

Summary
Pancreatic acinar cells are critical in gastrointestinal physiology and pancreatitis and may be involved in pancreatic cancer. Previously, a short rat pancreatic elastase promoter has been widely utilized to control acinar cell transgene expression. However, this partial sequence does not confer robust and stable expression. In this study, we tested the hypothesis that a transgene employing bacterial-artificial-chromosome (BAC) technology to express a tamoxifen-regulated Cre recombinase from a full-length mouse elastase gene (BAC-Ela-CreErT) would be more robust and stable. When founders were crossed with Rosa26 reporter mice nearly 100% of acini expressed β-galactosidase after tamoxifen treatment. The expression was specific for pancreatic acinar cells and these characteristics have remained stable for 2 years. However, because of high levels of expression in differentiated acinar cells, this construct is tamoxifen independent in ~50% of adult acinar cells. This model of pancreatic acinar specific Cre expression is a powerful tool for future transgenic and knockout studies.
Keywords: pancreas, elastase, tamoxifen, acinar cell, gene expression
Pancreatic acinar cells compose nearly 90% of the mass of the pancreas. These cells are the primary source of digestive enzymes necessary for the breakdown and absorption of nutrients from food and as such, are necessary for life. Several forms of pathologies also originate in pancreatic acinar cells. Expression of mutant genes such as trypsinogen (Whitcomb et al., 1996) or pancreatic serine protease inhibitor (Grigorescu and Grigorescu, 2005) leads to the development of hereditary chronic pancreatitis which is a high-risk factor for pancreatic cancer (Whitcomb et al., 1999). Furthermore, recent evidence suggests that pancreatic acinar cells may undergo metaplasia to form ductal pancreatic adenocarcinoma (Guerra et al., 2007). Thus, manipulation of gene expression in this cell type will be a valuable tool for investigations of both physiology and pathophysiology.
Cell specific manipulation of gene expression is a powerful tool for the study of the function of certain genes or for making animal models for a specific disease. One approach to cell specific gene expression is achieved using a differentiated cell specific promoter directly driving gene expression. For directing gene expression in pancreatic acinar cells the most popular system was originally developed based on the use of a short (250–513 bp) rat elastase gene enhancer/promoter (Rose et al., 1994). This approach has since been used very widely (Algul et al., 2007; Archer et al., 2006; Grippo et al., 2003; Guerra et al., 2007; Sandgren et al., 1990). However, this short genetic element demonstrates relatively low efficiency (10%–50%). The levels of expression from this promoter are also relatively low, although the level of expression can be improved using flanking regions such as the introns from growth hormone or metallothionine (Palmiter et al., 1991). However, even with these additional elements expression from this small elastase element is far less robust than expression from the endogenous gene (Quaife et al., 1987). Furthermore, this promoter is susceptible to unstable expression and infrequently may be lost in a few generations.
Another disadvantage of directly using the elastase promoter is that transgene expression from a differentiated cell specific gene promoter of this sort is likely to be greatly diminished if the expressed gene influences the differentiated state of the cells. A much more versatile approach is to use the Cre-loxP strategy to activate experimental gene expression from a constitutive promoter (Branda and Dymecki, 2004). For tissue specific gene expression using this system, a flanking loxP-stop-loxP cassette is added between a constitutive promoter and the experimental gene of interest (a so-called flox-stopped gene). The loxP-stop-loxP cassette prevents the experimental gene from being expressed until it is removed by Cre-recombinase. If the Cre-recombinase is expressed by a cell-specific promoter, then expression of the experimental gene takes place in a cell-specific manner even though the promoter driving gene expression is not cell-type specific. If the promoter used to drive the experimental transgene is not influenced by the state of cellular differentiation, then the experimental transgenes will continue being expressed even if the cells change phenotype. Another advantage of this system is that it can be combined with multiple flox-stopped-genes that exist in the scientific community, enabling economical use of available genetically modified models. This system can also be utilized for specific gene ablation when genes or relevant exons have been flanked by loxP sites allowing for cell-type specific deletion based on the expression of Cre recombinase.
