

Abstract
The Down syndrome cell adhesion molecule (Dscam) gene has essential roles in neural wiring and pathogen recognition in Drosophila melanogaster. Dscam encodes 38,016 distinct isoforms via extensive alternative splicing. The 95 alternative exons in Dscam are organized into clusters that are spliced in a mutually exclusive manner. The exon 6 cluster contains 48 variable exons and uses a complex system of competing RNA structures to ensure that only one variable exon is included. Here we show that the heterogeneous nuclear ribonucleoprotein hrp36 acts specifically within, and throughout, the exon 6 cluster to prevent the inclusion of multiple exons. Moreover, hrp36 prevents serine/arginine-rich proteins from promoting the ectopic inclusion of multiple exon 6 variants. Thus, the fidelity of mutually exclusive splicing in the exon 6 cluster is governed by an intricate combination of alternative RNA structures and a globally acting splicing repressor.
Pre-mRNA splicing is frequently used to regulate important biological processes. It is currently estimated that between 60% and 75% of human genes encode alternatively spliced pre-mRNAs1,2. Though most of these genes encode 2–3 different isoforms, there are several examples of genes that can generate much larger repertoires of mRNAs. For example, the human KCNMA1 gene can generate ~500 different isoforms, and the three NRXN genes together produce over 2,000 isoforms3,4. However, by far the most notable example is the D. melanogaster Dscam gene, which can generate 38,016 isoforms5.
Dscam encodes a diverse collection of transmembrane domain receptors that have an important role in neural wiring and pathogen recognition6. For example, in the nervous system, Dscam is required for the proper wiring of Bolwigs nerve5, olfactory receptor neurons7, projection neurons of the olfactory system8, mushroom body neurons9-11 and mechanosensory neurons12. In every case that has been examined, mutations in Dscam result in neural wiring defects. More recently, Dscam has been shown to play an important role in the insect immune system. In both D. melanogaster13 and the mosquito Anopheles gambiae14, Dscam is required to mount an effective immune response.
In D. melanogaster, Dscam contains 115 exons, of which 95 are alternatively spliced5 (Fig. 1a). These variable exons are organized in clusters; the exon 4, 6, 9 and 17 clusters contain 12, 48, 33 and 2 variable exons, respectively. Importantly, the exons within each cluster are spliced in a mutually exclusive manner—only one exon from each cluster is included in the mRNA. The exon 4, 6 and 9 clusters encode alternative versions of extracellular immunoglobulin repeats, whereas the exon 17 cluster encodes two different transmembrane domains, such that each of the 38,016 isoforms has the same overall domain structure.
Figure 1.
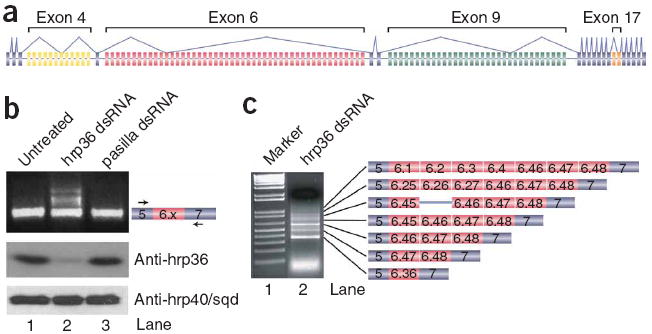
hrp36 is required for mutually exclusive splicing fidelity in the exon 6 cluster of Dscam. (a) Organization of D. melanogaster Dscam. (b) S2 cells were incubated alone or in the presence of hrp36 or pasilla double-stranded RNA, and splicing of the exon 6 cluster was examined by RT-PCR using primers in exons 5 and 7. Below, western blot showing that hrp36 levels are reduced in cells treated with hrp36 double-stranded RNA and that depletion of hrp36 is specific, as the levels of hrp40/sqd are unchanged. (c) A higher resolution gel, cloning and sequencing revealed that the larger bands correspond to products derived from mRNAs containing multiple exon 6 variants.
From a mechanistic view one of the most puzzling aspects of Dscam is how the splicing of the large clusters of exons occurs in a mutually exclusive manner. Thousands of eukaryotic genes contain regions that have two mutually exclusive exons, and a variety of mechanisms are known that ensure that only one exon is included in these mRNAs. These mechanisms involve steric hindrance, the use of both the major and minor spliceosomes, and the nonsense-mediated decay pathway15. However, none of these mechanisms explain how the variable clusters in Dscam can be spliced in a mutually exclusive manner.
Insight into the mechanism by which Dscam splicing occurs in a mutually exclusive manner was obtained from comparative genomics16,17. The exon 6 cluster contains two classes of conserved elements: the docking site, which is located in the intron downstream of constitutive exon 5, and the selector sequences, which are located upstream of each of the 48 exon 6 variants. Each selector sequence is complementary to a portion of the docking site, and this pairing juxtaposes one, and only one, alternative exon to the upstream constitutive exon. The mutually exclusive nature of the docking site-selector sequence interactions immediately suggests that the formation of these competing RNA structures is a central component of the mechanism guaranteeing that only one exon 6 variant is included in each Dscam mRNA. Notably, it seems as though a different mechanism is used to ensure that the exon 4 and 9 clusters are spliced in a mutually exclusive manner.
Although the mutually exclusive nature of docking site-selector sequence interactions suggests that they have a key role in ensuring that only one exon 6 variant is included in each mRNA, the mechanism by which these RNA structures do this is unknown. Moreover, these RNA structures alone cannot explain why the variable exons whose selector sequences do not interact with the docking site are not included. This has prompted us to search for proteins that have a role in ensuring the fidelity of exon 6 mutually exclusive splicing. Here, we show that the heterogeneous nuclear ribonucleo-protein (hnRNP) hrp36 has a key role in this process—depletion of hrp36 by RNA interference results in the inclusion of multiple exon 6 variants. The effects of hrp36 are cluster wide (hrp36 depletion affects most if not all exons within the exon 6 cluster) and cluster specific (hrp36 depletion has no effect on the mutually exclusive splicing of the exon 4, 9 or 17 clusters). Moreover, we find that hrp36 functions by preventing serine/arginine-rich (SR) proteins from promoting the ectopic inclusion of multiple exon 6 variants. These results add a new layer to the mechanism that ensures that only one exon 6 variant is included in the Dscam mRNA.
