydrophobic region of the active site and participates in aliphatic/aliphatic and in aliphatic/aromatic interactions with residues Val3, Phe5, His6, Ile9, Ala17 and Phe98.
Figure 2.

The binding mode of the inhibitor GK126 in the GIIA sPLA2 active site as calculated using GOLD.
As aforementioned, the inhibitor GK145 is the (R)-enantiomer of the inhibitor GK126. According to the structure for GK145-GIIA sPLA2 calculated by GOLD (Figure 3) the amide carbonyl of the 2-oxoamide group interacts with the calcium ion (NHCO…Ca2+ 2.30 Å). With regards to the inhibitor GK145, the 2-oxoamide group is flipped in comparison with that of GK126. As a result, the two hydrogen bonds with Gly29, in which participates the 2-oxoamide functionality of GK126, are not observed upon binding of GK145. This may be the reason that the inhibitor GK145 generates a lower GOLDscore Fitness and a higher IC50 value against GIIA sPLA2. On the other hand, the carboxylate group of the inhibitor interacts with the calcium ion (COO−…Ca2+ 2.65 Å), but does not participate in the hydrogen bond with Lys62 through the water molecule. Instead of the hydrogen bond with Lys62, a hydrogen bond of the carboxylate group with a water molecule near the hydrophilic region of the active site is observed. The side chain of (R)-leucine is placed near Leu2 of the active site and interacts in a similar mode as the (S)-leucine of inhibitor GK126. The long aliphatic 2-oxoacyl chain is accommodated in the hydrophobic region and interacts with the residues Val3, Phe5, His6, Ile9, Ala17 and Phe98 in a similar mode as the one of the enantiomeric GK126.
Figure 3.

The binding mode of the inhibitor GK145 in the GIIA sPLA2 active site as calculated using GOLD.
The inhibitor GK111 is based on the α-amino acid glycine. The binding of GK111 in the GIIA sPLA2 active site (Figure 4) reveals an interaction of the 2-carbonyl group of the inhibitor with the calcium ion (NHCOCO…Ca2+ 2.70 Å). The carboxylate group interacts with the calcium ion (COO− …Ca2+ 2.90 Å) and participates in two hydrogen bonds with Gly31 (COO−…H-N 2.40 Å, O…N 3.40 Å) and Lys62 through a water molecule (O…H-OH 1.91 Å, O…O 2.89 Å and H2O…H-N 1.89 Å, O…N 2.89 Å). In comparison with the binding of the inhibitor GK126, the two hydrogen bonds of the 2-oxoamide group with Gly29 are not observed. The lack of the (S)-leucine side chain, which interacts with Leu2 of the active site, also reduces the GOLDscore Fitness and increases the IC50 value against GIIA sPLA2. The long aliphatic 2-oxoacyl chain interacts with the residues Phe5, His6, Ile9, Ala17 and Phe98.
Figure 4.

The binding mode of the inhibitor GK111 in the GIIA sPLA2 active site as calculated using GOLD.
The binding of the inhibitor GK141 (Figure 5), which is based on the α-amino acid (S)-phenylalanine, indicates a hydrogen bond of the N-H of the 2-oxoamide group with Gly29 (NH…O 1.97 Å, N…O 2.98 Å). The 2-carbonyl group of the inhibitor interacts with the calcium ion (NHCOCO…Ca2+ 2.60 Å). The carboxylate group interacts with the calcium ion (COO−…Ca2+ 2.64 Å) and participates in two hydrogen bonds with Gyl31 (COO−…H-N 1.67 Å, O…N 2.60 Å) and with Lys62 through a water molecule (O…H-OH 1.60 Å, O…O 2.56 Å and H2O…H-N 1.89 Å, O…N 2.89 Å). The 2-carbonyl group of GK141 does not participate in the hydrogen bond with Gly29 as the one of the inhibitor GK126. The side chain of (S)-Phe seems to participate in aromatic/aliphatic and aromatic (π-π) stacking interactions with Leu2, Phe5, Tyr51 and His47. On the other hand, the region around the side chain of (S)-phenylalanine seems to be narrow and perhaps the phenyl ring clashes with the aforementioned residues; this may be the reason that GK141 gives the lowest GOLDscore Fitness among the studied 2-oxoamide inhibitors. The long aliphatic 2-oxoacyl chain participates in aliphatic/aliphatic and aliphatic/aromatic interactions with residues including Phe5, His6, Ile9 and Phe98.
Figure 5.

The binding mode of the inhibitor GK141 in the GIIA sPLA2 active site as calculated using GOLD.
Based on the molecular docking studies, it was possible to understand the structural characteristics that contribute in the enhancement of the inhibitory activity of the 2-oxoamide GK126 (Figure 6). The 2-oxoamide group is essential for the binding because it participates in hydrogen bonds with Gly29 and interacts with the calcium ion. The (R)-enantiomer displays a lower GOLDscore and a higher IC50 value, because the 2-oxoamide functionality is flipped and does not engage in hydrogen bonding with Gly29. The carboxylate group is also essential for binding because interacts with the calcium ion and participates in a hydrogen bond with Lys62 through a water molecule placed near the hydrophilic region of the active site. The (S)-leucine side chain contributes to the tight binding of GK126 by interacting with the side chain of the active site Leu2. The inhibitor GK111 lacks the (S)-leucine side chain and as a consequence its inhibitory activity is decreased. The long 2-oxoacyl aliphatic chain contributes to the tight binding by interacting with the hydrophobic region of the active site (Val3, Phe5, His6, Ile9, Ala17 and Phe98).
Figure 6.

The pharmacophore segments of the inhibitor GK126 and their interactions with the surrounding amino acids and the calcium ion of the active site.