A modification of Cre-recombinase that allows regulated activity has also been developed and has proven valuable in numerous studies (Guo et al., 2002). This Cre is modified by fusion to estrogen receptor elements that can bind the artificial estrogen, tamoxifen (CreErT). Under control conditions, in the absence of tamoxifen, CreErT binds to HSP90 which prevents the molecule from entering the nucleus and blocks its recombinase function. However, addition of tamoxifen releases the CreErT from HSP90 and allows movement to the nucleus where the CreErT is active. Several recent models have been developed using the short rat elastase enhancer/promoter to drive CreErT expression (Desai et al., 2007; Murtaugh et al., 2005; Strobel et al., 2007). The expression of CreErT from these models is highly specific but not highly efficient, with 10%–50% of the cells showing tamoxifen induced expression. In models with higher efficiency some tamoxifen-independent expression is also noted (Murtaugh et al., 2005). However, in general these are excellent models for acinar specificity and tamoxifen inducibility so long as high efficiency is not necessary. Unfortunately, high efficiency is often desirable, particularly when investigating the effects of Cre-mediated gene deletion.
To develop an improved system for pancreatic acinar cell specific transgene expression, we tested the hypothesis that a full-length pancreatic elastase gene construct developed using bacterial artificial chromosome (BAC) technology (Copeland et al., 2001) would prove superior to the short elastase promoter. The BAC, with the intact mouse elastase gene, contains all the regulatory components for elastase expression and the native surrounding gene locus which protects the transgene from being influenced by other local genes that may occur during random genomic insertion. This was predicted to allow high efficiency and specificity for genes expressed by this promoter.
For comparing the efficiency of Cre-recombination, we developed mouse models in which a tamoxifen-inducible Cre-recombinase was expressed by either the commonly utilized short rat elastase enhancer (Ela-CreErT) or a full-length mouse pancreatic elastase I gene promoter (BAC-Ela-CreErT) (Figs. 1a and 2a). Seven Ela-CreErT founders and two BAC-Ela-CreErT founders were identified by genotyping. Q-PCR based analysis indicated the presence of 10–15 copies of the transgene in Ela-CreErT founders and 2–3 copies in BAC-Ela-CreErT founders. To detect Cre recombination activity, these mice were crossed with Rosa26 reporter mice which carry a flox-stopped β-galactosidase gene. Double transgenic mice or single transgene littermates were injected daily with tamoxifen (3 mg/40 g body weight) for 3 days. The mice were sacrificed at 5 days and the pancreata were removed for X-gal staining. We found only one out of seven Ela-CreErT lines (1/7) showed β-galactosidase activity. This line displayed only 10%–15% X-gal positive cells in the pancreas when crossed with Rosa26 mice (Fig. 1b). In contrast, double transgenic mice developed from both BAC-Ela-CreErT founders crossed with Rosa26 displayed X-gal staining in nearly 100% of pancreatic acinar cells after tamoxifen treatment (Fig. 2b). These data indicate a major difference in efficiency for the full-length elastase gene compared with the short promoter.
FIG. 1.
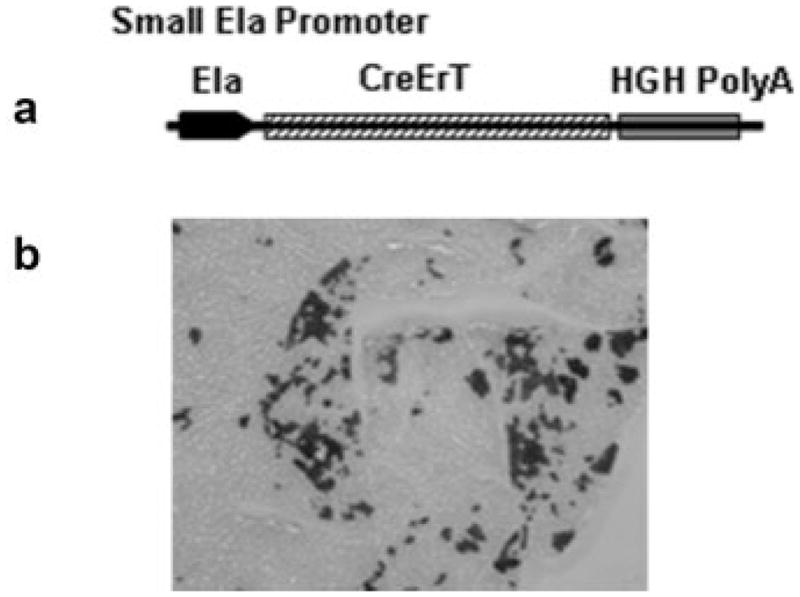
Gene expression driven by a short rat elastase enhancer was not efficient. (a) CreErT gene was inserted between a ~500 bp rat elastase enhancer and human growth hormone polyA signal. (b) Transgenic founders were crossed with Rosa26 reporter mice. Double transgenic mice were injected with tamoxifen at 3 mg/40 g body weight daily for 3 days and sections of the pancreata were stained with X-gal. Approximately 10–15% of acini were labeled in a mosaic pattern.
