

Abstract
Ribonucleotide reductase (RNR) is an enzyme for the de novo conversion of ribonucleotides to deoxyribonucleotides. The two human ribonucleotide reductase (RNR) small subunits hRRM2 and hp53R2 share 83% sequence homology but show distinct expression patterns and function. Structural analyses of the oxidized form of hRRM2 and hp53R2 indicate that both proteins contain a conserved Gln127-hp53R2/Gln165-hRRM2 close to the dinuclear iron center and the essential tyrosine residue Tyr124-hp53R2/Tyr162-hRRM2 and forms hydrogen bonds with the tyrosine and iron ligands, implying a critical role for the glutamine residue in assembling the dityrosyl-diiron radical cofactor. The present work also demonstrated that Tyr221 in hRRM2, which is replaced by Phe183 in hp53R2, forms hydrogen-bond with Tyr162 to extend the hydrogen-bond network from Gln165-hRRM2. Mutagenesis and spectroscopic experiments suggested that the tyrosine-to-phenylalanine switch at Phe183-hp53R2/Tyr221-hRRM2 could lead to differences in radical generation or enzymatic activity for hp53R2 and hRRM2. This study correlates the distinct catalytic mechanisms of the small subunits hp53R2 and hRRM2 with a hydrogen-bonding network and provides novel directions for designing and development of subunit-specific therapeutic agents for human RNR enzymes.
Keywords: human ribonucleotide reductase, dityrosyl-diiron radical cofactor center, enzyme structure and activity
Introduction
Ribonucleotide reductase uses free radicals to reduce ribonucleotides (NDPs) to deoxyribonucleotides (dNDPs), providing building blocks for DNA replication and repair (1). RNRs from different species utilize at least three different ways to initiate the radicals (2). Class I RNR enzymes found in most eukaryotic organisms are only active upon forming a complex consisting of the subunits R1 and R2. A stable tyrosyl radical is generated in the small subunit R2 (∼ 45 kDa) via a non-heme diiron-oxygen cluster when a substrate (NDP) is bound at the active site of the large subunit R1 (∼85 kDa). The radical is then transferred to the R1 catalytic site via a proton-coupled electron transfer (PCET) pathway (3, 4). Subsequently, the substrate is reduced at its C2′ position by abstracting a hydrogen atom to dNDP in the R1 catalytic center. This crucial rate-limiting step for DNA synthesis is highly regulated to maintain a balanced dNTP pool. As abnormal dNTP levels could lead to cell death or mutagenesis (5), RNR represents an important therapeutic target for a number of human diseases (6).
Most of our knowledge on RNR come from the study of E. coli RNR, the prototypic class I enzyme, which differs from the mammalian RNRs in multiple aspects (2, 7, 8). As in E. coli, the stable radical is initiated at the corresponding Tyr176 in human RNR small subunit with the assistance of the nearby iron cluster nearby and then transferred to R1 through the PCET pathway. In contrast to E. coli, our previous work showed that, in addition to Tyr176-hRRM2, Tyr162-hRRM2 is required for the initiation of the stable radical (9). We found that phenylalanine replacement of Tyr176 and Tyr162, two residues located at the opposite sides of the diiron center in hRRM2, deprives the mutants' radical content and the enzymatic activity. Our results implicate a dityrosyl-diiron radical cofactor system, Tyr176-diiron cluster-Tyr162, in the radical initiation by human RNR, which differs from the canonical mechanism based on the E. coli model.
The highly conserved diiron cluster active site - especially the radical harboring Tyr176-hRRM2 site - was extensively studied (10). Many residues close to the iron center and the tyrosine are invariant, which include the iron ligands that bind and neutralize the bound metal for the stabilization of the iron center and several hydrophobic residues (11). In this study, the residues in the vicinity of Tyr162-hRRM2 were carefully examined to extend our understanding of the dityrosyl-diiron cofactor in radical initiation and iron center stability. Based on the crystal structures of hRRM2 (PDB ID 2UW2) and hp53R2 (PDB ID 3HF1), which we recently reported and deposited at the Protein Data Bank, a highly conserved interior glutamine residue (Gln165-hRRM2) was found between Tyr162-hRRM2 and the iron center located 5.1 Å from Tyr-162 and 4.3 Å from Fe2. Mutagenesis study (Q to K, E, N or V) reveals that the glutamine residue is essential for the radical formation - probably through stabilizing the radical-iron cluster and by forming a hydrogen bonding network.
Human RNRs have two homologous small subunits, the canonical hRRM2 and tumor suppressor protein p53-regulated hp53R2 (12). p53R2 is essential as p53R2 knock-out transgenic mice are livable for only several weeks before succumbing to kidney failure (13, 14) and p53R2 plays an indispensable role in supplying dNTPs for DNA repair and mitochondrial DNA synthesis during the G0/G1 phase (15). Catalytic and regulatory mechanisms of human RNR enzymes with different small subunits have gained increasing interests. We have previously determined the functional differences of the two small subunits, i.e. enzymatic activity, protection against oxidation stress, chromosome location, transcription factor regulation, cell cycle dependency (16-20). The co-existence yet different functionalities of the two RNR small subunits sharing 83% homology remain enigmatic. A thorough understanding of the catalytic and regulation mechanisms of the two small subunits is key to developing potent and subunit-specific therapeutic agents.
The structural analysis identified that Phe183 of hp53R2, a distinct residue adjacent to the essential Tyr124-hp53R2/Tyr162-hRRM2, is replaced by Tyr221 in hRRM2. To study the impact of the structure change, the tyrosine was replaced by a phenylalanine (Y221F) in hRRM2 and the phenylalanine in hp53R2 by a tyrosine (F183Y) via site-direct mutagenesis. EPR and enzymatic assays showed that it alters the radical content and enzymatic activity, indicating that the tyrosine-to-phenylalanine switch (Phe183-hp53R2↔Tyr221-hRRM2) could be partly responsible for the different functionality of the two small subunits. It is likely that human RNR enzymes utilize this residue to regulate reductase activities through local and long-ranged interactions. Thus, the evidence that the functional differences of hp53R2 and hRRM2 could be affected by a single residue alternation in the small subunits is presented for the first time.
Results
Structure analyses
The published three-dimensional structures of the human RNR small subunits hRRM2 and hp53R2 were used for the structural and functional analysis. hp53R2 structure was solved and deposited at the Protein Data Bank (PDB ID 3HF1) recently by our group (21). The structure of hRRM2, which shares 83% homology in sequence with hp53R2, is also available (PDB ID 2UW2). Both structures are in a diferric (metR2) state where hp53R2 with dinuclear iron occupancy and hRRM2 with single iron (Fe2) in its active site. Oxidized R2 is in a stable inactive state that is structurally similar to the free radical active state R2 (2, 22). The overall structure of hRRM2 is similar to that of hp53R2 (21). In this study, we focused on the dityrosyl-diiron radical cofactor center, especially the vicinities of Tyr124-hp53R2/Tyr162-hRRM2. In hRRM2, the proposed radical cofactor contains the dinuclear iron center enclosed by six hydrophilic residues, Asp138, Glu169, His172, Glu232, Glu266, His269 from four helices (B, C, E, F helices) (1), and the two essential tyrosine residues, Tyr176, Tyr162 from C-helix (Figure 1A). The missing iron Fe1 in the diiron center of hRRM2 (2UW2) was modeled by superimposing Fe1 of the oxidized murine R2 (PDB ID 1W68). The two essential tyrosines Tyr176 and Tyr162 are located at the opposite sides of the diiron center, though not exactly symmetrical to the center (Figure 1A). Tyr176 is near Fe1 (7.0 Å) whereas Tyr162 is close to Fe2 (10.0 Å) (distances were measured between hydroxyl oxygen of the tyrosines to the Fe atoms). Tyr176 is close enough to the hydrophilic iron ligands to form hydrogen bonds (i.e. 2.0 Å to His172) whereas Tyr162 shows no direct access to the iron ligands (Figure 1B).