RESULTS
hrp36 ensures exon 6 mutually exclusive splicing fidelity
To identify proteins involved in controlling Dscam exon 6 mutually exclusive splicing, we performed an RNAi screen in which D. melanogaster S2 cells were treated with various double-stranded RNAs and Dscam splicing was monitored first by a fiber optic universal microarray18,19 containing 1,536 probes (including probes for all of the Dscam exons) and second by reverse transcription PCR (RT-PCR) using primers in exons 5 and 7 of Dscam followed by agarose gel electrophoresis. For the RNAi screen we used a library of ~400 double-stranded RNAs corresponding to genes encoding RNA-binding proteins, spliceosomal components and DEXH/D-box ATPases20. The microarray screen identified seven proteins as candidates for controlling the fidelity of mutually exclusive splicing, as they modulated the splicing of most if not all exons within at least one of the clusters (Supplementary Discussion and Supplementary Fig. 1 online). These include both subunits of U2AF (CG9998/dU2AF50 and CG3582/dU2AF38), dCAPER/dCC1.3 (CG11266/CC1.3), the spliceosomal DEXH/D-box ATPases dBrr2 (CG5931/Brr2), dPrp5 (CG6197/Prp5) and Rm62 (CG10279/Rm62/p68), and the hnRNP hrp36 (hrp36/Hrb87F/CG12749). However, our secondary RT-PCR screen using primers in exons 5 and 7 revealed that only hrp36 double-stranded RNA has an impact on exon 6 mutually exclusive splicing (Fig. 1b). We identified RT-PCR products larger than those containing only a single exon 6 variant when using RNA isolated from cells depleted of hrp36 (Fig. 1b, lane 2), but not when using RNA from untreated cells (Fig. 1b, lane 1) or from cells treated with a control double-stranded RNA (pasilla, encoded by ps) (Fig. 1b, lane 3). Cloning and sequencing of these products revealed that they are derived from RNAs containing multiple exon 6 variants spliced together (Fig. 1c). We identified products that include up to seven exon 6 variants, though larger products can be observed on gel, which suggests that vast numbers of exon 6 variants can be spliced together in the absence of hrp36. In most cases, adjacent exons are spliced together (for example, exon 6.45 is spliced to 6.46), though we also identified clones in which exons located far from one another in the pre-mRNA were spliced together (for example, exon 6.4 is spliced to exon 6.46). The observation that adjacent exons were frequently spliced together in the absence of hrp36 indicates that there are no inherent cis-acting physical barriers, such as intron size, that normally prevent adjacent exon 6 variants from being spliced to one another. Importantly, the correct exon 6 splice sites were used in all but one of the clones sequenced. In this case, the clone also contained the intron-separating exons 6.45 and 6.46. Consistent with the microarray results (Supplementary Fig. 1), most of the clones contained exons 6.46, 6.47 and 6.48, either alone or in combination with other exons. The effect of depleting hrp36 on exon 6 mutually exclusive splicing is not due to an off-target RNAi effect, as identical results are obtained using a second, nonoverlapping double-stranded RNA corresponding to the 3′ untranslated region of the hrp36 mRNA (Supplementary Fig. 2 online). Together, these data indicate that hrp36 has a key role in ensuring the fidelity of exon 6 mutually exclusive splicing in S2 cells.
hrp36 acts in a cluster-specific and cluster-wide manner
We next tested whether hrp36 has a role in ensuring the fidelity of mutually exclusive splicing in the other alternatively spliced clusters in Dscam (Fig. 2). Previously, we have shown that hrp36 has a role in alternative splicing of the exon 4 and 17 clusters of Dscam, but in these cases hrp36 controls which of the variable exons are selected rather than the number of exons included20. Consistent with these earlier results, when we used constitutive exon primers, we found no evidence that hrp36 RNAi results in the inclusion of multiple variable exons in the exon 4, 9 or 17 clusters (Fig. 2d; data not shown).
Figure 2.
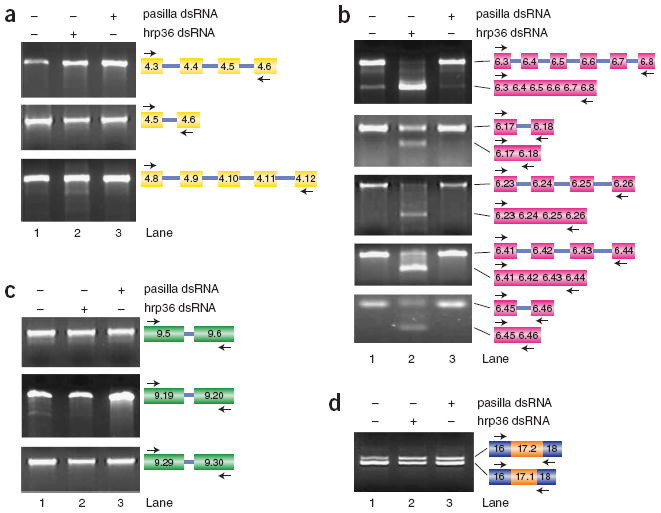
hrp36 functions in a cluster-specific manner. (a–d) RT-PCR was used to examine the role of hrp36 in mutually exclusive splicing of the exon 4 (a), exon 6 (b), exon 9 (c) and exon 17 (d) clusters. These experiments revealed no impact of hrp36 depletion on mutually exclusive splicing of the exon 4, exon 9 or exon 17 clusters. In contrast, depletion of hrp36 results in the splicing of multiple adjacent exon 6 variants throughout the exon 6 cluster. The primers used in each experiment and the identity of the products, which were confirmed by cloning and sequencing, are depicted.