FIG. 2.
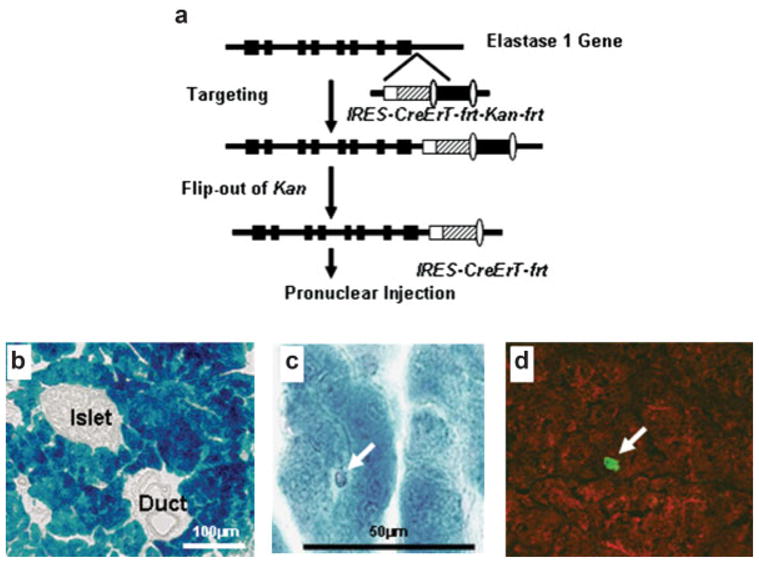
CreErT expression from a full length elastase gene promoter was highly efficient and specific. (a) CreErT gene was targeted between the last exon and polyA signal of the mouse elastase I gene in a bacterial artificial chromosome. (b). Founders were crossed with Rosa26 mice, injected with tamoxifen and stained with X-gal. Nearly 100% of acini were stained with no staining in ducts, islets, or blood vessels. (c) X-gal staining was negative in a centroacinar cell. (d). CreErT (red) was not colocalized with Hes1 (green), a centroacinar marker.
Consistent with the known distribution of endogenous elastase expression, X-gal staining in BAC-Ela-CreErT × Rosa26 double transgenic mice was exclusively localized to pancreatic acinar cells and not in duct or endocrine islets cells (Fig. 2b). Similar specificity was noted with the Ela-CreErT × Rosa26 mice (data not shown). An attempt was made to analyze expression of these constructs in centroacinar cells. A few centroacinar cells could be identified and these did not show X-gal staining (Fig. 2c). Similarly, immunofluorescent localization of CreErT and Hes-1, a marker for centroacinar cells, did not show colocalization (Fig. 2d). However, because of the difficulty of localizing centroacinar cells it was not possible to rule out the expression of CreErT in this cell type. Nonetheless, these data clearly indicate that the BAC transgene is at least as specific as the short elastase enhancer.