Figure 1.
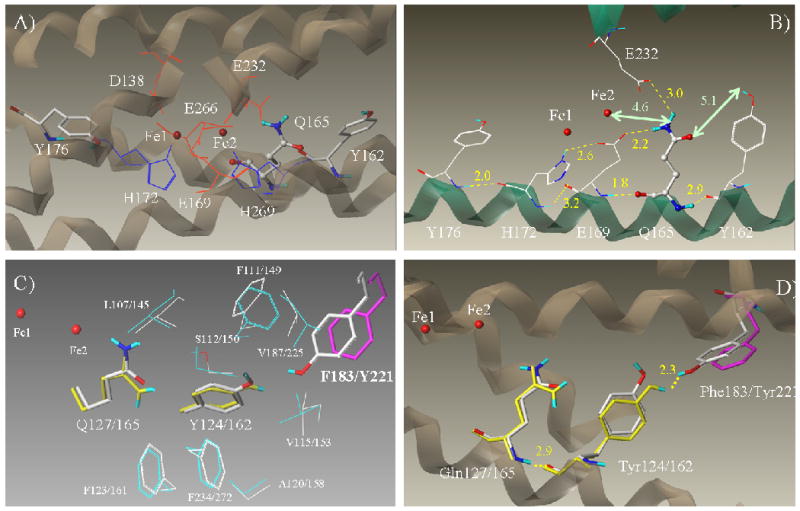
A) The dityrosyl-diiron center of hRRM2 (PDB ID 2UW2) is shown with the protein secondary structures at the background. Two iron atoms are colored red. The iron ligands and Q165 (highlighted as ball-and-stick) are shown. B) The hydrogen bond network of Q165 is depicted. Hydrogen bonds are colored yellow and labeled with bond lengths. Distances between Q165 to Y162 and Fe2 are marked in green. The secondary structures with the residues attached are shown. Five residues Y176, H172, E169, Q165, and Y162 are at five continuous helical turns on C-helix. C) Comparison of the residues surrounding Y124/162 of hp53R2/hRRM2. In hp53R2, Y124 and Q127 are colored yellow; F183 in magenta. The other residues are colored cyan. Residues in hRRM2 are colored by atom types (carbon – white, oxygen – red, hydrogen – cyan, and nitrogen – blue). Y124/Y162, Q127/Q165, F183/Y221 are highlighted in stick rendering. Residues were labeled as hp53R2/hRRM2. D) Hydrogen bond networks around Y124-hp53R2 and Y162-hRRM2.
Inspection of the asymmetrical dityrosyl-diiron radical cofactor center revealed a hydrophilic residue, glutamine 165 (glutamine 127 in hp53R2) of C-helix, located between Tyr162 and Fe2 in hRRM2. Interestingly, the two essential tyrosines Tyr176 and Tyr162, together with the other two iron ligands Glu169 and His172, reside in the same α-helix (C-helix) as Gln165, which forms five continuous helical turns (Figure 1B). As demonstrated in Figure1B, the side chain of Gln165 is located about 4.6 Å to Fe2, indicative of no bond formation between Gln165 and the iron atom. However, Gln165 is close enough to the two iron ligands, Glu169 from C-helix and Glu232 from E-helix (2.2 Å and 3.0 Å in side chains, respectively) to form hydrogen-bond. The backbone carbonyl group of Gln165 is also able to form a hydrogen-bond to the backbone amide group of Glu169 (1.8 Å in distance). On the other hand, the carboxyl oxygen of glutamine 165 is 5.1 Å away from the hydroxyl group of Tyr162, indicative of no hydrogen bonding by side chain in the conformation. However, the backbone amide group of Gln165 is within the range of forming hydrogen-bond to the backbone carbonyl group of Tyr162 (2.9 Å). Therefore, Gln165 is hydrogen-bonded to Tyr162 as well as the iron ligands Glu169 and Glu232, indicative of a hydrogen-bonding network connecting the tyrosine and the iron cluster by the glutamine.
The sequence alignment of four class I RNR small subunits (human p53R2, human RRM2, mouse RRM2, and E. coli RRM2) is displayed in Table 1, showing Tyr124, Gln127, Tyr138 and Phe183 of hp53R2 and the analogous residues in the other three. Figure 1C shows that Tyr162-hRRM2 and Tyr124-hp53R2 each are enclosed by ten mostly hydrophobic amino acids in hRRM2 and hp53R2, respectively. Nine out of the ten residues are conserved between the two proteins. The sole difference is tyrosine at position-221 in hRRM2, which is phenylalanine at the analogous position-183 in hp53R2 (Table 1). Phe183-hp53R2/Tyr221-hRRM2 sits at the end of the E-helix close to the protein surface, where the iron-ligand Glu232 is located. Unlike Tyr138-hp53R2/Tyr176-hRRM2, the Tyr124-hp53R2/Tyr162-hRRM2 side chain and the hydroxyl group are tilted away from the iron center (Figure 1). This ring conformation points the Tyr162 hydroxyl part toward the hydroxyl group of Tyr221 in hRRM2. Figure 1D demonstrated that the hydrogen-bonding pulls Tyr221 closer to Tyr162 in hRRM2 than Phe183 to Tyr124 in hp53R2. Hence, Tyr221 could be abridged to Gln165 via a hydrogen bonding network through Tyr162 in hRRM2. Since Phe183-hp53R2 lacks this hydroxyl group, the hydrogen bonding network from Gln127 to Tyr124 could not be extended to Phe183 in hp53R2.
Table 1.
Structure-based sequence alignment of ribonucleotide reductase small subunits from human hp53R2, hRRM2, mouse mRRM2, and E. coliM2 is shown. The key residues studied in this work are highlighted as bold, colored red and labeled by their positions in sequence.
E.coli insertion a – EMTSYWHLLGEGTHTVNGKTVTVSLRELK.