Although these data suggest that hrp36 does not impact the mutually exclusive splicing of the other clusters, we developed a more sensitive assay that uses primers within the alternatively spliced exons to more rigorously test this. For example, when primers in exons 6.3 and 6.8 are used in RT-PCR reactions with total RNA from untreated cells, or from cells treated with a control double-stranded RNA, we primarily observe products corresponding to unspliced pre-mRNA (presumably amplified from released lariat introns) (Fig. 2b, lanes 1 and 3). In contrast, when cells are treated with hrp36 double-stranded RNA, we observe a switch to a smaller product that contains exons 6.3 through 6.8 accurately spliced together (Fig. 2b, lane 2). Using this assay we find that hrp36 controls the fidelity of mutually exclusive splicing throughout the entire exon 6 cluster (Fig. 2b; data not shown). In contrast, cells depleted of hrp36 do not display defects in mutually exclusive splicing in the exon 4, 9 or 17 clusters (Fig. 2a,c,d). These results demonstrate that hrp36 acts in both a cluster-wide and cluster-specific manner to ensure the fidelity of mutually exclusive splicing.
We also tested whether hrp36 is the only hnRNP protein that regulates the fidelity of exon 6 mutually exclusive splicing in S2 cells. To do this we treated S2 cells with double-stranded RNAs to hrp36, hrp38, hrp40 and hrp48 either alone or in combination and examined exon 6 splicing by RT-PCR (Fig. 3a) and the extent of depletion of each protein by western blot (Fig. 3b). We found that depletion of only hrp36 (Fig. 3a,b, lane 2), but not the three other hrp proteins (Fig. 3a,b, lanes 3–5), results in the inclusion of multiple exons. Moreover, co-depletion of hrp36 and either hrp38, hrp40 or hrp48 did not enhance the effect of depleting hrp36 alone (Fig. 3a,b, lanes 6–8). Additionally, the simultaneous depletion of hrp38, hrp40 and hrp48 had no impact on exon 6 splicing (Fig. 3a,b, lane 12). Thus, hrp36 is the only major D. melanogaster hnRNP protein that has a role in the fidelity of exon 6 mutually exclusive splicing in S2 cells.
Figure 3.
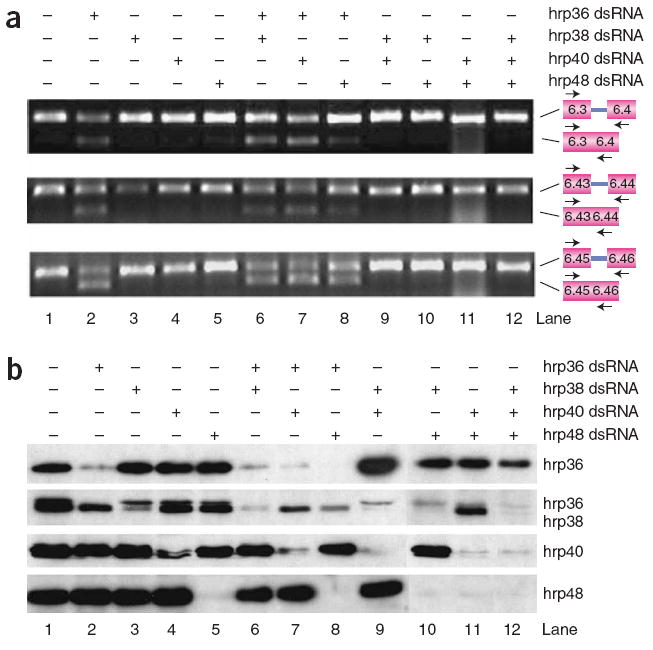
hrp36 is the only major hnRNP protein that regulates the fidelity of exon 6 mutually exclusive splicing. (a,b) D. melanogaster S2 cells were treated with hrp36, hrp38, hrp40 and hrp48 double-stranded RNAs as indicated. Exon 6 splicing was examined by RT-PCR with primers to detect the specified exons (a), and the extent and specificity of depletion was monitored by western blotting (b). Note that the hrp38 antibody cross-reacts with hrp36 to some extent.
hrp36 binds throughout the exon 6 cluster
To begin to examine possible mechanisms by which hrp36 could function to ensure the fidelity of exon 6 alternative splicing of the entire exon 6 cluster, we have examined the interaction between hrp36 and the Dscam pre-mRNA. First, we used a position-specific scoring matrix (PSSM) derived from systematic evolution of ligands by exponential enrichment (SELEX) experiments with hrp36 (M.B., R. Green, S. MacArthur, M. Eisen and D.C.R., unpublished data) to scan the entire D. melanogaster Dscam pre-mRNA for potential hrp36 binding sites. We identified numerous potential hrp36 binding sites throughout the Dscam gene. Within the exon 6 cluster we found that the hrp36 binding sites are significantly enriched in exons and depleted from introns (P = 0.0007) (Table 1). For example, 29 of the 48 exon 6 variants in D. melanogaster contain at least one hrp36 binding site, whereas only 14 of the 49 introns flanking the exon 6 variants contain a hrp36 site. Importantly, this trend is conserved in all 12 Drosophila species that have been sequenced (P = 0.0003). The most extreme case is the D. mojavensis Dscam gene, in which 40 of the 50 exon 6 variants contain at least one hrp36 site, whereas only 12 of the 51 introns do (Table 1). These results suggest that hrp36 may promote the fidelity of mutually exclusive splicing by binding to the exon 6 variants.
Table 1.