During the investigation of Cre activation in the animals crossed with Rosa26 it was noted that some acinar cells expressed β-galactosidase in the absence of tamoxifen treatments (Fig. 3). To understand this tamoxifen independent Cre-activity, we investigated β-galactosidase expression in animals of different ages. We observed that in the absence of tamoxifen no expression of β-galactosidase was observed in developing mice at 19.5 days post-coitus in the absence of tamoxifen treatment (Fig. 3a). Treatment of pregnant mice with tamoxifen (1 mg/day for 3 days) resulted in widespread activation of β-galactosidase in the pancreas of same aged embryos indicating the presence of the CreErT transgene at this early time (Fig. 3b). At day 1, after birth ~1% of cells showed activated β-galactosidase in the absence of tamoxifen (Fig. 3c). The percentage of labeled cells increased over-time (Fig. 3d–3f) with a dramatic increase after weaning (between day 14 and 29) such that ~50% of adult cells were labeled in the absence of tamoxifen (Fig. 3f). The spontaneous CreErT activity was only observed in easily recognizable acinar cells (Fig. 3c insert). This increase in Cre activity paralleled the normal increase in elastase gene expression that occurs in mice during this period, as during weaning there is known to be a spike in elastase expression (Han et al., 1986). Levels of CreErT paralleled those of endogenous elastase in this mouse model (Fig. 3j). Thus, the most likely explanation for this phenomenon is that CreErT is expressed at very high levels in the pancreas when driven from the full-length elastase gene. Indeed, we found that the CreErT expression in the BAC transgene was even higher than that of a CAGGS-CreErT transgene (Fig. 3j) which utilizes a CMV-chicken beta actin chimeric promoter to drive CreErT expression (Hayashi and McMahon, 2002). In adults, the spontaneous level of Cre-activity occurred in roughly 40%–50% of the acinar cells (Fig. 3g). Treatment with tamoxifen was able to induce the other 50%–60% of acinar cells (Fig. 3h,i).
FIG. 3.
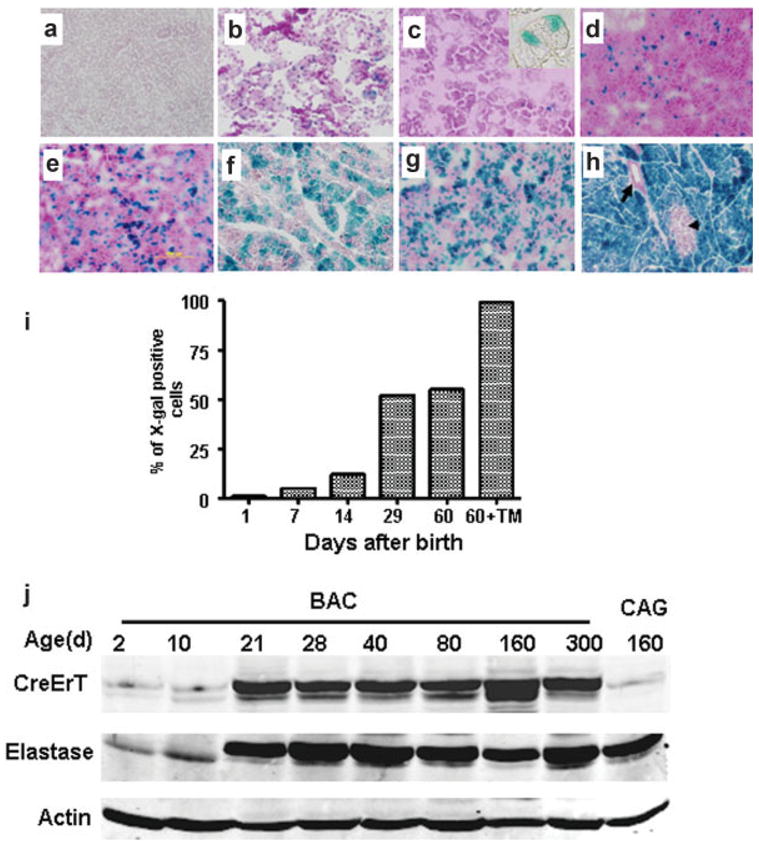
BAC-Ela-CreErT mice developed tamoxifen independence in acinar cells with high expression levels. Pancreas from a BAC-Ela-CreErT × Rosa26 embryo at 19.5 days post-coitus (dpc) was X-gal negative (a). Injection of pregnant mice with tamoxifen (3 daily injections with 1 mg/40 g) generated many X-gal positive cells in embryos removed at 19.5 dpc (b). In the absence of tamoxifen, the number of X-gal positive cells increased with age. Data shown are without tamoxifen on day 1 (c), day 7 (d), day 14 (e), day 29 (f), and day 60 (g) after birth. Injection of tamoxifen in animals after 8 weeks (three daily injections with 3 mg/40 g) led to the expected x-gal staining in nearly 100% of acini (h). Unstained cells are either islets (arrowhead) or ducts (arrow). Sections were counterstained with fast red. All were photographed at × 100 magnification. Quantitation of X-gal staining at the different times indicated the time-course of leakage of BAC-Ela-CreErT (i). The expression level of CreErT paralleled the level of endogenous elastase expression as indicated by Western blot (j).
