

Abstract
Women with inherited mutations in the BRCA1 gene have increased risk of developing breast cancer, but also exhibit a predisposition for the development of aggressive basal-like breast tumors. We report here that breast epithelial cells derived from patients harboring deleterious mutations in BRCA1 (BRCA1mut/+) give rise to tumors with increased basal differentiation relative to cells from BRCA1+/+ patients. Molecular analysis of disease-free breast tissues from BRCA1mut/+ patients revealed defects in progenitor cell lineage commitment even before cancer incidence. Moreover, we discovered that the transcriptional repressor Slug is an important functional regulator of human breast progenitor cell lineage commitment and differentiation and that it is aberrantly expressed in BRCA1mut/+ tissues. Slug expression is necessary for increased basal-like phenotypes prior to and following neoplastic transformation. These findings demonstrate that the genetic background of patient populations, in addition to affecting incidence rates, significantly impacts progenitor cell fate commitment and, therefore, tumor phenotype.
Keywords: BRCAl, Slug, differentiation, basal-like breast cancer
Introduction
Tumor suppressor genes, such as BRCA1, repress malignant transformation by ensuring the fidelity of DNA replication and chromosomal segregation in response to potentially deleterious events. The increased risk of breast cancer development in individuals with inherited mutations in BRCA1 has been attributed to compromised DNA damage repair activity (Welcsh and King, 2001). However, it has been unclear why mutations in BRCA1 are also preferentially associated with an increased propensity for developing a specific subtype of breast cancers, basal-like tumors, with a distinct molecular phenotype and a poor prognosis (Foulkes et al., 2004; Arnes et al., 2005). Recent evidence has indicated that BRCA1 might function to regulate mammary epithelial cell morphogenesis and differentiation (Furuta et al., 2005; Liu et al., 2008; Kubista et al., 2002). Whether these functions of BRCA1 directly relate to the increased development of basal-like breast cancer, however, is not known.
Human breast tissue contains two major specialized epithelial cell types: luminal cells with secretory functions surrounding the inner breast duct lumen and basal/myoepithelial cells with contractile functions that interface between luminal cells and the basement membrane. Corresponding to these cell states, human breast cancers are broadly classified into luminal-like or basal-like tumors based on their gene expression patterns (Peppercorn et al., 2008). Accordingly, it has been proposed that tumors with ‘luminal’ characteristics may result from the transformation of cells within the luminal lineage, while tumors exhibiting ‘basal-like’ differentiation may arise from basal cells. However, there is also a wealth of evidence indicating that breast tumors exhibiting luminal or basal-like differentiation have distinct constellations of genetic aberrations, which may also influence the tumor phenotype. For example, luminal tumors frequently express elevated levels of cyclin D1 (CCND1) and sustain mutations in phosphoinositide 3-kinase (PI3K) (Gauthier et al., 2007; Loi et al., 2009; Saal et al., 2005; Campbell et al., 2004), while dysregulated expression of ras isoforms, mutations in p53, loss of PTEN expression, and loss or silencing of BRCA1 are more commonly associated with basal-like tumors (Gluz et al., 2009; Rakha et al., 2008; Miyakis et al., 1998). Moreover, as mentioned above, inherited mutations in BRCA1 (BRCA1mut/+) strongly predispose for the formation basal-like tumors (Foulkes, 2003; Foulkes et al., 2004; Arnes et al., 2005).
In principle, the predisposition for basal-like tumors in BRCA1 mutation carriers could result either from the differentiation state of the precursor cells that become transformed or from the genetic alterations acquired during tumor formation. In this study, we examined the biology of disease-free breast tissues from patients harboring deleterious mutations in BRCA1. In doing so, we found a relationship between genetic alterations in perturbing mammary progenitor differentiation and their influence on tumor phenotype.
Results
Creation of human breast cancers in vivo exhibiting heterogeneous differentiation
To examine the connection between the role of BRCA1 in regulating breast progenitor cell differentiation and the susceptibility of BRCA1-mutation carriers to developing basal-like breast cancers, we used a recently described method for creating human breast tissues in vivo (Proia and Kuperwasser, 2006; Wu et al., 2009). This method involves three distinct temporal steps: (1) clearing of the murine mammary fat pad, (2) reconstitution of the mammary fat pad with human stromal cells and (3) introduction of lentiviral-infected organoids co-mixed with activated fibroblasts into the humanized fat pad. Because this system does not require any cell culture, the likelihood of genetic alterations or the selection of variant phenotypes during in vitro expansion is minimized.
In an attempt to generate tumors from patient-derived breast epithelial cells, we modified step (3) above by introducing oncogenes into dissociated single cell suspensions of epithelial cells before introducing them into humanized stroma (Figure 1a). We chose a set of oncogenes reflective of both the luminal and basal tumor classes, to reduce the potential for genetic bias towards either tumor subtype. We infected uncultured breast epithelial cell suspensions obtained from dissociated reduction mammoplasty tissues with lentiviruses harboring genes for a mutated form of p53 (p53R175H), wild type cyclin D1 (CCND1), a constitutively-activated form of P13K (PI3KCA), and an oncogenic form of K-ras (RasG12V). Breast tumors developed when all four genes were introduced simultaneously into the breast epithelial cells (Figure 1b,c).
Figure 1. Generation of human breast tumors in vivo.
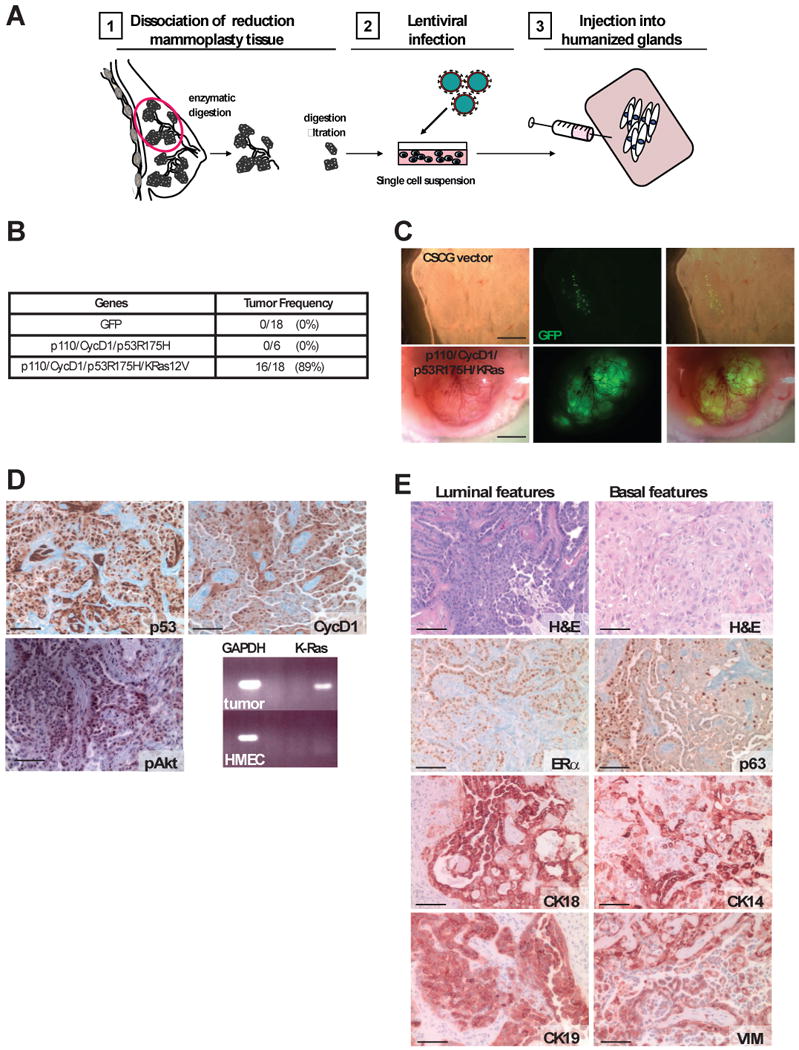
(A) Schematic depiction of the experimental strategy used to generate human breast tumors with limited ex-vivo culturing. (B,C) Tumor incidence table and GFP wholemount of unsorted breast epithelial cells infected with a GFP control virus or cells infected with the four oncogenes infected with GFP-containing viruses (constructs encoding K-ras and p53) (D) Immunoperoxidase staining of tumors for p53, cyclin D1, pAKT and express K-ras by RT-PCR (scale bar = 100 μm) (E) Tumor histopathology. Tumors generated from unsorted cells have a mixed phenotype, including areas that have characteristics of basal-type tumors including squamous appearance and immunoreactivity for cytokeratin 14 (CK14), vimentin (VIM) and p63, as well as areas of luminal phenotype that have a papillary growth pattern and reactivity for cytokeratins 8/18 (CK18), 19 (CK19) and estrogen receptor (ER) (scale bar = 100 μm). See also Figure S1.
Tumor formation with this procedure was observed with reduction mammoplasty tissues obtained from multiple patient samples. Expression of the introduced genes in the generated breast tumors was verified by immunostaining (for p53, cyclin D1, and p-Akt) and RT-PCR (for K-ras) (Figure 1d). Hematoxylin and eosin (H&E) stains of tumor sections revealed that the tumors were heterogeneous invasive carcinomas with regions of mixed squamous and papillary features, (Figure 1e, Figure S1). Immunostaining showed that cancer cells in squamous metaplastic regions expressed markers indicative of basal differentiation (cytokeratin 14 (CK14), p63, and vimentin (VIM)), and those within papillary regions expressed luminal markers (estrogen receptor (ER), CK8/18, and CK19) (Figure 1e).
We next applied this transformation protocol to mammary epithelial cells obtained from prophylactic mastectomy tissues from patients harboring deleterious mutations in BRCA1 (BRCA1mut/+) (Table S1, Figure S1). We observed that the identical set of oncogenes was sufficient to transform epithelial cells obtained from BRCA1mut/+ patients (Figure 2a). Although the introduced oncogenes were expressed to the same extent in wild-type and BRCA1 tumor tissues, immunostaining of tissue sections revealed strong expression of the basal epithelial markers CK14, p63, and vimentin in BRCA1mut/+ tumor cells (Figure 2b,c). In addition, while tumors arising from BRCA1+/+ epithelium exhibited regions that were CK8/18 and ER-positive, tumors arising from BRCA1mut/+ cells showed a statistically significant reduction in both CK8/18 and ER expression and increased CK14 expression, which is typical of basal-like tumors (Figure 2c).
