

Abstract
West Nile virus (WNV) is a re-emerging pathogen responsible for fatal outbreaks of meningoencephalitis in humans. Recent research using a mouse model of infection has indicated that specific chemokines and chemokine receptors help mediate the host response to WNV in the central nervous system, acting by at least two mechanisms: control of monocytosis in blood (Ccr2) and control of leukocyte movement from blood to brain (Cxcr3 and Ccr5). CCR5 also appears to be important in human infection, since individuals genetically deficient in this receptor have increased risk of symptomatic disease once infected. These findings provide detailed insight into non-redundant chemokine roles in organ-specific leukocyte recruitment during infection, and emphasize the importance of the balance between pathogen control and immunopathology in determining overall clinical outcome.
Keywords: leukocyte trafficking, flavivirus, chemoattractant, encephalitis
Introduction
Although the chemokine system as a whole is well-established as an important mediator of leukocyte trafficking and antimicrobial host defense, the full spectrum of protective chemokines is not known for any pathogen. This is a difficult challenge for human infections, particularly for recently emerging and re-emerging diseases where patients and clinical samples may be difficult to collect for systematic study. The emergence of West Nile virus (WNV) infection in the Western hemisphere is a good example of this, but, thanks to an excellent mouse model and the availability of archived WNV-seropositive serum samples, one in which significant progress has been made.
WNV is a 9 kilobase single-stranded RNA flavivirus that exists in nature as a zoonosis, cycling between mosquitoes and birds, with humans and other vertebrates acting as accidental dead-end hosts [1]. First identified in 1937 in a woman in Uganda, WNV had spread across and been limited to Africa and the Middle East until 1999 when it was first isolated in the United States from an infected flamingo in the Bronx Zoo in New York City. From there the virus spread rapidly across North America, causing annual outbreaks of severe neurologic illness and death in humans, horses and other mammals. Bird populations have also been affected, with high mortality, particularly for jays and crows. As of November 2009 there were 29,625 human cases of WNV reported to the CDC, including 1173 deaths (4.0%). Most states report cases each year, although the level of activity may be quite different from state to state. WNV has adapted to many mosquito species as vectors and to a large number of vertebrate hosts, which may partly explain its rapid spread in the Western Hemisphere. The lack of any previous exposure or immunity to the virus may partly explain the more intense epidemiologic features of the disease in the Western Hemisphere compared to those in Africa and Asia, where cases are typically milder and occur in episodic outbreaks.
WNV infection remains subclinical in most humans, but ~20% may develop symptoms of disease ranging from a mild flu-like illness, known as West Nile Fever, to more serious neurological complications, including meningitis and encephalitis. Post-neurologic sequelae are common [1]. In both mice and humans, WNV encephalitis is characterized by reaction of resident cells in the CNS and infiltration of inflammatory leukocytes, including monocytes and T cells, in the perivascular space and parenchyma [2,3]. Although increased age and immunosuppression are risk factors for severe WNV infection in man, little is known about its immunopathogenesis. An excellent mouse model of infection has been developed, and various components of the innate and adaptive immune response, including antibodies, T cells (particularly CD8+ T cells), macrophages and γδ T cells, have been identified that are critical for viral clearance. In this model CD4+ and CD8+ T cells, as well as NK cells and infiltrating macrophages accumulate within the CNS in aggregates that colocalize primarily with WNV-infected neurons.
Inflammatory chemokines and their cognate receptors (including Ccr1, Ccr2, Ccr5, Cxcr3 and Cx3cr1) are among the immunoregulatory factors that are most strongly produced in the WNV-infected mouse brain [4]. Inflammatory chemokines are typically produced in the model by microglia and astrocytes.
CCR5 and WNV infection
Ccr5 was the first chemokine receptor reported to play a clear functional role in the mouse model of WNV [4]. Approximately 20% each of CD4+ and CD8+ T cells, NK cells and macrophages in infected mouse brain express Ccr5, and infection is uniformly fatal in Ccr5-deficient mice as compared to wild type mice, in which the majority survive.