Distribution of hrp36 binding sites in the Dscam exon 6 cluster in 12 Drosophila species
| Species | Number of exon 6 variants | Number of exonic hrp36 sites | Number (%) of exons with ≥1 hrp36 site | Size of all exons (base pairs) | Exonic sites/kilobase | Number of intronic hrp36 sites | Number (%) of introns with ≥1 hrp36 site | Size of all introns (base pairs) | Intronic sites/kilobase | Fold enrichment exon/intron |
|---|---|---|---|---|---|---|---|---|---|---|
| D. melanogaster | 48 | 46 | 29 (60.4%) | 5,901 | 7.80 | 22 | 14 (28.6%) | 6,842 | 3.22 | 2.42 |
| D. simulans | 48 | 40 | 25 (52.1%) | 5,901 | 6.78 | 20 | 10 (20.4%) | 6,757 | 2.96 | 2.29 |
| D. yakuba | 41 | 43 | 25 (61.0%) | 5,039 | 8.53 | 15 | 9 (21.4%) | 6,692 | 2.24 | 3.81 |
| D. sechellia | 47 | 45 | 29 (61.7%) | 5,785 | 7.78 | 20 | 10 (20.8%) | 6,842 | 2.92 | 2.66 |
| D. erecta | 45 | 47 | 31 (68.9%) | 5,541 | 8.48 | 19 | 13 (26.3%) | 6,566 | 2.89 | 2.93 |
| D. ananassae | 46 | 55 | 28 (60.9%) | 5,666 | 9.71 | 14 | 6 (12.8%) | 6,168 | 2.27 | 4.28 |
| D. pseudoobscura | 48 | 69 | 34 (60.8%) | 5,913 | 11.67 | 30 | 11 (22.4%) | 6,881 | 4.36 | 2.68 |
| D. persimillis | 48 | 68 | 35 (72.9%) | 5,909 | 11.51 | 30 | 12 (24.5%) | 6,973 | 4.30 | 2.67 |
| D. willistoni | 49 | 38 | 29 (59.2%) | 6,028 | 6.30 | 7 | 3 (6.0%) | 5,927 | 1.18 | 5.34 |
| D. mojavensis | 50 | 60 | 40 (80.0%) | 6,156 | 9.75 | 17 | 12 (23.5%) | 7,685 | 2.21 | 4.41 |
| D. virilis | 52 | 53 | 36 (69.2%) | 6,548 | 8.09 | 20 | 10 (18.9%) | 7,692 | 2.60 | 3.11 |
| D. grimshawi | 53 | 49 | 32 (60.4%) | 6,522 | 7.51 | 18 | 14 (25.9%) | 8,040 | 2.24 | 3.36 |
To examine the potential interaction between hrp36 and the exon 6 variants, we performed gel mobility shift assays. For these experiments we tested exon 6.47, which contains two potential hrp36 binding sites, and exon 6.48, which contains one potential hrp36 binding site. 32P-labeled RNAs containing the entire exon sequence (123 nucleotides) were synthesized and incubated with recombinant hrp36, and the reactions were resolved on nondenaturing polyacrylamide gels. We found that hrp36 binds to both exon 6.47 and 6.48 (Fig. 4, lanes 1–5 and 11–15). However, point mutations that disrupt the integrity of the hrp36 binding sites prevent these interactions (lanes 6–10 and 16–20). Thus, hrp36 can physically interact with the exon 6 variants in a sequence-specific manner in vitro.
Figure 4.

hrp36 binds to exon 6 variants. Gel mobility shift experiments show that exons 6.47 (left) and 6.48 (right) bind to recombinant hrp36. However, mutations that disrupt the putative hrp36 binding sites eliminate the protein-RNA interaction.
To analyze whether hrp36 interacts with the Dscam pre-mRNA in vivo, we performed RNP immunoprecipitation-Chip (RIP-Chip) assays (M.B., R. Green, S. MacArthur, M. Eisen and D.C.R., unpublished data). Briefly, antibodies to hrp36 (and hrp38, hrp40 and hrp48 as controls) were used to immunoprecipitate RNP complexes from D. melanogaster S2 cell nuclear extracts. The RNAs isolated from these RNP complexes were then labeled and hybridized to 35-base-pair-resolution tiling arrays covering the entire D. melanogaster genome. The hybridization intensities of the RNP-associated RNAs were compared to the starting undepleted nuclear extract, and immuno-precipitations were performed using a nonspecific antibody. Although each hnRNP protein binds to the Dscam pre-mRNA to some extent, the pattern is distinct for each protein (Fig. 5). Most notably, hrp36 associated with the entire exon 4, 6 and 17 clusters, but not the exon 9 cluster. This is consistent with the idea that hrp36 has a role in regulating exon choice in the exon 4 and 17 clusters20 and mutually exclusive splicing of the exon 6 cluster while having no impact on the splicing of the exon 9 cluster. Thus, these results, in particular the association of hrp36 throughout the entire exon 6 cluster, are consistent with the observed effects of hrp36 on Dscam splicing. Moreover, the functional, bioinformatic, genomic and biochemical data together strongly suggest that hrp36 binds to multiple sites throughout the entire exon 6 cluster to repress the inclusion of multiple exon 6 variants, thus ensuring that only one exon is spliced into the mRNA.
Figure 5.
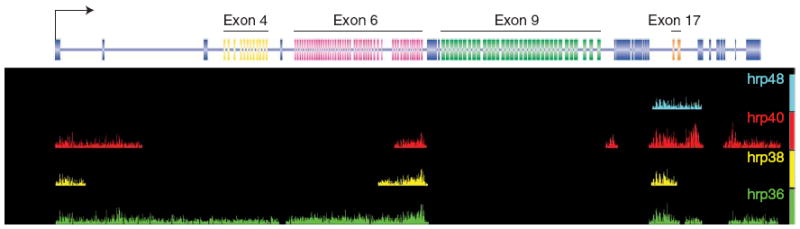
hrp36 binds to Dscam pre-mRNA throughout the entire exon 6 cluster. The distribution of the RNA sequences bound by hrp36, hrp38, hrp40 and hrp48 across the Dscam gene are shown. These maps were determined by labeling and hybridizing RNAs that coimmunoprecipitate with antibodies specific to each hnRNP protein to genome-wide Drosophila tiling arrays.
hrp36 prevents SR proteins from activating exon inclusion
Human hnRNP A1, which is closely related to hrp36, has been shown to repress splicing in some cases by binding throughout an exon21, where it prevents SR proteins from binding to the exons and promoting their inclusion. Based on this observation, we hypothesized that hrp36 may repress the inclusion of multiple exon 6 variants by preventing the ability of SR proteins to bind to the exons and promote their inclusion. A prediction of this model is that co-depleting an SR protein and hrp36 might reverse the effect of depleting hrp36 alone.
We tested this hypothesis by treating S2 cells with hrp36 double-stranded RNA either alone or in combination with different SR or SR-related protein double-stranded RNAs. Co-depletion of the SR protein B52 (Fig. 6a, lane 4) completely, or nearly completely, reversed the effect of depleting hrp36 (lane 2). Similar but weaker reversal was observed in cells treated with SC35 double-stranded RNA (lane 6), and to a lesser extent with SF2 (lane 7), RBP1-like (lane 3), SRm300 (lane 9) and SRm160 double-stranded RNAs (lane 10). No effect was observed in cells treated with RBP1 (lane 5) or SRp54 double-stranded RNAs (lane 8). These results are consistent with the hypothesis that in the absence of hrp36, SR proteins promote the inclusion of multiple exon 6 variants.