Figure 2. Human breast tumors derived from BRCA1mut/+ epithelial cells exhibit enhanced features of basal differentiation.
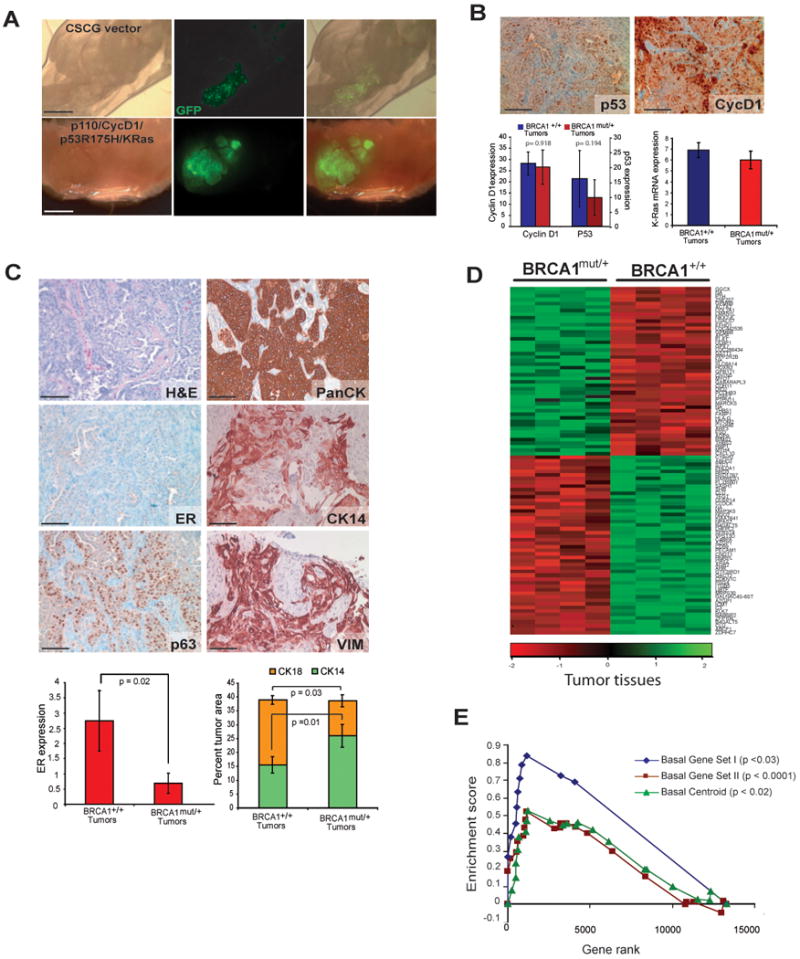
(A) Epithelial cells derived from morphologically normal prophylactic mastectomy tissues from BRCA1mut/+ carriers form tumors in mice after infection with p110/CycD1/ p53R175H/KRas lentiviruses (scale bar = 2mm). (B) Similar expression levels of p53, cyclin D1, pAKT and K-ras in BRCA1mut/+ and BRCA1+/+ tumors (scale bar = 100 μm). (C) BRCA1mut/+ tumor histopathology. Immunoperoxidase staining of tumors for breast epithelial characteristics (ER and pan cytokeratin) as well as basal-like tumor features (CK14, vimentin: VIM and p63) (scale bar = 100μm). (D) Heat map of hierarchical clustering of microarray data from tumors (n=4) arising from BRCA1+/+ epithelium and tumors (n=4) arising from BRCA1mut/+ epithelium. (scale bar = 100 μm) (E) GSEA analysis indicates the clustering is in part due to increased expression of genes associated with basal differentiation and with the basal-like breast cancer centroid. See also Tables S1, S2 and S3.
To evaluate more comprehensively whether the tumors generated from BRCA1mut/+ epithelium exhibited increased basal-like features, we performed global gene expression analyses (Table S2). Hierarchical clustering indicated that tumors arising from either BRCA1+/+ or BRCA1mut/+ epithelium could be segregated from one another based on global transcriptional profiles (Figure 2d). Gene Set Enrichment Analysis (GSEA) revealed that BRCA1mut/+ tumors exhibited a significant upregulation of genes associated with breast epithelial basal/myoepithelial cell differentiation compared to the tumors arising from BRCA1+/+ cells (Figure 2e: Basal Gene Set I, p<0.024; Basal Gene Set II, p<10-4, Table S3). In addition, GSEA indicated specific upregulation of genes in the human breast cancer ‘basal-like” centroid, which identifies the human basal-like tumor phenotype (Hu et al., 2006) in BRCA1mut/+ tumors (Basal Centroid, Figure 2e, p<0.033; Table S3) relative to BRCA1+/+ tumors. Collectively, these results indicated that compared to BRCA1+/+ tumors, BRCA1mut/+ tumors generated with identical transforming oncogenes exhibited increased basal-like differentiation.
Lineage differentiation defects in breast tissues from BRCA1-mutation carriers
As the BRCA1+/+ and BRCA1mut/+ tumors were generated with identical oncogenes, these results suggest that the predisposition of BRCA1mut/+ patients for developing basal-like tumors may result from cellular distinctions present prior to neoplastic transformation. We therefore purified breast epithelial cells from BRCA1+/+ and BRCA1mut/+ disease-free breast tissues and assessed the differentiation state of normal precursors in age-matched breast tissue samples. BRCA1+/+ and BRCA1mut/+ breast epithelial cells expressed similar levels of BRCA1 transcript and protein (Figure S2). However, gene-expression profiling indicated that many genes were differentially expressed between BRCA1+/+ and BRCA1mut/+ epithelial cells (Figure 3a, Table S4, Figure S2). Examination of gene ontology functional processes indicated that a number of genes associated with DNA transcription (repressor and activator), DNA binding, establishment and/or maintenance of chromatin architecture, and chromatin assembly or disassembly were differentially expressed in BRCA1mut/+ epithelium relative to BRCA1+/+ epithelium (Figure 3b).
Figure 3. BRCA1mut/+ breast epithelial cells exhibit defects in lineage differentiation.
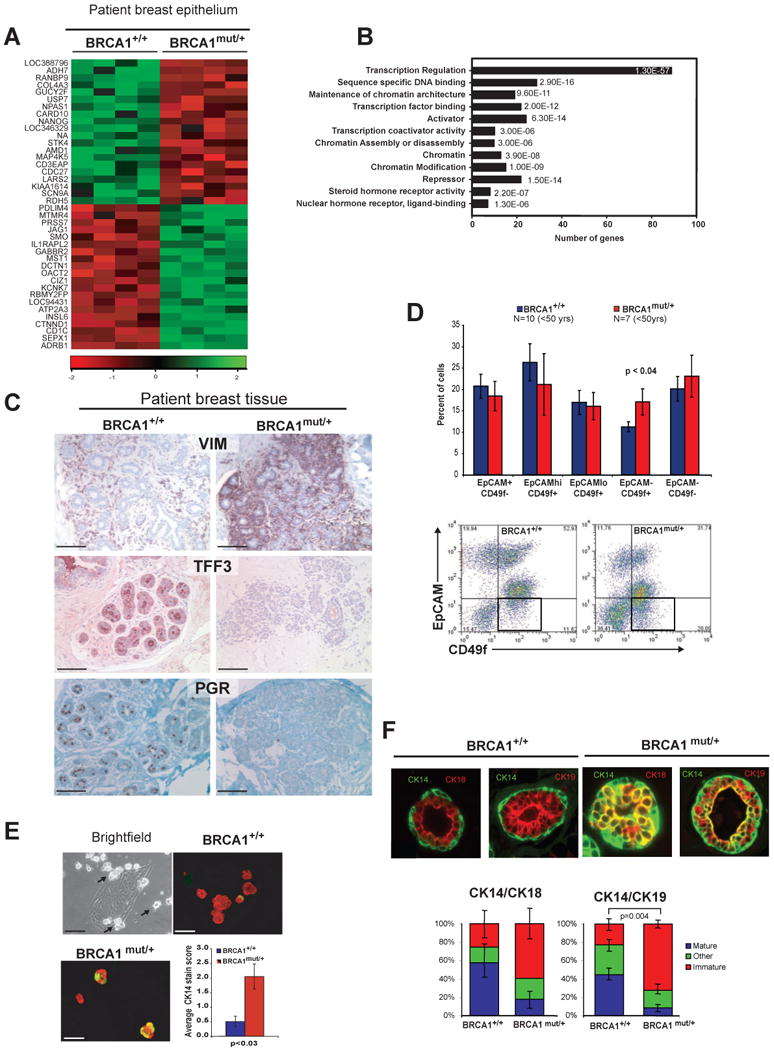
(A) Heat map of hierarchical clustering of microarray data from epithelial cells isolated from BRCA1+/+ breast patient samples (N=4) and BRCA1mut/+ patient samples (N=4). (B) Gene ontology biological process categories associated with BRCA1mut/+ breast epithelial cells. The DAVID Functional Annotation Tool was used to define categories with an enrichment score >1.5; and the number of genes represented in the list and the p value of genes differentially expressed in the microarray are shown. (C) Immunoperoxidase staining of normal human breast tissue from BRCA1+/+ and BRCA1mut/+ carriers with luminal-specific trefoil factor 3 (TFF3) and progesterone receptor (PGR) and basal-specific vimentin (VIM) antibodies (scale bar = 100 μm). Immunohistochemistry for TFF3, PGR and VIM was performed on age matched BRCA1+/+ (N=13) and BRCA1mut/+ (N=10) disease-free breast tissues. Differences in staining were observed primarily in lobules, not ducts. (D) Freshly dissociated, uncultured epithelial cells from age matched (<50 yrs) BRCA1+/+ (N=10) and BRCA1mut/+ (N=7) patients were analyzed for EpCAM and CD49f expression by flow cytometry. Representative dot plots of a BRCA1+/+ or BRCA1mut/+ patient are shown. (E) Human breast epithelial cells produce small (∼30-50 μm) luminal suspension spheres when grown under adherent conditions (indicated by arrows). Cytospun spheres were stained for CK 8/18 (red) and 14 (green)(scale bar = 100 μm). CK14 content in spheres was scored as described in methods. At least 30 spheres were scored for each patient sample. The average scores from 3 BRCA1+/+ and BRCA1mut/+ patient samples are shown in the graph. Error bars are +/- SEM and p-values were calculated by two-tailed t-test. (F) Acinar structures from patient-derived BRCA1+/+ (N=4) and BRCA1mut/+ patient (N=4) cells infected with GFP lentivirus to visualize outgrowth and grown in the HIM model. Tissue outgrowths were double stained for CK14 and CK8/18 or CK19 (representative photos, top). The staining was characterized as mature (CK14+ basal/ME layer and CK8/18 and/or 19+ luminal layer), immature (CK14+ basal/ME layer and CK14 and CK8/19 and/or 19+ luminal layer) or other (CK14 only, CK8/18/19 only etc.). The average number of the 3 categories of structures are shown in the graph (n = ≥ 85 acini). Error bars are +/- SEM and p-values were calculated by two-tailed t-test. See also Figures S2 and S3 as well as Tables S4 and S5.
