

Abstract
Carbocyclic nucleosides (−)-5′-homocarbovir and (+)-epi-4′-homocarbovir were prepared from an acylnitroso-derived hetero Diels–Alder cycloadduct. A kinetic enzymatic resolution generated an enantiopure aminocyclopentenol and Pd(0)-mediated decarboxylative allylations of allyl 2,2,2-trifluoroethyl malonates were used to install the 4′-hydroxyethyl groups. Late stage derivatization gave access to the cyclopropylamine analogs, (−)-5′-homoabacavir, and (+)-epi-4′-homoabacavir. All carbonucleoside target molecules were evaluated for antiviral activity.
Graphical abstract

1. Introduction
Carbocyclic nucleosides have been extensively targeted due to their potent antiviral and anti-cancer activities.1, (a), (b) Naturally occurring deoxynucleotides are needed to synthesize viral DNA. Carbovir (−)-1 and related compounds are analogs of natural deoxynucleotides and compete for incorporation into the growing viral DNA chain.2 Incorporation of dideoxynucleosides such as carbovir results in chain termination and disruption of the viral replication process.3
In particular, carbovir (−)-1 has shown significant in vitro anti-HIV activity through its triphosphate analog by inhibiting HIV-1 reverse transcriptase.2 Its congener, abacavir (−)-2, has a considerably improved toxicity profile and higher oral bioavailability.4 Abacavir is metabolized to the triphosphate of carbovir by a unique mechanism that circumvents formation of carbovir itself.5 Consequently, abacavir has in vitro activity and in vivo efficacy against HIV. It has been approved by the FDA for the treatment of HIV infection under the brand name Ziagen.6, (a), (b), (c)
Due to the important activity of carbocyclic nucleosides, there is continued interest in the syntheses of new analogs. One approach has been to extend the 4′-carbon side chain.7, (a), (b) Although the enantioselective syntheses of 5′-homocarbovir (−)-3a and 5′-homoabacavir (−)-3b have been reported8 (Fig. 1 ), only 5′-homocarbovir (±)-3a 9, (a), (b) has been evaluated in antiviral assays and showed poor activity in limited testing.9a To further investigate the activity profiles of (−)-3a as well as the previously untested (−)-3b, we developed a new synthetic route from an acylnitroso-derived hetero Diels–Alder cycloadduct, which utilizes decarboxylative rearrangements of allyl 2,2,2-trifluroethylmalonates to install the 4′-hydroxyethyl side chain. The modification of a single step in the sequence allowed for access to the unprecedented epi-4′ analogs (+)-4a and (+)-4b.
Fig. 1.

Carbocyclic nucleosides.
2. Results and discussion
Our research group’s continued interest in the syntheses of diverse carbocyclic nucleosides(b), 10, (a), (b), 11, (a) has led to the development of a reliable method for the synthesis of cyclopentenol (−)-7.12 We recently reported the Cp2TiCl-catalyzed reduction of Boc cycloadduct (±)-6.13 The reaction was performed on large scale (50 mmol) and an isolated yield of 79% was obtained after the resultant silylated alcohol was deprotected with a simple acidic work-up with 1 M HCl (Scheme 1 ). Kinetic enzymatic resolution of (±)-7 was achieved with commercially available immobilized Candida antarctica B lipase to ultimately form the enantiopure alcohol (−)-7.12
Scheme 1.

Synthesis of aminocyclopentenol (−)-7.
Cyclopentenol (−)-7 was coupled with 3-oxo-3-(2,2,2-trifluoroethoxy)propanoic acid 8 to form allyl 2,2,2-trifluoroethyl malonate (−)-9 in high yield (Scheme 2 ). As previously reported, malonate (−)-9 underwent facile decarboxylative rearrangement in the presence of Pd(dba)2 and dppe to provide homoallylic ester (−)-10 with complete diastereo- and regio-control.14 Formation of the π-allyl palladium complex proceeded on the less hindered face and anti attack by the in situ generated nucleophilic alkoxy ethenolate species afforded the product with retention of configuration. Reduction of (−)-10 with 2 equiv of diisobutylaluminum hydride (DIBAL-H) yielded the desired alcohol (−)-11.14 We then turned our attention to construction of the chloroguanine base.2, (a), 15
Scheme 2.

Synthesis of homoallylic ester (−)-10.