A variety of sPLA2 inhibitors have been reported in literature.5, 9, 37 Gelb and et al. have studied various inhibitors for their selectivity over the complete set of human and mouse sPLA2s (groups I, II, V, X, and XIIA sPLA2).38 They have reported a highly potent and selective indole-based inhibitor of GX sPLA2, specific inhibitors for GIIA and GIIE sPLA2 and a substituted 6,7-benzoindole that inhibits nearly all human and mouse sPLA2s in the low nanomolar range.39 In recent years, interest in sPLA2 inhibitors has increased because they seem to play an important role in the prevention of atherosclerotic cardiovascular disease.40, 41 In the present work, we demonstrate that a new 2-oxoamide is able to inhibit human GIIA sPLA2 in the submicromolar range and the mode of its interaction with the GIIA sPLA2 has been studied using molecular docking. These data indicate that this compound constitutes a new lead for the development of new GIIA sPLA2 inhibitors.
3. Conclusion
2-Oxoamide derivatives based on α-amino acids have been synthesized and tested for their in vitro inhibitory activity against three human sPLA2s (GIIA, GV and GX). Compound GK126, which is based on (S)-leucine, displayed inhibition of human and mouse GIIA sPLA2 (IC50 300 nM and 180 nM, respectively). It also inhibited the human GV sPLA2 with similar potency, while it did not display any measurable inhibition of GX sPLA2. Using a combination of simulated annealing and molecular docking calculations, it was possible to explore the inhibitor-enzyme complexes and rationalize the stereoelectronic characteristics that affect the inhibition potency of these compounds. The annealed structures resulted from the investigation of the conformational space of each inhibitor were docked in the enzyme active site. The inhibitor GK126, which is the most active compound among the inhibitors tested, is scored with the highest GOLDscore Fitness. The 2-oxoamide functionality is essential for the binding by interacting with the calcium ion and by participating in two hydrogen bonds with Gly29.
4. Experimental
4.1 Chemistry
Melting points were determined on a Buchi 530 apparatus and are uncorrected. Specific rotations were measured on a Perkin-Elmer 343 polarimeter using a 10 cm cell. NMR spectra were recorded on a Varian Mercury spectrometer. 1H and 13C NMR spectra were recorded at 200 MHz and 50 MHz respectively in CDCl3 or as specified. Chemical shifts are given in ppm, and peak multiplicities are described as follows: s, singlet, d, doublet, t, triplet and m, multiplet. Electron spray ionization (ESI) mass spectra were recorded on a Finnigan, Surveyor MSQ Plus spectrometer. TLC plates (Silica Gel 60 F254) and Silica Gel 60 (70–230 or 230–400 mesh) for column chromatography were purchased from Merck. Spots were visualised with UV light and/or phosphomolybdic acid and/or ninhydrin, both in EtOH. Dichloromethane was dried by standard procedures and stored over molecular sieves. All other solvents and chemicals were reagent grade and used without further purification.
The synthesis of inhibitor AX115 and of compounds 5a–c and 6a–c has been described elsewhere. 26
4.2 Synthesis of the 2-oxoamide inhibitors
4.2.1 General method for the coupling of 2-hydroxyhexadecanoic acid with amino components
To a stirred solution of 2-hydroxyhexadecanoic acid (1.0 mmol) and hydrochloride amino component (1.0 mmol) in CH2Cl2 (10 mL), Et3N (0.3 mL, 2.2 mmol) and subsequently 1-(3-dimethylaminopropyl)-3-ethyl carbodiimide hydrochloride (WSCI) (0.21 g, 1.1 mmol) and 1-hydroxybenzotriazole (HOBt) (0.14 g, 1.0 mmol) were added at 0 °C. The reaction mixture was stirred for 1 h at 0 °C and overnight at room temperature. The solvent was evaporated under reduced pressure and EtOAc (20 mL) was added. The organic layer was washed consecutively with brine, 1 N HCl, brine, 5% NaHCO3, and brine, dried over Na2SO4 and evaporated under reduced pressure. The residue was purified by column chromatography using CH2Cl2/MeOH 99:1 as eluent.
4.2.1.1 (2S)-Methyl 2-(2-hydroxyhexadecanamido)-4-methylpentanoate (2a)
Yield 79%; white solid; mp 47–49 °C; 1H NMR (200 MHz, CDCl3): δ 7.02-6.82 (m, 1H), 4.70-4.55 (m, 1H), 4.20-4.10 (m, 1H), 3.73 (s, 3H), 3.07 (s, 1H), 1.81-1.53 (m, 5H), 1.39-1.24 (m, 24H), 0.89 (m, 9H); 13C NMR (50 MHz, CDCl3): δ 174.15, 173.91, 72.05, 52.30, 50.27, 41.40, 34.74, 34.64, 31.89, 29.66, 29.63, 29.54, 29.36, 29.33, 24.82, 22.80, 22.66, 21.76, 21.67, 14.09. Anal. Calcd for C23H45NO4: C, 69.13; H, 11.35; N, 3.51. Found: C, 68.99; H, 11.48; N, 3.59.
4.2.1.2 (2R)-Ethyl 2-(2-hydroxyhexadecanamido)-4-methylpentanoate (2b)
Yield 64%; white solid; mp 34–35 °C; 1H NMR (200 MHz, CDCl3): δ 7.00-6.90 (m, 1H), 4.60-4.55 (m, 1H), 4.15-4.00 (m, 3H), 1.78-1.58 (m, 6H), 1.41-1.22 (m, 27H), 0.90 (m, 9H); 13C NMR (50 MHz, CDCl3): δ 174.27, 173.27, 72.08, 61.37, 50.37, 41.44, 34.74, 34.64, 31.86, 29.63, 29.52, 29.37, 29.29, 24.88, 24.83, 22.79, 22.62, 21.80, 21.71, 14.03. Anal. Calcd for C24H47NO4: C, 69.69; H, 11.45; N, 3.39. Found: C, 69.57; H, 11.56; N, 3.44.