Examination of genes associated with epithelial differentiation revealed that luminal genes and various hormone-related genes including progesterone and estrogen beta receptors (PGR, ESR2) (Table S4) were downregulated in BRCA1mut/ + cells, while genes associated with progenitor or basal cells were upregulated (Figure 3a, Table S4). We confirmed these differences in breast epithelial lineage differentiation using semi-quantitative immunohistochemistry (Allred scoring metric, see Methods) applied to disease-free prophylactic mastectomy tissues obtained from BRCA1mut/+ carriers and age-matched reduction mammoplasty tissues from BRCA1+/+ non-carriers. Consistent with the microarray results, progesterone receptor (PGR) expression was significantly reduced in luminal epithelial cells in 88% of BRCA1mut/+ tissues compared to only 11% of BRCA1+/+ breast tissues (Allred score >5, p<0.001) (Figure 3c, Table S5). In addition, trefoil factor 3 (TFF3), which is also associated with mature luminal differentiation, was nearly absent in 88% of BRCA1mut/+ tissues compared to only 36% of BRCA1+/+ tissues (Allred score <4, p < 0.0398; Figure 3c, Table S5). In contrast, 88% BRCA1mut/+ tissue samples exhibited moderate-to-high expression of the basal marker vimentin compared to 16% of BRCA1+/+ tissues (Figure 3c, Table S5, (p < 0.086).
We next used flow cytometry to asses the proportion of lineage-committed and progenitor epithelial cells in breast tissues. Cells expressing CD24 or high levels of EpCAM (ESA) enrich for cells of the luminal lineage, while cells expressing high levels of CD49f enrich for cells of the myoepithelial (ME)/basal lineage (Villadsen et al., 2007b; Shipitsin et al., 2007). Analysis of reduction mammoplasty breast tissues from BRCA1+/+ patients identified four populations of epithelial cells: EpCAMhi/CD49f- mature luminal cells, EpCAMhi/CD49f+ luminal progenitor cells, EpCAMlow/CD49f+ basal/myoepithelial (ME) cells, and EpCAM-/CD49f+ basal progenitor cells (Figure 3d, Figure S2, Keller et al., 2010; (Lim et al., 2009; Eirew et al., 2008).
Analysis of prophylactic mammoplasty tissues from BRCA1mut/+ (<50yrs) tissues revealed a statistically significant increase in the proportion of EpCAM-/CD49f+ basal progenitor cells (p<0.04; Figure 3d) and an appreciable but not statistically significant decrease in the number of EpCAMhi/CD49f+ luminal progenitor cells. These results indicate that BRCA1mut/+ tissues exhibit luminal and basal epithelial cell differentiation defects prior to any evidence of cancer.
Characterization of progenitor cells from BRCA1-mutation carriers
We next evaluated the breast progenitor activity of mammary epithelial cells obtained directly from breast tissues. We employed mammosphere (Dontu et al., 2003) and adherent colony-formation (Stingl et al., 2001) assays to assess progenitor activity, and evaluated whether they arose from luminal-committed, basal/ ME-committed or bipotent progenitors by staining for the differentiation markers CK14 and CK8/18. We found no significant differences in the formation of primary mammospheres suggesting that the total number of stem/progenitor cells may not differ between BRCA1+/+ and BRCA1mut/+ tissues (Figure S3). In addition, there was no significant difference in the distribution of CK8/18+ and CK14+ cells within both BRCA1+/+ and BRCA1mut/+ mammospheres (Figure S3).
Under adherent conditions, we found that human breast epithelial cells generated spherical colonies that grew in suspension as well as adherent colonies that grew on plastic (Figure 3e, Figures S3). While we did not observe a statistically significant difference in adherent progenitor colonies arising from BRCA1mut/+ cells, we did observe that spherical luminal colonies derived from BRCA1mut/+ cells expressed significantly higher levels of the basal maker CK14 in comparison to colonies from BRCA1+/+ cells which were more uniformly CK8/18-positive (Figure 3e).
We also assessed the in vivo outgrowth activity of progenitor cells from BRCA1mut/+ and BRCA1+/+ cells. Using the humanized cleared fat pad system, we found that BRCA1+/+ cells generated bilayered ductal/acinar outgrowths that contained an inner luminal layer of epithelial cells that stained predominantly for CK8/18 and 19, and an outer myoepithelial layer that stained for the basal/ME marker CK14 and SMA. In contrast, BRCA1mut/+ cells gave rise to immature ductal/acinar outgrowths that exhibited a significant increase in the numbers of bi-potent luminal cells expressing both CK19 and CK14 and to a lesser degree CK8/18 and CK14 (Figure3f, Figure S3). Taken together, these results reveal that luminal progenitor cells from BRCA1mut/+ tissue exhibit defects in full maturation and differentiation and retain features of basal differentiation.
Luminal cells give rise to tumors in BRCA1-mutation carriers
We next wanted to determine whether the increased basal differentiation observed following neoplastic transformation of BRCA1mut/+ cells was due to the increased numbers of EpCAM- basal cells or the increased basal differentiation state of luminal progenitor cells. Accordingly, we enriched for either luminal (EpCAM+) or basal/ME (CD10+) cells (Figure 4a) prior to lentiviral infection and injection into the mammary fat pad. Each subpopulation was isolated from breast tissues to >90% purity, as gauged by immunofluorescence (Figure 4b). The CD10+ subpopulation was enriched for basal/ME CK14+ cells, but CK14+ cells were depleted from the CD10-/EpCAM+ fraction (Figure 4c). Conversely, CK8/18+ luminal cells were enriched in the CD10-/EpCAM+ fraction compared to the CD10+ and parental unsorted populations (Figure 4c).
Figure 4. EpCAM+ luminal cells are able to recapitulate the tumor growth.
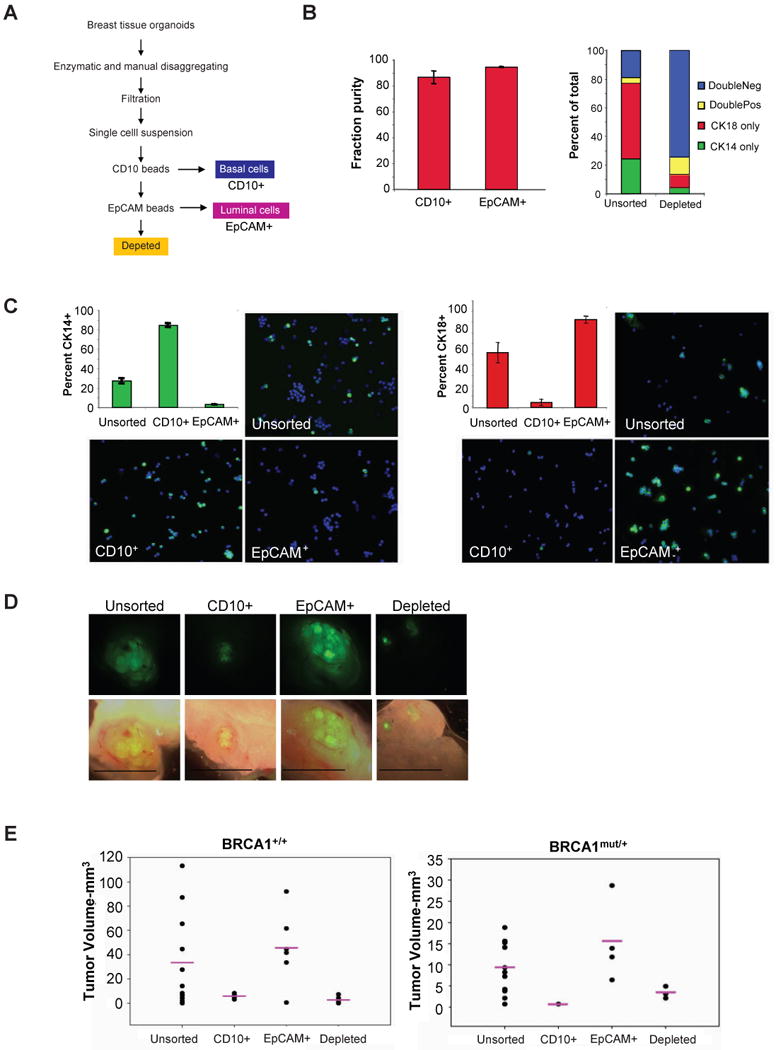
(A) Flow chart describing sorting scheme. (B) Assessment of the purity of cells following magnetic bead sorting. Quantification of double staining for the luminal marker CK8/18 and the basal marker CK14 following sorting indicates that the sorting strategy depletes cells positive for these markers. Secondary antibody labeling of immunocomplexes on bead-released sorted cells indicates purity of the fractions. (C) CK14 immunofluorescence (IF) staining and quantification of sorted fractions indicates basal cell enrichment in the CD10+ fraction and depletion in the CD10-/EpCAM+ fraction. CK8/18 IF staining of sorted fractions indicates luminal cell depletion in the CD10+ fraction and enrichment in the CD10-/EpCAM+ fraction. (D, E) Sorted epithelial cell fractions infected with identical oncogenes differ in their ability to form tumors. GFP wholemount micrographs of tumor outgrowths of sorted and infected breast epithelial cells from the four different fractions. BRCA1+/+ tumor data is compiled from three separate experiments with two different patient samples. Unsorted (n=14), CD10+ (n=4), CD10-/EpCAM+ (n=6), Depleted (n=8). BRCA1mut/+ tumor data is compiled from two experiments with one patient sample. BRCA1mut/+ Unsorted (n=8), CD10+ (n=1), CD10-/EpCAM+ (n=4), Depleted (n=4).
