

Abstract
The relationship between chronic pancreatitis (CP) and pancreatic ductal adenocarcinoma (PDAC) is unclear. CP is a risk factor for PDAC, CP is found within the vicinity of PDAC, and both share many similar genetic alterations. However, it has been long thought that PDAC arises only from duct cells. However, we have recently found that excessive activity within the Ras signaling pathway can lead to acinar cell death or metaplasia and is associated with the development of fibrosis resembling CP and the development of PDAC from acinar cells through the full complement of preneoplastic (pancreatic intraepithelial neoplasia) lesions. Therefore, it is time to reevaluate the relationship between CP and PDAC. We proposed a new model in which Ras activity is the direct link between these 2 diseases. Here we will briefly review the shared properties between CP and PDAC and describe the new model.
The nature of the relationship between chronic pancreatitis (CP) and pancreatic ductal adenocarcinoma (PDAC) has always been controversial. Largely this is because PDAC was thought to originate in duct cells, whereas CP clearly develops in acinar cells. However, recently it has become clear that acinar cells can also be a source of PDAC. Furthermore, we have recently found that excessive activity within the Ras signaling pathway can lead to acinar cell death or metaplasia and is associated with the development of fibrosis resembling CP. Therefore, it is time to reevaluate the relationship between these 2 diseases. We have recently proposed a new model in which Ras activity is a direct link between these 2 diseases.1,2 Here we will briefly review the shared properties between CP and PDAC and describe the new model. A more comprehensive coverage of the same issues will be available later.2
Previously Observed Relationships Between Chronic Pancreatitis and Pancreatic Ductal Adenocarcinoma
Chronic Pancreatitis and Pancreatic Ductal Adenocarcinoma Share a Prominent Stroma
Both CP and PDAC develop an extensive fibrotic response often referred to as desmoplasia that leads to similar histopathologic features. Both diseases possess extensive fibrotic stroma infiltrated with a variety of leukocytes, acinar cell atrophy, and distorted and blocked ducts.3,4 The only important histologic difference between CP and PDAC is the presence of carcinoma in PDAC. However, cancer cells typically make up only a small fraction (~10%) of the volume of the tumors.5,6 Furthermore, focal areas of fibrosis lacking cancer that resemble CP are inevitably present surrounding PDAC, such that PDAC always occurs in the presence of CP. Also, precursors to PDAC are often observed within areas of CP. These observations suggest strong linkage between the diseases but do not clearly indicate the relationship between these diseases.
Chronic Pancreatitis Is a Risk Factor for Pancreatic Ductal Adenocarcinoma
CP is a major risk factor for PDAC.7–10 Although the vast majority of CP patients do not progress to PDAC, the cumulative risk of pancreatic cancer in subjects with CP was reported to be 4% after 20 years, with a standardized incidence ratio of 14.10 This is at least 10-fold greater risk than for those without CP. For patients with a rare form of CP, hereditary pancreatitis, the risk is even greater at 53 times the normal and a cumulative lifetime risk of 40%, which is the highest of any known genetically associated risk factor.11 However, the mutations associated with hereditary CP are not observed in PDAC from patients without hereditary CP.12 Furthermore, the vast majority of patients with CP do not progress to PDAC. Therefore, significant barriers must exist between the mechanisms responsible for CP and the development of PDAC.
Both Chronic Pancreatitis and Pancreatic Ductal Adenocarcinoma Possess Preneoplastic Lesions
Pathologic studies have suggested 2 important precursors to PDAC, pancreatic intraepithelial neoplasias (PanINs) and intraductal papillary mucinous neoplasms.13,14 PanINs are present in nearly all patients with CP.15–17 It has also been observed that chromosomal instability and genomic damage are present in pancreatic duct cells from patients with CP, similar to what is observed in PDAC.18 The specific mechanisms responsible for the genetic instability observed in CP are unclear. Nonetheless, these observations provide a potential explanation for the relationship between CP and PDAC.