| 108 | 111 | 122 | 193 | |||||
| ↓ | ↓ | ↓ | ↓ | |||||
| E.coliM2 | ETW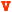 ET ET AFS AFS |
ETIHSRS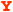 TH TH |
IIRNIVNDPS | VVFDDIVTNE | QIQKRAEGIS | SYYDELI(a) | KK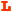 YLCLMSV YLCLMSV |
|
| 163 | 166 | 177 | 222 | |||||
| ↓ | ↓ | ↓ | ↓ | |||||
| mRRM2 | RCFGF IAM IAM |
ENIHSEsMSL |
LIDTYIKDPK | EREYLFNAIE | TMPCVKKKAD | WALRWIGDKE | ATGERVVAF |
|
| 162 | 165 | 176 | 221 | |||||
| ↓ | ↓ | ↓ | ↓ | |||||
| hRRM2 | RCFGFIAM |
ENIHSEMSL |
LIDTYIKDPK | EREFLFNAIE | TMPCVKKKAD | WALRWIGDKE | ATGERVVAF |
|
| 124 | 127 | 138 | 183 | |||||
| ↓ | ↓ | ↓ | ↓ | |||||
| hp53R2 | RCFGFILI |
ENVHSEMSL |
LIDTYIRDPK | KREFLFNAIE | TMPYVKKKAD | WALRWIADRK | ST GERVVAF GERVVAF |
|
Mutagenesis study on Gln127-hp53R2/Gln165-hRRM2
The structure analysis suggests that Gln127-hp53R2/Gln165-hRRM2 might play a key role in iron cluster and radical stability. A panel of hp53R2 and hRRM2 mutants were designed based on the charge state (Q to K and E), size (Q to N and K), and lipophilicity property (Q to V) to study the structural and functional roles of this conserved glutamine residue (Table 2). The wild-type and mutant proteins were purified from the cell lysates through a single chromatography. Circular dichroism measurements were conducted to ensure that the proteins were properly folded (Figure 2). The CD spectra were recorded at 37°C in 50 mM Tris buffer (pH 7.4). All of the glutamine mutants exhibited similar secondary structure signatures as the native proteins, confirming that the mutants form the same structures as the native proteins (Figure 2A and 2B). A standard [3H] CDP reduction assay was performed to measure the catalytic activity of the wild type RNR in the presence of an excess of purified recombinant human M1 protein (see Experimental Section). The relative enzyme activity (as determined by the dCDP conversion rate) was used to assess changes incurred by the mutation and was converted to the specific activity for overall comparison. hp53R2 or hRRM2 reconstituted with M1 exhibited a full enzymatic activity (Table 2). Yet only a minimal activity was detected with all of the Gln127-hp53R2/Gln165-hRRM2 mutants. The replacement of glutamine (Q) with the bulky basic lysine (K) yielded mutants (Q127K-hp53R2/Q165K-hRRM2) without any detectable activity. The valine, asparagine, and glutamic acid mutants, Q127V/Q165V, Q127N/Q165N, and Q127E/Q165E of hp53R2/hRRM2, also yielded a low activity. The reductase activity for relatively conserved substitutions, Q127E/Q165E and Q127N/Q165N are as low as that for Q127V/Q165V, suggesting that human RNRs require the precise configuration of the residue's functional groups i.e. size, charge state and hydrophilicity, to satisfy the structural and functional requirement at this position.
Table 2. Characteristics of the Ribonucleotide Reductase Small Subunits Proteins.
Ribonucleotide Reductase activity was presented as formation of dCDP (see Materials and Methods), and was measured in the presence of 0.059 μM (5 μg) of R1 protein and 0.057 μM (2.5 μg) of hRRM2 or hp53R2 proteins with a total sample volume of 100 μl. Each value is the average of two to three determinations with deviations < 0.3. ND: not detectable. Iron quantification is performed on freshly purified protein using atomic absorption spectrometry with lower limit > 0.01. The radical content was quantified against a standard solution of 1mmol/L CuSO4 in 50mmol/L EDTA by double integration of spectra registered at no saturating microwave levels using standard Bruker method. The lowest limit of detection is 0.001.
| Proteins | Specific Activity (nmol/min/mg R2) | Iron Content (mol/mol R2, %) | Radical Content (mol/mol R2) |
|---|---|---|---|
| hp5 3R2 | |||
| wild type | 4152 ± 96 | 1.53 ± 0.26 100 | 0.82 |
| F183Y | 4592 ± 176 | 1.82 ± 0.27 119 | 0.90 |
| Y124W | 1352 ± 104 | 0.84 ± 0.10 55 | 0.27 |
| Y124W/F183Y | 192 ± 24 | 0.56 ± 0.07 37 | 0.04 |
| Q127V | 384 ± 88 | 0.40 ± 0.16 26 | 0.08 |
| Q127K | ND | 0.24 ± 0.11 16 | ND |
| Q127E | 376 ± 24 | 0.68 ± 0.12 44 | 0.07 |
| Q127N | 160 ± 48 | 0.80 ± 0.02 52 | 0.03 |
| hRRM2 | |||
| wild type | 6000 ± 96 | 3.10 ± 0.58 100 | 1.23 |
| Y221F | 4456 ± 248 | 1.45 ± 0.42 47 | 0.91 |
| Y162W | 208 ± 16 | 0.72 ± 0.13 23 | 0.04 |
| Y162W/Y221F | 1304 ± 152 | 0.95 ± 0.07 31 | 0.27 |
| Q165V | 232 ± 48 | 0.62 ± 0.14 20 | 0.05 |
| Q165K | ND | 0.34 ± 0.04 11 | ND |
| Q165E | 480 ± 136 | 0.70 ± 0.12 22 | 0.10 |
| Q165N | 400 ± 24 | 0.50 ± 0.06 16 | 0.08 |
Figure 2.
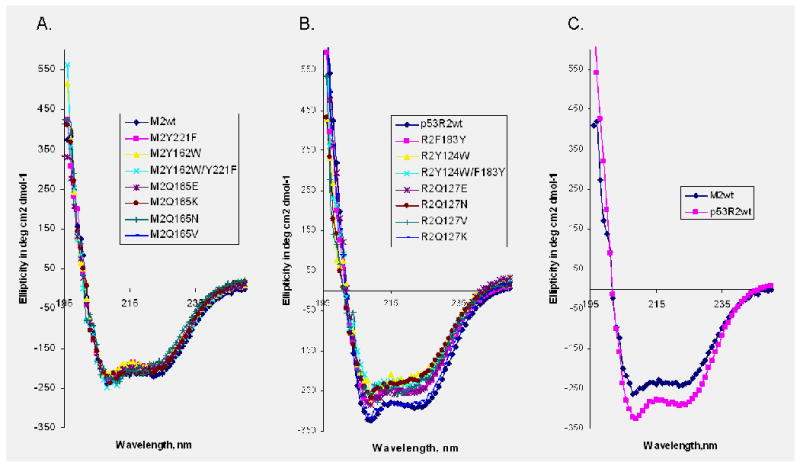
Circular dichroism spectra of human RRM2 and mutants (A), p53R2 and the mutants (B), comparison of hRRM2 and hp53R2 spectrum (C), recorded at 37°C in 50 mM Tris buffer (pH 7.4) using a 0.01 cm path length cell. Each spectrum shown represents the average of five.
The amount of iron bound to the wild-type and mutant proteins treated with nitric acid was determined using atomic absorption spectroscopy (20). The bacterial transformed wild-type and mutant plasmids were grown in normal Lennox L Broth (LB) media with no additional supplement iron insertion. The RNR proteins purified were active without reassembling the iron center. The iron content of all the above mutants is significantly less than that of the wild-type enzyme (mostly < 50%) (Table 2), suggesting that the glutamine plays an important role in iron stabilization. The most dramatic change occurred with the lysine substitution, which caused the largest disturbance by reducing the iron content to mere 16% and 11% of the wild-type hp53R2 and hRRM2, respectively. However, considering that all mutants retained minimal activity, it suggests that the glutamine's role may not be limited to stabilizing the iron cluster.
EPR spectroscopy was used to monitor the stability of the tyrosyl radical. The 9 GHz EPR spectra of wild-type hRRM2 and hp53R2 (Figure 3A) are virtually identical to those reported (22-24). Since the EPR spectra were similar throughout the measurements upon normalization, the amplitudes of the EPR signals thus reflect the spin concentration. Correlating with the minimal reductase activity, no stable radical was detected in any of the Gln127-hp53R2/Gln165-hRRM2 mutants (Figure 3A and Table 2), indicating that the glutamine residue is involved in maintaining a stable radical in addition to stabilizing the iron center. Thus, the mutagenesis studies confirmed that Gln127-hp53R2/Gln165-hRRM2 plays a role in the iron and radical stability, therefore the enzyme activity, as predicted in the structural analyses.