Basal/ME-enriched (CD10+), luminal/progenitor-enriched (CD10-/EpCAM+), and marker-depleted (CD10-/EpCAM-) cells were infected with the p53R175H, CCND1, PI3KCA, and RasG12V oncogenes and injected into humanized murine mammary glands. The luminal-enriched CD10-/EpCAM+ fraction consistently formed tumors with growth kinetics, frequencies and histopathology similar to tumors arising from unsorted cells from either BRCA1mut/+ or BRCA1+/+ derived tissues (Figure 4d,e). Thus, basal/ME (CD10+) or depleted (CD10-/EpCAM-) cells from either BRCA1mut/+ or BRCA1+/+ breast epithelial cell populations were not preferentially transformed with this combination of oncogenes. Rather, these results indicate that the target cell for transformation likely resides within the luminal EpCAM+/CD10- population. Collectively, these results imply that the increased basal phenotype of BRCA1-associated tumors results from the pre-existing increased basal differentiation state of the luminal progenitor population.
Slug suppresses breast progenitor cell lineage commitment
To investigate the molecular mechanism BRCA1 effects on progenitor cell differentiation, we classified the breast epithelial gene-expression signature described above based on signaling pathways that were differentially expressed in BRCA1mut/+ cells. Remarkably, the most significantly represented signaling pathways identified in BRCA1mut/+ breast epithelial signature were the Wnt, Notch, and melanogenesis pathways (Figure S4).
Notably, the transcriptional repressor Slug, which is an established regulator of melanocyte development, is a downstream target of both Wnt and Notch signaling (Niessen et al., 2008; DiMeo et al., 2009). This connection prompted us to examine Slug expression in breast epithelial tissues and cells harboring mutations in BRCA1. We did not find differences in SLUG mRNA expression, consistent with the microarray data, but we did observe abundant Slug protein in 87% of disease-free BRCA1mut/+ prophylactic mastectomy tissues, while its expression was reduced in tissues from reduction mammoplasty BRCA1+/+ tissues (Allred score >1, p <0.01) (Figure 5a).
Figure 5. Slug regulates breast epithelial differentiation and lineage commitment.
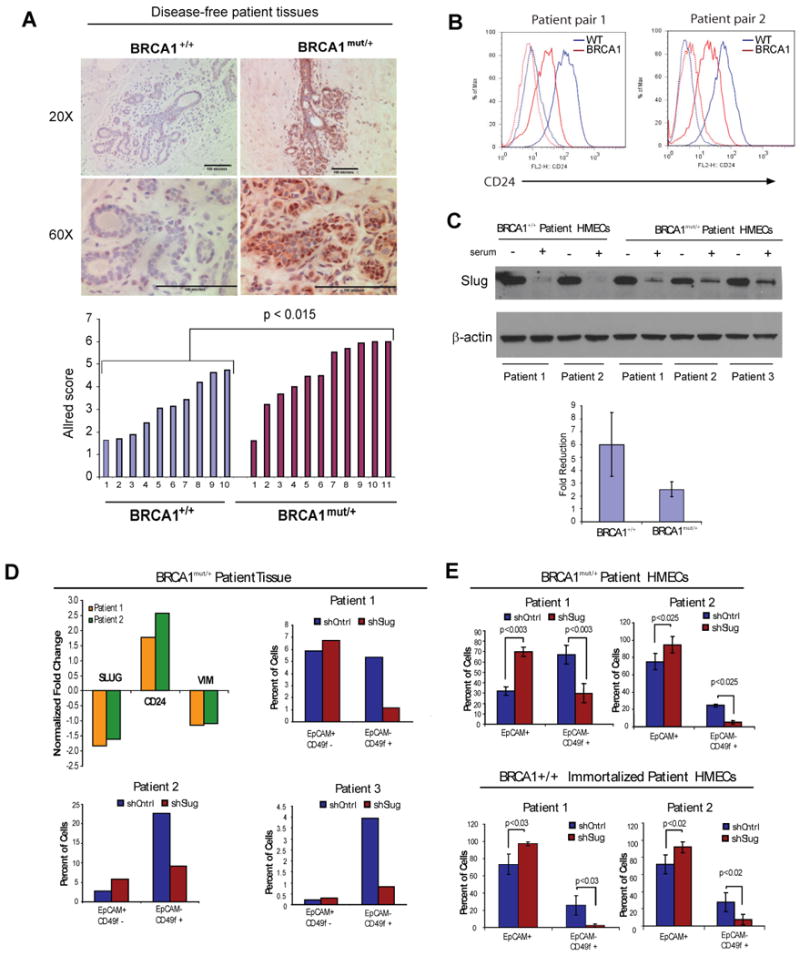
(A) IHC staining of PM and RM tissues for Slug protein; staining was quantified by Allred scoring (see Supplemental Methods); two-tailed t-test used to derive p-value. (B) Flow cytometry analysis of CD24 expression in immortalized BRCA1+/+ and BRCA1mut/+ epithelial cells derived from 4 different patient tissues following serum-induced differentiation (C) Slug protein expression in immortalized BRCA1+/+ and BRCA1mut/+ epithelial cells derived from patient breast tissues following serum-induced differentiation. Quantification of fold reduction in Slug protein expression upon serum treatment from 3 different experiments (p=0.24). (D) Flow cytometry analysis of EpCAM and CD49f expression in patient-derived breast epithelial cells from breast tissues of 3 different BRCA1mut/+ patients following Slug knockdown. (E) Flow cytometry analysis of EpCAM and CD49f expression in patient-derived breast epithelial cells from immortalized BRCA1mut/+ epithelial cells derived from BRCA1mut/+ tissues following Slug knockdown. See also Figures S4 and S5.
As Slug is a transcriptional repressor, we next investigated whether Slug expression might be affecting breast progenitor lineage commitment and differentiation. As serum addition can promote cellular differentiation of immortalized human mammary epithelial cells (HMECs), which are a model for bi-potent breast progenitor cells (Keller et al,. 2010; Zhao et al., 2010), we treated HMECs from patient-derived BRCA1+/+ and BRCA1mut/+ tissues with serum and assessed epithelial differentiation. Treatment of BRCA1+/+ HMECs with serum resulted in luminal differentiation, as measured an increase EpCAM+/CD24+ luminal cells as well as increased CD24 expression and increased CK8/18 expression (Figure 5b, data not shown). However, addition of serum to BRCA1mut/+ HMEC cells failed to induce complete luminal differentiation, consistent with defects in luminal lineage commitment (Figure 5b, c). Luminal differentiation was accompanied by a reduction in Slug protein level in both BRCA1mut/+ and BRCA1+/+ cells, although the overall reduction was somewhat reduced in BRCA1mut/+ cells (Figure 5c).
To investigate whether Slug directly inhibits breast epithelial lineage commitment and differentiation, we used lentiviral-mediated short hairpin-inhibition of Slug expression in primary prophylactic mastectomy cells isolated from three different patients with deleterious BRCA1 mutations. Slug knockdown led to a reduction in the proportion of EpCAM-/CD49f+ progenitor cells and a concomitant increase in the proportion of EpCAM+, CD44lo, and CD24+ luminal cells (Figure 5d, Figure S5). Furthermore, expression of the basal marker vimentin was greatly reduced, while expression of the luminal marker CD24 was increased (Figure 5c, Figure S5). We also examined the effects of Slug inhibition on lineage commitment of immortalized HMECs derived from BRCA1mut/+ patients. As with primary cells, inhibition of Slug expression resulted in a decrease in EpCAM-/CD49f+ basal progenitor cells and an increase in EpCAM+ luminal cells (Figure 5e). Given these findings, we also examined whether inhibition of Slug might also be important for luminal differentiation in BRCA1+/+ cells. Indeed, inhibition of Slug in BRCA1+/+ cells also led to a reduction in the proportion of EpCAM-/CD49f+ basal progenitor cells and an increase in luminal cells. Taken together, these findings indicate that Slug is a regulator of human breast progenitor cell differentiation and its expression blocks luminal differentiation.
BRCA1 regulation of Slug protein stability
To examine whether BRCA1 regulates Slug expression, we used short interfering RNAs (siRNA) to inhibit BRCA1 expression in human breast MCF10A cells, which express wild-type BRCA1 (Elstrodt et al., 2006). Quantitative RT-PCR and western blotting confirmed knockdown of BRCA1 expression (Figure 6a). Knockdown of BRCA1 by siRNA led to a modest but highly reproducible 2-fold increase in Slug protein expression, in the absence of increased mRNA expression (Figure 6a). These results suggest that loss of BRCA1 may lead to increased Slug protein expression by a post-translational mechanism. We therefore examined the stability of Slug protein in cells following siRNA-inhibition of BRCA1 as well as in cells with mutations in BRCA1. We confirmed that Slug protein is highly unstable in the BRCA1+/+ MCF10A cells (Figure 6b,c). BRCA1mut cells (SUM149, SUM1315) and siBRCA1-MCF10A cells were collected at regular time intervals subsequent to cyclohexamide (CHX) treatment and subjected to western blot analysis. While Slug protein levels were turned over in siControl-MCF10A cells, Slug protein was still detected up to 6 hours following CHX treatment in siBRCA1-MCF10A cells and in cancer lines harboring mutations in BRCA1 (Figure 6c). Importantly, the difference in stability noted in Slug protein in BRCA1mut SUM149 and SUM1315 cells was not due to a defect in proteasome activity as cyclin D1 protein was still degraded. Taken together, these results indicate that BRCA1 regulates Slug protein stability.