4.2.1.3 (S)-Ethyl 4-methyl-2-palmitamidopentanoate
Yield 79%; white solid; mp 51–52 °C; 1H NMR (200 MHz, CDCl3): δ 6.00 (d, J=8.4 Hz, 1H), 4.67-4.52 (m, 1H), 4.15 (q, J=7.1 Hz, 2H), 2.18 (t, J=7.5 Hz, 2H), 1.57 (m, 5H), 1.24 (m, 27H), 1.00-0.85 (m, 9H); 13C NMR (50 MHz, CDCl3): δ 173.25, 172.83, 61.12, 50.51, 41.71, 36.47, 31.83, 29.59, 29.56, 29.51, 29.41, 29.26, 29.14, 25.54, 24.80, 22.72, 22.58, 21.91, 14.03, 14.00. Anal. Calcd for C24H47NO3: C, 72.49; H, 11.91; N, 3.52. Found: C, 72.34; H, 12.09; N, 3.41.
4.2.1.4 (2S)-tert-Butyl 2-(2-hydroxyhexadecanamido)-3-phenylpropanoate (5d)
Yield 57%; white solid; mp 55–56 °C; 1H NMR (200 MHz, CDCl3): δ 7.30-7.2 (m, 5H Ar), 4.80-4.68 (m, 1H), 4.07-4.01 (m, 1H), 3.15-2.97 (m, 2H), 1.80-1.46 (m, 2H), 1.37 (s, 9H), 1.23 (s, 24H), 0.84 (t, J=6.7 Hz, 3H); 13C NMR (50 MHz, CDCl3): δ 173.60, 170.63, 135.78, 129.09, 128.03, 126.61, 82.13, 71.74, 52.90, 37.87, 34.47, 31.59, 29.37, 29.22, 29.12, 29.04, 27.57, 24.61, 22.36, 13.80; MS (ESI) m/z (%): (48) 476.3 (M+H)+, 420.3 (100) (M-tBu+H+H)+. Anal. Calcd for C29H49NO4: C, 73.22; H, 10.38; N, 2.94. Found: C, 73.05; H, 10.46; N, 3.04.
4.2.2 General method for the saponification of 2-hydroxyamides
To a stirred solution of a 2-hydroxyamide ester (1.00 mmol) in a mixture of dioxane-H2O (9:1, 10 mL) was added 1 N NaOH (1.1 mL, 1.1 mmol), and the mixture was stirred for 12 h at room temperature. The organic solvent was evaporated under reduced pressure, and H2O (5 mL) was added. The aqueous layer was washed with EtOAc, acidified with 1 N HCl, and extracted with EtOAc (3 × 6 mL). The combined organic layers were washed with brine, dried over Na2SO4, and evaporated under reduced pressure.
4.2.2.1 (2S)-2-(2-Hydroxyhexadecanamido)-4-methylpentanoic acid (3a)
Yield 89%; white solid; 1H NMR (200 MHz, CDCl3): δ 7.80 (br s, 1H), 7.40-7.30 (m, 1H), 4.44 (m, 1H), 4.08 (m, 1H), 1.80-1.50 (m, 6H), 1.45-1.10 (m, 24H), 0.89 (m, 9H); 13C NMR (50 MHz, CDCl3): δ 176.59, 175.40, 72.19, 71.98, 51.21, 40.56, 40.39, 34.47, 31.95, 29.74, 29.68, 29.48, 29.37, 25.27, 25.15, 24.97, 23.02, 22.68, 21.44, 21.26, 14.10. Anal. Calcd for C22H43NO4: C, 68.53; H, 11.24; N, 3.63. Found: C, 68.41; H, 11.42; N, 3.51.
4.2.2.2 (2R)-2-(2-Hydroxyhexadecanamido)-4-methylpentanoic acid (3b)
Yield 54%; white solid; 1H NMR (200 MHz, CDCl3): δ 7.80 (br s, 1H), 7.40-7.30 (m, 1H), 4.46 (m, 1H), 4.07 (m, 1H), 1.80-1.50 (m, 6H), 1.45-1.10 (m, 24H), 0.89 (m, 9H); 13C NMR (50 MHz, CDCl3): δ 176.57, 175.38, 72.20, 71.98, 51.15, 40.55, 40.37, 34.45, 31.92, 29.73, 29.67, 29.47, 29.36, 25.26, 25.13, 24.96, 23.02, 22.67, 21.43, 21.25, 14.09. Anal. Calcd for C22H43NO4: C, 68.53; H, 11.24; N, 3.63. Found: 68.43; H, 11.32; N, 3.51.
4.2.2.3 (S)-4-Methyl-2-palmitamidopentanoic acid (GK144)
Yield: 100%; white solid; mp 93–95 °C; 1H NMR (200 MHz, CDCl3): δ 10.46 (s, 1H), 6.22 (d, J=8.2 Hz, 1H), 4.70-4.55 (m, 1H), 2.24 (t, J=7.6 Hz, 2H), 1.80-1.45 (m, 5H), 1.40-1.10 (m, 24H), 1.00-0.85 (m, 9H); 13C NMR (50 MHz, CDCl3) δ 176.47, 174.23, 50.83, 41.24, 36.44, 31.89, 29.66, 29.48, 29.32, 29.18, 25.63, 24.86, 22.79, 22.64, 21.85, 14.06. Anal. Calcd for C22H43NO3: C, 71.50; H, 11.73; N, 3.79. Found: C, 71.31; H, 11.89; N, 3.70.
