

Abstract
Some therapeutic monoclonal antibodies function by focusing the killing power of the immune system on particular cellular targets, a process known as antibody-dependent cell-mediated cytotoxicity (ADCC). There is considerable interest in developing small molecules therapeutics that utilize this mechanism of action and so act as a kind of antibody surrogate. Progress and challenges in this interesting field are reviewed here.
Therapeutic monoclonal antibodies operate by one of two general mechanisms. Some antagonize protein-protein interactions critical for cellular signaling. For example, Humira and Remicade, which are used to inhibit inflammation in patients with rheumatoid arthritis, bind to tumor necrosis factor alpha (TNF-α) and prevent it from binding to its cognate receptor. Avastin is an anti-Vascular Endothelial Growth Factor VEGF antibody that sequesters this hormone from the VEGF Receptor 2 (VEGFR2), thus inhibiting angiogenesis, an important process in the development of “wet” macular degeneration and the spread of some cancers. Many other such examples exist. But other antibodies employ a more complex mechanism of action known as antibody-dependent cell-mediated cytotoxicity (ADCC). [1] In this case, the Fc region (Fig. 1) of certain antibody isotypes can recruit effector molecules and cells to a target cell by virtue of recognition of a cell surface marker. These include the complement system, natural killer cells and other pieces of the armamentarium of the immune system. In this way, binding of antibodies to a receptor displayed at sufficient density on a target cell can result in the destruction of that cell type. This process is called antibody-dependent cell-mediated cytotoxicity (ADCC) and is an important therapeutic strategy. For example Rituximab, an anti-CD20 antibody, has been used in the treatment of a variety of autoimmune disorders [2]. CD20 expression is restricted to B cells, including memory cells, and Rituximab mediates the destruction of most or all CD20+ B cells through ADCC. When the immune system “reboots” and new B cells are made, it seems to be the case that the cells with autoantigen reactivity are often not reconstituted. Remarkably, most patients tolerate a lack of B cells quite well. Rituximab is employed for the treatment of a variety of autoimmune diseases, including multiple sclerosis [3-5], and is under study for several others, such as Type I diabetes [6]••.
Figure 1.
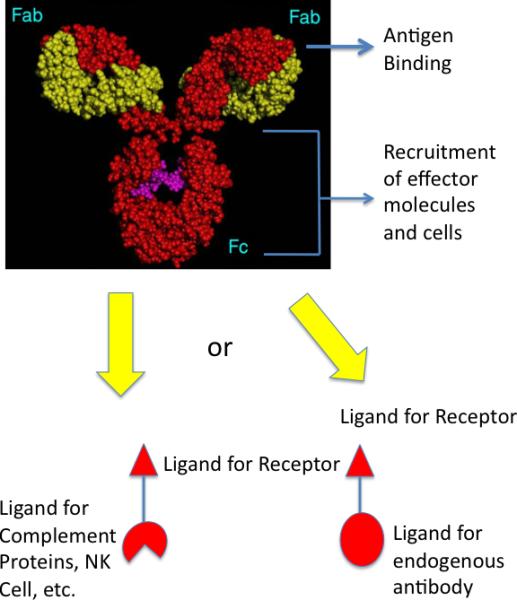
Comparison of a native IgG antibody and a hypothetical synthetic model. Native antibodies have two antigen binding pockets in their Fab regions and a constant region (Fc) capable of interacting with effector molecules and cells, such as the complement proteins, macrophages, natural killer cells, etc. Antibodies could be made by joining a high affinity and selectivity protein-binding molecule to one or more ligands for effector molecules or cells (left). Alternatively, the receptor ligand could be coupled to a molecule bound tightly by an endogenous antibody, whose Fc region would then act to recruit the effector molecules (right; also see Fig. 2)
Antibody drugs that operate via ADCC are thus quite different from the vast majority of small molecule therapeutics. They are neither classical agonists nor antagonists, but rather act as matchmakers between unique receptors on the surface of target cells and the various effector molecules and cells of the immune system. Some investigators have become intrigued with the idea of creating synthetic antibody surrogates capable of attacking pathogenic cells via ADCC as a new class of drugs. In theory, this could be achieved by linking two kinds of small molecules: a ligand that displays a high affinity and selectivity for a given cell surface receptor on the target cell of interest with a ligand for a receptor on the surface of natural killer (NK) cells, or macrophages or a ligand for one of the complement proteins (Fig. 1). A slightly less ambitious, but closely related, approach would be to link a cell receptor ligand to a molecule capable of binding to an endogenous antibody (Fig. 1) whose Fc domain would then do the job of recruiting immune effectors to the cell targeted by the first small molecule (Fig. 2).