Chronic Pancreatitis and Pancreatic Ductal Adenocarcinoma Possess K-Ras Mutations
In PDAC, mutations in the proto-oncogene K-Ras are found in nearly all cases.19 For this reason, this mutation has been extensively explored as a potential diagnostic marker.20–23 Unfortunately for use of K-Ras mutations as a diagnostic aid, K-Ras mutations have also been observed in ~30% of samples from patients with CP, although highly variable levels have been reported.24 –27 This is likely due to differences in sampling, DNA extraction, or polymerase chain reaction methods. Nonetheless, it is clear that K-Ras mutations are very often found in CP. Furthermore, CP generally contains early PanINs that often possess mutations in K-Ras.28 –31 Therefore, K-Ras mutations are not specific for invasive PDAC, and their presence in normal individuals32 suggests that they can precede CP. This is particularly interesting in light of the observation that elevated levels of K-Ras activity in acinar cells generate CP in a mouse model.33 Taken together, the data indicate that the qualitative presence of K-Ras mutations is not an accurate predictor of pancreatic cancer, because this mutation can clearly precede other required alterations.
New Evidence for the Role of Ras Activity in Chronic Pancreatitis and Cancer Development
Ras Activity in Acinar Cells Generates Both Fibrosis and Cancer
Mutations in K-Ras are nearly universal in PDAC. Therefore, a number of models based on expression of mutant K-Ras have been developed. Several studies have shown that when mutant K-Ras expression is directed by promoters active during pancreatic development such as PDX1,34,35 p48,34 nestin,36 or elastase,37 it results in development of PanINs that progress to invasive PDAC. On the other hand, adult mice were reported to be refractory to adult acinar cell37 or duct cell38 expression of mutant K-Ras and did not develop PanINs or PDAC.37 However, if mice with adult acinar cell expression of mutant K-Ras were challenged with an inflammatory stimulus, then they developed the full spectrum of PanINs and invasive PDAC.37 These observations have been interpreted to suggest that during adulthood, PDAC is initiated by a combination of genetic (eg, somatic K-Ras mutations) and non-genetic (eg, tissue damage) events.37 However, another interpretation of this observation is that low levels of mutant K-Ras expression are not sufficient to meet a threshold of pathologic Ras pathway signaling, but that when further activated by exogenous stimulants, such a threshold can be met.
Evidence for the existence of a threshold of Ras pathway activity in pancreatic pathology comes from studies expressing higher than endogenous levels of mutant K-Ras. Although the expression of endogenous levels of mutant K-Ras results in only a minor change in K-Ras activity within the targeted cells,35 higher levels of expression generate Ras pathway activity that mimics what is observed in PDAC.1 In this model, elevated, but not endogenous, mutant K-Ras expression in adult acinar cells caused a rapid development of CP with abundant PanINs and intraductal papillary mucinous neoplasms that progressed to invasive and meta-static PDAC. Therefore, this model confirms the potential of acinar cells to form PDAC as previously reported37 and suggests that at least in mouse models, elevated levels of Ras activity are necessary and sufficient to cause both CP and PDAC. It is important to note that in this model increased activity of Ras was the cause, rather than the result, of CP.
These data suggest that the activity level of the K-Ras pathway, rather than the presence of mutations, is the biologically relevant parameter. Currently nothing is known about the activity level of K-Ras in CP. However, it is known that K-Ras is activated by several stimuli including CCK treatments,39 expression of cyclooxygenase-2,40 and expression of transforming growth factor–α,41 and each of these has been shown to induce CP-like fibrosis when examined in animal models.40,42,43 Furthermore, the addition of CCK44 or transforming growth factor–α45 to animals with endogenous levels of mutant K-Ras expression has been found to accelerate tumor development. Therefore, these data suggest the possibility that increases of Ras activity in pancreatic acinar cells brought about by various etiologic stimulations might mediate fibrosis and inflammation.
A Model for Ras Activity as a Common Cause of Inflammation in Chronic Pancreatitis and Pancreatic Ductal Adenocarcinoma
With our current understanding of the influence of inflammation on tumorigenesis, we can describe a model that explains many of the known links between CP and PDAC (Figure 1). The initial event common to CP and PDAC is inflammation. The causes of pancreatic inflammation in CP can be the effects of ethanol, bile, blockage of ducts, hereditary alterations of trypsinogen, or a variety of other etiologies.4 Most of these stimuli are known to activate the Ras pathway, and our recent data indicate that high levels of Ras activity are sufficient for initiating an inflammatory response. Therefore, we suggest that elevated levels of Ras pathway activity are the key event in both diseases. If these stimuli are sufficiently strong, acinar cells are lost, and pancreatic stellate cells are recruited and activated, resulting in the formation of focal histologic CP. Multiple repetitions of this cycle lead to full-blown clinical CP. During this initiation period, increases in Ras activity likely occur primarily through the extrinsic pathway without K-Ras mutations. However, the chronic inflammatory microenvironment found in CP increases genetic instability18 and cell proliferation,17 thus greatly accelerating the rate of gene mutations and therefore the probability of K-Ras mutations. Thus, longer durations of CP are more highly correlated with mutations of K-Ras. If K-Ras becomes mutated, then this feeds into the Ras pathway activity, and if, in combination, the intrinsic and extrinsic pathways of Ras activation reach a sufficient level of activity, it results in the development of early PanINs and increases the likelihood of developing PDAC.23 However, the development of PDAC requires further genetic events beyond Ras mutation, which partially explains why most CP does not lead to PDAC. Alternatively, spontaneous preexisting mutations in K-Ras, or other molecules impinging on the K-Ras signaling pathway, might increase responses to extrinsic factors, leading to higher levels of Ras signaling pathway activity and the resulting inflammation. Thus, K-Ras mutations might predispose to pancreatitis.