4.2.3 General method for the oxidation of 2-hydroxyamides (Method A)
To a solution of 2-hydroxyamide (1.0 mmol) in a mixture of toluene (3 mL) and EtOAc (3 mL), a solution of NaBr (0.11 g, 1.1 mmol) in water (0.5 mL) was added followed by AcNH-TEMPO (2.2 mg, 0.01 mmol). To the resulting biphasic system, which was cooled at 0 °C, an aqueous solution of 0.35 M NaOCl (3.1 mL, 1.1 mmol) containing NaHCO3 (0.25 g, 3 mmol) was added dropwise under vigorous stirring, at 0 °C over a period of 1 h. After the mixture had been stirred for a further 15 min at 0 °C, EtOAc (10 mL) and H2O (10 mL) were added. The aqueous layer was separated and washed with EtOAc (2 × 10 mL). The combined organic layers were washed consecutively with 5% aqueous citric acid (10 mL) containing KI (0.04 g), 10% aqueous Na2S2O3 (10 mL), and brine and dried over Na2SO4. The solvents were evaporated under reduced pressure and the residue was purified by column chromatography using petroleum ether/EtOAc 2:8 as eluent.
4.2.3.1 (S)-4-Methyl-2-(2-oxohexadecanamido)pentanoic acid (GK126)
Yield 29%; yellow oil; 1H NMR (200 MHz, CDCl3): δ 7.46 (m, 1H), 4.51 (m, 1H), 2.91 (t, J = 7.0 Hz, 2H), 1.80-1.40 (m, 5H), 1.40-1.10 (m, 22H), 0.90 (m, 9H); 13C NMR (50 MHz, CDCl3): δ 198.68, 170.47, 160.36, 51.40, 40.83, 36.87, 31.91, 29.68, 29.49, 29.42, 29.36, 29.08, 24.86, 23.07, 22.92, 22.67, 21.53, 14.09; MS (ESI) m/z (%): 384.3 (M+H)+. Anal. Calcd for C22H41NO4: C, 68.89; H, 10.77; N, 3.65. Found: C, 68.75; H, 10.89; N, 3.75.
4.2.3.2 (R)-4-Methyl-2-(2-oxohexadecanamido)pentanoic acid (GK145)
Yield 35%; yellow oil; 1H NMR (200 MHz, CDCl3): δ 7.38 (m, 1H), 4.56 (m, 1H), 2.91 (t, J = 7.0 Hz, 2H), 1.80-1.45 (m, 5H), 1.40-1.10 (m, 22H), 0.90 (m, 9H); 13C NMR (50 MHz, CDCl3): δ 198.58, 177.46, 160.29, 51.18, 40.82, 36.85, 34.74, 31.90, 29.67, 29.47, 29.40, 29.34, 29.05, 28.87, 27.27, 24.85, 23.05, 22.88, 22.66, 21.50, 14.09; MS (ESI) m/z (%): 384.3 (M+H)+. Anal. Calcd for C22H41NO4: C, 68.89; H, 10.77; N, 3.65. Found: C, 68.72; H, 10.89; N, 3.76.
4.2.4 General method for the oxidation of 2-hydroxyamides (Method B)
To a solution of 2-hydroxyamide (1 mmol) in dry CH2Cl2 (10 mL) Dess-Martin periodinane was added (0.64 g, 1.5 mmol) and the mixture was stirred for 1 h at room temperature. The organic solvent was evaporated under reduce pressure and Et2O (30 mL) was added. The organic phase was washed with saturated aqueous NaHCO3 (20 mL) containing Na2S2O3 (1.5 g, 9.5 mmol), H2O (20 mL), dried over Na2SO4 and the organic solvent was evaporated under reduced pressure. The residue was purified by column chromatography using petroleum ether (bp 40–60 °C)/EtOAc 9:1 as eluent.
4.2.4.1 (S)-tert-Butyl 2-(2-oxohexadecanamido)-3-phenylpropanoate (6d)
Yield 63%; white solid; mp 46–47 °C; [α]D = +29.0° (c = 1, CH2Cl2); 1H NMR (200 MHz, CDCl3): δ 7.34 (d, J=8.4 Hz, 1H) 7.30-7.08 (m, 5H Ar), 4.72-4.62 (m, 1H), 3.08 (d, J=6.3 Hz, 2H), 2.84 (t, J=7.3 Hz, 2H), 1.62-1.45 (m, 2H), 1.37 (s, 9H), 1.23 (s, 22H), 0.84 (t, J=6.7 Hz, 3H); 13C NMR (50 MHz, CDCl3): δ 198.23, 169.48, 159.45, 135.61, 129.25, 128.35, 126.97, 82.45, 53.49, 37.96, 36.56, 31.81, 29.54, 29.48, 29.32, 29.25, 29.21, 28.93, 27.77, 23.06, 22.58, 14.01; MS (ESI) m/z (%): 472.5 (M−H)−. Anal. Calcd for C29H47NO4: C, 73.53; H, 10.00; N, 2.96. Found: C, 73.41; H, 10.16; N, 3.09.
4.2.5 General method for the cleavage of tert-butyl ester
A solution of the tert-butyl ester derivative (1 mmol) in 50% TFA/CH2Cl2 (0.5 M) was stirred for 1 h at room temperature. The organic solvent was evaporated under reduced pressure. The residue was purified by recrystallization [EtOAc/petroleum ether (bp 40–60 °C)].