Figure 2.
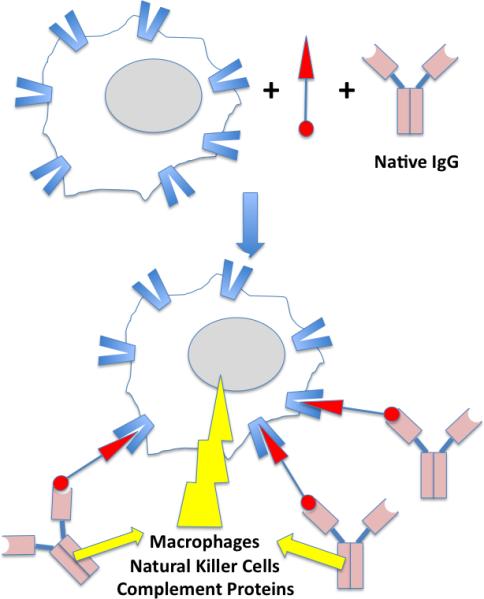
Illustration of ADCC (antibody-dependent cell-mediated cytotoxicity) mediated by a bifunctional molecule capable of binding to a receptor on the surface of the target cell as well as an endogenous IgG antibody. The yellow lightening blot represents the attack of the effector proteins and cells on the target cell. See text for details.
Characterization of target selectivity
Any biochemist that has ever employed an antibody in her or his research knows the joy of a “good” antibody that, for example, “lights up” one and only one band on a Western blot. Unfortunately, we are all too familiar with the frustrations of poor antibodies that provide a smeary mess of several bands in the same protocol. The former can be used to derive clear-cut results as a molecular probe and the latter cannot. Obviously, only exceedingly good monoclonal antibodies are candidates for therapeutic applications, especially in cases where the therapeutic effect is realized via ADCC. Off target effects clearly need to be avoided given that one is initiating an immune attack on cells to which the antibody binds. This is a potentially major problem in the development of synthetic antibody surrogates. Synthetic molecules rarely exhibit protein-binding properties that rival those of a good antibody. Achievement of the low nM or even pM dissociation constants typical of a good antibody-antigen complex generally requires a great deal of labor-intensive optimization of a lead compound in the synthetic molecule world. Perhaps even more difficult is the issue of protein target selectivity. Off target effects are common in classical pharmacology. Indeed it is problematic to even determine the selectivity of synthetic protein-binding agents in most cases. There is no small molecule analogue of a Western blot that allows one to examine binding of the molecule to a significant fraction of the proteome simultaneously and at least semi-quantitatively. The reason that such protocols are easy to carry out for antibody-antigen complexes is that: 1) The kinetic dissociation rates of these complexes are slow, allowing them to be taken out of equilibrium conditions, washed and analyzed, and 2) antibodies contain a highly conserved constant region that is recognized by anti-IgG antibodies (commonly known as secondary antibodies), making them easy to track. While some small molecules bind very tightly and thus dissociate slowly from their targets, most lack an easily detectable “handle” that is not involved in protein binding, akin to the Fc region of an antibody. So to even begin to think about the development of small molecules with antibody-like selectivities, good assays for measuring selectivity are needed.
The most straightforward approach is to add a linker arm to a molecule of interest that allows it to be immobilized and used as an affinity resin. A good example is the synthesis of K-Trap (Fig. 3), a derivative of the natural product trapoxen, by Taunton, Schreiber and coworkers [7,8]. Whereas trapoxen itself could not be immobilized in a straightforward fashion, they employed K-Trap to construct an affinity column that led to the identification of a histone deacetylase (HDAC) as a major target of this molecule and was a critical element in the discovery of this class of important chromatin modifying proteins. Unfortunately, in many cases, it is not clear where one can modify a molecule without compromising its activity and even when possible, this almost always requires significant synthetic effort.
Figure 3.