Upon deprotection of the Boc group with 12 M HCl, aminoalcohol (−)-11 underwent nucleophilic aromatic substitution with 2-amino-4,6-dichloropyrimidine to afford alcohol (+)-12 in 50% yield over two steps (Scheme 3 ). Azo coupling of (+)-12 with 4-chlorobenzenediazonium chloride followed by reduction of the resulting aryl azo compound with Zn in acetic acid afforded triaminopyrimidine (−)-13. 2-Propanol was selected as the co-solvent over ethanol due to the formation of side products from reaction with ethanol.16 Cyclization of (−)-13 with triethyl orthoformate in the presence of catalytic acid provided chloroguanine (−)-14. Careful monitoring of the reaction time was necessary to prevent substitution of the 6-Cl group with ethanol, which formed an inseparable byproduct.17 Chloroguanine (−)-14 was refluxed in 0.33 M NaOH for 2 h to produce 5′-homocarbovir (−)-3a in 60% yield (Scheme 4 ). Alternatively, (−)-14 was stirred in 2-propanol at 70 °C in the presence of excess cyclopropylamine to form 5′-homoabacavir (−)-3b in 80% yield.
Scheme 3.

Synthesis of intermediate (−)-14.
Scheme 4.

Synthesis of target carbonucleosides (−)-3b and (−)-3b.
trans-Carbocyclic nucleosides have been reported in the literature.(a), 18 The synthetic routes towards these molecules were specifically designed around formation of the necessary anti-1,4-aminocyclopentene. We envisioned a route that required minimal modification from our method to (−)-3a and (−)-3b. Cyclopentenol (−)-7 was treated with 3-oxo-3-(2,2,2-trifluoroethoxy)propanoic acid 8 under Mitsunobu conditions to form allyl 2,2,2-trifluoroethyl malonate (−)-15 in high yield with the expected inversion of configuration at C-4′ (Scheme 5 ). Pd(0)-mediated decarboxylative rearrangement of allyl 2,2,2-trifluoroethyl malonate (−)-15 proceeded with complete diastereo- and regio-control. The retention of configuration during this process is again noteworthy. Reduction of (+)-16 with 2 equiv of diisobutylaluminum hydride (DIBAL-H) yielded the desired alcohol (+)-17. With the new C-4′ stereochemistry established, construction of the chloroguanine base proceeded as described for the synthesis of the syn-1,4-disubstituted cyclopentene derivative. Chloroguanine (+)-20 was synthesized in five steps from alcohol (+)-17.
Scheme 5.

Synthesis of intermediate (+)-20.
Chloroguanine (+)-20 was refluxed in 0.33 M NaOH for 2 h to produce epi-4′-homocarbovir (+)-4a in 60% yield (Scheme 6 ). Alternatively, (+)-20 was stirred in 2-propanol at 70 °C in the presence of excess cyclopropylamine to form epi-4′-homoabacavir (+)-4b in 90% yield. The epi-4′ derivatives proved to be slightly more polar compounds than (–)-3a and (−)-3b but were amenable to purification by column chromatography.
Scheme 6.
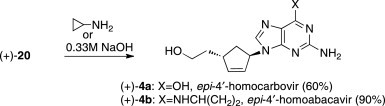
Synthesis of target carbonucleosides (+)-4a and (+)-4b.
Compounds (−)-3a, (−)-3b, (+)-4a, and (+)-4b were subjected to comprehensive screening for antiviral activity. They were tested for antiviral activity in human embryonic lung (HEL) [herpes simplex virus-1 (KOS), herpes simplex virus-2 (G), vaccinia virus, vesicular stomatitis virus, and herpes simplex virus-1 (TK-KOS ACVr), cytomegalovirus, varicella-zoster virus] cell cultures and Crandell-Rees feline kidney (CRFK) [anti-feline corona virus, anti-Feline herpes virus] cell cultures. The compounds were also evaluated for anti-HIV-1 and anti-HIV-2 activity in human T-lymphocyte (CEM) cells. None of the compounds exhibited significant antiviral activity or cytotoxicity.19
3. Conclusion
The decarboxylative allylations of allyl 2,2,2-trifluoroethyl malonates were the key steps en route to 5′-homo-carbocyclic nucleoside core structures. Inversion of the C-4′ stereochemistry at the appropriate stage allowed for the synthesis of new epi-4′-homo-carbocyclic nucleoside analogs without an increase in the number of synthetic transformations. Compounds (−)-3a, (−)-3b, (+)-4a, and (+)-4b were evaluated for antiviral activity.