Ccr5 appears to control infection by mediating recruitment of leukocytes to the infected CNS. After the virus is deposited in the skin, it travels to draining lymph nodes, blood and spleen, where it is normally cleared before entering the brain. In infected Ccr5-deficient mice, the distribution of leukocyte subsets in the spleen and cytokine levels in spleen and blood are similar to those in infected wild type mice, and virus is cleared normally from spleen and blood. In contrast, Ccr5-deficient mice are unable to normally control virus replication in the CNS, and this is associated with markedly reduced accumulation of CD4+ and CD8+ T cells, NK cells and macrophages in the brain. WNV-infected mice lacking Ccr5 can be rescued from fatal outcome by adoptive transfer of splenocytes from WNV-infected wild type mice, but not by splenocytes from WNV-infected Ccr5-deficient mice, and this is associated with increased accumulation of leukocytes in the CNS. Together, these data strongly suggest that in the mouse model the presence of WNV in the CNS triggers Ccr5-dependent influx of leukocytes needed for viral clearance and recovery.
Interestingly, Ccr5 deficiency results in less leukocyte accumulation in the CNS than can be accounted for by the percentage of Ccr5+ leukocytes in infected brain from wild type mice, suggesting the possibility of a positive feedback loop for leukocyte recruitment, potentially involving cytokines or chemokines released by these cells. Other functional effects of Ccr5, such as effects on T cell proliferation, apoptosis, differentiation, and activation, must also be considered but have not yet been addressed experimentally.
There is genetic evidence that human CCR5 is also important in WNV pathogenesis [5–8]. This has been tested through genotype:phenotype association analysis of CCR5Δ32, a complete loss-of-function allele of CCR5, in 4 separate groups of WNV-infected Caucasian subjects. CCR5Δ32 is very common in Caucasians (10% allele frequency, 1% homozygous frequency in the US), but rarely found in other racial groups.
Homozygous CCR5Δ32 was markedly increased in all four groups relative to expectation, as defined by the genotype frequency in a cohort of healthy US Caucasian random blood donors from the US National Institutes of Health Department of Transfusion Services, whose blood samples were obtained prior to the WNV epidemic in the US, and in a group of WNV-seronegative individuals who had had a symptomatic illness in which WNV had been considered in the differential diagnosis. The Odds Ratios were large and similar in magnitude in all four groups (overall OR=4.2). This value is similar to that reported previously for association of the homozygous CCR5Δ32 genotype with the HIV exposed-uninfected phenotype, which is generally accepted as very strong.
Although only 5% of symptomatic WNV cases were CCR5Δ32 homozygotes in these studies, the data suggested that most if not all infected CCR5Δ32 homozygotes in the catchment area of the state public health departments became symptomatic. This conclusion is based on the assumption that ~20% of WNV-infected individuals wil become symptomatic in the North American epidemic, and the generally accepted 1% frequency of CCR5Δ32 homozygotes among North American Caucasians. This analysis assumes that CCR5 deficiency does not affect the risk of WNV infection and that the fraction of symptomatic WNV cases reported to state public health services is high. With these caveats in mind, the calculation is consistent with the mouse model in which CCR5 deficiency resulted in 100% susceptibility to severe symptomatic disease and ultimately death. An association of CCR5 deficiency with fatal outcome was found only in one group, from Arizona. This was not as strong an association as would be predicted from the uniform lethality of WNV in the mouse model, possibly because of differences in infecting virus strain, inoculum size and/or supportive care, among other factors.