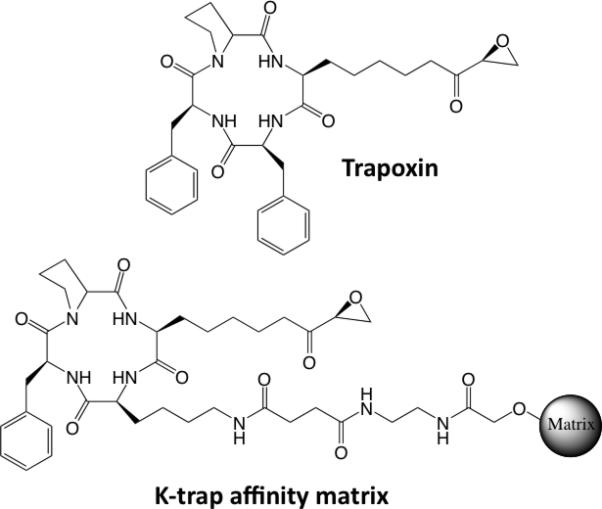
Structures of trapoxen, a naturally occurring HDAC inhibitor, and K-trap, a trapoxen derivative tethered to a solid support. The latter was a key reagent in the identification of HDACs as the target of trapoxen.
In the absence of a convenient linker arm, it is sometimes possible to probe target selectivity by competition with immobilized molecules. A nice example was the work of Bantscheff, et al. [9]• in which derivatives of seven promiscuous kinase inhibitors were synthesized that could be attached to beads. When exposed to a cellular extract, these “kinobeads” retained a large number of protein kinases as well as many other ATP-binding proteins, as determined by mass spectrometric analysis of the retained material. To evaluate the selectivity of unmodified compounds, the kinobead-bound material was challenged with a particular small molecule protein kinase inhibitor and the proteins displaced from the column by a given concentration of that molecule were identified. In this way, for example, it was found that in the presence of 1 μM of Imatinib, 13 proteins exhibited > 50% depletion form the beads, including the BCR-Abl kinase that is thought to be the critical functional target of Imatinib. By carrying out a titration of the soluble drug, an IC50 for protein displacement from the column could be calculated for each protein target of Imatinib. While this approach clearly cannot provide proteome-wide coverage of the binding selectivity of a small molecule, it is extremely useful for probing a particular sub-type of the proteome. It is useful to point out that one might have imagined that immobilized ATP could have been used in place of the immobilized promiscuous kinase inhibitors and ATP columns have certainly been reported. But in the hands of Bantshceff, et al., the ATP-binding material was so dominated by heat shock proteins that they could not carry out the quantitative assessment of kinase-binding selectivity with this matrix [9]•.
Another kind of competition assay that provides a powerful method to probe subproteomes employs activity-based protein profiling (ABPP) [10]. In this technique, a crude extract is treated with a bivalent molecule that includes a tag tethered to a moiety capable of mechanism-based coupling to an entire class of enzymes. A particularly well-developed example of this is a tagged fluorophosphonate that will react with almost any serine peptidase or protease (Fig. 4) through its active site residue [11]. If the tag is a fluorescent molecule then, after treatment of an extract with the reagent, a gel can be run and fluorescent bands corresponding to the levels of active serine hydrolases will be observed on the gel. More recently, alkyne tags have been appended to the ABPP reagent, allowing proteins modified by them to be captured and enriched by “clicking” onto them an azide-containing biotin moiety or some other element that allows efficient protein retention [12]. Mass spectrometry can then be employed to catalogue the captured proteins semi-quantitatively. To determine the selectivity of a serine hydrolase inhibitor, one can compare the results of this assay in the presence and absence of the compound. If the compound hits only the intended target, then only this hydrolase will disappear from the activity profile (Fig. 4). However, if other hydrolases are also inhibited by the compound (i.e. it has off target effects) then these proteins will be lost to some degree from the profile as well [13,14]••. Thus, ABPP is a powerful tool for the analysis of selectivity within a given mechanistic class of proteins. As the number of useful reagents for ABPP increases, it will be possible to analyze the selectivity of drug candidates and discover unanticipated off target effects with much greater efficiency than is the case now.
Figure 4.