Figure 1.
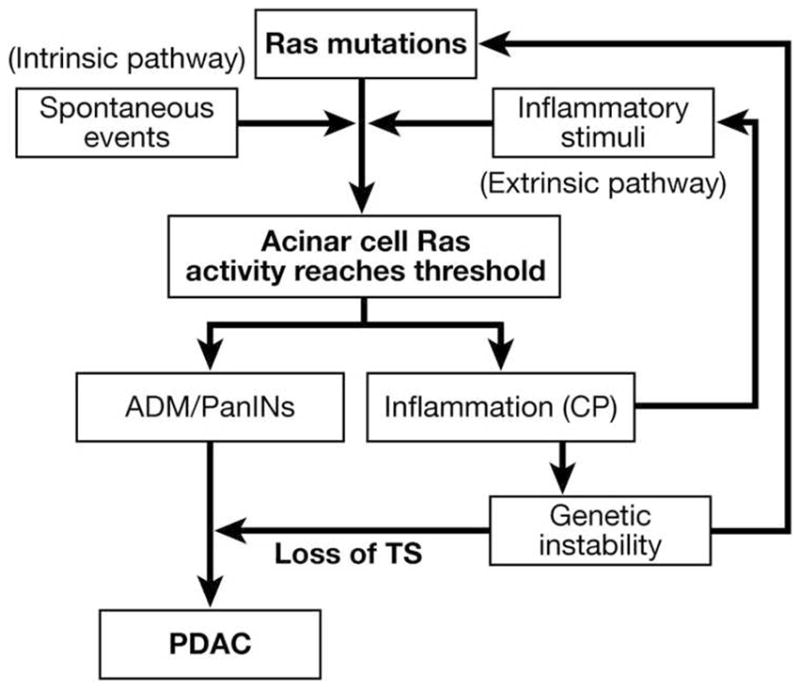
Ras activity levels control the development of pancreatic diseases. For the initiation of pathologies, Ras activity levels must be elevated beyond a threshold. This level of activity can be achieved by high levels of extrinsic Ras activators (extrinsic pathway), by intrinsic alterations of Ras activity including activating mutations of K-Ras (intrinsic pathway), or by a combination of these 2 pathways. Pathologic levels of Ras activity cause acinar cells to undergo acinarductal metaplasia (ADM) and to form PanINs. High levels of Ras activity also lead to the generation of inflammation resembling CP. Inflammation induced genetic instability, and increased proliferation likely increases the probability of genetic alterations including mutations in K-Ras and loss of tumor suppressors (TS), which are required for the development of PDAC. (Modified from Ji et al1).
Genetic instability mediated by increased inflammation also increases the rate of other spontaneous genetic alterations, including silencing of p16 and other genetic and epigenetic changes that bring about the progression to advanced PanINs and eventually invasive carcinoma.46 Thus, CP-generated inflammation explains it as a risk factor for PDAC. Increased genetic instability explains the presence of many early genetic mutations found in CP and low-grade PanINs that are shared with PDAC. The common histology between CP and PDAC is explained by the inflammatory influences of both CP and PDAC on the surrounding stromal cells. The shared microenvironment explains the commonality in gene expression observed between CP and PDAC. Further understanding of these processes will be necessary for prevention of this cancer and might guide the development of new therapies.
This model for the relationship between CP and PDAC does not change greatly on the basis of the cell of origin of PDAC. However, if the cell of origin of PDAC is metaplastic acinar cells, then both CP and PDAC originate in the same cell type, and these 2 diseases are likely consequences of the same mechanisms, with CP forming a direct precursor stage to PDAC. In this case, there would be a continuum of gene expression changes in the same cell lineage directly linking these 2 diseases.