4. Experimental section
4.1. General
Commercially available reagents and anhydrous solvents were used without further purification unless otherwise specified. Tetrahydrofuran (THF) was distilled from sodium and benzophenone prior to use. Methylene chloride (CH2Cl2) was distilled from calcium hydride prior to use. Glassware was oven dried for 16 h or flame-dried under vacuum and purged with Ar prior to use. Reactions were monitored by thin-layer chromatography (TLC) on silica gel 60 F254 (0.2 mm) precoated aluminum foil and visualized with an ethanolic solution of KMnO4. Flash chromatography was performed with silica gel 60 (230–400 mesh). 1H NMR and 13C NMR spectra were recorded at ambient temperature on a Varian DirectDrive 600 MHz instrument with the residual solvent peaks as internal standards. The line positions of multiplets are given in parts per million (δ) and the coupling constants (J) are given as absolute values in hertz. Infrared spectra were recorded using an FT-IR spectrometer and are reported in cm−1. Melting points were obtained using a Thomas Hoover UniMelt capillary melting point apparatus and are uncorrected. Optical rotations were measured on a Rudolph Research Autopol III. A Bruker MicrOTOF-II equipped with an electrospray ionization source was used for obtaining high resolution accurate mass measurements via flow-injection analyses.
Compound (−)-7 was obtained by dynamic enzymatic resolution as previously described.12 Compounds (±)-7, (−)-9, (−)-10, (−)-11, (−)-15, and (+)-16 were obtained by methods previously described.14
4.1.1. (−)-2-Amino-9-((1R,4R)-4-(2-hydroxyethyl)cyclopent-2-enyl)-9H-purin-6-ol (3a)
In a clean oven dried 15 mL sealed tube equipped with a Teflon screw cap, a solution of (−)-14 (18 mg, 0.064 mmol) in 0.33 M NaOH (1 mL) was stirred at 100 °C for 2 h. The reaction mixture was concentrated, adsorbed on silica gel, and purified by column chromatography (silica gel; eluted with 10–12% CH3OH/CH2Cl2) to give 10 mg of a (−)-3a as a pure white powder (60%). Mp 216–217 °C, 220 °C dec; [α]D 20 −14 (c 0.1, DMSO); 1H NMR (600 MHz, DMSO-d 6) δ 1.42–1.51 (m, 2H), 1.64–1.71 (m, 2H), 2.66 (ddd, J=8.4, 8.4, 13.2 Hz, 1H), 2.77–2.84 (m, 1H), 3.41–3.51 (m, 1H), 4.45 (t, J=4.8, 4.8 Hz, 1H), 5.27–5.33 (m, 1H), 5.83 (ddd, J=1.8, 1.8, 6.0 Hz, 1H), 6.14 (ddd, J=1.8, 1.8, 6.0 Hz, 1H), 6.43 (br s, 2H), 7.55 (s, 1H), 10.54 (br s, 1H); 13C NMR (150 MHz, DMSO-d 6) δ 38.2, 38.6, 41.4, 56.6, 59.3, 116.8, 128.4, 134.8, 140.4, 150.8, 153.4, 156.8; IR (thin film) 1476, 1531, 1604,1683, 2927, 3108, 3307 cm−1; HRMS (ESI) m/z [M+H]+: calcd for C12H16N5O2 +, 262.1299; found, 262.1285; HRMS (ESI) m/z [M+Na]+: calcd for C12H15N5O2Na+, 284.1118; found, 284.1107.
4.1.2. (−)-2-((1R,4R)-4-(2-Amino-6-(cyclopropylamino)-9H-purin-9-yl)cyclopent-2-enyl)ethanol (3b)
In a clean oven dried 15 mL sealed tube equipped with a Teflon screw cap, a solution of (−)-14 (20 mg, 0.0715 mmol) and cyclopropylamine (0.049 mL, 0.715 mmol) in i-PrOH (1 mL) was stirred at 70 °C for 4 h. The reaction mixture was concentrated, adsorbed on silica gel, and purified by column chromatography (silica gel; eluted with 5–7% CH3OH/CH2Cl2) to give 17 mg of (−)-3b as a clear oil (80%). [α]D 20 −27 (c 0.2, CH3OH); 1H NMR (600 MHz, DMSO-d 6) δ 0.55–0.59 (m, 2H), 0.63–0.67 (m, 2H), 1.43–1.52 (m, 2H), 1.65–1.71 (m, 1H), 2.67 (ddd, J=8.4, 8.4, 13.2 Hz, 1H), 2.77–2.85 (m, 1H), 3.41–3.51 (m, 2H), 4.46 (t, J=4.8, 4.8 Hz, 1H), 5.32–5.37 (m, 1H), 5.83 (br s, 2H), 5.84 (ddd, J=2.4, 2.4, 5.4 Hz, 1H), 6.14 (ddd, J=2.4, 2.4, 5.4 Hz, 1H), 7.29 (br s, 1H), 7.56 (s, 1H); 13C NMR (150 MHz, DMSO-d 6) δ 6.4, 23.8, 38.2, 38.6, 41.3, 58.2, 59.3, 113.7, 128.7, 134.6, 140.1, 151.9, 155.9, 160.1; IR (thin film) 1488, 1590, 3203, 3322 cm−1; HRMS (ESI) m/z [M+H]+: calcd for C15H21N6O+, 301.1771; found, 301.1761.