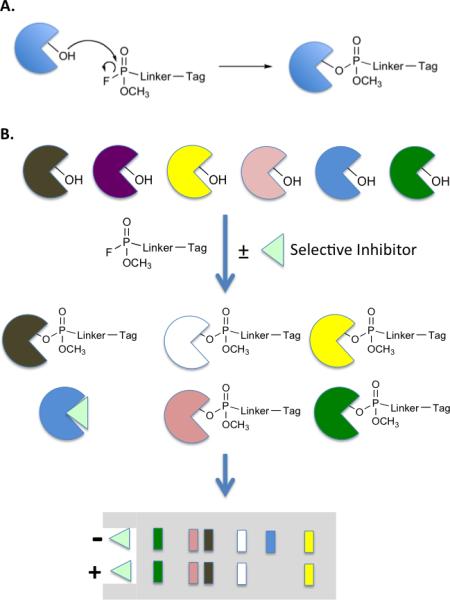
Activity-based protein profiling (ABPP) as a tool for monitoring inhibitor selectivity. A. Fluorophosphonate chemistry for ABPP of serine hydrolases. B. Protocol for testing the selectivity of a serine hydrolase inhibitor (light green triangle). The figure depicts an ideal situation in which six serine hydrolases (different colored pies) are treated with a tagged fluorophosphonate in the presence or absence of the inhibitor. If the inhibitor is specific for the blue hydrolase, then its signal in the profile will disappear, but that of the other proteins will be unaffected. The figure depicts the use of gel-based analysis with blotting specific for the tag to monitor protein labeling.
Incorporating selectivity filters into screening protocols
It seems obvious to propose that if the selectivity of a protein-targeted therapeutic is a major issue, then one should worry about it when designing a screening experiment. However, this is usually not the case. Most high-throughput screens are focused on potency and selectivity is an issue usually dealt with later. As a result, much time and effort can be wasted post-screening weeding out poorly selective compounds that nonetheless bind the desired target with high affinity.
Some groups have attempted to rectify this situation by building in a high demand for selectivity in order for a compound to be chosen as a hit [15,16]. A good example that is particularly relevant to the goal of creating drugs that work via ADCC is a screen that our group carried out against the Vascular Endothelial Growth Factor Receptor 2 (VEGFR2) (Fig. 5) [17]••. The screen employed two cell types. Cells that do not express VEGR2 were labeled with green quantum dots. VEGFR2 was then expressed in the same cell type and these cells were labeled with red quantum dots. Therefore, the only difference between the red and green labeled cells is the presence or absence of the target receptor. The differentially labeled cells were then mixed together in a 1:1 ratio and exposed to the approximately 260,000 peptoid-displaying beads. A bead was scored as a hit only if it bound almost entirely red, not green, cells. In other words, we demanded that the peptoid ignore all of the other molecules on the surface of the cell besides VEGFR2. A poorly selective VEGFR2 ligand, or a ligand for some other cell surface molecule, will bind both red and green cells.
Figure 5.
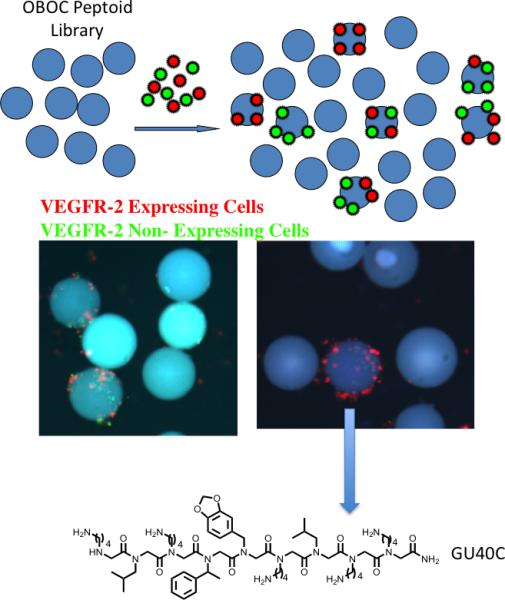
A screening protocol for the identification of highly selective ligands for an integral membrane protein [17]••. See text for details. The fluorescent micrographs show the results from a screen employing cells that do (red-labeled) or do not (green-labeled) express VEGFR2. When an OBOC library of peptoids containing about 260,000 compounds was employed, several thousand beads displaying bound red and green cells were observed (left micrograph). Only five beads that bound almost red cells almost exclusively were observed. The structure of the peptoid, GU40C, displayed by the bead pictured is shown.