These studies only addressed the association of CCR5 deficiency with symptomatic cases of WNV infection diagnosed by serologic criteria, and did not distinguish whether the mechanisms involved effects on initial infection or disease progression or both. This question was addressed in a recent study that took advantage of the American Red Cross comprehensive WNV screening program [7]. Since this program began in 2003, almost 35 million blood donations were screened in the US through July 2008. Of these, samples were initially judged reactive or non-reactive by a screening PCR test. Repeated testing of all reactive samples by both WNV-specific PCR and IgM serology was carried out to define WNV true positives and WNV false positives, and the latter was used as a control group for genotype comparison. The frequencies of homozygous CCR5Δ32 were similar in the two groups, consistent with CCR5 deficiency not being a risk factor for initial infection by WNV. However, when the genotype data were stratified by whether the subjects reported symptoms consistent with WNV infection within two weeks of donating blood, a large difference was observed, with all homozygotes skewed into the symptomatic WNV true positive cases, while being homogeneously distributed between the symptomatic and asymptomatic WNV false positive individuals in the control group. Symptoms were self-reported by all participants in the study through a questionnaire, and as expected fewer WNV false positive subjects than true positive subjects reported symptoms. Limitations of this study include potential bias in the makeup of the control group, the subjective nature of the symptom scores and the lack of independent replication of the results. With these caveats the data are consistent with the notion that CCR5 deficiency is a risk factor for development of early clinical manifestations after WNV infection and for severe symptomatic disease, but not for initial infection.
The precise mechanism of action of CCR5 in human WNV infection has not yet been delineated. Its role in leukocyte trafficking to brain in the mouse model is an important clue, but additional studies directly measuring the distribution of CCR5 ligands and CCR5+ leukocyte subsets in autopsy samples of brain and other tissues from patients with WNV infection are needed. It is also important to note that although the association is very strong, most symptomatic WNV cases (~95%) were not associated with homozygous CCR5Δ32, suggesting that other genetic susceptibility factors may also affect outcome. There are several obvious candidates that merit analysis, including functional polymorphisms in the genes for the chemokine ligand CCL5, and variation in the copy number of CCL3 genes that are inherited. Both of these genetic factors have already been shown to affect the rate of HIV/AIDS disease progression.
Blocking CCR5 with drugs, which is now possible due to FDA approval in 2008 of Maraviroc for HIV/AIDS, could increase the risk of symptomatic WNV disease, should the treated patient become exposed to the virus. The net effect on risk may depend on many factors, including the level of activity of the epidemic, the exposure of treated individuals to mosquito bites, the physical distribution of the blocking agent, and the level of receptor blockade required for clinical benefit. Intermediate levels of CCR5 may be adequate to confer WNV resistance, since heterozygous CCR5Δ32 was not associated with symptomatic WNV infection [5]. However, strategies to treat or prevent HIV infection by targeting CCR5 generally aim to completely block all receptors. CCR5 antagonists developed as topical microbicides, such as the chemokine analogue PSC-RANTES, would be unlikely to place patients at increased risk of WNV infection.
Since drug resistance is an important problem for HIV, developing new classes of anti-retroviral agents that act at novel targets such as CCR5 should continue to be high priority while the potential effect of CCR5 blocking agents on WNV risk is assessed prospectively. Moreover, the findings should serve as a catalyst to reinforce advice from physicians and foster compliance among patients with regard to sound general infectious disease prevention practices, including avoidance of mosquito bites by wearing protective barriers and using chemical repellants.
CXCR3
The Th1-specific chemokine Cxcl10 and its receptor Cxcr3 are both strongly upregulated by WNV in the mouse model. Loss-of-function tests by immunodepletion and genetic inactivation have revealed an important role in the model for Cxcl10, and this has been supported using Cxcr3 knockout mice [9,10]. Loss of either resulted in decreased CD4+ and CD8+ T cell accumulation in the CNS, increased viral burden and death. Perforin-mediated CD8+ T cell responses are critical for effective clearance of WNV from infected neurons. Consistent with this, enhancing migration of CD8+ T cells into the parenchyma of the WNV-infected CNS has been reported to increase viral clearance and survival.
CCR2
Monocytes play an important role during CNS injury and infection as precursors of macrophages and microglial cells [11]. Lethal intranasal delivery of a non-neurotropic strain of WNV has been reported to induce accumulation of monocytes in the CNS, >90% of which are Ly6chi and capable of differentiating into both microglia and macrophages. Administration of neutralizing anti-Ccl2 antibody has been reported to delay migration of these cells into the brain and prolong survival, suggesting a Ccr2-dependent, pathogenic role for monocytes in the brain in the model. In contrast, monocyte depletion in vivo using clodronate-loaded liposomes has been reported to increase mortality of mice challenged i.p. with a neurotropic strain of WNV, supporting a protective role for these cells [12]. Thus, monocytes may be beneficial or harmful depending on the context and details of the model.