Figure 6.

hrp36 prevents ectopic exon 6 inclusion by SR proteins. To test whether the effect of depleting hrp36 could be reversed by co-depleting various SR proteins, S2 cells were treated with double-stranded RNAs to hrp36 and/or the indicated SR proteins. (a) The inclusion of multiple exon 6 variants induced by depletion of hrp36 can be reversed to various extents by co-depleting different SR proteins. (b) Depletion of the SR proteins alone has no effect on exon 6 mutually exclusive splicing in the presence of hrp36.
An alternative explanation for these observations is that the depletion of SR proteins compromises the general efficiency of splicing and consequently decreases the probability that multiple exon 6 variants would be included. If this were the case, we should observe exon 6 skipping in cells that contain hrp36 but are depleted of SR proteins by RNAi. To test this, we examined exon 6 splicing in cells treated with the SR protein double-stranded RNAs, but not with hrp36 double-stranded RNA, by RT-PCR with primers in exons 6.3 and 6.4 (Fig. 6b, top) or exons 5 and 7 (Fig. 6b, bottom). Importantly, we did not observe any defects in the exon 6 mutually exclusive splicing mechanism in cells treated with any of the SR protein double-stranded RNAs alone. This suggests that treating cells with the SR protein double-stranded RNAs does not result in a general decrease in splicing efficiency. Together, our results suggest that hrp36 ensures the fidelity of exon 6 mutually exclusive splicing by preventing the ability of SR proteins to promote exon 6 inclusion.
DISCUSSION
We have shown that the hnRNP protein hrp36 serves to prevent the inclusion of multiple exon 6 variants. There are several noteworthy features about the manner by which hrp36 functions to ensure the fidelity of mutually exclusive splicing. First, hrp36 acts throughout the entire exon 6 cluster and simultaneously controls the splicing of a large collection of exons. Second, the role of hrp36 in mutually exclusive splicing is restricted to the exon 6 cluster; hrp36 has no effect on the mutually exclusive splicing of the exon 4, 9 or 17 clusters. Finally, hrp36 binds to most if not all of the exons within the exon 6 cluster. The fact that the exon 6 variants are accurately spliced together in the absence of hrp36 indicates that there are no cis-acting physical barriers that prevent this from occurring under normal circumstances. Thus, our data suggest that the binding of hrp36 to the exon 6 variants represses the exon 6 variants from being spliced together.
We have further shown that hrp36 prevents SR proteins from promoting ectopic exon 6 inclusion; depleting different SR proteins, B52 in particular, could in fact restore normal mutually exclusive splicing of the exon 6 cluster to cells that were also depleted of hrp36. Given the well-established antagonism between hnRNP proteins and SR proteins21,22, the simplest explanation for these data is that when hrp36 is bound to the exon 6 variants, it prevents SR proteins from activating their splicing, perhaps by preventing the SR proteins from binding to the exons. Alternatively, as SR proteins are known to have exon-independent functions23, it is also possible that B52 functions without binding to the exons.
To examine the possibility that B52 promotes exon 6 inclusion by binding to the exon 6 variants, we searched the exon 6 cluster for matches to the B52 consensus sequence determined by SELEX (ref. 24). No matches to this sequence were found in the entire Dscam pre-mRNA. However, the properties of the B52 SELEX consensus are different from the SELEX consensus sequences determined for other SR proteins23. Thus, the B52 SELEX consensus is not likely to represent the full repertoire of potential B52 binding sites. Further work will therefore be needed to determine whether B52 binds to the exon 6 variants in the absence of hrp36.
Depleting hrp36 in S2 cells has a profound effect on exon 6 splicing, and little if any functional Dscam protein should be produced under such circumstances. As Dscam is essential for viability5, it is surprising that flies lacking hrp36 are viable25,26 and that exon 6 splicing occurs normally in hrp36-null flies (S.O. and B.R.G., unpublished data). This apparent discrepancy is not due to an RNAi off-target effect, as two nonoverlapping hrp36 double-stranded RNAs have the same effect on both exon 6 mutually exclusive splicing and hrp36 levels in S2 cells (Supplementary Fig. 1). Thus, there is a true difference in the requirement for hrp36 in exon 6 mutually exclusive splicing between S2 cells and flies. There are many possible reasons that Dscam splicing occurs normally in hrp36 mutants. One possibility is that D. melanogaster individuals express one or more proteins that are functionally redundant with hrp36 that are not expressed in S2 cells. Alternatively, as the effect of depleting hrp36 can be reversed by co-depleting SR proteins, such as B52, it could be that the balance between these factors is either different in flies and S2 cells, or is compensated for in the hrp36 mutant flies. Additionally, the requirement of hrp36 for Dscam splicing may be tissue-specific; some cells or tissues may require hrp36, whereas hrp36 may be dispensable in other cells and tissues. Nonetheless, our results clearly demonstrate that hrp36 is essential for Dscam splicing in S2 cells. Further experiments will be required to analyze the role of hrp36 in Dscam splicing in D. melanogaster in more detail and to identify other proteins that functionally substitute for this function of hrp36.
The results reported here support and extend our previously proposed model for the mechanism of exon 6 mutually exclusive splicing17 (Fig. 7). We propose that hrp36 binds to most if not all of the exon 6 variants and represses their inclusion. When a selector sequence interacts with the docking site, something about that interaction causes the hrp36 molecules bound to the downstream exon to either dissociate from the exon or become unable to repress splicing of that exon. This would then allow for SR proteins to bind the exon and promote its inclusion. As the docking site-selector sequence interaction would only act on the selected exon, all of the downstream exons would remain repressed by hrp36 and could not be included. This model explains why multiple exons are included when hrp36 is depleted: in the absence of hrp36, all of the exon 6 variants can be included because none of the exons are repressed.
Figure 7.