To examine the stability of the CreErT expression in the transgenic models, we examined the efficiency of β-galactosidase expression in the animals again after 2 years of breeding (eight generations). We found that the BAC transgene maintained expression in ~100% of pancreatic acinar cells when treated with tamoxifen (Fig. 3h). In contrast, the animals with the short elastase promoter lost nearly all CreErT expression (Data not shown)..
In summary, we have shown that a full-length elastase gene in a BAC provides for highly efficient and stable development of pancreatic acinar directed transgene expression. Using this system, a new transgenic mouse line has been developed that expresses CreErT in pancreatic acinar cells. This model is highly efficient, specific, and stable. The high levels of CreErT expression achieved in adult acinar cells allow for some tamoxifen-independent expression. However, tamoxifen administration is still able to induce rapid induction of gene expression in the remaining acinar cells. This model will be highly useful for a variety of studies on pancreatic physiology and pathophysiology.
METHODS
Mice
All mouse colonies were maintained in a specific pathogen-free barrier facility at the University of Michigan and the University of Texas M. D. Anderson Cancer Center approved by the Association for Assessment and Accreditation of Laboratory Animal Care International, and procedures conformed to the Institutional Animal Care and Use Committee protocols. CAGGS-CreErT mice were provided by Dr. Andrew P. McMahon (Harvard Medical School, Boston, MA).
Construction of Ela-CreErT
CreErT was released from pBS with Apa I and EcorRI, blunt ligated to a 531bp rat elastase promoter (a gift from Dr. Raymond MacDonald, UT Southwestern University, Dallas, TX) containing a human growth hormone poly A sequence. The Ela-CreErT-polyA cassette was released and used for producing transgene.
Construction of BAC-Ela-CreErT
A mouse BAC (clone RP23-359O19, size 222 kb) carrying the intact elastase I gene was used. Alignment according to the BAC end sequencing with sp6 and T7 primer, this BAC is localized between 100,425,585 and 100,648,109 on chromosome 15. The BAC was modified to express CreErT with an E. coli-based chromosome engineering system developed by Dr. Copeland (NCI) (Copeland et al., 2001). pIGCN-Cre-ErT was produced by replacing the EGFP-Cre with Cre-ErT (provided by Dr. Andrew P. McMahon, Harvard Medical School, Boston, MA) in plasmid pICGN21. For recombination mediated targeting, arms homologous to the sequence of elastase I gene between the last exon and the polyA signal (in lower case) were added to either side of the IRES-Cre-ErT-frt-Kan-frt cassette by PCR with primers (Forward: cgacttccggagtccagtggcctccccaagatggctcttagctttgcGCCAA GCTATCGAATTCCGCC, Reverse:ctgagcgataacctcagctcta actccttcttgcttacttcaggtctcCTATTCCAGAAGTAGTGAGGA). Using homologous recombination, the IRES-Cre-ErT-frt-Kan-frt cassette was inserted into the elastase I gene. The Kan selection marker was then removed by flp-mediated recombination and the insertion site and orientation were verified by sequencing.
Generation Of Transgenic Mice
The subcloned DNA were purified and transgenic mice (Ela-CreErT) were developed at the University of Michigan Transgenic Animal Model Core by pronuclear microinjection. The BAC-Ela-CreErT mice were produced through the University of Texas M. D. Anderson Cancer Center Genetically Engineered Mouse Facility.
Genotyping
Tail biopsies were performed at 3 weeks of age. DNA was prepared for PCR with REDExtract-N-Amp™ Tissue PCR Kit (Sigma, St. Louis, MO) according to manufacture’s protocol. Primers (Forward: gcctgcattaccggtcga, Reverse: tatcctggcagcgatcgc) were used to detect the Cre gene. Transgene copy numbers were determined by Q-PCR using beta globin (Forward: ccaatctgctcacacag gatagagagggcagg, Reverse ccttgaggctgtccaagtgattcaggc catcg) as a standard.