Figure 3.
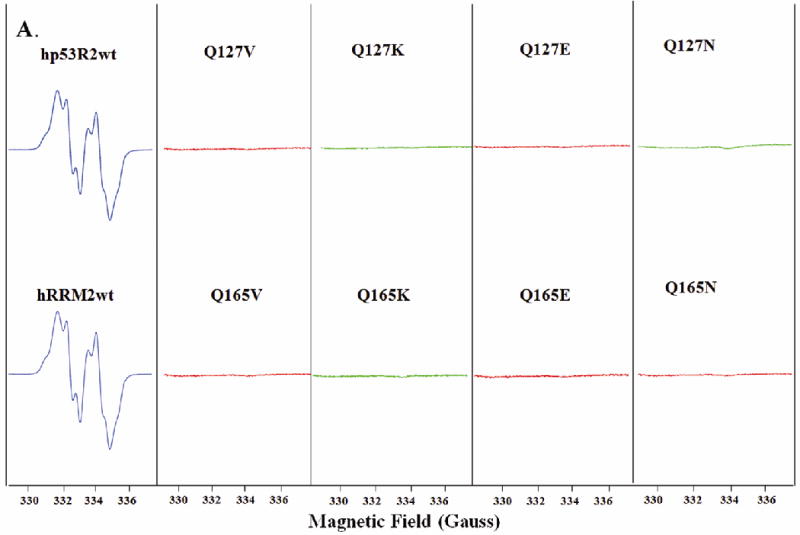
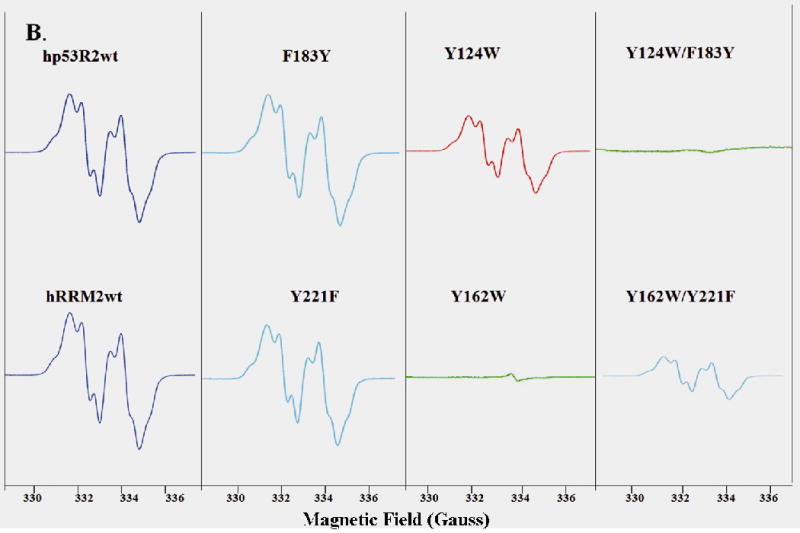
A) EPR spectra of hp53R2 and hRRM2 wild types and the mutants Q127V, Q127K, Q127E, Q127N, and Q165V, Q165K, Q165E, Q165N. B) EPR spectra of wide types and mutants F183Y, Y124W, Y124W/F183Y in hp53R2 and Y221F, Y162W, Y162W/Y221F in hRRM2. EPR spectra of human RRM2 and p53R2 proteins recorded at 20K in a Bruker 300E spectrometer with microwave power 0.1 mW, microwave frequency 9.375 GHz, and modulation amplitude 2.0 G.
Phe183-hp53R2 to Tyr221-hRRM2 Substitution alters the enzymatic activity of the two RNR small subunits
The CD spectra indicate that the two human RNR small subunits share highly similar structural features (Figure 2C). However, human RNR with hRRM2 exhibits a higher enzymatic activity than that with hp53R2 (9), i.e. 6000 vs. 4152 mmol/min/mg R2 in specific activity or 97.1% vs. 80.5% turnover rate, respectively (Table 2). The sequence alignment (Table 1) shows that Tyr221 in hRRM2 (Tyr222 in mouse RRM2) is replaced by a leucine in E.coli and a phenylalanine in hp53R2. Structural analysis indicates Phe183-hp53R2/Tyr221-hRRM2 represents the only variable in the conserved structural settings surrounding Tyr124-hp53R2/Tyr162-hRRM2 (Figure 1C). To assess the significance of this residue, site-direct mutagenesis was performed to substitute tyrosine for a phenylalanine (Y221F) in hRRM2 and phenylalanine for a tyrosine hp53R2 to (F183Y). CD spectra were recorded at 37°C assured the proper folding by the mutants (Figure 2A and 2B). Intriguingly, the F183Y-hp53R2 mutant enhanced the enzyme activity to 4592 (89.1% turn over rate) close to that of hRRM2. On the other hand, Y221F mutation in hRRM2 lowered the enzyme activity from 6000 (97.1%) to 4456 (72.1%) towards the level of hp53R2.
Tyr124-hp53R2/Tyr162-hRRM2 was mutated to tryptophan (Y to W) for its radical generation property. The double mutants, Y162W/Y221F-hRRM2 and Y124W/F183Y-hp53R2, were also generated to assess the tyrosine-to-phenylalanine switch (Table 2). The tryptophan substitution Y124W-hp53R2 retained enzyme activity of 1352 mmol/min/mg R2 (26.2% of wild-type) while Y162W-hRRM2 mutant exhibited a minimal activity (208, i.e., 3.4% of wild-type). The double mutants reversed their activities (Table 2). Y124W/F183Y-hp53R2 displayed a minimal activity (192, i.e., 3.8% of the wild-type) while Y162W/Y221F-hRRM2 exhibited 1304 (21.1%, i.e., the activity level of Y124W-p53R2). Therefore, the tyrosine-to-phenylalanine exchange between two subunits, F183Y-hp53R2/Y221F-hRRM2, could convert the enzymatic activity of both the wild type and mutant proteins.
The effect of Phe-to-Tyr-switch on the stability of the iron cluster was studied (Table 2). Under the experimental condition employed, hp53R2 holds 1.53 irons per dimer while hRRM2 may take ∼ 3.10 irons per dimer, indicative of the different iron-binding capability of the two small subunits. The iron contents of the wide-type and mutant proteins showed the same trend as the enzyme activity following the F-to-Y substitution. F183Y-hp53R2 wss able to hold more iron (1.82 irons per dimer) than the wild-type hp53R2 and the ability of Y221F-hRRM2 mutant to hold iron was significantly lower (1.45 irons per dimer) than the wild type (Table 2), suggesting that the tyrosine/phenylalanine residue affects the iron cluster stability. The correlation of the enzyme activity with the iron content indicates a long-range enzymatic activity regulation through the iron center by the tyrosine-phenylalanine substitution. In contrast, the Y162W-hRRM2 mutants exhibited minimal activity yet with 23% iron content, suggesting that the tryptophan substitution does more than destabilizing the iron center and the tyrosine could play a role in radical generation and stability. Similar activity and iron content were observed with the Y124W/F183Y-hp53R2 double mutants, again demonstrating the substitution, F183Y-hp53R2 vs. Y221F-hRRM2, regulates enzyme functions.