4.2.5.1 2-(2-Oxohexadecanamido)acetic acid (GK111)
Yield 90%; white solid; 1H NMR (200 MHz, CDCl3): δ 5.00 (m, 1H), 3.95 (d, J = 4.0 Hz, 2H), 2.85 (t, J = 7.0 Hz, 2H), 1.80-1.50 (m, 4H), 1.40-1.15 (m, 20H), 0.90 (m, 3H); 13C NMR (50 MHz, CD3OD): δ 197.85, 172.79, 171.34, 40.44, 40.30, 38.31, 36.58, 31.91, 29.61, 29.44, 29.37, 29.32, 29.01, 23.20, 23.07, 22.57, 13.30; MS (ESI) m/z (%): 326.4 (M−H)−. Anal. Calcd for C18H33NO4: C, 66.02; H, 10.16; N, 4.28. Found: C, 65.89; H, 10.34; N, 4.15.
4.2.5.2 3-(2-Oxohexadecanamido)propanoic acid (GK112)
Yield 75%; white solid; 1H NMR (200 MHz, CDCl3): δ 4.90 (b, 1H), 3.48 (t, J=7.0 Hz, 2H), 2.82 (t, J=7.2 Hz, 2H), 2.54 (t, J=7.0 Hz, 2H), 1.62–1.55 (m, 2H), 1.50-1.05 (m, 22H), 0.88 (t, J=7.0 Hz, 3H); 13C NMR (50 MHz, CDCl3) δ 198.32, 173.91, 160.26, 38.17, 36.59, 34.94, 33.35, 32.98, 31.91, 29.61, 29.42, 29.32, 29.02, 23.36, 23.11, 22.57, 13.29; MS (ESI) m/z (%): 340.3 (100) [M−H]−. Anal. Calcd for C19H35NO4: C, 66.83; H, 10.33; N, 4.10. Found: C, 66.68; H, 10.48; N, 4.19.
4.2.5.3 5-(2-Oxohexadecanamido)pentanoic acid (GK122)
Yield 91%; white solid; mp 103–105 °C; 1H NMR (200 MHz, CDCl3): δ 7.06 (t, J = 5.6 Hz, 1H), 3.39-3.26 (m, 2H), 2.91 (t, J = 7.4 Hz, 2H), 2.40 (t, J = 7.0 Hz, 2H), 1.78-1.48 (m, 6H), 1.26 (s, 22H), 0.88 (t, J = 6.6 Hz); 13C NMR (50 MHz, CDCl3) δ 199.34, 178.56, 160.27, 38.83, 36.73, 33.27, 31.89, 29.62, 29.57, 29.42, 29.32, 29.04, 28.58, 23.16, 22.66, 21.79, 14.09; MS (ESI) m/z (%): 368.3 (100) [M−H]−. Anal. Calcd for C21H39NO4: C, 68.25; H, 10.64; N, 3.79. Found: C, 68.11; H, 10.80; N, 3.84.
4.2.5.4 (S)-2-(2-Oxohexadecanamido)-3-phenylpropanoic acid (GK141)
Yield 95%; white solid; mp 104–105 °C; [α]D = +8.7° (c = 1, CH3OH); 1H NMR (200 MHz, CD3OD): δ 7.31-7.15 (m, 5H Ar), 4.72-4.64 (m, 1H), 3.26-3.23 (m, 1H), 3.12-2.96 (m, 1H), 2.75 (t, J=7.0 Hz, 2H), 1.59-1.46 (m, 2H), 1.29 (s, 22H), 0.90 (t, J=6.7 Hz, 3H); 13C NMR (50 MHz, CD3OD): δ 199.14, 174.29, 162.39, 138.14, 130.29, 129.45, 127.88, 54.54, 39.65, 38.04, 37.74, 33.09, 30.78, 30.68, 30.58, 30.50, 30.13, 24.29, 23.75, 14.46; MS (ESI) m/z (%): 418.2 (100) (M+H)+. Anal. Calcd for C25H39NO4: C, 71.91; H, 9.41; N, 3.35. Found: C, 71.81; H, 9.53; N, 3.22.
4.3 In vitro sPLA2 assay
A continuous fluorimetric assay described previously was used to determine the inhibitory activity of compounds GK111, GK112, GK122, GK126, GK141, GK144, GK145, and AX115.29
4.3.1 IC50 Value Determination
IC50 values for compounds GK111, GK112, GK122, GK126 and GK145 are reported as the mean of duplicate or triplicate analysis with standard deviations. All other compounds were screened at single doses and reported as >1.66 µM, or a % inhibition at 16.6 µM is given. IC50 values were determined by nonlinear regression analysis of a semi-log plot of percent inhibition versus log of inhibitor concentration. Curves were fit to a variable slope sigmoidal dose-response curve using the Kaleidagraph software. Five inhibitor concentrations ranging from 10% to 90% inhibition were used for each IC50 value determination.