β-Galactosidase Reporter Assay
To determine the temporal and spatial expression profiles of the CreErT transgenes, we crossed the CreErT transgenic mice with Rosa26 reporter mice. The reporter mice express β-galactosidase in the presence of active Cre recombinase (Soriano, 1999). After three daily injections of tamoxifen 3 mg per 40 g body weight, the pancreas was embedded in OCT and sectioned for X-gal staining. Sections were postfixed with 0.25% glutaraldehyde in PBS and briefly washed with rinse solution (0.1 M phosphate buffer, pH 7.3, 0.1% deoxycholic acid, 0.2% NP-40, and 2 mM MgCl2). X-gal staining was performed by incubating samples in staining buffer (2.5 mg/ml X-gal, 5 mM potassium ferricyanide, and 5 mM potassium ferrocyanide) overnight at 37°C and in some cases followed by counterstaining with nuclear fast red.
Immunofluorescence
Frozen sections of the pancreas 8 μm thick were fixed in freshly prepared 4% paraformaldehyde for 30 min. The sections were then permeablized with 0.5% Triton X-100 for 30 min and blocked with 5% bovine serum albumin in phosphate-buffered saline containing 0.05% Triton X100. Polyclonal antibodies against Cre (1:1,000, EMD Chemicals, Gibbstown, NJ) and Hes1 (1:50, Santa Cruz Biotechnology, Santa Cruz, CA) were added overnight at 4°C before Alexa Fluor 594 donkey anti-rabbit IgG and Alexa Fluor 488 labeled donkey anti-goat IgG secondary antibodies were applied (Vector Laboratories, Burlingame, CA). Fluorescent imaging was taken on a Zeiss LSM510 confocal scanning microscope (Zeiss, Thornwood, NY).
Western Blotting
Western blotting was performed as previously described with modification (Ji et al., 2001). Briefly, pancreata were harvested and homogenized in lysis buffer (50 mM/l HEPES, pH 7.5, 150 mM/l NaCl, 2.5 mM/l EGTA, 0.1% Tween-20, 1 mM/l dithiothreitol, 1 mM/l, NaF, 0.1 mM/l sodium orthovanadate, 0.1 mM/l phenyl-methylsulfonyl fluoride, 2 μg/ml aprotinin, and 5 μg/ml leupeptin) and centrifuged to remove cellular debris. Samples of 30 μg of protein denatured with Laemmli buffer were subjected to electrophoresis on sodium do-decyl sulfate-polyacrylamide gels and transferred by electroblotting to Hybond membranes. The blots were incubated with primary antibodies anti-Cre (1:10,000, EMD Chemicals Inc. Gibbstown, NJ) and anti-elastase (Abcam, Cambridge, MA). Fluorescent dye labeled secondary antibody was used for detection with Odyssey Infrared Imaging System (LI-COR Biotechnology, Lincoln, Nebraska).
Acknowledgments
Contract grant sponsor: NIH, Contract grant number: DK52067-05 (C.D.L.), Contract grant sponsor: Pilot Feasibility Project of Michigan Gastrointestinal Peptide Research Center, Contract grant numbers: 5P30 DK34933 and R21 DK068414-01 (B.J.).
The authors thank Drs. John A. Williams and Grzegorz T. Gurda at University of Michigan for their critical comments.
LITERATURE CITED
- Algul H, Wagner M, Lesina M, Schmid RM. Overexpression of ErbB2 in the exocrine pancreas induces an inflammatory response but not increased proliferation. Int J Cancer. 2007;121:1410–1416. doi: 10.1002/ijc.22779. [DOI] [PubMed] [Google Scholar]
- Archer H, Jura N, Keller J, Jacobson M, Bar-Sagi D. A mouse model of hereditary pancreatitis generated by transgenic expression of R122H trypsinogen. Gastroenterology. 2006;131:1844–1855. doi: 10.1053/j.gastro.2006.09.049. [DOI] [PubMed] [Google Scholar]
- Branda CS, Dymecki SM. Talking about a revolution: The impact of site-specific recombinases on genetic analyses in mice. Dev Cell. 2004;6:7–28. doi: 10.1016/s1534-5807(03)00399-x. [DOI] [PubMed] [Google Scholar]
- Copeland NG, Jenkins NA, Court DL. Recombineering: A powerful new tool for mouse functional genomics. Nat Rev Genet. 2001;2:769–779. doi: 10.1038/35093556. [DOI] [PubMed] [Google Scholar]