EPR studies showed that F183Y-hp53R2 and Y221F-hRRM2 mutants have no direct impact on the radical content (Figure 3B). On the other hand, Y162W-hRRM2 and Y124W-hp53R2 mutants showed different effects on the radical content in correlation with the enzymatic activity. Only a minimal radical signal was detected in Y162W-hRRM2 whereas Y124W-hp53R2 retained about one-third of the radical content of the wild-type protein, showing that the tryptophan substitution destabilized the radical cluster. Interestingly, the double mutant Y162W/Y221F-hRRM2 gained the EPR inactive mutant Y162W-hRRM2 a radical signal with similar profile but lower radical content of the mutant Y124W-hp53R2, while Y124W/F183Y-hp53R2 was EPR silent. This observation indicates that the tyrosine/phenylalanine could regulate the radical stability via Tyr124-hp53R2/Tyr162-hRRM2.
Discussion
R2 residues at the highly conserved diiron radical cluster and surroundings have been extensively studied - especially, by the Sjöberg's laboratory (2, 10, 25). The first sphere of the iron site includes residues directly contacting irons (iron ligands). The second sphere of the iron site includes mostly hydrophobic residues that contact the iron ligands and some aromatic residues in the vicinity of the iron cluster. The iron ligands, which bind and neutralize the bound metal were found to be structurally and functionally significant and are mostly invariant. Mutations at the second sphere and some of its aromatic residues may alter the cluster assembly if they are involved in radical generation, stabilization or reduction (24-30). We have previously identified that tyrosine residue Tyr124-hp53R2/Tyr162-hRRM2, distant from the diiron center (10.0 Å from Fe2 in hRRM2) in human RNR small subunits, is essential for radical generation and stabilization, which is distinct from the radical harboring tyrosine residue Tyr138-hp53R2/Tyr176-hRRM2 (9). We have found that both Y138F and Y124F mutants of hp53R2, and Y176F and Y162F mutants of hRRM2, lack in radical content and enzymatic activity (9) similar to Y122F, which was shown to harbor the stable free radical of E. coli R2 (31). When compared with Tyr138-hp53R2/Tyr176-hRRM2 (6.4 Å from Fe1 in hRRM2) and the aromatic residue Trp64-hp53R2/Trp102-hRRM2 (7.3Å from Fe1 in hRRM2) located in the vicinity of the active site and the members of the PCET pathway, Tyr124-hp53R2/Tyr162-hRRM2 is more distant from the iron cluster and lacks direct contact with the iron ligands. This finding strongly suggested that Tyr124-hp53R2/Tyr162-hRRM2 could represent another aromatic residue oxidized by the intermediate X (Fe3+/Fe4+ species) in addition to Tyr138-hp53R2/Tyr176-hRRM2 and Trp64-hp53R2/Trp102-hRRM2 in the vicinity of the active site (9).
The functional roles of Gln127-hp53R2/Gln165-hRRM, a residue in the second sphere between Tyr124-hp53R2/Tyr162-hRRM2 and the iron cluster, were investigated. Mutagenesis studies demonstrated that the glutamine is crucial in maintaining the stability of iron center as well as the radical cluster and the reductase activity, suggesting that this residue probably involved in catalysis. The inactive mutant Q127N-hp53R2/Q165N-hRRM2 suggests little structural flexibility at this position, implying that the glutamine involved in such functions may require specific structural arrangement. This is consistent with our analysis that the glutamine may be involved in the hydrogen-bond network connecting the iron ligands with Tyr124-hp53R2/Tyr162-hRRM2. Interestingly, that the glutamine is located in the same α-helix (C-helix) buried in the protein that also contains the two tyrosines Tyr176 and Tyr162 (using hRRM2 as an example, the same features found in hp53R2) as well as the two iron ligands Glu169 and His172. And the five C-helix residues are facing the same side of the helix comprised of five consecutive helical turns. Most importantly, structure inspection revealed that hydrogen-bonds were formed between these residues either through their side chains or backbone (Figure 1B). The fact that the two essential tyrosines Tyr176 and Tyr162 are located at the same α-helix with hydrophilic iron ligands and that Gln165 is capable of forming hydrogen-bonds suggests that the two tyrosines are potentially connected via a hydrogen-bond network. This structure feature may provide the rational for the function role of the second essential tyrosine Tyr162 in the radical initiation and the correlation of the two tyrosines. Gln127-hp53R2/Gln165-hRRM2 therefore represents an important residue bridging the correlation.
Residues at the Fe2 site of the diiron cluster differ significantly between the murine and E. coli R2 (8). The structure-based sequence alignment shows that Gln127-hp53R2/Gln165-hRRM2 is conserved in mammalian and yeast RNRs, but is replaced by a tryptophan (Trp111) in E. coli R2. Trp111 was proposed to be connected to the iron center through a hydrogen-bond network and form a transient tryptophan radical in Y122F mutant (26-28). Also, the E. coli R2 lacks a tyrosine at position 108; instead, it is replaced by a valine at this position (Table 1). It was reported that transient radical could form at the nearby Trp107 in Y122F E. coli mutant (24, 26). The two essential residues, Gln127/Gln165 and Tyr124/Tyr162 in hp53R2/hRRM2, are Trp111 and Val108 in E. coli small subunit. Co-substitution of the two residues in the RNR small subunit during the evolution from E. coli to mammals underscores related roles of the two residues in forming of the hydrogen-bond network, which is probably coupled with electron transfer for the function and mechanism of the dityrosyl-diiron radical cofactor.
It has been shown that the enzymatic activities of human RNRs with the two small subunits are different, but the underlying mechanism was unclear (9, 12, 20). Higher organisms like human might utilize multiplicity of small subunits to fine tune the highly regulated dNTPs pool for DNA synthesis. The two small subunits hRRM2 and hp53R2 show different iron uptake capacity (32, 33). Our experiments indicate that hp53R2 hold only half of the hRRM2 iron content. And hRRM2 could hold bound-iron longer than hp53R2. These observations suggest that hp53R2 may possess a more flexible iron center than hRRM2. In addition, it was reported that p53R2 is a selective binder of Fe (II) while RRM2 could bind Fe (II) or Fe (III) with a similar affinity (32). The lower iron-affinity of hp53R2 could partly be due to the oxidation of the bound irons in the oxygen-rich environment where hp53R2 binds the oxidized iron ions, i.e., Fe (III) poorly.
Despite the structural similarity between Phe183-hp53R and Tyr221-hRRM2, a subtle impact on the radical cluster stabilization caused by the different hydrogen bonding pattern of the two residues was observed (Figure 3B). Local electrostatic and structural alterations caused by the Phe-to-Tyr switch could be transmitted to the dityrosyl-diiron clusters through the hydrogen bond network, which includes Tyr124-hp53R2/Tyr162-hRRM2 and Gln127-hp53R2/Gln165-hRRM2. Hydrogen-bond constitutes an important factor that influences enzymatic activity and redox potentials - in particular, the radical proteins (34, 35). It is likely that human RNRs utilize the distinct hydrogen-bonding environments of the second tyrosine in hp53R2 and hRRM2 to regulate their enzymatic activities. It is interesting to point out that the RNR tyrosyl radical was found to be hydrogen-bonded in mice and yeasts, though the hydrogen bond is absent in the E. coli enzyme (36, 37). The sequence alignment shows that both mouse and yeast have a tyrosine at the corresponding position for Tyr221-hRRM2, while E. coli contains a leucine residue instead (Table 1). Conformational differences and perturbed electronic structures around its iron center and the consequent functional differences have been observed in hp53R2 and hRRM2 (32, 34). The Phe-to-Tyr switch could be partly responsible for the different functionality of the two small subunits. Our study correlates the distinct catalytic mechanisms of human RNR containing small subunits hp53R2 or hRRM2 to a hydrogen-bonding network and provides new insights on the structure-function relationship in mammalian RNRs.