Summary
Pancreatic cancer and CP share many features. On the basis of new evidence from genetic mouse models, a new explanation for this has arisen. In this model, pancreatic cancer and CP might have many features in common because both are part of a spectrum of alterations that occurs in pancreatic acinar cells. The activity of the Ras signaling pathway appears to be the key mechanism controlling these changes. Activation of this pathway, either by genetic mutations or by alterations in the levels of signaling molecules in the microenvironment, can trigger alterations in acinar cells, leading to their death or metaplasia in a background of fibrosis and inflammation. This concept fits with most previous observations made in humans and in mouse models on these 2 diseases and provides new hypotheses for further testing.
Acknowledgments
Funding
This research was supported by NIH DK052067, 5R21DK068414, M.D. Anderson Support Core grant CA16672, M.D. Anderson Pancreatic Specialized Programs of Research Excellence (SPORE) grant P20 CA101936, and by the Lockton Endowment.
Abbreviations used in this paper
- CP
chronic pancreatitis
- PanIN
pancreatic intraepithelial neoplasia
- PDAC
pancreatic ductal adenocarcinoma
Footnotes
Conflicts of interest
The authors disclose no conflicts.
References
- 1.Ji B, Tsou L, Wang H, et al. Ras activity levels control the development of pancreatic diseases. Gastroenterology. 2009;137:1072–1082. doi: 10.1053/j.gastro.2009.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Logsdon CD, Ji B, Hwang R. Novel molecular relationships between chronic pancreatitis and pancreatic cancer. In: Neoptolemos JPU, Abbruzzese R, Büchler MW, editors. Handbook of pancreatic cancer. New York: Springer; (in press) [Google Scholar]
- 3.Steer ML, Waxman I, Freedman S. Chronic pancreatitis. N Engl J Med. 1995;332:1482–1490. doi: 10.1056/NEJM199506013322206. [DOI] [PubMed] [Google Scholar]
- 4.Witt H, Apte MV, Keim V, et al. Chronic pancreatitis: challenges and advances in pathogenesis, genetics, diagnosis, and therapy. Gastroenterology. 2007;132:1557–1573. doi: 10.1053/j.gastro.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 5.Seymour AB, Hruban RH, Redston M, et al. Allelotype of pancreatic adenocarcinoma. Cancer Res. 1994;54:2761–2764. [PubMed] [Google Scholar]
- 6.Mutema G, Fenoglio-Preiser C. Pathology and natural history of pancreatic cancer. In: Abbruzzese JL, editor. Gastrointestinal oncology. New York: Oxford University Press; 2004. [Google Scholar]
- 7.Ekbom A, McLaughlin JK, Karlsson BM, et al. Pancreatitis and pancreatic cancer: a population-based study. J Natl Cancer Inst. 1994;86:625–627. doi: 10.1093/jnci/86.8.625. [DOI] [PubMed] [Google Scholar]
- 8.Malka D, Hammel P, Maire F, et al. Risk of pancreatic adenocarcinoma in chronic pancreatitis. Gut. 2002;51:849–852. doi: 10.1136/gut.51.6.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bansal P, Sonnenberg A. Pancreatitis is a risk factor for pancreatic cancer. Gastroenterology. 1995;109:247–251. doi: 10.1016/0016-5085(95)90291-0. [DOI] [PubMed] [Google Scholar]
- 10.Lowenfels AB, Maisonneuve P, Cavallini G, et al. Pancreatitis and the risk of pancreatic cancer: International Pancreatitis Study Group. N Engl J Med. 1993;328:1433–1437. doi: 10.1056/NEJM199305203282001. [DOI] [PubMed] [Google Scholar]
- 11.Lowenfels AB, Maisonneuve P, DiMagno EP, et al. Hereditary pancreatitis and the risk of pancreatic cancer. International Hereditary Pancreatitis Study Group. J Natl Cancer Inst. 1997;89:442–446. doi: 10.1093/jnci/89.6.442. [DOI] [PubMed] [Google Scholar]
- 12.Whitcomb DC. Inflammation and cancer: V—chronic pancreatitis and pancreatic cancer. Am J Physiol Gastrointest Liver Physiol. 2004;287:G315–G319. doi: 10.1152/ajpgi.00115.2004. [DOI] [PubMed] [Google Scholar]