4.3.2 Assay Procedure
To seven wells of a 96-well microtiter plate was added 100 µL of solution A (27 µM bovine serum albumin, 50 mM KCl, 1 mM CaCl2, 50 mM Tris-HCl, pH 8.0) followed by the desired concentration of inhibitor (1 µL in DMSO from serial-diluted stock solutions) or 1 µL of DMSO for control reactions. To the first well was added an additional 100 µL of solution A as a negative control. Solution B was prepared by mixing the appropriate amount of sPLA2, depending on the specific activity, to Solution A. This solution was prepared immediately prior to each set of assays, to avoid loss of enzymatic activity due to sticking to the walls of the container. Solution B was delivered in 100 µL portions to all seven wells except the first well. Quickly after the addition of Solution B, the assay was initiated by the addition of 100 µL of Solution C (4.2 µM 1-hexadecanoyl-2-(1-pyrenedecanoyl)-sn-glycero-3-phosphoglycerol (pyrPG, Molecular Probes) vesicles in assay buffer (50 mM KCl, 1 mM CaCl2, 50 mM Tris-HCl, pH 8.0)) to all seven wells. The fluorescence (excitation = 342 nm, emission = 395 nm) was read with a microtiter plate spectrophotometer (Fluorocount, Packard Instruments). Control reactions without enzyme or inhibitor were run with each assay of seven wells and the percent inhibition calculated from the initial slopes of fluorescence versus time. The amount of enzyme used per well are as follows: hGIIA, 6 ng; hGV, 12.5 ng; hGX, 17.7 ng; mGIIA, 3.4 ng; mGV, 12.7 ng; mGX, 10.7 ng. All of the recombinant mouse and human sPLA2s were prepared as previously described.38
4.4. Computational methods
4.4.1 Preparation of the GIIA sPLA2 crystal structure
The crystal structures of the GIIA sPLA2 enzyme which were deposited in the RCSB protein data bank with PDB IDs: 1DB4 holo form 2.20 Å X-ray resolution,10 1DB5 holo form 2.80 Å X-ray resolution,10 1J1A holo form 2.20 Å X-ray resolution,11 1KVO holo form 2.00 Å X-ray resolution12 were downloaded. The objective was to judge which crystal structure is appropriate for the docking calculations. This was determined by using the following procedure: (i) using “Superposition panel” of Maestro 9.042 all the crystal structures were superimposed based on all the backbone atoms including beta carbons. The crystal structure with PDB ID: 1DB4 was chosen as a reference structure for the superposition (RMSD between: 1DB4–1DB5: 0.147 Å, 1DB4-1J1A: 0.746 Å, 1DB4-1KVO: 0.487 Å). No significant structural differences were observed as indicated by the RMSD values (Figure 1 in Supplementary Data); (ii) the active site region was examined to determine if the superimposed ligands can fit into the reference site without steric clashes. No significant steric clashes were observed; (iii) the active site region of all the crystal structures, in turn, was examined in order to determine whether any residues in the superimposed protein differ appreciably in position or conformation from those in the reference site. No significant differences were observed. Even though all crystal structures are suitable, the 1DB4 PDB file was chosen for the molecular docking calculations because this file contains a single unit of the GIIA sPLA2 enzyme co-crystallized with an indole inhibitor. The 1DB4 crystal structure was prepared using the “Protein Preparation Wizard” panel43 of Schrödinger Suite 2009. Using the “Preprocess and analyze structure” tool, the bond orders were assigned, all the hydrogen atoms were added, the calcium ion was treated in order to have the correct geometry and the correct formal charge (+2), the disulfide bonds were assigned, and all the water molecules at a distance greater than 5 Å from any het group were deleted. Using Epik 2.0,44, 45 a prediction of het groups ionization and tautomeric states was performed. An optimization of the hydrogen bonding network was performed using the “H-bond assignment” tool. Finally, using the “Impref utility” the positions of the hydrogen atoms were optimized by keeping all the heavy atoms in place. The prepared structure was saved in PDB file format and was used as the input file in the docking calculations.
4.4.2 Preparation of the ligands
The crystallographic ligands and the 2-oxoamide ligands were sketched using the SYBYL 8.0 molecular sketcher.36 All the hydrogen atoms were added to define the correct ionization and tautomeric states, and the oxygen atoms of the carboxylate and phosphonate groups were considered as equivalent with atom type for the oxygen atoms of O.co2. The molecules were subjected to energy minimization using the standard Tripos46 molecular mechanics force field of the SYBYL 8.0 molecular modeling package, and the Powell47 energy minimization algorithm with a gradient of 0.01 kcal mol−1 Å−1 was used for the minimization procedure.
4.4.3 Simulated annealing method
The investigation of the conformational space of each inhibitor, using the method of simulated annealing48 was performed. Each inhibitor was heated at temperature 2000 K for 2000 fs and was cooled at a temperature 0 K for 10000 fs for 100 cycles. For the simulation, the Tripos46 standard molecular mechanic force field was used. The dielectric function was selected to be distance dependent with a value of 1 for the distance-dependent dielectric constant and nonbonded cutoffs of 8.0 Å. The annealed structures were minimized using the Powell energy minimization algorithm with a gradient of 0.01 kcal mol−1 Å−1. For the simulated annealing the SYBYL 8.036 was used.
4.4.4 Molecular docking procedure
For the molecular docking calculation the genetic algorithm GOLD 4.131 was used along with the standard GOLD scoring function GOLDscore.49 The active site of the GIIA sPLA2 was set as 6.0 Å around the native ligand. All the critical amino acids (Leu2, Val3, Phe5, His6, Ile9, Ala17, Ala18, Tyr21, His27, Gly29, Gly31, Asp48, Tyr51, Lys62 and Phe98) including the calcium ion and the His47/Asp91 catalytic dyad were considered in the active site. For the five water molecules, near the active site, the state “Toggle” was specified, and the orientation of the hydrogen atoms of each water molecule was automatically optimized using the “Spin” option. The geometry of the calcium ion was simulated by GOLD as octahedral with RMSD of 0.465 Å. For highest accuracy of the docking calculations, long search settings were used for the GOLD GA: 100 dockings with 100 000 GA operations per docking; the algorithm was not allowed to terminate early when the same solution was produced repeatedly. The GOLD algorithm supports flexible docking for the ligand, which is not depended on its initial conformation, and provides partial flexibility for the receptor.