Figure 6. BRCA1-mutation promotes increased Slug protein stability.
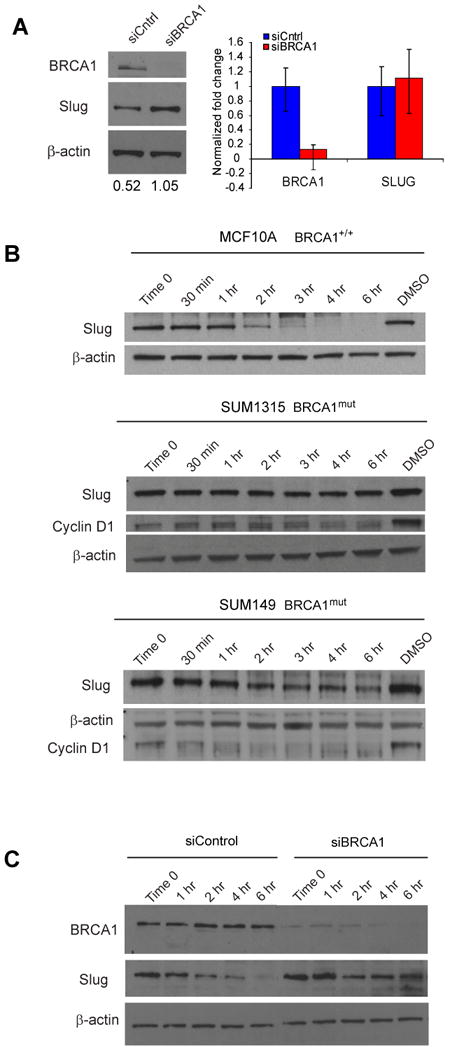
(A) Loss of BRCA1 leads to increased Slug protein but not mRNA expression in MCF10A cells. QRT-PCR and Western blot analysis of BRCA1 and Slug expression in BRCA1+/+ MCF10A cells transfected with siBRCA1 or siControl. QRT-PCR data was normalized to GAPDH and to siControl. (B) BRCA1+/+ (MCF10A) and BRCA1mut (SUM1315, SUM149) cells were treated with cycloheximide (CHX) to prevent further protein synthesis at indicated time intervals. Western blot analysis demonstrates that Slug protein is highly unstable in MCF10A cells while it had a significantly longer half-life in SUM149 and SUM1315 cells. Cyclin D1 and Actin were used as controls. (C) BRCA1+/+ (MCF10A) were transfected with siBRCA1 or siControl and treated with cycloheximide (CHX) to prevent further protein synthesis at indicated time intervals. Western blot analysis demonstrates that Slug protein is turned over in siControl MCF10A cells while it remained expressed in siBRCA1 MCF10A cells. See also Figure S6.
To begin to understand the mechanism involved, we looked at whether the ubiquitin ligase function of BRCA1 might be important for regulating Slug protein stability. BRCA1 associates with the BRCA1-associated RING domain-1 protein (BARD1) to form a heterodimer with ubiquitin E3 ligase activity. Therefore, we examined whether BARD1 knock down might also result in increased Slug protein stability. We used siRNAs to inhibit BARD1 expression in human breast MCF10A cells and collected cell lysates at regular time intervals after cyclohexamide (CHX) treatment. Although BARD1 protein was inhibited to nearly undetectable levels, Slug protein stability was similar to that of control cells, indicating that the ubiquitin-ligase functions of BARD1 was likely not regulating Slug protein stability (Figure S6). We next examined direct interactions between BRCA1 and Slug proteins. However, co-immunoprecipciaton of Slug with BRCA1 failed to demonstrate an interaction (Figure S6), although we did observe interaction between BRCA1 and BARD1. Further studies will be needed to determine what which BRCA1functions are involved in regulating Slug protein stability.
Slug regulation of basal-like breast cancer phenotype
To study the role of Slug in basal-like breast cancer phenotype, we examined Slug expression in sporadic and BRCA1-associated breast tumor tissues. Slug protein was preferentially expressed in ER-negative tumors derived from BRCA1-mutation carriers as well as ER-negative sporadic breast cancers, but its levels were higher in BRCA1-associated breast cancers (p<0.007) (Figure 7a). Furthermore, Slug protein was expressed in cell lines derived from basal-like breast cancers and elevated in cancer cell lines that harbored mutations in BRCA1 (Figure 7b).
Figure 7. Slug regulation of breast cancer phenotype in BRCA1-mutaion carriers.
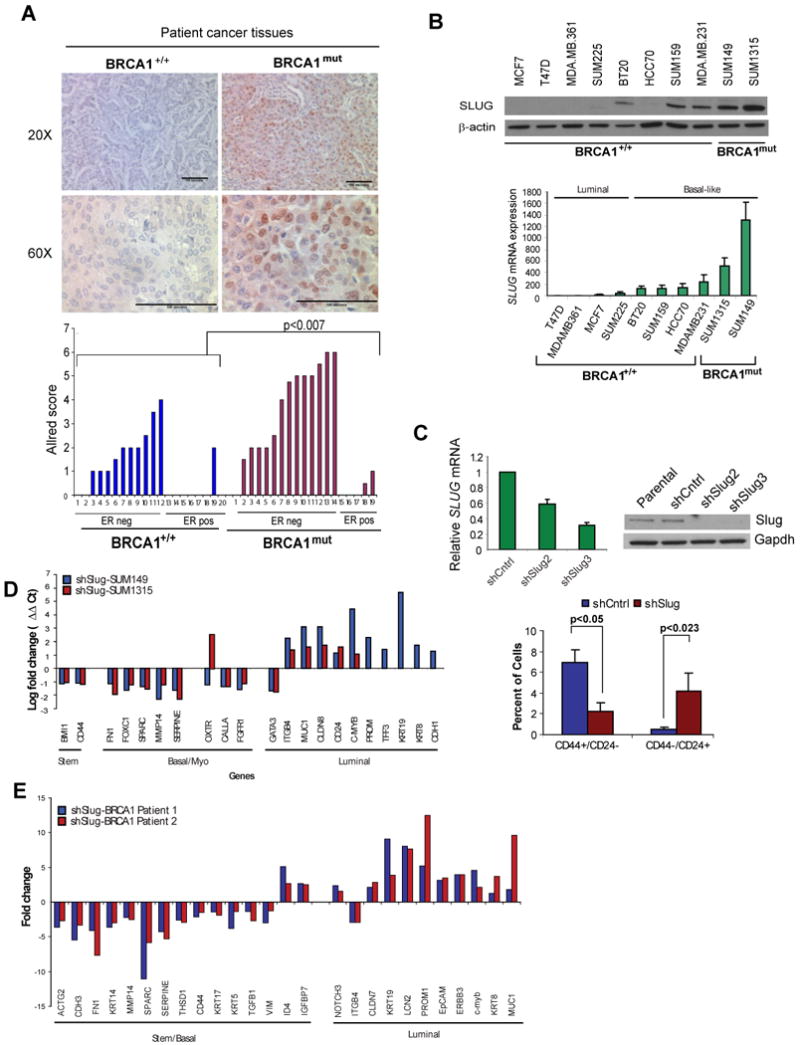
(A) Immunohistochemistry of Slug protein in breast carcinomas with known BRCA1-mutation status; two-tailed t-test used to derive p-values of Allred scores. (B) Western blot analysis of Slug and BRCA1 expression in breast cancer cell lines. SLUG mRNA levels were normalized to GAPDH and to BRCA1 levels in the cell lines. (C) SUM149 BRCA1mut breast cancer cells infected with lentiviruses targeting Slug (shSlug2 and shSlug3) or a scrambled sequence (shCntrl). Flow cytometry analysis of CD44 and CD24 expression in patient-derived SUM149 cell following Slug knockdown. (D) Quantitative RT-PCR array against a panel of 86 genes expressed in breast luminal, basal and stem cells was performed on SUM1315-shSlug and SUM149-shSlug and their respective scrambled controls. Genes differentially expressed in both cell lines in the shSlug cells compared to the scrambled controls are plotted. PROM, TFF3, KRT19, KRT8, and CDH1 genes are not expressed in SUM1315 cells. (E) BRCA1mut/+ epithelial cells infected with oncogenes in the presence of Slug knockdown leads to cells with features of luminal-like breast cancers. BRCA1mut/+ patient-derived breast epithelial cells were infected with lentiviruses encoding mutant p53, cyclin D1, and K-ras with shSlug or a scrambled sequence (shCntrl) and a quantitative RT-PCR array was performed. Genes differentially expressed >2-fold in both patient samples compared to the scrambled controls are plotted. See also Figure S7 and Table S6.
To test whether Slug is necessary for regulating the basal-like tumor phenotype, we used lentiviral-mediated short hairpin-inhibition of Slug in breast cancer cells derived from primary patient BRCA1mut breast cancers. We found that shSlug reduced endogenous Slug mRNA levels between 40-80% in BRCA1mut SUM149 and SUM1315 cancer cell lines and reduced protein to nearly undetectable levels (Figure 7c). Slug inhibition resulted in a ∼6-fold reduction in the proportion of CD44+/CD24- stem-like basal cells in SUM149, and a ∼4-fold increase in the proportion of CD44-/CD24+ luminal cells, consistent with increased differentiation and luminal lineage commitment (Figure 7c). Similarly, reducing Slug expression in BRCA1mut SUM1315 cells increased the proportion of CD24+ luminal cells by nearly 3-fold (Figure S7). We also performed quantitative mRNA expression profiling using a custom qRT-PCR array targeting 86 genes associated with basal/ME, luminal or stem cell differentiation (Table S6). Consistent with the changes observed by flow cytometry, inhibition of Slug expression resulted in upregulation of luminal genes in tumor cells including CK19, CK8, E-cadherin, MUC1, CD24, and TFF3, and repression of basal, mesenchymal and stem cell genes in both SUM149 and SUM1315 lines (Figure 7d).