When this experiment was carried out, thousands of beads that bound both colored cells were observed, but only five beads that bound almost exclusively red cells were identified. Subsequent analysis of these peptoids supported the idea that they are indeed highly selective VEGFR2 ligands. A high affinity dimeric version of one of the hits, GU40C4, was labeled red and incubated with living cells that either did or did not express VEGFR2. Fluorescence microscopy was then used to visualize binding after a brief wash. As seen in Fig. 6, clear binding of the red-labeled peptoid to cells that express VEGFR2 was observed, whereas no detectable red fluorescence could be detected on cells lacking this receptor, a result similar to what one would see using a good antibody for immunofluorescence. Synthetic ligands with this high level of receptor binding selectivity should be useful reagents for the creation of antibody surrogates.
Figure 6.
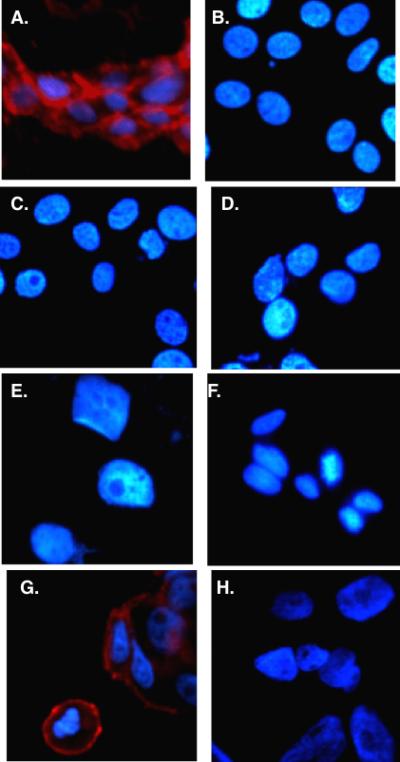
Live cell fluorescence imaging of cells that do (panels A and G) or do not (all other panels) express VEGFR2 incubated with biotinylated GU40C4 (a dimeric version of GU40C (Fig. 5) and red streptavidin-coated quantum dots. The nucleus of the cell was stained blue in each case.
Towards antibody mimics capable of mediating effector functions
As mentioned in the introduction, antibodies are capable of far more than simply binding antigens. The Fc region is capable of recruiting the complement, macrophages, natural killer cells and other components of the armamentarium of the immune system to mount a chemical attack against “foreign” cells. It would of considerable interest to develop completely synthetic molecules that allow this aspect of antibody function to be mimicked. To do so, one would have to link a molecule that mimics the antigen-binding pocket of an antibody (i.e., a high affinity and selectivity protein ligand) to a molecule that mimics the function of the Fc region (see Fig. 1). The latter would be a ligand for complement proteins, for a receptor on macrophages or the like, allowing the chimeric molecule to match make an association between, for example, a target cell expressing a particular receptor and the armamentarium of the immune system.
Important steps in this direction have been reported. Since molecules that mimic the Fc region of an antibody do not yet exist, in each case, an intermediate strategy was employed where a protein ligand was fused to a molecule that would attract to it a bona fide antibody. For example Barbas, Lerner and colleagues showed that a catalytic IgG aldolase antibody can be linked quite stably to almost any 1,3-diketone [18,19]. If the 1,3-diketone is fused to a receptor-binding molecule, the catalytic antibody and its Fc region can be brought to cells that express the target of the small molecule. Of course, the small molecule acquires the PK properties of the antibody. This can be advantageous, particularly for compounds that display high affinity, but are cleared rapidly from the circulation.
An even more attractive idea is to take advantage of certain native IgG antibodies that are present at high levels in a large percentage of the population. For example, most people have antibodies against DNP (dinitrophenol) [20]. Spiegel and co-workers recently reported promising ex vivo experiments in which small molecules that bind to prostate-specific membrane antigen (PSMA), a cell surface marker for prostate cancer cells, fused with DNP [21]••. They first demonstrated that this compound, which they called an ARM (antibody-recruiting small molecule) (Fig. 2), could mediate the formation of complexes between anti-DNP antibodies and prostate cancer cells expressing PSMA. Cells that did not express PSMA did not form complexes with the antibody in the presence of the ARM. More importantly, when the chimeric ARM was mixed with anti-DNP antibodies, PSMA-expressing cells and peripheral blood mononuclear cells (PBMCs; NK cells, macrophages, etc.), high levels of targeted cytotoxicity were observed. Again, this effect required the presence of PSMA on the cell surface, as cells lacking this marker were not targeted for destruction by the hybrid molecule and the anti-DNP antibodies.