4.1.3. (+)-2-Amino-9-((1R,4S)-4-(2-hydroxyethyl)cyclopent-2-enyl)-9H-purin-6-ol (4a)
In a clean oven dried 15 mL sealed tube equipped with a Teflon screw cap, a solution of (+)-20 (30 mg, 0.107 mmol) in 0.33 M NaOH (1 mL) was stirred at 100 °C for 2 h. The reaction mixture was concentrated, adsorbed on silica gel and purified by column chromatography (silica gel; eluted with 15% CH3OH/CH2Cl2) to give 17 mg of (+)-4a as a white powder (60%). Mp 169−170 °C; [α]D 20 +80 (c 0.05, DMSO); 1H NMR (600 MHz, DMSO-d 6) δ 1.42–1.49 (m, 1H), 1.59–1.66 (m, 1H), 1.97–2.08 (m, 2H), 3.02–3.08 (m, 1H), 3.43–3.52 (m, 2H), 4.64 (t, J=5.4, 5.4 Hz, 1H), 5.32–5.37 (m, 1H), 5.85 (ddd, J=2.4, 2.4, 5.4 Hz, 1H), 6.19 (ddd, J=1.8, 1.8, 6.0 Hz, 1H), 6.42 (br s, 2H), 7.48 (s, 1H), 10.53 (br s, 1H); 13C NMR (150 MHz, DMSO-d 6) δ 38.4, 38.5, 41.5, 58.9, 59.9, 117.3, 128.7, 135.3, 142.2, 151.1, 153.9, 157.3; IR (thin film) 1406, 1477, 1530, 1604, 1674, 2926, 3109, 3307 cm−1; HRMS (ESI) m/z [M+H]+: calcd for C12H16N5O2 +, 262.1299; found, 262.1307.
4.1.4. (+)-2-((1S,4R)-4-(2-Amino-6-(cyclopropylamino)-9H-purin-9-yl)cyclopent-2-enyl)ethanol (4b)
In a clean oven dried 15 mL sealed tube equipped with a Teflon screw cap, a solution of (+)-20 (25 mg, 0.0893 mmol) and cyclopropylamine (0.061 mL, 0.893 mmol) in i-PrOH (1.5 mL) was stirred at 70 °C for 4 h. The reaction mixture was concentrated, adsorbed on silica gel, and purified by column chromatography (silica gel; eluted with 6–8% CH3OH/CH2Cl2) to give 21 mg of (+)-4b as a clear oil (90%). [α]D 20 +33 (c 0.1, CH3OH); 1H NMR (600 MHz, CD3OD) δ 0.58–0.62 (m, 2H), 0.82–0.86 (m, 2H), 1.57–1.64 (m, 1H), 1.74–1.81 (m, 1H), 2.13–2.16 (m, 2H), 2.88–2.95 (m, 1H), 3.10–3.18 (m, 1H), 3.60–3.70 (m, 2H), 5.50–5.54 (m, 1H), 5.91 (ddd, J=2.4, 2.4, 5.4 Hz, 1H), 6.28 (ddd, J=1.8, 1.8, 6.0 Hz, 1H), 7.59 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 7.75, 24.5, 39.3, 39.7, 42.9, 60.6, 61.6, 115.1, 129.2, 137.1, 143.6, 151.7, 157.7, 162.1; IR (thin film) 1485, 1596, 3200, 3325 cm−1; HRMS (ESI) m/z [M+H]+: calcd for C15H21N6O+, 301.1771; found, 301.1788.