To further demonstrate the role of Slug in the development of basal-like breast cancers in BRCA1mut/+ cells, mammary epithelial cells from disease-free prophylactic mastectomy tissues from three different BRCA1mut/+ patients were transduced with lentiviruses harboring p53R175H, CCND1, PI3KCA, and Ras oncogenes with or without targeting Slug expression. Patient-derived BRCA1mut/+ cells expressing shSlug showed increased expression of genes associated with luminal tumors including CK19, CK8, MUC1, EpCAM, and TFF3 with concomitant repression of many genes associated with basal-like breast cancers including SPARC, SERPINE, CD44, CK14, CK5, CK17, and vimentin compared to control patient-derived BRCA1mut/+ cells (Figure 7e).
Finally, to demonstrate the connection between Slug expression and BRCA1-mutation before transformation, we examined whether the genes that were induced following Slug-inhibition were differentially expressed based on BRCA1 status in disease-free tissues (Slug Gene Set, Table S3). We used GSEA to evaluate the expression of these Slug transcriptional targets in BRCA1mut/+ epithelium from four different patient samples. Six out of eight Slug target genes were repressed in BRCA1mut/+ cells relative to BRCA1+/+ cells (Figure S7), yielding significant enrichment by GSEA (p<0.0207). Collectively, these results show that upregulation of Slug blocks luminal lineage commitment and increases the propensity for basal breast tumor formation.
Discussion
A fundamental difference between breast cancers arising in BRCA1-mutation carriers compared to sporadic cancers is their tendency toward a basal subtype. By using an in vivo model that minimized cell culture, we were able to create human breast cancers that recapitulated many features of clinically relevant tumors to validate the previously untested idea that the predisposition for basal-like tumors in BRCA1-mutation carriers arises from perturbations in breast epithelial differentiation caused by compromised BRCA1 function (Foulkes, 2003). Our results show that breast epithelial cells isolated from BRCA1-mutation carriers preferentially form tumors with increased basal differentiation compared to cells isolated from non-carrier tissues. In addition, we found that that EpCAM+/CD10- luminal cells from both BRCA1+/+ and BRCA1mut/+ tissues enriched for tumor forming ability in this model system, but that the latter exhibited increased features of basal differentiation prior to transformation. However, since basal progenitor cells were also expanded in disease-free breast tissues from BRCA1mut/+ tissues, it is possible that these cells might also serve as targets of neoplastic transformation in patients. Nevertheless, our findings are consistent with the notion that tumor phenotype can be significantly impacted by the pre-existing differentiation state of the normal precursor (“cell of origin”) targeted for neoplastic transformation (Gupta et al., 2005; Ince et al., 2007). However, since mutations in a single allele of BRCA1 can alter the differentiation potential of the same cellular targets of transformation, leading to tumors with different phenotypes, this indicates that the initiating genetic mutation (“mutation of origin”) is a critical factor in defining tumor subtype. Future studies will be needed to determine whether other combinations of cooperating oncogenes give rise to BRCA1-associated basal-like tumors in basal/ME cells and whether mutations in other tumor suppressor genes or oncogenes also affect the differentiation potential of progenitor cells that drive tumor phenotypes.
While we have not excluded the possibility that LOH of the wild-type BRCA1 allele is necessary for basal-like tumor formation, tumors in this model system were driven by ectopic oncogenes suggesting that LOH was not necessarily a rate-limiting step. Furthermore, LOH is a stochastic event in BRCA1mut/+ patients, affecting the mutant or wild-type alleles at similar frequencies (Clarke et al., 2006). Since the analysis of prophylactic mastectomy tissues showed differentiation defects in significant proportions of the breast tissue, this suggests that LOH was not likely responsible for the perturbations in breast epithelial differentiation or basal tumor phenotype. Our findings, combined with those of others (Burga et al., 2009; Lim et al., 2009) indicate that haploinsufficiency of BRCA1 affects breast epithelial differentiation and progenitor cells in patients.
The present study also provides several additional lines of evidence that breast epithelial differentiation is altered in the presence of BRCA1 mutations. First, genes involved in epigenetic functions including DNA transcription and chromatin modification are overrepresented in the transcriptional signature of BRCA1-mutant cells. Interestingly, many of the upregulated genes are involved in the establishment and/or maintenance of chromatin structure, including demethylases, methyltransferases, histones, acetyltransferases and several components of the ubiquitin pathway. These observations are consistent with the idea that BRCA1 mutations affect large-scale chromatin unfolding (Ye et al., 2001), underscoring its role as an integral component of multi-protein complexes that modulate gene expression (Narod and Foulkes, 2004).
Second, the distinct transcriptional profile of BRCA1mut/+ cells may reflect activation of signaling pathways associated with progenitor/basal cells, increased basal differentiation and decreased luminal differentiation. Previous results suggest that reduction in BRCA1 levels leads to a failure of luminal lineage commitment and increased expansion of an uncommitted progenitor EpCAM- population (Liu et al., 2008). Consistent with this observation found that BRCA1mut/+ breast tissue exhibited an increase in the proportion of EpCAM-/CD49f+ basal progenitor cells. However, in contrast to other findings (Lim et al., 2009), we did not find an expansion of EpCAM+/CD49f+ luminal progenitor cells; although we did observe defects in luminal progenitor differentiation. This difference might reflect the genetic differences between the BRCA1 patient populations in the two studies. Nonetheless, overall these studies reinforce the idea that BRCA1 is a critical regulator of breast epithelial progenitor lineage commitment.
All of the BRCA1-mutation carrier samples used in this study harbored frameshift mutations that compromise at a minimum the C-terminal BRCT domain (Figure S2), which could destabilize protein-protein interactions between BRCA1 and its C-terminal binding partners. However, the overall levels of BRCA1 expression were surprisingly not affected. In addition, as perturbations in differentiation and Slug expression could be detected without changes in BRCA1 expression level, the effects of BRCA1 mutation may be at the level of protein-protein interactions rather than overall expression level. Consistent with this notion, reduction of BRCA1 in MCF10A cells by RNA interference impaired differentiation and could be rescued by expression of a wild-type or a RAD51 mutant of BRCA1 but not with a BRCA1 C-terminal BRCT domain mutant (Furuta et al., 2005). Future studies will be necessary to fully dissect the precise domains and mechanism by which BRCA1 regulates breast epithelial differentiation. In addition, further experiments will be needed to determine whether certain mutations in BRCA1 affect differentiation and regulate progenitor cell fate differently than others and whether different mutations alter the propensity for the development of basal-like tumors.
The observation that BRCA1mut/+ epithelial cells express genes involved in melanogenesis and stem cell biology prompted us to examine Slug and its requirement for maintaining progenitor cells and basal differentiation. We found that haploinsufficiency or knockdown of BRCA1 was associated with increased expression and stability of the transcriptional repressor Slug and that Slug was shown to be a regulator of the basal phenotype. This suggests that perturbations in luminal differentiation due in part to Slug expression, is likely responsible for the increased propensity for the development of tumors with basal-like features. Although our results do not address whether Slug is sufficient to induce basal differentiation, Slug is expressed in breast stem/progenitor cells and has can promote a basal-like phenotype in the luminal MCF-7 breast cancer cell line and increased basal differentiation marker expression in the non-tumorigenic MCF10A breast line (Sarrio et al., 2008). Moreover, the fact that Slug is expressed in basal-like breast cancers not associated with BRCA1 mutations (Storci et al., 2008) implies that acquisition of its expression enables basal differentiation.
Materials and Methods
Detailed Methods are described in Supplemental Materials
BRCA1-mutation carrier tissues
All human breast tissue procurement for these experiments was obtained in compliance with the laws and institutional guidelines, as approved by the institutional IRB committee from Beth Israel Deaconess Medical Center (BIDMC) and Tufts University School of Medicine. Disease-free prophylactic mastectomy (n=31; 12 fresh, 19 formalin-fixed paraffin embedded) and tumor tissues (n=19) derived from women carrying a known deleterious BRCA1 heterozygous mutation were obtained with patient consent from the Surgical Pathology files or immediately following prophylactic mastectomy surgery at BIDMC. Tissues in which BRCA1 mutation was confirmed but not known were submitted for sequence/genotyping at Myriad Genetic Laboratories. Non-BRCA1 tumor tissues (n=20) were obtained from discarded material at Tufts Medical Center and non-cancerous breast tissue was obtained from patients undergoing elective reduction mammoplasty at Tufts Medical Center or BIDMC (n=38; 18 fresh, 24 formalin-fixed paraffin embedded). BRCA1 mutation status and clinical information are listed in Table S1. The range of patient ages for fresh BRCA1+/+ tissue used in this study was 30-54 with a median age of 40; the range of patient ages for fresh BRCA1mut/+ tissue used in this study was 35-53 with a median age of 44. All disease-free breast tissues were verified by surgical pathologists prior to use in these studies.
Cell Lines and Tissue Culture
SUM cell lines were obtained from Dr. Stephen Ethier (Kramanos Institute, MI), while the MCF10A cell lines were purchased from ATCC. MCF10A cells were cultured in DMEM with 10% calf serum. SUM149PT cells were cultured in Ham's F12 with 5% calf serum, insulin (5 μg/mL), and hydrocortisone (1 μg/mL) while SUM1315MO2 were in Ham's F12 with 5% calf serum, insulin (5 μg/mL), and EGF (10 ng/mL). All cell lines were grown at 37°C and 5% CO2. BRCA1mut/+ HMECs were immortalized with the catalytic subunit of human telomerase as previously described (Elenbaas et al, 2000). BHME cells were cultured in MEGM (Lonza) supplemented with bovine pituitary extract (BPE), insulin (5 μg/mL), EGF (10 ng/mL) and hydrocortisone (1 μg/mL).