- 13.Takaori K, Hruban RH, Maitra A, et al. Current topics on precursors to pancreatic cancer. Adv Med Sci. 2006;51:23–30. [PubMed] [Google Scholar]
- 14.Maitra A, Fukushima N, Takaori K, et al. Precursors to invasive pancreatic cancer. Adv Anat Pathol. 2005;12:81–91. doi: 10.1097/01.pap.0000155055.14238.25. [DOI] [PubMed] [Google Scholar]
- 15.Rosty C, Geradts J, Sato N, et al. p16 inactivation in pancreatic intraepithelial neoplasias (PanINs) arising in patients with chronic pancreatitis. Am J Surg Pathol. 2003;27:1495–1501. doi: 10.1097/00000478-200312000-00001. [DOI] [PubMed] [Google Scholar]
- 16.Volkholz H, Stolte M, Becker V. Epithelial dysplasias in chronic pancreatitis. Virchows Arch A Pathol Anat Histol. 1982;396:331–349. doi: 10.1007/BF00431392. [DOI] [PubMed] [Google Scholar]
- 17.Hermanova M, Nenutil R, Kren L, et al. Proliferative activity in pancreatic intraepithelial neoplasias of chronic pancreatitis resection specimens: detection of a high-risk lesion. Neoplasma. 2004;51:400–404. [PubMed] [Google Scholar]
- 18.Brentnall TA, Chen R, Lee JG, et al. Microsatellite instability and K-ras mutations associated with pancreatic adenocarcinoma and pancreatitis. Cancer Res. 1995;55:4264–4267. [PubMed] [Google Scholar]
- 19.Almoguera C, Shibata D, Forrester K, et al. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988;53:549–554. doi: 10.1016/0092-8674(88)90571-5. [DOI] [PubMed] [Google Scholar]
- 20.Ren YX, Xu GM, Li ZS, et al. Detection of point mutation in K-ras oncogene at codon 12 in pancreatic diseases. World J Gastroenterol. 2004;10:881–884. doi: 10.3748/wjg.v10.i6.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sawada Y, Gonda H, Hayashida Y. Combined use of brushing cytology and endoscopic retrograde pancreatography for the early detection of pancreatic cancer. Acta Cytol. 1989;33:870–874. [PubMed] [Google Scholar]
- 22.Kubota Y, Takaoka M, Tani K, et al. Endoscopic transpapillary biopsy for diagnosis of patients with pancreaticobiliary ductal strictures. Am J Gastroenterol. 1993;88:1700–1704. [PubMed] [Google Scholar]
- 23.Arvanitakis M, Van Laethem JL, Parma J, et al. Predictive factors for pancreatic cancer in patients with chronic pancreatitis in association with K-ras gene mutation. Endoscopy. 2004;36:535–542. doi: 10.1055/s-2004-814401. [DOI] [PubMed] [Google Scholar]
- 24.Lohr M, Maisonneuve P, Lowenfels AB. K-Ras mutations and benign pancreatic disease. Int J Pancreatol. 2000;27:93–103. doi: 10.1385/IJGC:27:2:093. [DOI] [PubMed] [Google Scholar]
- 25.Berthelemy P, Bouisson M, Escourrou J, et al. Identification of K-ras mutations in pancreatic juice in the early diagnosis of pancreatic cancer. Ann Intern Med. 1995;123:188–191. doi: 10.7326/0003-4819-123-3-199508010-00005. [DOI] [PubMed] [Google Scholar]
- 26.Hsiang D, Fries H, Buchler MW, et al. Absence of K-ras mutations in the pancreatic parenchyma of patients with chronic pancreatitis. Am J Surg. 1997;174:242–246. doi: 10.1016/s0002-9610(97)00133-5. [DOI] [PubMed] [Google Scholar]
- 27.Nakaizumi A, Uehara H, Takenaka A, et al. Diagnosis of pancreatic cancer by cytology and measurement of oncogene and tumor markers in pure pancreatic juice aspirated by endoscopy. Hepatogastroenterology. 1999;46:31–37. [PubMed] [Google Scholar]
- 28.Lohr M, Kloppel G, Maisonneuve P, et al. Frequency of K-ras mutations in pancreatic intraductal neoplasias associated with pancreatic ductal adenocarcinoma and chronic pancreatitis: a meta-analysis. Neoplasia. 2005;7:17–23. doi: 10.1593/neo.04445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deramaudt T, Rustgi AK. Mutant KRAS in the initiation of pancreatic cancer. Biochim Biophys Acta. 2005;1756:97–101. doi: 10.1016/j.bbcan.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 30.Klein WM, Hruban RH, Klein-Szanto AJ, et al. Direct correlation between proliferative activity and dysplasia in pancreatic intra-epithelial neoplasia (PanIN): additional evidence for a recently proposed model of progression. Mod Pathol. 2002;15:441–447. doi: 10.1038/modpathol.3880544. [DOI] [PubMed] [Google Scholar]