4.1.5. (+)-2-((1R,4R)-4-(2-Amino-6-chloropyrimidin-4-ylamino)cyclopent-2-enyl)ethanol (12)
To an EtOH (6.5 mL) solution of (−)-11 (720 mg, 3.17 mmol) was added 12 M HCl (0.33 mL, 3.80 mmol) and the reaction was stirred at 80 °C for 4 h. The reaction mixture was concentrated to a light brown oil. To a solution of the oil and 2-amino-4,6-dichloropyrimidine (574 mg, 3.33 mmol) in n-BuOH (6.5 mL) was added triethylamine (1.10 mL, 7.92 mmol). The reaction was stirred in a sealed tube at 110 °C for 4 days. The reaction mixture was concentrated to a solid. The crude material was adsorbed on silica gel, and purified by column chromatography (silica gel; eluted with 1–2% CH3OH/CH2Cl2 to remove unreacted 2-amino-4,6-dichloropyrimidine, then 5% CH3OH/CH2Cl2 to elute product) to afford 375 mg of (+)-12 as a tan solid (50%). Mp 85−87 °C; [α]D 20 +180 (c 0.05, CH3OH); 1H NMR (600 MHz, CDCl3) δ 1.27 (ddd, J=6.6, 6.6, 13.2 Hz, 1H), 1.39 (br s, 1H), 1.57–1.64 (m, 3H), 1.75–1.81 (m, 1H), 2.64 (ddd, J=7.8, 7.8, 13.2 Hz, 1H), 2.78–2.84 (m, 1H), 3.68–3.78 (m, 2H), 4.82 (br s, 2H), 5.73 (ddd, J=1.8, 1.8, 5.4 Hz, 1H), 5.8 (s, 1H), 5.92–5.96 (m, 1H); 13C NMR (150 MHz, CDCl3) δ 38.4, 39.0, 41.3, 57.1, 61.2, 130.5, 138.6, 162.3, 163.5; IR (thin film) 1574, 3317 cm−1; HRMS (ESI) m/z [M+H]+: calcd for C11H16ClN4O+, 255.1013; found, 255.1013; HRMS (ESI) m/z [M+Na]+: calcd for C11H15ClN4ONa+, 277.0832; found, 277.0827.
4.1.6. (−)-2-((1R,4R)-4-(2,5-Diamino-6-chloropyrimidin-4-ylamino)cyclopent-2-enyl)ethanol (13)
A solution of p-chloroaniline (125 mg, 0.981 mmol) in 3 N HCl (2 mL) was stirred at 0 °C. To this solution was added a solution of sodium nitrite (68 mg, 0.981 mmol) in water (0.8 mL) and the mixture was allowed to stir at 0 °C for 10 min. The resultant diazonium salt solution was added to a mixture of cyclopentene (+)-12 (200 mg, 0.785 mmol), acetic acid (4 mL), water (4 mL), and sodium acetate trihydrate (1.6 g). The reaction mixture was stirred overnight at rt. The yellow precipitate was filtered and washed with cold water until the eluting filtrate was pH 7. The solids were dried under vacuum to yield 260 mg of product (84%). A portion (180 mg) of the material was dissolved in water/i-PrOH (5 mL/5 mL) and Zn dust (296 mg, 4.53 mmol) was added. The mixture was stirred at 90 °C for 4 h. The reaction mixture was concentrated to a solid. The crude material was adsorbed on silica gel and purified by column chromatography (silica gel; eluted with 2–5% CH3OH/CH2Cl2 to remove p-chloroaniline and other impurities, then 6–7% CH3OH/CH2Cl2 to elute product) to afford 93 mg of (−)-13 as a pink oil (76%). [α]D 20 −36 (c 0.05, CH3OH); 1H NMR (600 MHz, CD3OD) δ 1.28 (ddd, J=6.6, 6.6, 13.2 Hz, 1H), 1.54–1.61 (m, 1H), 1.75–1.81 (m, 1H), 2.66 (ddd, J=7.8, 7.8, 13.2 Hz, 1H), 2.73–2.80 (m, 1H), 3.60–3.68 (m, 2H), 5.09–5.13 (m, 1H), 5.77 (ddd, J=1.8, 1.8, 5.4 Hz, 1H), 5.91 (ddd, J=1.8, 1.8, 5.4 Hz, 1H); 13C NMR (150 MHz, CD3OD) δ 39.7, 40.5, 42.6, 58.3, 61.6, 114.6, 132.7, 138.6, 143.0, 157.3, 158.2; IR (thin film) 1440, 1502, 1571, 2925, 3329 cm−1; HRMS (ESI) m/z [M+H]+: calcd for C11H17ClN5O+, 270.1132; found, 270.1122.
4.1.7. (−)-2-((1R,4R)-4-(2-Amino-6-chloro-9H-purin-9-yl)cyclopent-2-enyl)ethanol (14)
A mixture of (−)-13 (93 mg, 0.345 mmol), triethyl orthoformate (2 mL), and 12 M HCl (0.087 mL) was stirred overnight at rt. The suspension was concentrated under high vacuum. A dilute solution of hydrochloric acid (0.5 M, 4 mL) was added and the mixture was stirred at rt for 1 h. The mixture was neutralized to pH 8 with 1 M sodium hydroxide and concentrated to solids. The crude material was adsorbed on silica gel and purified by column chromatography (silica gel; eluted with 8% CH3OH/CH2Cl2) to afford 70 mg of (−)-14 as white powder (72%). Mp 70–71 °C; [α]D 20 −112 (c 0.1, CH3OH); 1H NMR (600 MHz, CD3OD) δ 1.59–1.66 (m, 2H), 1.81–1.87 (m, 1H), 2.85 (ddd, J=7.8, 7.8, 13.2 Hz, 1H), 2.92–2.99 (m, 1H), 3.60–3.69 (m, 2H), 5.55–5.60 (m, 1H), 5.90 (ddd, J=2.4, 2.4, 5.4 Hz, 1H), 6.24 (ddd, J=2.4, 2.4, 5.4 Hz, 1H), 8.03 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 39.5, 39.7, 43.3, 61.5, 61.6, 125.4, 129.0, 142.5, 142.6, 151.6, 155.1, 161.7; IR (thin film) 1402, 1468, 1507, 1562, 1612, 2928, 3211, 3331 cm−1; HRMS (ESI) m/z [M+Na]+: calcd for C12H14ClN5ONa+, 302.0772; found, 302.0785.