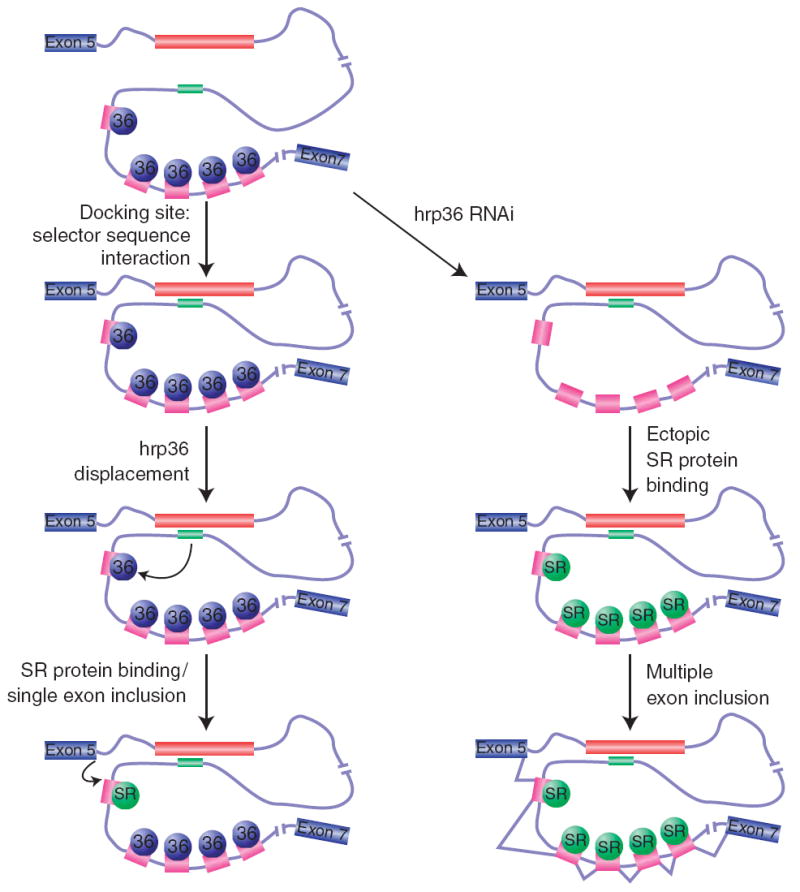
Model for the mechanism by which hrp36 prevents the inclusion of multiple exon 6 variants. hrp36 (blue circles) binds to all of the exon 6 variants (magenta) and represses their inclusion. When the selector sequence upstream of a specific exon interacts with the docking site, this results in the de-repression of hrp36 on the exon immediately downstream, but not for the other exon 6 variants. In this way, only a single exon 6 variant is included. hrp36 competes with SR proteins (green circles) for binding to the exon 6 variants. In the absence of hrp36, these activators could bind to all of the exon 6 variants and enhance their splicing to one another.
While this model explains many aspects of how splicing of the exon 6 cluster occurs in a mutually exclusive manner, there remain many questions. First of all, how does the docking site-selector sequence interaction relieve the repression exerted by hrp36 on the downstream exon? What determines which of the 48 selector sequences interacts with the docking site in a given pre-mRNA? Is this process highly regulated or does it occur in a random or stochastic manner? Microarray experiments have revealed little if any regulation of exon 6 splicing11,27, which is consistent with the biology of Dscam. In the nervous system, it seems that the actual identity of the Dscam isoforms that are expressed is unimportant, provided that each neuron expresses a different set of isoforms than the neighboring neurons11,28-31. Similarly, within the immune system, where Dscam functions in a manner analogous to vertebrate antibodies13,16, hemocytes must be capable of expressing most if not all of the potential Dscam isoforms to mount an effective immune defense against invading pathogens. The simplest way to accomplish these tasks in both the nervous and immune systems would be via a stochastic mechanism. It is therefore noteworthy that each exon 6 variant is repressed by hrp36 and contains a selector sequence that can interact with the docking site. Thus, the exon 6 cluster is organized such that even if exon selection occurs randomly, splicing would still occur in a mutually exclusive manner. In addition, the stochastic splicing of the exon 6 cluster could easily be overridden in a developmental-, tissue- or cell-specific manner by the expression of specific positive or negative regulators that could either enhance or repress the inclusion of specific exons.
METHODS
RNAi microarray screen
Transcription templates were generated by PCR from a plasmid library containing complementary DNA fragments from ~400 genes encoding mRNA processing factors20. Double-stranded RNA was synthesized by in vitro transcription (Epicentre T7 flash kit) and annealed by incubating at 100 °C for 2 min and slowly cooling the reactions to room temperature (20–22 °C) over a 40-min time period. 40 μg of double-stranded RNA was added to each well of a 6-well culture dish containing 2 × 106 D. melanogaster S2 cells in D. melanogaster SFM medium (Invitrogen) supplemented with l-glutamine and Pen/strep. The cells were grown at 27 °C for 2 d, after which an additional 40 μg of double-stranded RNA was added to each well, and the incubation continued for 2 more d. Total RNA was isolated from each sample with Trizol (Invitrogen) according to the manufacturer’s instructions.
Microarray experiments were performed using the DASL assay18 with a pool of oligonucleotides complementary to 1,536 constitutive or alternative exons in ~200 D. melanogaster pre-mRNAs. Between 2 and 10 biological replicates per double-stranded RNA were performed. After normalization of the data and averaging of replicate samples with a correlation coefficient greater than 0.9, Z-scores were calculated for each exon in each sample. Details of these experiments and a global analysis of the results will be described elsewhere.
RT-PCR
Total RNA was isolated from S2 cells using Trizol reagent (Invitrogen) following the manufacturer’s protocol. Reverse transcription was performed on each RNA sample using 200 units of Superscript II (Invitrogen) and 5 μg of RNA in a 20 μl reaction, at 42 °C for 1 h. PCR reactions were then set up to analyze alternative splicing of various regions of Dscam. Primer sequences are available upon request.