- 31.Klimstra DS, Longnecker DS. K-ras mutations in pancreatic ductal proliferative lesions. Am J Pathol. 1994;145:1547–1550. [PMC free article] [PubMed] [Google Scholar]
- 32.Andea A, Sarkar F, Adsay VN. Clinicopathological correlates of pancreatic intraepithelial neoplasia: a comparative analysis of 82 cases with and 152 cases without pancreatic ductal adenocarcinoma. Mod Pathol. 2003;16:996–1006. doi: 10.1097/01.MP.0000087422.24733.62. [DOI] [PubMed] [Google Scholar]
- 33.Ji B, Song J, Tsou L, et al. Activation of K-RAS in pancreatic acinar cells causes chronic pancreatitis in transgenic mice. Gastroenterology. 2007;132:A116–A117. [Google Scholar]
- 34.Hingorani SR, Petricoin EF, Maitra A, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 35.Aguirre AJ, Bardeesy N, Sinha M, et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003;17:3112–3126. doi: 10.1101/gad.1158703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carriere C, Seeley ES, Goetze T, et al. The Nestin progenitor lineage is the compartment of origin for pancreatic intraepithelial neoplasia. Proc Natl Acad Sci U S A. 2007;104:4437–4442. doi: 10.1073/pnas.0701117104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guerra C, Schuhmacher AJ, Canamero M, et al. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007;11:291–302. doi: 10.1016/j.ccr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 38.Brembeck FH, Schreiber FS, Deramaudt TB, et al. The mutant K-ras oncogene causes pancreatic periductal lymphocytic infiltration and gastric mucous neck cell hyperplasia in transgenic mice. Cancer Res. 2003;63:2005–2009. [PubMed] [Google Scholar]
- 39.Duan RD, Zheng CF, Guan KL, et al. Activation of MAP kinase kinase (MEK) and Ras by cholecystokinin in rat pancreatic acini. Am J Physiol. 1995;268:G1060–G1065. doi: 10.1152/ajpgi.1995.268.6.G1060. [DOI] [PubMed] [Google Scholar]
- 40.Muller-Decker K, Furstenberger G, Annan N, et al. Preinvasive duct-derived neoplasms in pancreas of keratin 5-promoter cyclo-oxygenase-2 transgenic mice. Gastroenterology. 2006;130:2165–2178. doi: 10.1053/j.gastro.2006.03.053. [DOI] [PubMed] [Google Scholar]
- 41.Wagner M, Greten FR, Weber CK, et al. A murine tumor progression model for pancreatic cancer recapitulating the genetic alterations of the human disease. Genes Dev. 2001;15:286–293. doi: 10.1101/gad.184701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pandol SJ, Gukovsky I, Satoh A, et al. Animal and in vitro models of alcoholic pancreatitis: role of cholecystokinin. Pancreas. 2003;27:297–300. doi: 10.1097/00006676-200311000-00004. [DOI] [PubMed] [Google Scholar]
- 43.Colby JK, Klein RD, McArthur MJ, et al. Progressive metaplastic and dysplastic changes in mouse pancreas induced by cycloox-ygenase-2 overexpression. Neoplasia. 2008;10:782–796. doi: 10.1593/neo.08330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carriere C, Young AL, Gunn JR, et al. Acute pancreatitis markedly accelerates pancreatic cancer progression in mice expressing oncogenic Kras. Biochem Biophys Res Commun. 2009;382:561–565. doi: 10.1016/j.bbrc.2009.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Siveke JT, Einwachter H, Sipos B, et al. Concomitant pancreatic activation of Kras(G12D) and Tgfa results in cystic papillary neoplasms reminiscent of human IPMN. Cancer Cell. 2007;12:266–279. doi: 10.1016/j.ccr.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 46.Maitra A, Hruban RH. Pancreatic cancer. Annu Rev Pathol. 2008;3:157–188. doi: 10.1146/annurev.pathmechdis.3.121806.154305. [DOI] [PMC free article] [PubMed] [Google Scholar]