4.1.8. (+)-tert-Butyl (1R,4S)-4-(2-hydroxyethyl)cyclopent-2-enylcarbamate (17)
A solution of (+)-16 (390 mg, 1.16 mmol) in anhydrous THF (3.5 mL) was cooled to –78 °C. The mixture was treated dropwise with DIBAL-H (1 M in THF)(3.5 mL, 3.5 mmol) and stirred for 3 h while warming to rt. To the mixture was added 50 mL of satd Rochelle salt solution and 20 mL of EtOAc and the mixture was stirred for 1 h. The organic layer was separated and the aqueous phase was washed with EtOAc (3×20 mL). The combined organics were then washed with satd NaCl, dried over anhydrous sodium sulfate and concentrated to an oil. Purification by column chromatography (silica gel; eluted with 60% EtOAc/hexanes) provided 190 mg of (+)-17 as a clear oil (75%). [α]D 20 +138 (c 0.05, CH2Cl2); 1H NMR (600 MHz, CDCl3) δ 1.20–1.27 (m, 1H), 1.44 (s, 9H), 1.52–1.60 (m, 1H), 1.66–1.72 (m, 1H), 1.80–1.87 (m, 1H), 1.88–1.94 (m, 1H), 2.89–2.96 (m, 1H), 3.64–3.75 (m, 2H), 4.44–4.51 (m, 1H), 4.69–4.77 (m, 1H), 5.70 (ddd, J=2.4, 2.4, 5.4 Hz, 1H), 5.89 (dddd, J=1.8, 1.8, 1.8, 5.4 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 28.4, 38.4, 38.6, 40.7, 56.2, 61.5, 79.3, 131.1, 138.6, 155.3; IR (thin film) 1521, 1684, 2929, 3330 cm−1; HRMS (ESI) m/z [M+H]+: calcd for C12H22NO3 +, 228.1600; found, 228.1597; HRMS (ESI) m/z [M+Na]+: calcd for C12H21NO3Na+, 250.1419; found, 250.1414.
4.1.9. (+)-2-((1S,4R)-4-(2-Amino-6-chloropyrimidin-4-ylamino)cyclopent-2-enyl)ethanol (18)
To an EtOH (1.6 mL) solution of (+)-17 (180 mg, 0.791 mmol) was added 12 M HCl (0.079 mL, 0.791 mmol) and the reaction was stirred at 80 °C for 4 h. The reaction mixture was concentrated to a light brown oil. To a solution of the oil and 2-amino-4,6-dichloropyrimidine (143 mg, 0.831 mmol) in n-BuOH (2 mL) was added triethylamine (0.275 mL, 1.98 mmol). The reaction was stirred in a sealed tube at 110 °C for 4 days. The reaction mixture was concentrated to a solid. The crude material was adsorbed on silica gel and purified by column chromatography (silica gel; eluted with 1–3% CH3OH/CH2Cl2 to remove unreacted 2-amino-4,6-dichloropyrimidine, then 3–4% CH3OH/CH2Cl2 to elute product) to afford 125 mg of (+)-18 as a tan oil (60%). [α]D 20 +42 (c 0.1, CH2Cl2); 1H NMR (600 MHz, CDCl3) δ 1.54–1.65 (m, 3H), 1.68–1.77 (m, 1H), 1.87–1.99 (m, 3H), 2.94–3.03 (m, 1H), 3.67–3.77 (m, 2H), 4.80 (br s, 2H), 5.76 (ddd, J=2.4, 2.4, 5.4 Hz, 1H), 5.79 (s, 1H), 5.98–6.01 (m, 1H); 13C NMR (150 MHz, CDCl3) δ 38.4, 38.5, 41.2, 57.0, 61.8, 130.4, 140.1, 162.5, 163.8; IR (thin film) 1576, 2921 cm−1; HRMS (ESI) m/z [M+Na]+: calcd for C11H16ClN4O+, 255.1013; found, 255.1013.