Lentiviral Constructs and virus production
Lentiviral constructs used for gene transduction into human mammary epithelial cells were created using standard cloning techniques into the self-inactivating CS-CG (Miyoshi et al., 1998) viral vector generously provided by Inder Verma (Salk Institute, La Jolla, CA). pLENTI-KRASG12V and pLenti-CMV-PIK3CA-myr+CMV-CCND1 were obtained from from Min Wu (Aveo Pharmaceuticals, Cambridge, MA). A wild-type human p53 cDNA clone was generously provided by Josh LaBaer (Harvard Institute of Proteomics, Harvard Medical School, Cambridge, MA). Site directed mutagenesis was employed to change amino acid residue 175 from R to H. The VSV-G-pseudotyped lentiviral vectors were generated by transient co-transfection of the vector construct with the VSV-G-expressing construct pCMV-VSV-G (Miyoshi et al., 1998) and the packaging construct pCMV ΔR8.2Δvpr (Miyoshi et al., 1998) generously provided by Inder Verma, into 293T cells with the FuGENE 6 transfection reagent (Roche). High-titer stocks of the virus were prepared by ultracentrifugation at 100,000 × g. Lentiviral shRNA constructs targeting Slug (Addgene plasmids 10904 and 10905) were prepared as previously described (Gupta et al., 2005).
Breast tissues were minced and enzymatically digested overnight with a mixture of collagenase and hyaluronidase as previously described (Kuperwasser et al., 2004; Proia and Kuperwasser, 2006) and dissociated to a single cell suspension. Immediately after dissociation, cells were resuspended with lentiviruses expressing the genes of interest and injected into cleared and humanized mammary fat pads.
Animals and Surgery
All animal procedures were conducted in accordance with a protocol approved by the Tufts University IACUC committee. A colony of immunocompromised NOD/SCID mice was maintained in house under aseptic sterile conditions. Mice were administered autoclaved food and water ad libitum. Surgeries were performed under sterile conditions, and animals received antibiotics in the drinking water up to two weeks after all surgical procedures. Animals were humanized and injected as previously described (Kuperwasser et al., 2004; Proia and Kuperwasser, 2006).
Immunohistochemistry
Immunohistochemistry was performed on formalin-fixed, paraffin-embedded tissue sections with sodium citrate antigen retrieval, followed by visualization with the ABC Elite peroxidase kit and NovaRed substrate (Vector labs) for detection of αSMA (1:500, clone ♋sm-1), CK14 (1:500, clone LL002), CK8/18 1:500, clone DC-10), Vimentin (1:500, clone V9), CK19 (1:500, clone b170) (all, Vector Labs), TFF3 (Abnova, clone 3D9, 1:200)) and Slug (1:200, Cell Signaling). Staining for pan-cytokeratin (Ventana Medical Systems), p53 (Ventana Medical Systems), cyclin D1 (NeoMarkers), pAKT (Cell Signaling, 1:100), ER (Ventana Medical Systems), p63 (Ventana Medical Systems), and PR was performed by the Histology Special Procedures Laboratory at Tufts Medical Center. IHC results were semi-quantitatively analyzed (see Supplmental Materials for details).
Mammospheres and Adherent colony forming assays
Viable cells dissociated from organoids derived from BRCA1+/+ (n=4) and BRCA1+/- (n=4) patients were plated at 35,000 cells per well in 6-well plates for adherent colony growth in MEGM media (Lonza) or at 20,000 cells per ml in 6-well ultra-low attachment plates (Corning) in MEGM media minus the addition of bovine pituitary extract for mammosphere growth. Colonies and mammospheres were allowed to form for 8 days after which non-adherent suspension colonies from adherent culture and mammospheres from non-adherent culture were collected for analysis.
Mammospheres collected from non-adherent culture and suspension colonies collected from adherent culture were cytospun onto glass slides and fixed in methanol for immunofluorescence analysis. Quantification of mammosphere and suspension colony numbers was accomplished using a Multisizer 3 COULTER COUNTER (Beckman-Coulter).
Immunomagnetic Bead sorting
Epithelial organoids from BRCA1+/+ and BRCA1+/mut patients were dissociated to a single cell suspension and sorted with CELLection pan-mouse IgG magnetic beads according to the manufacturers instructions and as described previously (Allinen et al., 2004) (Dynal, Invitrogen) conjugated to an anti-CD10 antibody (DAKO). CD10+ cells were released from the beads by DNase treatment per the manufacturer's instructions. Cells that did not bind to the CD10-immunobeads were further sorted with magnetic beads conjugated to an anti-EpCAM antibody (Abcam).
Flow cytometry and FACS
Uncultured cells from BRCA1+/+ (n=10) or BRCA1mut/+ (n=7) organoid preparations were dissociated to a single-cell suspension as described above and filtered through a 20 μm nylon mesh (Millipore). Endothelial, lymphocytic, monocytic, and fibroblastic lineages were depleted with antibodies to CD31, CD34 and CD45 (all Thermo/LabVision) and Fibroblast Specific Protein/IB10 (Sigma) and a cocktail of Pan-mouse IgG and IgM Dynabeads (Dynal, Invitrogen) according to the manufacturers instructions and as described previously (Villadsen et al., 2007a) and in Supplemental Methods.
Non-confluent cultures of SUM149, SUM1315, and immortalized HMECs cells were trypsinized into single cell suspension, counted, washed with PBS, and stained with antibodies specific for human cell CD24 (PE) and CD44 (APC) (BD Biosciences). Antibody-bound cells were washed and resuspended at 1 × 106 cells/ml in FB and run on a FACSCalibur flow cytometer (BD Biosciences) or sorted on a BD Influx FACS sorter (BD Biosciences). Flow cytometry data was analyzed with the Flowjo software package (TreeStar).
Microarray and RT-PCR analysis
RNA was extracted from the tissues and cell lines with the RNAeasy Mini Kit (Qiagen). Standard RT-PCR to confirm expression of KRAS lentiviral construct-specific transcript, and quantitative real time PCR was used for detecting Slug, BRCA1, and Vimentin transcript in cell lines. See Supplemental Methods for all primer sequences and details. Custom qRT-PCR arrays (Table S6) were obtained from SA Biosciences.
Total RNA for microarray expression studies was isolated from fibroblast-depleted single cell suspensions of uncultured BRCA1+/+ or BRCA1mut/+ cells or from tumors generated from infected BRCA1+/+ or BRCA1mut/+ cells using the RNeasy Mini kit (Qiagen). Synthesis of cDNA from total RNA and hybridization/scanning of microarrays were performed with Affymetrix GeneChip products (HGU133A) as described in the GeneChip manual. Raw data files (.CEL) were converted into probe set values by RMA normalization. See Supplemental Methods for hierarchical clustering and GSEA analysis.
Supplementary Material
Acknowledgments
We thank Gerbug Wulf, Steve Come and Susan Troyan at Beth Israel Deaconess Medical Center for assistance with BRCA1mut/+ tissue procurement and Kai Tao and Lisa Arendt for technical assistance. We thank Myriad Genetic Laboratories for gene-specific BRACAnalysis. We thank Annette Shepard-Barry at Tufts Medical Center in the Histology-Special Procedures Lab for histological and immunohistochemical staining. We thank Stephen Kwok for assistance with cell sorting and Supriya Gupta for assistance with gene expression experiments and data collection. We thank Josh LaBaer at Harvard Medical School for generously providing us with human cyclin D1, ras, p53 and PI3K cDNAs. This work was supported by grants from the American Cancer Society-New England Division-Broadway on Beachside Postdoctoral Fellowship (P.K), the Raymond and Beverly Sackler Foundation (PK. and C.K.), the Breast Cancer Research Foundation (CK, TD), the Department of Defense Breast Cancer Research Program (BC073866) (PK, TD, CK) and the NIH/NCI (CA125554, CA125554) CK, IK). C.K. is a Raymond and Beverly Sackler Foundation Scholar.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allinen M, Beroukhim R, Cai L, Brennan C, Lahti-Domenici J, Huang H, Porter D, Hu M, Chin L, Richardson A, Schnitt S, Sellers WR, Polyak K. Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell. 2004;6:17–32. doi: 10.1016/j.ccr.2004.06.010. [DOI] [PubMed] [Google Scholar]
- Arnes JB, Brunet JS, Stefansson I, Begin LR, Wong N, Chappuis PO, Akslen LA, Foulkes WD. Placental cadherin and the basal epithelial phenotype of BRCA1-related breast cancer. Clin Cancer Res. 2005;11:4003–4011. doi: 10.1158/1078-0432.CCR-04-2064. [DOI] [PubMed] [Google Scholar]
- Burga LN, Tung NM, Troyan SL, Bostina M, Konstantinopoulos PA, Fountzilas H, Spentzos D, Miron A, Yassin YA, Lee BT, Wulf GM. Altered proliferation and differentiation properties of primary mammary epithelial cells from BRCA1 mutation carriers. Cancer Res. 2009;69:1273–1278. doi: 10.1158/0008-5472.CAN-08-2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell IG, Russell SE, Choong DY, Montgomery KG, Ciavarella ML, Hooi CS, Cristiano BE, Pearson RB, Phillips WA. Mutation of the PIK3CA gene in ovarian and breast cancer. Cancer Res. 2004;64:7678–7681. doi: 10.1158/0008-5472.CAN-04-2933. [DOI] [PubMed] [Google Scholar]