Western blots
Protein was harvested from S2 cells in 1× SDS-PAGE buffer, heated to 100 °C and then resolved on a 12.5% (w/v) SDS-PAGE gel. Proteins were then electoblotted to a PVDF membrane (Biorad) and blocked in 5% (w/v) nonfat dry milk in Tris-buffered saline/Tween (TBST) at room temperature for 1 h. The primary antibody was added directly to the blocking reagent and incubated overnight at room temperature. Blots were washed in TBST and incubated with the secondary antibody linked to horseradish peroxidase for 1 h at room temperature. Western blots were developed using Western Lightning chemiluminescence kit (Perkin Elmer).
Gel-shift assays
RNAs corresponding to exons 6.47 and 6.48, or variants of these exons in which point mutations were generated to disrupt the putative hrp36 binding sites, were synthesized by in vitro transcription with α-32P-UTP and gel purified. The RNAs were incubated with the indicated quantities of hrp36 that were expressed in and purified from Escherichia coli. After incubation for 15 min at room temperature, heparin was added to a final concentration of 0.5 mg ml−1, and the reactions were resolved on a 5% (w/v) (29:1) acrylamide gel. The gel was then dried and visualized by autoradiography.
Identification of hrp36 binding sites in Dscam
A PSSM for the hrp36 binding site derived from SELEX experiments was used to search the Dscam gene from 12 Drosophila species17. We used a raw score of 5.0 as a cutoff for putative hrp36 binding sites. The location of each site within the exon 6 cluster was used to determine the frequency of sites within exons and introns for each species. The statistical significance of the findings was evaluated by performing a chi-squared test on the differential enrichment of the hrp36 sites in exons versus introns.
RIP-Chip assays
Nuclear RNP fractions from S2 cells were prepared using a modified version the mammalian RNP protocol32. Briefly, 16 l of exponentially growing S2 cells were harvested, and their nuclei were isolated by hypotonic lysis following the Dignam extract protocol33. The nuclei were resuspended in 2.5 packed-cell volumes with ice-cold (2–4 °C) HNEB2 (10 mM Tris-HCl pH 7.5, 100 mM NaCl, 2.5 mM MgCl2, 0.5% (v/v) Triton X-100, 0.2 mM PMSF) and sonicated 3 times for 30 s. An RNP-enriched fraction was isolated by centrifugation of the sonicated nuclei over a 30% (w/v) sucrose cushion (HNEB2 plus 30% (w/v) sucrose) for 15 min at 4,800g. Immunopurification of the hnRNP-containing RNP complexes was done by mixing 1 ml of the S2 RNP fraction with ~200 ng of affinity-purified rabbit polyclonal antibodies in a total volume of 10 ml of ice-cold IPB1 (10 mM Tris-HCl pH 7.5, 150 mM NaCl, 2.5 mM MgCl2, 0.05% (v/v) Triton X-100, 0.5 mM PMSF, 1× protease inhibitor cocktail). The antibody/RNP fraction was rocked for 15 min at 4 °C, and the solution was filtered through a 0.2-mm Milex GV filter (Millipore) followed by a 15-min incubation at 4 °C with 0.25 ml of a 50% (v/v) slurry of protein A agarose CL-4B (Sigma). The beads were recovered and washed 4 times with 10 ml of cold (2–4 °C) IPB2 (10 mM Tris-Cl pH 7.5, 150 mM NaCl, 2.5 mM MgCl2, 0.25% (v/v) Triton X-100) buffer. The RNA was eluted with 350 μl of buffer RLT1 (Qiagen) with β-mercaptoethanol (Qiagen) and purified following the RNeasy midi kit protocol (Qiagen). Total RNA purified from a single immunopurification was amplified and labeled following the GeneChip WT amplified double-stranded cDNA synthesis kit (Affymetrix) and hybridized on a single GeneChip Drosophila 1.0R whole-genome tiling array following the manufacturer’s recommendations (Affymetrix). These arrays were compared to arrays hybridized with sample isolated from total RNA purified directly from the starting RNP fraction. Downstream data analysis was performed using the TiMAT application suite (http://bdtnp.lbl.gov/TiMAT/), and the data were visualized using the Affymetrix Integrated Genome Browser (http://www.affymetrix.com/support/developer/tools/download_igb.affx).
Supplementary Material
Note: Supplementary information is available on the Nature Structural & Molecular Biology website.
Acknowledgments
We thank members of the Graveley lab for discussions and comments on the manuscript. This work was funded by the Crick-Jacobs Center for Theoretical and Computational Biology (G.W.Y.), a long-term fellowship from the Human Frontier Science Program to M.B., and grants from the US National Institutes of Health to D.C.R. (GM61987) and B.R.G. (GM62561 and GM67842).
Footnotes
COMPETING INTERESTS STATEMENT The authors declare competing financial interests: details accompany the full-text HTML version of the paper at http://www.nature.com/nsmb/.