- Desai BM, Oliver-Krasinski J, De Leon DD, Farzad C, Hong N, Leach SD, Stoffers DA. Preexisting pancreatic acinar cells contribute to acinar cell, but not islet beta cell, regeneration. J Clin Invest. 2007;117:971–977. doi: 10.1172/JCI29988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigorescu M, Grigorescu MD. Genetic factors in pancreatitis. Rom J Gastroenterol. 2005;14:53–61. [PubMed] [Google Scholar]
- Grippo PJ, Nowlin PS, Demeure MJ, Longnecker DS, Sandgren EP. Preinvasive pancreatic neoplasia of ductal phenotype induced by acinar cell targeting of mutant Kras in transgenic mice. Cancer Res. 2003;63:2016–2019. [PubMed] [Google Scholar]
- Guerra C, Schuhmacher AJ, Canamero M, Grippo PJ, Verdaguer L, Perez-Gallego L, Dubus P, Sandgren EP, Barbacid M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007;11:291–302. doi: 10.1016/j.ccr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- Guo C, Yang W, Lobe CG. A Cre recombinase transgene with mosaic, widespread tamoxifen-inducible action. Genesis. 2002;32:8–18. doi: 10.1002/gene.10021. [DOI] [PubMed] [Google Scholar]
- Han JH, Rall L, Rutter WJ. Selective expression of rat pancreatic genes during embryonic development. Proc Natl Acad Sci USA. 1986;83:110–114. doi: 10.1073/pnas.83.1.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: A tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol. 2002;244:305–318. doi: 10.1006/dbio.2002.0597. [DOI] [PubMed] [Google Scholar]
- Ji B, Bi Y, Simeone D, Mortensen RM, Logsdon CD. Human pancreatic acinar cells lack functional responses to cholecystokinin and gastrin. Gastroenterology. 2001;121:1380–1390. doi: 10.1053/gast.2001.29557. [DOI] [PubMed] [Google Scholar]
- Murtaugh LC, Law AC, Dor Y, Melton DA. β-Catenin is essential for pancreatic acinar but not islet development. Development. 2005;132:4663–4674. doi: 10.1242/dev.02063. [DOI] [PubMed] [Google Scholar]
- Palmiter RD, Sandgren EP, Avarbock MR, Allen DD, Brinster RL. Heterologous introns can enhance expression of transgenes in mice. Proc Natl Acad Sci USA. 1991;88:478–482. doi: 10.1073/pnas.88.2.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quaife CJ, Pinkert CA, Ornitz DM, Palmiter RD, Brinster RL. Pancreatic neoplasia induced by ras expression in acinar cells of transgenic mice. Cell. 1987;48:1023–1034. doi: 10.1016/0092-8674(87)90710-0. [DOI] [PubMed] [Google Scholar]
- Rose SD, Kruse F, Swift GH, MacDonald RJ, Hammer RE. A single element of the elastase I enhancer is sufficient to direct transcription selectively to the pancreas and gut. Mol Cell Biol. 1994;14:2048–2057. doi: 10.1128/mcb.14.3.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandgren EP, Luetteke NC, Palmiter RD, Brinster RL, Lee DC. Overexpression of TGF alpha in transgenic mice: Induction of epithelial hyperplasia, pancreatic metaplasia, and carcinoma of the breast. Cell. 1990;61:1121–1135. doi: 10.1016/0092-8674(90)90075-p. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Strobel O, Dor Y, Alsina J, Stirman A, Lauwers G, Trainor A, Castillo CF, Warshaw AL, Thayer SP. In vivo lineage tracing defines the role of acinar-to-ductal transdifferentiation in inflammatory ductal metaplasia. Gastroenterology. 2007;133:1999–2009. doi: 10.1053/j.gastro.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitcomb DC, Applebaum S, Martin SP. Hereditary pancreatitis and pancreatic carcinoma. Ann N Y Acad Sci. 1999;880:201–209. doi: 10.1111/j.1749-6632.1999.tb09524.x. [DOI] [PubMed] [Google Scholar]
- Whitcomb DC, Gorry MC, Preston RA, Furey W, Sossenheimer MJ, Ulrich CD, Martin SP, Gates LK, Jr, Amann ST, Toskes PP, Liddle R, McGrath K, Uomo G, Post JC, Ehrlich GD. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat Genet. 1996;14:141–145. doi: 10.1038/ng1096-141. [DOI] [PubMed] [Google Scholar]