4.1.10. (+)-2-((1S,4R)-4-(2,5-Diamino-6-chloropyrimidin-4-ylamino)cyclopent-2-enyl) ethanol (19)
A solution of p-chloroaniline (125 mg, 0.981 mmol) in 3 N HCl (2 mL) was stirred at 0 °C. To this solution was added a solution of sodium nitrite (68 mg, 0.981 mmol) in water (0.8 mL) and the mixture was allowed to stir at 0 °C for 10 min. The resultant diazonium salt solution was added to a mixture of cyclopentene (+)-18 (200 mg, 0.785 mmol), acetic acid (4 mL), water (4 mL), and sodium acetate trihydrate (1.6 g). The reaction mixture was stirred overnight at rt. The yellow precipitate was filtered and washed with cold water until the eluting filtrate was pH 7. The solids were dried under vacuum to yield 220 mg of the aryl azo compound (71%). A portion (210 mg) of the material was dissolved in water/i-PrOH (6 mL/6 mL) and Zn dust (349 mg, 5.33 mmol) was added. The mixture was stirred at 90 °C for 1.5 h. The reaction mixture was concentrated to solids. The crude material was adsorbed on silica gel and purified by column chromatography (silica gel; eluted with 3% CH3OH/CH2Cl2 to remove p-chloroaniline and other impurities, then 5% CH3OH/CH2Cl2 to elute product) to afford 115 mg of (+)-19 as an oil (80%). [α]D 20 +104 (c 0.1, CH3OH); 1H NMR (600 MHz, CD3OD) δ 1.51–1.58 (m, 1H), 1.65–1.72 (m, 1H), 1.91–2.01 (m, 2H), 2.95–3.02 (m, 1H), 3.59–3.66 (m, 2H), 5.14–5.18 (m, 1H), 5.81 (ddd, J=1.8, 2.1, 6.0 Hz, 1H), 5.98 (ddd, J=1.8, 2.1, 6.0 Hz, 1H); 13C NMR (150 MHz, CD3OD) δ 39.3, 39.8, 42.4, 58.0, 61.8, 114.7, 132.1, 139.9, 142.9, 157.3, 158.2; IR (thin film) 1443, 1565, 2864, 2926, 3359 cm−1; HRMS (ESI) m/z [M+H]+: calcd for C11H17ClN5O+, 270.1122; found, 270.1118.
4.1.11. (+)-2-((1S,4R)-4-(2-Amino-6-chloro-9H-purin-9-yl)cyclopent-2-enyl)ethanol (20)
A mixture of (+)-19 (115 mg, 0.426 mmol), triethyl orthoformate (2.5 mL), and hydrochloric acid (12 M, 0.11 mL) was stirred overnight at rt. The mixture was concentrated to a tan solid. A dilute solution of hydrochloric acid (0.5 M, 4 mL) was added and the mixture was stirred at rt for 1 h. The mixture was neutralized to pH 8 with 1 M sodium hydroxide and concentrated to tan solids. The crude material was adsorbed on silica gel and purified by column chromatography (silica gel; eluted with 8% CH3OH/CH2Cl2) to afford 85 mg of (+)-20 as an off-white oil (72%). [α]D 20 +68 (c 0.1, CH3OH); 1H NMR (600 MHz, CD3OD) δ 1.57–1.64 (m, 1H), 1.78 (dddd, J=6.6, 7.2, 7.2, 13.2 Hz, 1H), 2.14 (ddd, J=6.6, 8.1, 14.1 Hz, 1H), 2.19 (ddd, J=3.0, 7.5, 14.1 Hz, 1H), 3.16–3.23 (m, 1H), 3.61–3.70 (m, 2H), 5.59–5.63 (m, 1H), 5.91 (ddd, J=2.4, 2.4, 5.4 Hz, 1H), 6.29 (ddd, J=1.8, 1.8, 6.0 Hz, 1H), 7.94 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 39.2, 39.2, 43.0, 61.4, 61.6, 125.4, 128.7, 142.7, 144.0, 151.5, 155.0, 161.6; IR (thin film) 1447, 2859, 2929, 3305 cm−1; HRMS (ESI) m/z [M+Na]+: calcd for C12H14ClN5ONa+, 302.0779; found, 302.0785.
Acknowledgements
We would like to thank Dr. Jed Fisher (University of Notre Dame) for helpful discussions, Dr. Bill Boggess (University of Notre Dame) and Nonka Sevova (University of Notre Dame) for mass spectroscopic analyses, and Dr. Jaroslav Zajicek (University of Notre Dame) for NMR assistance. We acknowledge the NIH (GM068012) for support of this work. L.P.T. acknowledges a Wolf Fellowship (2009, UND).
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.tet.2010.11.097.