While DNP itself is probably not a good choice for a component of a real world pharmaceutical because of the presence of the nitro aromatic moiety, it should be possible to identify high affinity and selectivity ligands for anti-DNP antibodies with better PK properties by high-throughput screening [22]. By linking such a “DNP surrogate” to appropriate receptor-binding molecules, many different target cells types could, in theory, be targeted for elimination by the immune system (Fig. 2)
A particularly interesting potential application of this type of ADCC-like therapeutic strategy would be to target autoimmune T or B cells for destruction without affecting the normal and desired functions of the immune system. In order to accomplish this, one would need to fuse DNP or some suitable surrogate to a compound capable of selective recognition of the antigen-specific T cell receptor (TCR) on autoimmune T cells or the antigen-specific B cell Receptor (BCR) on autoimmune B cells. Our laboratory has recently published a screening protocol that allows the discovery of such molecules [23]••. It is an elaboration of the two-color screen employed in the VEGFR2 study discussed above (see Fig. 5). In this case, the entire population of CD4+ T cells from a healthy mouse was isolated and labeled with green quantum dots. Likewise, the entire CD4+ T cell population from mice with Experimental Autoimmune Encephalomyelitis (EAE), a multiple sclerosis-like condition, were isolated and labeled with red quantum dots. The hypothesis was that the main difference in these two populations should be a preponderance of autoimmune T cells with TCRs specific for the autoantigen in the diseased animal. A screen was done in which a 1:1 ratio of the two T cell populations was exposed to a large OBOC peptoid library and beads that only bound red T cells detectably were identified. Two such hits were obtained. We went on to show that one of the peptoids isolated in this screen was indeed a modest affinity, but highly selective ligand for the autoimmune TCR. This novel technology now opens the door for screens against human autoimmune cells and suggests the possibility of developing reagents capable of ablating particular, antigen-specific autoimmune responses.
Acknowledgements
The work from my own laboratory described in this review was funded by a contract from the NHLBI (NO1-HV-28185) and the NIH Director's Pioneer Award (DP10D00066301).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hale G, Clark M, Waldmann H. Therapeutic potential of rat monoclonal antibodies: isotype specificity of antibody-dependent cell-mediated cytotoxicity with human lymphocytes. J Immunol. 1985;134:3056–3061. [PubMed] [Google Scholar]
- 2.Matthews R. The B cell slayer. Science. 2007;318:1232–1233. doi: 10.1126/science.318.5854.1232. [DOI] [PubMed] [Google Scholar]
- 3.De Palma R, Sementa A. Rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358:2645–2646. doi: 10.1056/NEJMc080557. author reply 2646-2647. [DOI] [PubMed] [Google Scholar]
- 4.Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, Bar-Or A, Panzara M, Sarkar N, Agarwal S, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358:676–688. doi: 10.1056/NEJMoa0706383. [DOI] [PubMed] [Google Scholar]
- 5.Chaudhuri A, Behan PO. Rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358:2646. author reply 2646-2647. [PubMed] [Google Scholar]
- ••6.Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, Becker DJ, Gitelman SE, Goland R, Gottlieb PA, Marks JB, McGee PF, Moran AM, et al. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N Engl J Med. 2009;361:2143–2152. doi: 10.1056/NEJMoa0904452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taunton J, Collins JL, Schreiber SL. Synthesis of natural and modified trapoxins, useful reagents for exploring histone deacetylase function. J. Amer. Chem. Soc. 1996;118:10412–10422. [Google Scholar]
- 8.Taunton J, Hassig CA, Schreiber SL. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science. 1996;272:408–411. doi: 10.1126/science.272.5260.408. [DOI] [PubMed] [Google Scholar]