Two major monocyte subsets have been identified in both humans and mice: ‘resident’ monocytes, which traffic into peripheral tissues under non-inflammatory conditions, and ‘inflammatory’ monocytes, which rapidly respond to tissue injury and inflammation [13]. High expression of Ccr2 is associated with the ‘inflammatory’ (Ly6chi) monocyte subset and correlates with their function of rapid recruitment into inflamed tissues. Conversely, ‘resident’ (Ly6clo) monocytes express high levels of Cx3cr1, not Ccr2. Ccr2 is expressed primarily on monocytes and monocyte-derived cells, but is also expressed on a subset of T cells. Ccr2 is activated by the selective ligand Ccl2, and the nonselective ligands Ccl7 and Ccl12. Ccr2-deficient mice are monocytopenic and tissue recruitment of monocytes is impaired in these mice in multiple mouse models of bacterial and fungal infection [14–20].
mRNA for all Ccr2 ligands is induced by WNV infection of the CNS [4,21]. Compared to control mice infected with WNV, Ccr2-deficiency has been reported to result in markedly increased mortality (~20% survival) and increased viral load [21]. Ccl2 and Ccl7 protein levels are significantly elevated in knockout mouse brain over wild type mouse brain post-infection, suggesting that Ccr2 may also control local clearance of its ligands.
After infection of Ccr2−/− mice, similar numbers of CD4+ and CD8+ T cells, and NK1.1+ cells accumulate in the brain compared to wild type control mice, whereas accumulation of inflammatory monocytes (Ly6chiCD11b+) is severely deficient. In wild type mice infection is associated with a highly selective monocytosis in the blood that peaks just before the time when monocytes begin to accumulate in the brain. This is entirely dependent on Ccr2 since Ccr2 deficient mice, which are constitutively monocytopenic, remain monocytopenic throughout the course of WNV infection. Ccr2 deficiency in contrast does not affect the dynamics of other subsets of leukocytes tested in the blood, where they typically decrease, or in the brain, where they accumulate.
Competitive repopulation analysis of differentially labeled Ccr2+/+ and Ccr2−/− Ly6chi monocytes injected into WNV-infected Ccr2−/− recipient mice after infection has suggested that the main determinant of brain monocyte accumulation in Ccr2 knockout mice is monocytopenia [21]. In particular, Ccr2 knockout monocytes are clearly able to traffic into the brain and there does not appear to be a clear advantage of monocytes from wild type mice to do so. The analysis is complicated by the rapid reduction in the ratio of knockout to wild type monocytes in the blood after transfer into knockout mice, whether they are infected with WNV or not. This suggests that Ccr2 is not only important for egress of monocytes from bone marrow, but also for retaining monocytes in the blood once they are there. These studies also suggest that monocytes lacking Ccr2 may traffic back to the bone marrow and preferentially accumulate, unable to egress. Thus the mechanism of monocytopenia in uninfected Ccr2−/− mice may involve both decreased monocyte egress from bone marrow to blood and increased monocyte return from blood to bone marrow, among other possibilities. Thus, Ccr2 appears to be involved very early after infection with WNV, increasing monocyte numbers in the periphery. Once monocytes are in circulation, it appears that another receptor is critical for their migration into the infected CNS, perhaps Ccr5. Additional work will be needed to define whether there is also a deficiency of T cell subsets and other leukocyte subsets known to express Ccr2, such as highly differentiated Th1 cells, that are compensated by subsets that do not express the receptor.
The highly selective monocytosis induced by WNV in this model is interesting and highly unusual. As with WNV, other infectious agents have been reported that fail to overcome the monocytopenia in Ccr2-deficient mice, but in these diseases a specific monocytosis has not been described as a typical feature. Additional work will be needed to define whether Ccr2 and its ligands are needed for trafficking, organization and/or activation of monocytes within the brain, once the cells have crossed the blood brain barrier. Although previous work has shown that both Ccl2 and Ccl7 are important determinants of the normal monocyte count in unstressed wild type mice, it is not yet known which Ccr2 ligands induce monocytosis in WNV-infected mice, and whether Ccr2+ monocyte deficiency actually accounts for increased mortality in the model.