Supplementary data
The following is the Supplementary data related to this article:
References and notes
- 1.(a) Agrofoglo L., Suhas E., Farese A., Condom R., Challand S.R., Earl R.A., Guedj R. Tetrahedron. 1994;50:10611. [Google Scholar]; (b) Crimmins M.T. Tetrahedron. 1998;54:9229. [Google Scholar]
- 2.Vince R., Hua M. J. Med. Chem. 1990;33:17. doi: 10.1021/jm00163a004. [DOI] [PubMed] [Google Scholar]
- 3.Orr D.C., Figueiredo H.T., Mo C.-L., Penn C.R., Cameron J.M. J. Biol. Chem. 1992;267:4177. [PubMed] [Google Scholar]
- 4.Daluge S.M., Good S.S., Faletto M.B., Miller W.H., Clair S., M.H., Boone L.R., Tisdale M., Parry N.R., Reardon J.E., Dornsife R.E., Averett D.R., Krenitsky T.A. Antimicrob. Agents Chemother. 1997;41:1082. doi: 10.1128/aac.41.5.1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fung E.N., Cai Z., Burnette T.C., Sinhababu A.K. J. Chromatogr., B. 2001;754:285. doi: 10.1016/s0378-4347(00)00619-8. [DOI] [PubMed] [Google Scholar]
- 6.(a) Foster R.H., Failds D. Drugs. 1998;55:729. doi: 10.2165/00003495-199855050-00018. [DOI] [PubMed] [Google Scholar]; (b) Melroy J., Nair V. Curr. Pharm. Des. 2005;11:3847. doi: 10.2174/138161205774580642. [DOI] [PubMed] [Google Scholar]; (c) Yuen G.J., Weller S., Pakes G.E. Clin. Pharm. 2008;47:351. doi: 10.2165/00003088-200847060-00001. [DOI] [PubMed] [Google Scholar]
- 7.(a) Yang M., Schneller S.W. Bioorg. Med. Chem. Lett. 2005;15:149. doi: 10.1016/j.bmcl.2004.10.019. [DOI] [PubMed] [Google Scholar]; (b) Roberts S.M., Jones M.F. J. Chem. Soc., Perkin Trans. 1. 1988:2927. [Google Scholar]
- 8.Olivo H.E., Yu J. Tetrahedron: Asymmetry. 1997;8:3785. [Google Scholar]
- 9.(a) Rhee H., Yoon D., Jung M.E. Nucleosides, Nucleotides Nucleic Acids. 2000;19:619. doi: 10.1080/15257770008035012. [DOI] [PubMed] [Google Scholar]; (b) Akella A.B., Vince R. Tetrahedron. 1996;52:2789. [Google Scholar]
- 10.(a) Jiang MX.-W., Jin B., Gage J.L., Priour A., Savela G., Miller M.J. J. Org. Chem. 2006;71:4164. doi: 10.1021/jo060224l. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Li F., Brogan J.B., Gage J.L., Zhang D., Miller M.J. J. Org. Chem. 2004;69:4538. doi: 10.1021/jo0496796. [DOI] [PubMed] [Google Scholar]
- 11.(a) Cesario C., Miller M.J. J. Org. Chem. 2009;74:5730. doi: 10.1021/jo900861x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cesario C., Tardibono L., Miller M.J. Tetrahedron Lett. 2010;51:3053. doi: 10.1016/j.tetlet.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mulvihill M.J., Gage J.L., Miller M.J. J. Org. Chem. 1998;63:3357. [Google Scholar]
- 13.Cesario C., Tardibono L., Miller M.J. J. Org. Chem. 2009;74:448. doi: 10.1021/jo802184y. For N–O bond reductions of cycloadducts, see: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tardibono L., Patzner J., Cesario C., Miller M. J. Org. Lett. 2009;11:4076. doi: 10.1021/ol901518g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.For examples of guanine base formation, see:; (a) Balo C., Blanco J.M., Fernandez F., Lens E., Lopez C. Tetrahedron. 1998;54:2833. [Google Scholar]
- 16.2-((1R,4R)-4-((2,5-Diamino-6-ethoxypyrimidin-4-yl)amino)cyclopent-2-en-1-yl)ethanol was identified (by LC–MS) as a byproduct when the reaction was conducted in EtOH.
- 17.2-((1R,4R)-4-(2-Amino-6-ethoxy-9H-purin-9-yl)cyclopent-2-en-1-yl)ethanol was identified (by LC–MS) as a byproduct when prolonged reaction times were allowed.
- 18.(a) Grumann A., Marley H., Taylor R.J.K. Tetrahedron Lett. 1995;36:7767. [Google Scholar]
- 19.Detailed antiviral assay data for (−)-3a, (−)-3b, (+)-4a, (+)-4b, and related intermediates can be found in the Supplementary data.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.