- •9.Bantscheff M, Eberhard D, Abraham Y, Bastuck S, Boesche M, Hobson S, Mathieson T, Perrin J, Raida M, Rau C, et al. Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat Biotechnol. 2007;25:1035–1044. doi: 10.1038/nbt1328. [DOI] [PubMed] [Google Scholar]
- 10.Cravatt BF, Wright AT, Kozarich JW. Activity-based protein profiling: from enzyme chemistry to proteomic chemistry. Annu Rev Biochem. 2008;77:383–414. doi: 10.1146/annurev.biochem.75.101304.124125. [DOI] [PubMed] [Google Scholar]
- 11.Kidd D, Liu Y, Cravatt BF. Profiling serine hydrolase activities in complex proteomes. Biochemistry. 2001;40:4005–4015. doi: 10.1021/bi002579j. [DOI] [PubMed] [Google Scholar]
- 12.Speers AE, Cravatt BF. Profiling enzyme activities in vivo using Click chemistry methods. Chem. & Biol. 2004;11:535–546. doi: 10.1016/j.chembiol.2004.03.012. [DOI] [PubMed] [Google Scholar]
- ••13.Ahn K, Johnson DS, Mileni M, Beidler D, Long JZ, McKinney MK, Weerapana E, Sadagopan N, Liimatta M, Smith SE, et al. Discovery and characterization of a highly selective FAAH inhibitor that reduces inflammatory pain. Chem Biol. 2009;16:411–420. doi: 10.1016/j.chembiol.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••14.Bachovchin DA, Brown SJ, Rosen H, Cravatt BF. Identification of selective inhibitors of uncharacterized enzymes by high-throughput screening with fluorescent activity-based probes. Nat Biotechnol. 2009;27:387–394. doi: 10.1038/nbt.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peng L, Liu R, Marik J, Wang X, Takada Y, Lam KS. Combinatorial chemistry identifies high-affinity peptidomimetics against alpha4beta1 integrin for in vivo tumor imaging. Nature Chem. Biol. 2006;2:381–389. doi: 10.1038/nchembio798. [DOI] [PubMed] [Google Scholar]
- 16.Oyama T, Sykes KF, Samli KN, Minna JD, Johnston SA, Brown KC. Isolation of lung tumor specific peptides from a random peptide library: generation of diagnostic and cell-targeting reagents. Cancer Lett. 2003;202:219–230. doi: 10.1016/j.canlet.2003.08.011. [DOI] [PubMed] [Google Scholar]
- ••17.Udugamasooriya DG, Dineen SP, Brekken RA, Kodadek T. A peptoid “antibody surrogate” that antagonizes VEGF receptor 2 activity. J. Amer. Chem. Soc. 2008;130:5744–5752. doi: 10.1021/ja711193x. [DOI] [PubMed] [Google Scholar]
- 18.Li LS, Rader C, Matsushita M, Das S, Barbas CF, 3rd, Lerner RA, Sinha SC. Chemical adaptor immunotherapy: design, synthesis, and evaluation of novel integrin-targeting devices. J Med Chem. 2004;47:5630–5640. doi: 10.1021/jm049666k. [DOI] [PubMed] [Google Scholar]
- 19.Rader C, Sinha SC, Popkov M, Lerner RA, Barbas CF., 3rd Chemically programmed monoclonal antibodies for cancer therapy: adaptor immunotherapy based on a covalent antibody catalyst. Proc Natl Acad Sci U S A. 2003;100:5396–5400. doi: 10.1073/pnas.0931308100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ortega E, Kostovetzky M, Larralde C. Natural DNP-binding immunoglobulins and antibody multispecificity. Mol Immunol. 1984;21:883–888. doi: 10.1016/0161-5890(84)90143-3. [DOI] [PubMed] [Google Scholar]
- ••21.Murelli RP, Zhang AX, Michel J, Jorgensen WL, Spiegel DA. Chemical control over immune recognition: a class of antibody-recruiting small molecules that target prostate cancer. J Am Chem Soc. 2009;131:17090–17092. doi: 10.1021/ja906844e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Astle JM, SImpson LS, Huang Y, Reddy MM, Wilson R, Connell S, Wilson J, Kodadek T. Seamless bead to microarray screening: Rapid identification of the highest affinity protein ligands from large combinatorial libraries. Chem. & Biol. 2010;17:38–45. doi: 10.1016/j.chembiol.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••23.Gocke AR, Udugamasooriya DG, Archer CT, Lee J, Kodadek T. Isolation of antagonists of antigen-specific autoimmune T cell proliferation. Chem. & Biol. 2009;16:1133–1139. doi: 10.1016/j.chembiol.2009.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]