At present, there are no data delineating leukocyte dynamics in patients with WNV infection. If factors limiting monocytosis result in poor outcome in patients, novel therapeutics able to induce monocytosis, such as GM-CSF or AMD3100 (a CXCR4 antagonist), could be efficacious. AMD3100 is currently approved for stem cell mobilization, and can also mobilize monocytes in both humans and mice [22,23]. In this regard, a study by Klein and colleagues showed that AMD3100 can significantly improve survival from WNV infection in mice [24].
Monocytosis could also potentially be induced in humans using agonists to CCR2, since the ‘inflammatory’ monocyte subset in humans (CD14+CD16− monocytes) that is analogous to the Ccr2+ Ly6Chi mouse monocyte subset that we studied also uniformly expresses CCR2. At present, CCR2 agonists are not available clinically and are in general limited to chemokine ligands which have very short half-lives. Unless modified, CCR2 ligands may not be ideal for such an application.
A genetic approach may also be available for translating findings on Ccr2 to human WNV disease, since several common polymorphisms have been described for CCR2, CCL2 and CCL7. Understanding the mechanisms of Ccr2 action in monocyte migration during WNV infection in mice is an important first step towards these goals.
Conclusions
In conclusion, Ccr2, Ccr5 and Cxcr3 (and its ligand Cxcl10) have all been identified as functionally important and protective in a mouse model of WNV infection, whereas Ccr1 and Cx3cr1, which are also expressed in the model, do not appear to play important roles at the level of overall mortality. Ccr2 may be most important in monocyte recruitment to CNS, acting mainly by regulating monocytosis in the blood. Cxcr3 appears to control CD4+ and CD8+ T lymphocyte accumulation in the brain. Ccr5 deficiency is associated with defects in accumulation of macrophages, CD4+ and CD8+ T cells and NK cells. The importance of Ccr5 has been translated to human disease through genetic association with CCR5Δ32 allele, and this has important implications regarding potential risk of CCR5 blocking agents in patients infected with WNV.
Figure 1.

Outcome is heterogeneous after WNV infection.
Figure 2.
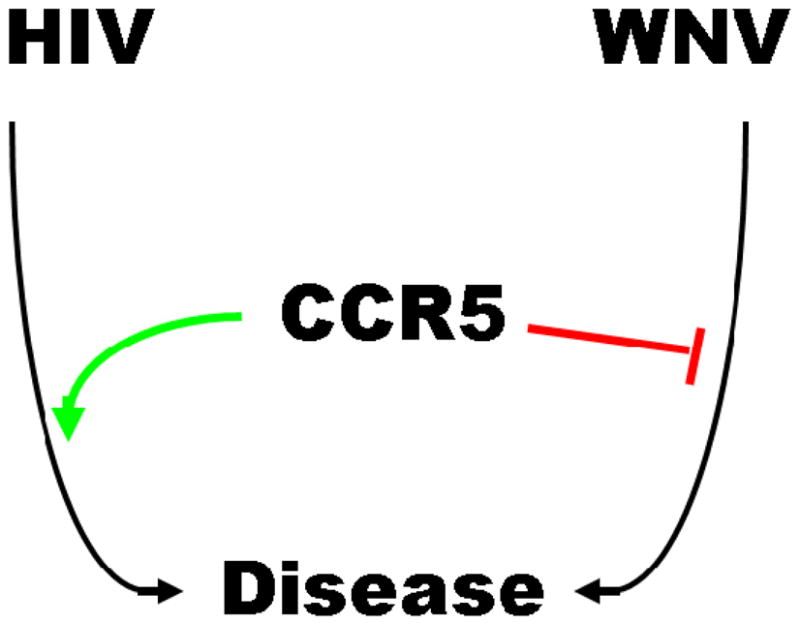
CCR5 is beneficial in host defense against WNV infection but harmful in facilitating HIV infection.