In summary, this study demonstrated the essential role of Gln127-hp53R2/Gln165-hRRM2 as mutating of the residue diminished the radical signal and the enzymatic activity. It is possible that this residue is involved in a hydrogen-bond network connecting the iron cluster with Tyr124-hp53R2/Tyr162-hRRM2 and, therefore, plays a critical role in the assembly of the dityrosyl-diiron radical cofactor. The present work also demonstrated the significant role of Phe183-hp53R2/Tyr221-hRRM2 in the enzymatic activity regulation. Based on this study, we conclude that the Tyr-to-Phe switch at the Phe183-hp53R2/Tyr221-hRRM2 site could lead to the differences in radical generation and enzymatic activity of hp53R2 and hRRM2. It is likely that human RNR enzymes utilize this residue to regulate reductase activities through local and long-ranged interactions. The circular dichroism study confirmed that our conclusions are based on the structure-function relationship of the corresponding residues but not the distortion in protein conformation caused by the mutations. Our study that combined three-dimensional structure and enzymatic function analyses contribute to the understanding of the catalytic and regulatory mechanisms of different human RNRs. The subunit-specific structural features may lead to a new direction for the design and development of subunit-specific therapeutic agents in addition to conventional approach targeting active sites or the PCET pathway.
Experimental Section
Plasmids
The T7 RNA polymerase-responsive vector pET28a containing the cDNA encoding for hRRM2 and hp53R2 proteins was used as a source for site-directed mutagenesis.
Oligonucleotide-Directed Mutagenesis
Oligonucleotides were synthesized using an Applied Biosystems DNA/RNA synthesizer (Model 392). Mutations of Tyr221 in hRRM2 to Phe221 and Phe183 in hp53R2 to Tyr183 were carried out by PCR using pET-hRRM2 or pET-p53R2 as a template. The primer for hRRM2 Y221F was 5′-GGGGACAAAGAGGCTACCTTTGGTGAACGTGTTGTAGCC -3′, and for hp53R2 F183Y 5′ GATAGCAGATAGAAAATCTACTTATGGGGAAAGAGTGGTGGCC -3′ (boldface letters denote the mutated codon). PCR was conducted by pre-heating a mixture containing 10ng of pET-hRRM2, 187ng of primer, and 0.2 mM dNTPs (Amersham Pharmacia Biotech) in a buffer recommended for Pfu polymerase (Stratagene) to 95°C for 1 min; then followed by 18 cycles of incubation at 95°C for 30 s, 55°C for 30 s, and 68°C for 12 min. One microliter of restriction enzyme Dpn1 was added to the mixture after cooling to room temperature, which was followed by incubation at 37 °C for 1 hour. Each construct (pET-hRRM2Y221F, pET-p53R2F183Y, pET-hRRM2Y162W, pET-hp53R2Y124W, pET-hRRM2Y162W/Y221F, pET-hp53R2Y124W/F183Y, pET-hRRM2Q165V, pET-hRRM2Q165K, pET-hRRM2Q165E, pET-hRRM2Q165N, pET-hp53R2Q127V, pET-hp53R2Q127K, pET-hp53R2Q127E, pET-hp53R2Q127N) was transfected into the E. coli strain BL21 (DE3) (Stratagene, La Jolla, CA), which enables isopropyl-1-thio-β-D-galactopyranoside (IPTG) - induced overexpression of hp53R2 and hRRM2 mutants.
Protein Expression and Purification
Native and mutant hRRM2 and hp53R2 were expressed in BL21 (DE3) bacteria. In a typical preparation, a 20 ml culture of transformed bacteria grown overnight was added to 1 L of LB medium containing 30 μg/ml kanamycin and grown at 37°C for 2-3 h (O.D600 did not exceed 0.9). Protein expression was induced by 1 mM IPTG, and bacterial growth continued for 16 h at 25°C. Harvested cell pellets were disrupted by applying vigorous agitation and incubation with Bugburster and Benonase (Novagen) at 4°C for 60 min. Immobilized Ni (II) affinity chromatography resin (Ni-NTA, Qiagen) was incubated with the lysate supernatant at 4°C for 1 hour. To avoid contamination, new resins were used for all the purification processes. Then the resin was loaded onto a gravity column and washed with at least 30 bed volumes of a wash buffer containing 50 mM NaH2PO4, 800 mM NaCl, 50 mM imidazole, 0.1% Triton-X 100, and 10 mM β-mercaptoethanol (pH 7.0). The protein eluted with 50 mM NaH2PO4, 300 mM NaCl, and 125 mM imidazole (pH 7.0). The eluates were dialyzed overnight at 4°C against 2000 volumes of 25 mM Tris-HCl (pH 7.4) and 100mM KCl. All the samples were tested to ensure the enzymatic activity after the process. Expression and purification of hRRM1 was conducted in a similar way except that this protein was eluted without β-mercaptoethanol.
Qualitative and Quantitative Analysis of Protein Preparations
Protein concentration was determined using NanoDrop Spectrophotometer ND-1000, OD280. Protein purity was determined by densitometric scanning of the coomassie-stained SDS-PAGE gels.
In vitro Activity Assay
The activities of hRRM2/hRRM1 and hp53R2/hRRM1 were measured using a modified [3H] CDP reduction assay. The reaction mixture contained 0.125 μM [3H] CDP (0.3 μCi) (ViTrax Co., Placentia, CA), 50 mM of HEPES (pH 7.2), 6 mM DTT, 4 mM MgOAC, 2 mM ATP, 0.05 mM CDP, 100 mM KCl, 0.24 mM NADPH. The enzyme activity was measured in the presence of 0.059 μM of R1 protein and 0.057 μM of hRRM2 or hp53R2 proteins with a total sample volume of 100 μl. After incubation at 37°C for 15-30 min and dephosphorylation with phosphodiesterase, the supernatant was analyzed by HPLC using a C-18 reversed phase column to separate [3H] cytidine and [3H] deoxycytidine. Radioactivity of the HPLC aliquots was measured with a β-RAM-2B flow-through radioisotope beta/gamma detector (IN/US Systems, Tampa, FL). The specific enzyme activity was reported as nmol of dCDP formed/min/mg protein (33).
Electron Paramagnetic Resonance Spectra
X-band EPR spectra were measured with a Bruker EMX spectrometer equipped with an Oxford helium cryostat. Purified samples of hRRM2 and hp53R2 proteins were frozen in liquid nitrogen prior to insertion in the cavity. Instrumental parameters: T = 20 K, microwave frequency = 9.376 GHz, microwave power = 0.5 mW, modulation amplitude 4 gauss, modulation frequency 100 KHz. The radical content was quantified against a standard solution of 1 mmol/L CuSO4 in 50 mmol/L EDTA by double integration of spectra registered at nonsaturating microwave levels by standard Bruker software.