Supplementary Material
Acknowledgments
The project is co-funded by the European Social Fund and National Resources (EPEAEK II) (G.K.) and by a grant from the National Institutes of Health (HL36235) (M.H.G.). V. D. Mouchlis was funded by the Research Promotion Foundation (RPF) (bilateral agreement CY-SLO/0407/06).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Burke JE, Dennis EA. Cardiovasc. Drugs Ther. 2009;23:49. doi: 10.1007/s10557-008-6132-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leslie CC. Prostaglandins, Leukotrienes Essent. Fatty Acids. 2004;70:373. doi: 10.1016/j.plefa.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 3.Bonventre JV, Huang Z, Taheri MR, O'Leary E, Li E, Moskowitz MA, Sapirstein A. Nature. 1997;390:622. doi: 10.1038/37635. [DOI] [PubMed] [Google Scholar]
- 4.Uozumi N, Kume K, Nagase T, Nakatani N, Ishii S, Tashiro F, Komagata Y, Maki K, Ikuta K, Ouchi Y, Miyazaki J, Shimizu T. Nature. 1997;390:618. doi: 10.1038/37622. [DOI] [PubMed] [Google Scholar]
- 5.Lambeau G, Gelb MH. Annu. Rev. Biochem. 2008;77:495. doi: 10.1146/annurev.biochem.76.062405.154007. [DOI] [PubMed] [Google Scholar]
- 6.Kramer RM, Hession C, Johansen B, Hayes G, McGray P, Chow EP, Tizard R, Pepinsky RB. J. Biol. Chem. 1989;264:5768. [PubMed] [Google Scholar]
- 7.Scott DL, White SP, Browning JL, Rosa JJ, Gelb MH, Sigler PB. Science. 1991;254:1007. doi: 10.1126/science.1948070. [DOI] [PubMed] [Google Scholar]
- 8.Wery JP, Schevitz RW, Clawson DK, Bobbitt JL, Dow ER, Gamboa G, Goodson T, Jr, Hermann RB, Kramer RM, McClure DB, Mihelich ED, Putnam JE, Sharp JD, Stark DH, Teater C, Warrick MW, Jones ND. Nature. 1991;352:79. doi: 10.1038/352079a0. [DOI] [PubMed] [Google Scholar]
- 9.Magrioti V, Kokotos G. Expert Opin. Ther. Pat. 2010;20:1. doi: 10.1517/13543770903463905. [DOI] [PubMed] [Google Scholar]
- 10.Schevitz RW, Bach NJ, Carlson DG, Chirgadze NY, Clawson DK, Dillard RD, Draheim SE, Hartley LW, Jones ND, Mihelich ED, Olkowski JL, Snyder DW, Sommers C, Wery J-P. Nat. Struct. Biol. 1995;2:458. doi: 10.1038/nsb0695-458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hansford KA, Reid RC, Clark CI, Tyndall JD, Whitehouse MW, Guthrie T, McGeary RP, Schafer K, Martin JL, Fairlie DP. Chembiochem. 2003;4:181. doi: 10.1002/cbic.200390029. [DOI] [PubMed] [Google Scholar]
- 12.Cha SS, Lee D, Adams J, Kurdyla JT, Jones CS, Marshall LA, Bolognese B, Abdel-Meguid SS, Oh BH. J. Med. Chem. 1996;39:3878. doi: 10.1021/jm960502g. [DOI] [PubMed] [Google Scholar]
- 13.Mouchlis VD, Mavromoustakos TM, Kokotos G. J. Comput.-Aided Mol. Des. 2010;24:107. doi: 10.1007/s10822-010-9319-7. [DOI] [PubMed] [Google Scholar]
- 14.Tomoo K, Yamane A, Ishida T, Fujii S, Ikeda K, Iwama S, Katsumura S, Sumiya S, Miyagawa H, Kitamura K. Biochim. Biophys. Acta. 1997;1340:178. doi: 10.1016/s0167-4838(97)00041-1. [DOI] [PubMed] [Google Scholar]
- 15.Mouchlis VD, Mavromoustakos TM, Kokotos G. J. Chem. Inf. Model. 2010;50:1589. doi: 10.1021/ci100217k. [DOI] [PubMed] [Google Scholar]
- 16.Pintore M, Mombelli E, Wechman C, Chretien JR. Anti-Inflammatory Anti-Allergy Agents Med. Chem. 2006;5:175. [Google Scholar]
- 17.Taylor RD, Jewsbury PJ, Essex JW. J. Comput.-Aided Mol. Des. 2002;16:151. doi: 10.1023/a:1020155510718. [DOI] [PubMed] [Google Scholar]
- 18.Halperin I, Ma B, Wolfson H, Nussinov R. Proteins. 2002;47:409. doi: 10.1002/prot.10115. [DOI] [PubMed] [Google Scholar]
- 19.Wei D, Jiang X, Zhou L, Chen J, Chen Z, He C, Yang K, Liu Y, Pei J, Lai L. J. Med. Chem. 2008;51:7882. doi: 10.1021/jm8010096. [DOI] [PubMed] [Google Scholar]
- 20.Kokotos G, Kotsovolou S, Six DA, Constantinou-Kokotou V, Beltzner CC, Dennis EA. J. Med. Chem. 2002;45:2891. doi: 10.1021/jm025538p. [DOI] [PubMed] [Google Scholar]
- 21.Kokotos G, Six DA, Loukas V, Smith T, Constantinou-Kokotou V, Hadjipavlou-Litina D, Kotsovolou S, Chiou A, Beltzner CC, Dennis EA. J. Med. Chem. 2004;47:3615. doi: 10.1021/jm030485c. [DOI] [PubMed] [Google Scholar]
- 22.Constantinou-Kokotou V, Peristeraki A, Kokotos CG, Six DA, Dennis EA. J. Pept. Sci. 2005;11:431. doi: 10.1002/psc.628. [DOI] [PubMed] [Google Scholar]