Table 1.
West Nile virus at a glance.
| Family | Flaviviridae (includes YFV, Dengue, TBE, JEV, MVE) |
| Biology | arboviral CNS pathogen |
| Host range | diverse, including many mammals and birds |
| Vector | Culex spp. mosquitoes |
| Epidemiology | First isolated in Uganda (1938); Re-emergent in USA (1999) |
| Genome | 11 kb (+) single stranded RNA |
| Structure | enveloped icosahedral nucleocapsid ~50 nm diameter sphere |
| Tissue tropism | neurotropic |
| Treatment | no specific agents |
| Prevention | no human vaccines |
Acknowledgments
This work was supported by the Division of Intramural Research of the National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hayes EB, Gubler DJ. West Nile virus: epidemiology and clinical features of an emerging epidemic in the United States. Annu Rev Med. 2006;57:181–194. doi: 10.1146/annurev.med.57.121304.131418. [DOI] [PubMed] [Google Scholar]
- 2.Kelley TW, Prayson RA, Ruiz AI, Isada CM, Gordon SM. The neuropathology of West Nile virus meningoencephalitis. A report of two cases and review of the literature. Am J Clin Pathol. 2003;119:749–753. doi: 10.1309/PU4R-76JJ-MG1F-81RP. [DOI] [PubMed] [Google Scholar]
- 3.Sampson BA, Ambrosi C, Charlot A, Reiber K, Veress JF, Armbrustmacher V. The pathology of human West Nile Virus infection. Hum Pathol. 2000;31:527–531. doi: 10.1053/hp.2000.8047. [DOI] [PubMed] [Google Scholar]
- 4.Glass WG, Lim JK, Cholera R, Pletnev AG, Gao JL, Murphy PM. Chemokine receptor CCR5 promotes leukocyte trafficking to the brain and survival in West Nile virus infection. J Exp Med. 2005;202:1087–1098. doi: 10.1084/jem.20042530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Glass WG, McDermott DH, Lim JK, Lekhong S, Yu SF, Frank WA, Pape J, Cheshier RC, Murphy PM. CCR5 deficiency increases risk of symptomatic West Nile virus infection. J Exp Med. 2006;203:35–40. doi: 10.1084/jem.20051970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lim JK, Louie CY, Glaser C, Jean C, Johnson B, Johnson H, McDermott DH, Murphy PM. Genetic deficiency of chemokine receptor CCR5 is a strong risk factor for symptomatic West Nile virus infection: a meta-analysis of 4 cohorts in the US epidemic. J Infect Dis. 2008;197:262–265. doi: 10.1086/524691. [DOI] [PubMed] [Google Scholar]
- 7.Lim JK, McDermott DH, Lisco A, Foster GA, Krysztof D, Follmann D, Stramer SL, Murphy PM. CCR5 deficiency is a risk factor for early clinical manifestations of West Nile virus infection but not for viral transmission. J Infect Dis. 2008;201:178–185. doi: 10.1086/649426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lim JK, Glass WG, McDermott DH, Murphy PM. CCR5: no longer a “good for nothing” gene--chemokine control of West Nile virus infection. Trends Immunol. 2006;27:308–312. doi: 10.1016/j.it.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 9.Klein RS, Lin E, Zhang B, Luster AD, Tollett J, Samuel MA, Engle M, Diamond MS. Neuronal CXCL10 directs CD8+ T-cell recruitment and control of West Nile virus encephalitis. J Virol. 2005;79:11457–11466. doi: 10.1128/JVI.79.17.11457-11466.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang B, Chan YK, Lu B, Diamond MS, Klein RS. CXCR3 mediates region-specific antiviral T cell trafficking within the central nervous system during West Nile virus encephalitis. J Immunol. 2008;180:2641–2649. doi: 10.4049/jimmunol.180.4.2641. [DOI] [PubMed] [Google Scholar]