Atomic Absorption Spectrometry
Measurement of iron content was performed using a Perkin-Elmer atomic absorption spectrometer equipped with a HGA850 graphite furnace at the City of Hope Pharmacology Analytic Core Facility. Equal amounts of purified wild type and mutant proteins (100μg) were mixed with equal volume of 6N HNO3. After completely dehydrating by speed vacuum, the pellet was diluted with 0.00005N HNO3. PE Pure Iron standard solution (Perkin-Elmer, Norwalk, CT) was 2-fold serially diluted from 50-1000ug/L to construct the standard curve (y = 0.0054x + 0.0119; x, concentration; y, absorbance; r = 0.99859). Iron content was expressed in μM of iron per μM of RNR small subunit protein.
Circular Dichroism Spectrometry
Circular dichroism spectra (Model 62 DS, AVIV Lakewood, NJ) of hRRM2 and hp53R2 and the mutants (0.2 mg/ml) were recorded from 250 nm to 195 nm in 50 mM Tris buffer (pH 7.4). All measurements were carried out at 37°C using a 0.01 cm path length quartz curette. The data pitch was 0.1 nm with a 1 nm bandwidth at a scan speed of 1.0 nm/Sec. Each spectrum shown represents the average of five. All CD data were expressed as the mean residue ellipticity, [θ], in units of degrees square centimeter per decimole (38).
Structure Models of hRRM2 and hp53R2
Structures of human small subunit hRRM2 and hp53R2 were downloaded from Protein Data Bank (PDB ID 2UW2 and 3HF1, respectively). The X-ray structure was relaxed and prepared using SYBYL 8.0 protein preparation tool (Tripos, Inc., St. Louis, MO). The structure models were optimized and evaluated using SYBYL Protable module if necessary, where Ramachandran plot, local geometry and the location of buried polar residue and exposed non-polar residues were examined. The structures were refined by a serial of energy minimization steps using AMBER all-atom force field with the cutoff of nonbonded (NB) interactions at 8.0Å and the distance-dependent dielectric constant set at 4.0r following the gradient termination of the Powell method with RMS of 0.005 kcal/mol. Hydrogen bond was considered with minimum angle of 115° and maximum distance 3.0Å.
Acknowledgments
We thank Dr. Yong Ba from Department of Chemistry and Biochemistry, California State University at Los Angeles and Dr. Angelo Di Bilio from California Institute of Technology for EPR experiments, and Dr. Mark E Davis and James Van Deventer from California Institute of Technology for CD experiments. Research was supported by NCI R01 Grant CA72767 and the Sino-American Cancer Foundation.
This work was supported by NCI grant CA 127541-01 for each author.
Abbreviations
- RNR
Ribonucleotide Reductase
- hRRM2
human RNR small subunit M2
- hp53R2
human RNR small subunit R2
- metR2
diferric bound RNR small subunits
- NDP
ribonucleotide
- dNDP
deoxyribonucleotides
- PCET
Proton-Coupled Electron Transfer
- EPR
Electron Paramagnetic Resonance
- CD
Circular Dichroism
Footnotes
No potential conflicts of interest were disclosed
References
- 1.Eklund H, Uhlin U, Färnegårdh M, Logan DT, Nordlund P. Structure and function of the radical enzyme ribonucleotide reductase. Prog Biophys Mol Biol. 2001;77:177–268. doi: 10.1016/s0079-6107(01)00014-1. [DOI] [PubMed] [Google Scholar]
- 2.Kolberg M, Strand KR, Graff R, Andersson KK. Structure, function, and mechanism of ribonucleotide reductases. Biochim Biophys Acta. 2004;1699:1–34. doi: 10.1016/j.bbapap.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 3.Sjöberg BM. Ribonucleotide reductases - A group of enzymes with different metallosites and a similar reaction mechanism. Struct Bonding. 1997;88:139. [Google Scholar]
- 4.Stubbe J, Nocera DG, Yee CS, Chang MCY. Radical initiation in the class I ribonucleotide reductase: Long-range proton-coupled electron transfer? Chem Rev. 2003;103:2167–2201. doi: 10.1021/cr020421u. [DOI] [PubMed] [Google Scholar]
- 5.Gon S, Beckwith J. Ribonucleotide reductases: influence of environment on synthesis and activity. Antioxid Redox Signal. 2006;8:773–80. doi: 10.1089/ars.2006.8.773. [DOI] [PubMed] [Google Scholar]
- 6.Cerqueira NM, Fernandes PA, Ramos MJ. Ribonucleotide reductase: a critical enzyme for cancer chemotherapy and antiviral agents. Recent Pat Anticancer Drug Discov. 2007;2:11–29. doi: 10.2174/157489207779561408. [DOI] [PubMed] [Google Scholar]
- 7.Strand KR, Karlsen S, Kolberg M, Røhr AK, Görbitz CH, Andersson KK. Crystal structural studies of changes in the native dinuclear iron center of ribonucleotide reductase protein R2 from mouse. J Biol Chem. 2004;279:46794–46801. doi: 10.1074/jbc.M407346200. [DOI] [PubMed] [Google Scholar]
- 8.Kauppi B, Nielsen BB, Ramaswamy S, et al. The three-dimensional structure of mammalian ribonucleotide reductase protein R2 reveals a more-accessible iron-radical site than Escherichia coli R2. J Mol Biol. 1996;262:706–720. doi: 10.1006/jmbi.1996.0546. [DOI] [PubMed] [Google Scholar]
- 9.Zhou B, Su L, Shao J, et al. A dityrosyl-diiron radical cofactor center is essential for human ribonucleotide reductases. Mol Cancer Ther. 2005;4:1830–1836. doi: 10.1158/1535-7163.MCT-05-0273. [DOI] [PubMed] [Google Scholar]
- 10.Andersson ME, Högbom M, Rinaldo-Matthis A, et al. Structural and mutational studies of the carboxylate cluster in iron-free ribonucleotide reductase R2. Biochemistry. 2004;43:7966–72. doi: 10.1021/bi036088l. [DOI] [PubMed] [Google Scholar]
- 11.Uhlin U, Eklund H. Structure of ribonucleotide reductase protein R1. Nature. 1994;370:533. doi: 10.1038/370533a0. [DOI] [PubMed] [Google Scholar]
- 12.Tanaka H, Arakawa H, Yamaguchi T, et al. A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage. Nature. 2000;404:42–49. doi: 10.1038/35003506. [DOI] [PubMed] [Google Scholar]
- 13.Powell DR, Desai U, Sparks MJ, et al. Rapid development of glomerular injury and renal failure in mice lacking p53R2. Pediatr Nephrol. 2005;20:432–440. doi: 10.1007/s00467-004-1696-5. [DOI] [PubMed] [Google Scholar]
- 14.Kimura T, Takeda S, Sagiya Y, Gotoh M, Nakamura Y, Arakawa H. Impaired function of p53R2 in Rrm2b-null mice causes severe renal failure through attenuation of dNTP pools. Nat Genet. 2003;34:440–445. doi: 10.1038/ng1212. [DOI] [PubMed] [Google Scholar]
- 15.Wang X, Zhenchuk A, Wiman KG, Albertioni F. Regulation of p53R2 and its role as potential target for cancer therapy. Cancer Lett. 2009;276:1–7. doi: 10.1016/j.canlet.2008.07.019. [DOI] [PubMed] [Google Scholar]