Published online at http://www.nature.com/nsmb
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions
References
- 1.Blencowe BJ. Alternative splicing: new insights from global analyses. Cell. 2006;126:37–47. doi: 10.1016/j.cell.2006.06.023. [DOI] [PubMed] [Google Scholar]
- 2.Xing Y, Lee C. Alternative splicing and RNA selection pressure–evolutionary consequences for eukaryotic genomes. Nat Rev Genet. 2006;7:499–509. doi: 10.1038/nrg1896. [DOI] [PubMed] [Google Scholar]
- 3.Black DL. Protein diversity from alternative splicing: a challenge for bioinformatics and post-genome biology. Cell. 2000;103:367–370. doi: 10.1016/s0092-8674(00)00128-8. [DOI] [PubMed] [Google Scholar]
- 4.Graveley BR. Alternative splicing: increasing diversity in the proteomic world. Trends Genet. 2001;17:100–107. doi: 10.1016/s0168-9525(00)02176-4. [DOI] [PubMed] [Google Scholar]
- 5.Schmucker D, et al. Drosophila Dscam is an axon guidance receptor exhibiting extraordinary molecular diversity. Cell. 2000;101:671–684. doi: 10.1016/s0092-8674(00)80878-8. [DOI] [PubMed] [Google Scholar]
- 6.Zipursky SL, Wojtowicz WM, Hattori D. Got diversity? Wiring the fly brain with Dscam. Trends Biochem Sci. 2006;31:581–588. doi: 10.1016/j.tibs.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 7.Hummel T, et al. Axonal targeting of olfactory receptor neurons in Drosophila is controlled by Dscam. Neuron. 2003;37:221–231. doi: 10.1016/s0896-6273(02)01183-2. [DOI] [PubMed] [Google Scholar]
- 8.Zhu H, et al. Dendritic patterning by Dscam and synaptic partner matching in the Drosophila antennal lobe. Nat Neurosci. 2006;9:349–355. doi: 10.1038/nn1652. [DOI] [PubMed] [Google Scholar]
- 9.Wang J, et al. Transmembrane/juxtamembrane domain-dependent Dscam distribution and function during mushroom body neuronal morphogenesis. Neuron. 2004;43:663–672. doi: 10.1016/j.neuron.2004.06.033. [DOI] [PubMed] [Google Scholar]
- 10.Wang J, Zugates CT, Liang IH, Lee CH, Lee T. Drosophila Dscam is required for divergent segregation of sister branches and suppresses ectopic bifurcation of axons. Neuron. 2002;33:559–571. doi: 10.1016/s0896-6273(02)00570-6. [DOI] [PubMed] [Google Scholar]
- 11.Zhan XL, et al. Analysis of Dscam diversity in regulating axon guidance in Drosophila mushroom bodies. Neuron. 2004;43:673–686. doi: 10.1016/j.neuron.2004.07.020. [DOI] [PubMed] [Google Scholar]
- 12.Chen BE, et al. The molecular diversity of Dscam is functionally required for neuronal wiring specificity in Drosophila. Cell. 2006;125:607–620. doi: 10.1016/j.cell.2006.03.034. [DOI] [PubMed] [Google Scholar]
- 13.Watson FL, et al. Extensive diversity of Ig-superfamily proteins in the immune system of insects. Science. 2005;309:1874–1878. doi: 10.1126/science.1116887. [DOI] [PubMed] [Google Scholar]
- 14.Dong Y, Taylor HE, Dimopoulos G. AgDscam, a hypervariable immunoglobulin domain-containing receptor of the Anopheles gambiae innate immune system. PLoS Biol. 2006;4:e229. doi: 10.1371/journal.pbio.0040229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith CW. Alternative splicing–when two’s a crowd. Cell. 2005;123:1–3. doi: 10.1016/j.cell.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 16.Anastassiou D, Liu H, Varadan V. Variable window binding for mutually exclusive alternative splicing. Genome Biol. 2006;7:R2. doi: 10.1186/gb-2006-7-1-r2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Graveley BR. Mutually exclusive splicing of the insect Dscam pre-mRNA directed by competing intronic RNA secondary structures. Cell. 2005;123:65–73. doi: 10.1016/j.cell.2005.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fan JB, et al. A versatile assay for high-throughput gene expression profiling on universal array matrices. Genome Res. 2004;14:878–885. doi: 10.1101/gr.2167504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yeakley JM, et al. Profiling alternative splicing on fiber-optic arrays. Nat Biotechnol. 2002;20:353–358. doi: 10.1038/nbt0402-353. [DOI] [PubMed] [Google Scholar]
- 20.Park JW, Parisky K, Celotto AM, Reenan RA, Graveley BR. Identification of alternative splicing regulators by RNA interference in Drosophila. Proc Natl Acad Sci USA. 2004;101:15974–15979. doi: 10.1073/pnas.0407004101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu J, Mayeda A, Krainer AR. Exon identity established through differential antagonism between exonic splicing silencer-bound hnRNP A1 and enhancer-bound SR proteins. Mol Cell. 2001;8:1351–1361. doi: 10.1016/s1097-2765(01)00409-9. [DOI] [PubMed] [Google Scholar]
- 22.Matlin AJ, Clark F, Smith CW. Understanding alternative splicing: towards a cellular code. Nat Rev Mol Cell Biol. 2005;6:386–398. doi: 10.1038/nrm1645. [DOI] [PubMed] [Google Scholar]
- 23.Graveley BR. Sorting out the complexity of SR protein functions. RNA. 2000;6:1197–1211. doi: 10.1017/s1355838200000960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi H, Hoffman BE, Lis JT. A specific RNA hairpin loop structure binds the RNA recognition motifs of the Drosophila SR protein B52. Mol Cell Biol. 1997;17:2649–2657. doi: 10.1128/mcb.17.5.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haynes SR, Johnson D, Raychaudhuri G, Beyer AL. The Drosophila Hrb87F gene encodes a new member of the A and B hnRNP protein group. Nucleic Acids Res. 1991;19:25–31. doi: 10.1093/nar/19.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zu K, Sikes ML, Haynes SR, Beyer AL. Altered levels of the Drosophila HRB87F/hrp36 hnRNP protein have limited effects on alternative splicing in vivo. Mol Biol Cell. 1996;7:1059–1073. doi: 10.1091/mbc.7.7.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neves G, Zucker J, Daly M, Chess A. Stochastic yet biased expression of multiple Dscam splice variants by individual cells. Nat Genet. 2004;36:240–246. doi: 10.1038/ng1299. [DOI] [PubMed] [Google Scholar]
- 28.Hattori D, et al. Dscam diversity is essential for neuronal wiring and self-recognition. Nature. 2007;449:223–227. doi: 10.1038/nature06099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hughes ME, et al. Homophilic Dscam interactions control complex dendrite morphogenesis. Neuron. 2007;54:417–427. doi: 10.1016/j.neuron.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matthews BJ, et al. Dendrite self-avoidance is controlled by Dscam. Cell. 2007;129:593–604. doi: 10.1016/j.cell.2007.04.013. [DOI] [PubMed] [Google Scholar]
- 31.Soba P, et al. Drosophila sensory neurons require Dscam for dendritic self-avoidance and proper dendritic field organization. Neuron. 2007;54:403–416. doi: 10.1016/j.neuron.2007.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Piñol-Roma S, Swanson MS, Matunis MJ, Dreyfuss G. Purification and characterization of proteins of heterogeneous nuclear ribonucleoprotein complexes by affinity chromatography. Methods Enzymol. 1990;181:326–331. doi: 10.1016/0076-6879(90)81133-f. [DOI] [PubMed] [Google Scholar]
- 33.Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Note: Supplementary information is available on the Nature Structural & Molecular Biology website.