- 11.Getts DR, Terry RL, Getts MT, Muller M, Rana S, Shrestha B, Radford J, Van Rooijen N, Campbell IL, King NJ. Ly6c+ “inflammatory monocytes” are microglial precursors recruited in a pathogenic manner in West Nile virus encephalitis. J Exp Med. 2008;205:2319–2337. doi: 10.1084/jem.20080421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ben-Nathan D, Huitinga I, Lustig S, van Rooijen N, Kobiler D. West Nile virus neuroinvasion and encephalitis induced by macrophage depletion in mice. Arch Virol. 1996;141:459–469. doi: 10.1007/BF01718310. [DOI] [PubMed] [Google Scholar]
- 13.Tacke F, Randolph GJ. Migratory fate and differentiation of blood monocyte subsets. Immunobiology. 2006;211:609–618. doi: 10.1016/j.imbio.2006.05.025. [DOI] [PubMed] [Google Scholar]
- 14.Jia T, Serbina NV, Brandl K, Zhong MX, Leiner IM, Charo IF, Pamer EG. Additive roles for MCP-1 and MCP-3 in CCR2-mediated recruitment of inflammatory monocytes during Listeria monocytogenes infection. J Immunol. 2008;180:6846–6853. doi: 10.4049/jimmunol.180.10.6846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol. 2006;7:311–317. doi: 10.1038/ni1309. [DOI] [PubMed] [Google Scholar]
- 16.Huffnagle GB, Traynor TR, McDonald RA, Olszewski MA, Lindell DM, Herring AC, Toews GB. Leukocyte recruitment during pulmonary Cryptococcus neoformans infection. Immunopharmacology. 2000;48:231–236. doi: 10.1016/s0162-3109(00)00222-8. [DOI] [PubMed] [Google Scholar]
- 17.Traynor TR, Kuziel WA, Toews GB, Huffnagle GB. CCR2 expression determines T1 versus T2 polarization during pulmonary Cryptococcus neoformans infection. J Immunol. 2000;164:2021–2027. doi: 10.4049/jimmunol.164.4.2021. [DOI] [PubMed] [Google Scholar]
- 18.Peters W, Scott HM, Chambers HF, Flynn JL, Charo IF, Ernst JD. Chemokine receptor 2 serves an early and essential role in resistance to Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2001;98:7958–7963. doi: 10.1073/pnas.131207398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scott HM, Flynn JL. Mycobacterium tuberculosis in chemokine receptor 2-deficient mice: influence of dose on disease progression. Infect Immun. 2002;70:5946–5954. doi: 10.1128/IAI.70.11.5946-5954.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsou CL, Peters W, Si Y, Slaymaker S, Aslanian AM, Weisberg SP, Mack M, Charo IF. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest. 2007;117:902–909. doi: 10.1172/JCI29919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lim JK, Rivollier A, Lionakis M, McDermott DH, Farber JM, Kelsall BK, Murphy PM. J Immunol. doi: 10.1161/CIRCRESAHA.111.242578. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hubel K, Liles WC, Broxmeyer HE, Rodger E, Wood B, Cooper S, Hangoc G, Macfarland R, Bridger GJ, Henson GW, Calandra G, Dale DC. Leukocytosis and Mobilization of CD34+ Hematopoietic Progenitor Cells by AMD3100, a CXCR4 Antagonist. Support Cancer Ther. 2004;1:165–172. doi: 10.3816/SCT.2004.n.008. [DOI] [PubMed] [Google Scholar]
- 23.Capoccia BJ, Shepherd RM, Link DC. G-CSF and AMD3100 mobilize monocytes into the blood that stimulate angiogenesis in vivo through a paracrine mechanism. Blood. 2006;108:2438–2445. doi: 10.1182/blood-2006-04-013755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McCandless EE, Zhang B, Diamond MS, Klein RS. CXCR4 antagonism increases T cell trafficking in the central nervous system and improves survival from West Nile virus encephalitis. Proc Natl Acad Sci USA. 2008;105:11270–11275. doi: 10.1073/pnas.0800898105. [DOI] [PMC free article] [PubMed] [Google Scholar]