- 16.Liu X, Zhou B, Xu L, et al. Metastasis-suppression potential of ribonucleotide reductase small subunit p53R2 in human cancer cells. Clinical Cancer Research. 2006;12:6337–6344. doi: 10.1158/1078-0432.CCR-06-0799. [DOI] [PubMed] [Google Scholar]
- 17.Xue L, Zhou B, Liu X, et al. Ribonucleotide reductase small subunit p53R2 facilitates p21 induction of G1 arrest under UV irradiation. Cancer Res. 2007;67:16–21. doi: 10.1158/0008-5472.CAN-06-3200. [DOI] [PubMed] [Google Scholar]
- 18.Xue L, Zhou B, Liu X, et al. Structurally dependent redox property of ribonucleotide reductase subunit p53R2. Cancer Res. 2006;66:1900–1905. doi: 10.1158/0008-5472.CAN-05-2656. [DOI] [PubMed] [Google Scholar]
- 19.Liu X, Zhou B, Xue L, et al. Ribonucleotide reductase subunits M2 and p53R2 are potential biomarkers for metastasis of colon cancer. Clin Colorectal Cancer. 2007;6:374–381. doi: 10.3816/CCC.2007.n.007. [DOI] [PubMed] [Google Scholar]
- 20.Shao J, Zhou B, Zhu L, et al. Determination of the potency and subunit-selectivity of ribonucleotide reductase inhibitors with a recombinant-holoenzyme-based in vitro assay. Biochem Pharmacol. 2005;69:627–634. doi: 10.1016/j.bcp.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 21.Smith P, Zhou B, Ho N, Yuan YC, Su L, Tsai SC, Yen Y. 2.6Å X-ray crystal structure of human p53R2, a p53-inducible ribonucleotide reductase. Biochemistry. 2009;48:11134–41. doi: 10.1021/bi9001425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Andersson KK, Gräslund A. Diiron-oxygen proteins. In: Sykes AG, editor. Adv Inorg Chem. Vol. 43. Academic press; Orlando: 1995. pp. 353–408. [Google Scholar]
- 23.Guittet O, Håkansson P, Voevodskaya N, Fridd S, Gräslund A, Arakawa H, Nakamura Y, Thelander L. Mammalian p53R2 protein forms an active ribonucleotide reductase in vitro with the R1 protein, which is expressed both in resting cells in response to DNA damage and in proliferating cells. J Biol Chem. 2001;276:40647–51. doi: 10.1074/jbc.M106088200. [DOI] [PubMed] [Google Scholar]
- 24.Katterle B, Sahlin M, Schmidt PP, et al. Kinetics of transient radicals in Escherichia coli ribonucleotide reductase. Formation of a new tyrosyl radical in mutant protein R2. J Biol Chem. 1997;272:10414–21. doi: 10.1074/jbc.272.16.10414. [DOI] [PubMed] [Google Scholar]
- 25.Ormö M, Regnström K, Wang Z, Que L, Jr, Sahlin M, Sjöberg BM. Residues important for radical stability in ribonucleotide reductase from Escherichia coli. J Biol Chem. 1995;270:6570–6. doi: 10.1074/jbc.270.12.6570. [DOI] [PubMed] [Google Scholar]
- 26.Kolberg M, Logan DK, Bleifuss G, et al. A new tyrosyl radical on Phe208 as ligand to the diiron center in Escherichia coli ribonucleotide reductase, mutant R2-Y122H. Combined x-ray diffraction and EPR/ENDOR studies. J Biol Chem. 2005;280:11233–11246. doi: 10.1074/jbc.M414634200. [DOI] [PubMed] [Google Scholar]
- 27.Ekberg M, Birgander P, Sjöberg BM. In vivo assay for low-activity mutant forms of Escherichia coli ribonucleotide reductase. J Bacteriol. 2003;185:1167–73. doi: 10.1128/JB.185.4.1167-1173.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sahlin M, Lassmann G, Potsch S, Slaby A, Sjöberg BM, Graslund A. Tryptophan radicals formed by iron/oxygen reaction with Escherichia coli ribonucleotide reductase protein R2 mutant Y122F. J Biol Chem. 1994;269:11699–11702. [PubMed] [Google Scholar]
- 29.Potsch S, Lendzian F, Ingemarson R, et al. The iron-oxygen reconstitution reaction in protein R2-Tyr-177 mutants of mouse ribonucleotide reductase. EPR and electron nuclear double resonance studies on a new transient tryptophan radical. J Biol Chem. 1999;274:17696–17704. doi: 10.1074/jbc.274.25.17696. [DOI] [PubMed] [Google Scholar]
- 30.Regnström K, Aberg A, Ormö M, Sahlin M, Sjöberg BM. The conserved serine 211 is essential for reduction of the dinuclear iron center in protein R2 of Escherichia coli ribonucleotide reductase. J Biol Chem. 1994;269:6355–61. [PubMed] [Google Scholar]
- 31.Larsson A, Sjoberg BM. Identification of the stable free radical tyrosine residue in ribonucleotide reductase. Embo J. 1998;5:2037–2040. doi: 10.1002/j.1460-2075.1986.tb04461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lendzian F. Structure and interactions of amino acid radicals in class I ribonucleotide reductase studied by ENDOR and high-field EPR spectroscopy. Bioch Biophy Acta. 2005;1707:67–90. doi: 10.1016/j.bbabio.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 33.Ekberg M, Pötsch S, Sandin E, et al. Preserved catalytic activity in an engineered ribonucleotide reductase R2 protein with a nonphysiological radical transfer pathway. The importance of hydrogen bond connections between the participating residues. J Biol Chem. 1998;273:21003–21008. doi: 10.1074/jbc.273.33.21003. [DOI] [PubMed] [Google Scholar]
- 34.Schmidt P, Andersson KK, Barra AL, Thelander L, Gräslund A. High field EPR studies of mouse ribonucleotide reductase indicate hydrogen bonding of the tyrosyl radical. J Bio Chem. 1996;271:23615–23618. doi: 10.1074/jbc.271.39.23615. [DOI] [PubMed] [Google Scholar]
- 35.Bar G, Bennati M, Nguyen HHT, Ge J, Stubbe J, Griffin RG. High-frequency (140-GHz) time domain EPR and ENDOR spectroscopy: the tyrosyl radical-diiron cofactor in ribonucleotide reductase from yeast. J Am Chem Soc. 2001;123:3569–3576. doi: 10.1021/ja003108n. [DOI] [PubMed] [Google Scholar]
- 36.Wei PP, Tomter AB, Røhr AK, Andersson KK, Solomon EL. Circular dichroism and magnetic circular dichroism studies of the active site of p53R2 from human and mouse: iron binding and nature of the biferrous site relative to other ribonucleotide reductases. Biochemistry. 2006;45:14043–14051. doi: 10.1021/bi061127p. [DOI] [PubMed] [Google Scholar]
- 37.Shao J, Zhou B, Zhu L, Qiu W, Yuan YC, Xi B, Yen Y. In vitro characterization of enzymatic properties and inhibition of the p53R2 subunit of human ribonucleotide reductase. Cancer Res. 2004;64:1–6. doi: 10.1158/0008-5472.can-03-3048. [DOI] [PubMed] [Google Scholar]
- 38.Greenfield NJ. Using circular dichroism spectra to estimate protein secondary structure. Nature protocols. 2006;1(6):2876–2890. doi: 10.1038/nprot.2006.202. [DOI] [PMC free article] [PubMed] [Google Scholar]