- 23.Yaksh TL, Kokotos G, Svensson CI, Stephens D, Kokotos CG, Fitzsimmons B, Hadjipavlou-Litina D, Hua XY, Dennis EA. J. Pharmacol. Exp. Ther. 2006;316:466. doi: 10.1124/jpet.105.091686. [DOI] [PubMed] [Google Scholar]
- 24.Stephens D, Barbayianni E, Constantinou-Kokotou V, Peristeraki A, Six DA, Cooper J, Harkewicz R, Deems RA, Dennis EA, Kokotos G. J. Med. Chem. 2006;49:2821. doi: 10.1021/jm050993h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Six DA, Barbayianni E, Loukas V, Constantinou-Kokotou V, Hadjipavlou-Litina D, Stephens D, Wong AC, Magrioti V, Moutevelis-Minakakis P, Baker SF, Dennis EA, Kokotos G. J. Med. Chem. 2007;50:4222. doi: 10.1021/jm0613673. [DOI] [PubMed] [Google Scholar]
- 26.Antonopoulou G, Barbayianni E, Magrioti V, Cotton N, Stephens D, Constantinou-Kokotou V, Dennis EA, Kokotos G. Bioorg. Med. Chem. 2008;16:10257. doi: 10.1016/j.bmc.2008.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barbayianni E, Stephens D, Grkovich A, Magrioti V, Hsu YH, Dolatzas P, Kalogiannidis D, Dennis EA, Kokotos G. Bioorg. Med. Chem. 2009;17:4833. doi: 10.1016/j.bmc.2009.03.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dess DB, Martin JC. J. Am. Chem. Soc. 1991;113:7277. [Google Scholar]
- 29.Smart BP, Pan YH, Weeks AK, Bollinger JG, Bahnson BJ, Gelb MH. Bioorg. Med. Chem. 2004;12:1737. doi: 10.1016/j.bmc.2004.01.022. [DOI] [PubMed] [Google Scholar]
- 30.Burke JE, Babakhani A, Gorfe AA, Kokotos G, Li S, Woods VL, Jr, McCammon JA, Dennis EA. J. Am. Chem. Soc. 2009;131:8083. doi: 10.1021/ja900098y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.GOLD 4.1. Cambridge, UK: CCDC; 2009. [Google Scholar]
- 32.Verdonk ML, Chessari G, Cole JC, Hartshorn MJ, Murray CW, Nissink JW, Taylor RD, Taylor R. J. Med. Chem. 2005;48:6504. doi: 10.1021/jm050543p. [DOI] [PubMed] [Google Scholar]
- 33.Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. Proteins. 2003;52:609. doi: 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
- 34.Scott DL, Sigler PB. Adv. Protein Chem. 1994;45:53. doi: 10.1016/s0065-3233(08)60638-5. [DOI] [PubMed] [Google Scholar]
- 35.Li B, Liu Z, Zhang L. J. Chem. Inf. Model. 2009;49:1725. doi: 10.1021/ci900044j. [DOI] [PubMed] [Google Scholar]
- 36.SYBYL, molecular modeling software packages, version 8.0. 1699 South Hanley Rd., St. Louis, MO 63144-2917: Tripos Inc.; 2007. [Google Scholar]
- 37.Reid RC. Curr. Med. Chem. 2005;12:3011. doi: 10.2174/092986705774462860. [DOI] [PubMed] [Google Scholar]
- 38.Singer AG, Ghomashchi F, Le Calvez C, Bollinger J, Bezzine S, Rouault M, Sadilek M, Nguyen E, Lazdunski M, Lambeau G, Gelb MH. J. Biol. Chem. 2002;277:48535. doi: 10.1074/jbc.M205855200. [DOI] [PubMed] [Google Scholar]
- 39.Oslund RC, Cermak N, Gelb MH. J. Med. Chem. 2008;51:4708. doi: 10.1021/jm800422v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rosenson RS. Cardiovasc. Drugs Ther. 2009;23:93. doi: 10.1007/s10557-008-6148-1. [DOI] [PubMed] [Google Scholar]
- 41.Garcia-Garcia HM, Serruys PW. Curr. Opin. Lipidol. 2009;20:327. doi: 10.1097/MOL.0b013e32832dd4c7. [DOI] [PubMed] [Google Scholar]
- 42.Maestro, version 9.0. New York, NY: Schrödinger, LLC; p. 2009. [Google Scholar]
- 43.Schrödinger Suite 2009 Protein Preparation Wizard; Epik version 2.0. New York, NY: Schrödinger, LLC; 2009. [Google Scholar]; Impact version 5.5. New York, NY: Schrödinger, LLC; 2009. [Google Scholar]; Prime version 2.1. New York, NY: Schrödinger, LLC; 2009. [Google Scholar]
- 44.Epik, version 2.0. New York, NY: Schrödinger, LLC; 2009. [Google Scholar]
- 45.Shelley JC, Cholleti A, Frye LL, Greenwood JR, Timlin MR, Uchimaya M. J. Comput.-Aided Mol. Des. 2007;21:681. doi: 10.1007/s10822-007-9133-z. [DOI] [PubMed] [Google Scholar]
- 46.Clark M, Crammer DR, III, Van Opdenbosch N. J. Comput. Chem. 1989;10:982. [Google Scholar]
- 47.Powell DJM. Math. Program. 1977;12:241. [Google Scholar]
- 48.Kirkpatrick S, Gelatt CD, Jr, Vecchi MP. Science. 1983;220:671. doi: 10.1126/science.220.4598.671. [DOI] [PubMed] [Google Scholar]
- 49.Jones G, Willett P, Glen RC, Leach AR, Taylor R. J. Mol. Biol. 1997;267:727. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.