- Clarke CL, Sandle J, Jones AA, Sofronis A, Patani NR, Lakhani SR. Mapping loss of heterozygosity in normal human breast cells from BRCA1/2 carriers. Br J Cancer. 2006;95:515–519. doi: 10.1038/sj.bjc.6603298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMeo TA, Anderson K, Phadke P, Feng C, Perou CM, Naber S, Kuperwasser C. A Novel Lung Metastasis Signature Links Wnt Signaling with Cancer Cell Self-Renewal and Epithelial-Mesenchymal Transition in Basal-like Breast Cancer. Cancer Res. 2009 doi: 10.1158/0008-5472.CAN-08-4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eirew P, Stingl J, Raouf A, Turashvili G, Aparicio S, Emerman JT, Eaves CJ. A method for quantifying normal human mammary epithelial stem cells with in vivo regenerative ability. Nat Med. 2008;14:1384–1389. doi: 10.1038/nm.1791. [DOI] [PubMed] [Google Scholar]
- Elstrodt F, Hollestelle A, Nagel JH, Gorin M, Wasielewski M, van den OA, Merajver SD, Ethier SP, Schutte M. BRCA1 mutation analysis of 41 human breast cancer cell lines reveals three new deleterious mutants. Cancer Res. 2006;66:41–45. doi: 10.1158/0008-5472.CAN-05-2853. [DOI] [PubMed] [Google Scholar]
- Foulkes WD. BRCA1 functions as a breast stem cell regulator. J Med Genet. 2003:1–5. doi: 10.1136/jmg.2003.013805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foulkes WD, Brunet JS, Stefansson IM, Straume O, Chappuis PO, Begin LR, Hamel N, Goffin JR, Wong N, Trudel M, Kapusta L, Porter P, Akslen LA. The prognostic implication of the basal-like (cyclin E high/p27 low/p53+/glomeruloid-microvascular-proliferation+) phenotype of BRCA1-related breast cancer. Cancer Res. 2004;64:830–835. doi: 10.1158/0008-5472.can-03-2970. [DOI] [PubMed] [Google Scholar]
- Furuta S, Jiang X, Gu B, Cheng E, Chen PL, Lee WH. Depletion of BRCA1 impairs differentiation but enhances proliferation of mammary epithelial cells. Proc Natl Acad Sci U S A. 2005;102:9176–9181. doi: 10.1073/pnas.0503793102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier ML, Berman HK, Miller C, Kozakeiwicz K, Chew K, Moore D, Rabban J, Chen YY, Kerlikowske K, Tlsty TD. Abrogated response to cellular stress identifies DCIS associated with subsequent tumor events and defines basal-like breast tumors. Cancer Cell. 2007;12:479–491. doi: 10.1016/j.ccr.2007.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gluz O, Liedtke C, Gottschalk N, Pusztai L, Nitz U, Harbeck N. Triple-negative breast cancer--current status and future directions. Ann Oncol. 2009;20:1913–1927. doi: 10.1093/annonc/mdp492. [DOI] [PubMed] [Google Scholar]
- Gupta PB, Kuperwasser C, Brunet JP, Ramaswamy S, Kuo WL, Gray JW, Naber SP, Weinberg RA. The melanocyte differentiation program predisposes to metastasis after neoplastic transformation. Nat Genet. 2005;37:1047–1054. doi: 10.1038/ng1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Fan C, Oh DS, Marron JS, He X, Qaqish BF, Livasy C, Carey LA, Reynolds E, Dressler L, Nobel A, Parker J, Ewend MG, Sawyer LR, Wu J, Liu Y, Nanda R, Tretiakova M, Ruiz OA, Dreher D, Palazzo JP, Perreard L, Nelson E, Mone M, Hansen H, Mullins M, Quackenbush JF, Ellis MJ, Olopade OI, Bernard PS, Perou CM. The molecular portraits of breast tumors are conserved across microarray platforms. BMC Genomics. 2006;7:96. doi: 10.1186/1471-2164-7-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ince TA, Richardson AL, Bell GW, Saitoh M, Godar S, Karnoub AE, Iglehart JD, Weinberg RA. Transformation of different human breast epithelial cell types leads to distinct tumor phenotypes. Cancer Cell. 2007;12:160–170. doi: 10.1016/j.ccr.2007.06.013. [DOI] [PubMed] [Google Scholar]
- Keller PJ, Lin AF, Arendt LM, Klebba I, Jones AD, Rudnick JA, Dimeo TA, Gilmore H, Jefferson DM, Graham RA, Naber SP, Schnitt S, Kuperwasser C. Mapping the cellular and molecular heterogeneity of normal and malignant breast tissues and cultured cell lines. Breast Cancer Res. 2010 Oct 21;12(5):R87. doi: 10.1186/bcr2755. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubista M, Rosner M, Kubista E, Bernaschek G, Hengstschlager M. Brca1 regulates in vitro differentiation of mammary epithelial cells. Oncogene. 2002;21:4747–4756. doi: 10.1038/sj.onc.1205580. [DOI] [PubMed] [Google Scholar]
- Kuperwasser C, Chavarria T, Wu M, Magrane G, Gray JW, Richardson A, Weinberg RA. Reconstruction of functionally normal and malignant human breast tissues in mice. Proc Natl Acad Sci U S A. 2004;101:4966–4971. doi: 10.1073/pnas.0401064101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim E, Vaillant F, Wu D, Forrest NC, Pal B, Hart AH, sselin-Labat ML, Gyorki DE, Ward T, Partanen A, Feleppa F, Huschtscha LI, Thorne HJ, Fox SB, Yan M, French JD, Brown MA, Smyth GK, Visvader JE, Lindeman GJ. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med. 2009;15:907–913. doi: 10.1038/nm.2000. [DOI] [PubMed] [Google Scholar]
- Liu S, Ginestier C, Charafe-Jauffret E, Foco H, Kleer CG, Merajver SD, Dontu G, Wicha MS. BRCA1 regulates human mammary stem/progenitor cell fate. Proc Natl Acad Sci U S A. 2008;105:1680–1685. doi: 10.1073/pnas.0711613105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loi S, Haibe-Kains B, Lallemand F, Pusztai L, Bardelli A, Gillett C, Ellis P, Piccart-Gebhart MJ, Phillips WA, McArthur A, Sotiriou C. Correlation of PIK3CA mutation-associated gene expression signature (PIK3CA-GS) with deactivation of the PI3K pathway and with prognosis within the luminal-B ER+ breast cancers. J Clin Oncol. 2009;27:533. [Google Scholar]
- Miyakis S, Sourvinos G, Spandidos DA. Differential expression and mutation of the ras family genes in human breast cancer. Biochem Biophys Res Commun. 1998;251:609–612. doi: 10.1006/bbrc.1998.9527. [DOI] [PubMed] [Google Scholar]
- Miyoshi H, Blomer U, Takahashi M, Gage FH, Verma IM. Development of a self-inactivating lentivirus vector. J Virol. 1998;72:8150–8157. doi: 10.1128/jvi.72.10.8150-8157.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narod SA, Foulkes WD. BRCA1 and BRCA2: 1994 and beyond. Nat Rev Cancer. 2004;4:665–676. doi: 10.1038/nrc1431. [DOI] [PubMed] [Google Scholar]
- Niessen K, Fu Y, Chang L, Hoodless PA, McFadden D, Karsan A. Slug is a direct Notch target required for initiation of cardiac cushion cellularization. J Cell Biol. 2008;182:315–325. doi: 10.1083/jcb.200710067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peppercorn J, Perou CM, Carey LA. Molecular subtypes in breast cancer evaluation and management: divide and conquer. Cancer Invest. 2008;26:1–10. doi: 10.1080/07357900701784238. [DOI] [PubMed] [Google Scholar]
- Proia D, Kuperwasser C. Reconstruction of human mammary tissues in a mouse model. Nature Protocols. 2006;1:206–211. doi: 10.1038/nprot.2006.31. [DOI] [PubMed] [Google Scholar]
- Rakha EA, Reis-Filho JS, Ellis IO. Basal-like breast cancer: a critical review. J Clin Oncol. 2008;26:2568–2581. doi: 10.1200/JCO.2007.13.1748. [DOI] [PubMed] [Google Scholar]
- Saal LH, Holm K, Maurer M, Memeo L, Su T, Wang X, Yu JS, Malmstrom PO, Mansukhani M, Enoksson J, Hibshoosh H, Borg A, Parsons R. PIK3CA mutations correlate with hormone receptors, node metastasis, and ERBB2, and are mutually exclusive with PTEN loss in human breast carcinoma. Cancer Res. 2005;65:2554–2559. doi: 10.1158/0008-5472-CAN-04-3913. [DOI] [PubMed] [Google Scholar]
- Sarrio D, Rodriguez-Pinilla SM, Hardisson D, Cano A, Moreno-Bueno G, Palacios J. Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res. 2008;68:989–997. doi: 10.1158/0008-5472.CAN-07-2017. [DOI] [PubMed] [Google Scholar]
- Shipitsin M, Campbell LL, Argani P, Weremowicz S, Bloushtain-Qimron N, Yao J, Nikolskaya T, Serebryiskaya T, Beroukhim R, Hu M, Halushka MK, Sukumar S, Parker LM, Anderson KS, Harris LN, Garber JE, Richardson AL, Schnitt SJ, Nikolsky Y, Gelman RS, Polyak K. Molecular definition of breast tumor heterogeneity. Cancer Cell. 2007;11:259–273. doi: 10.1016/j.ccr.2007.01.013. [DOI] [PubMed] [Google Scholar]
- Stingl J, Eaves CJ, Zandieh I, Emerman JT. Characterization of bipotent mammary epithelial progenitor cells in normal adult human breast tissue. Breast Cancer Res Treat. 2001;67:93–109. doi: 10.1023/a:1010615124301. [DOI] [PubMed] [Google Scholar]
- Storci G, Sansone P, Trere D, Tavolari S, Taffurelli M, Ceccarelli C, Guarnieri T, Paterini P, Pariali M, Montanaro L, Santini D, Chieco P, Bonafe M. The basal-like breast carcinoma phenotype is regulated by SLUG gene expression. J Pathol. 2008;214:25–37. doi: 10.1002/path.2254. [DOI] [PubMed] [Google Scholar]
- Villadsen R, Fridriksdottir AJ, Ronnov-Jessen L, Gudjonsson T, Rank F, Labarge MA, Bissell MJ, Petersen OW. Evidence for a stem cell hierarchy in the adult human breast. J Cell Biol. 2007b;177:87–101. doi: 10.1083/jcb.200611114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welcsh PL, King MC. BRCA1 and BRCA2 and the genetics of breast and ovarian cancer. Hum Mol Genet. 2001;10:705–713. doi: 10.1093/hmg/10.7.705. [DOI] [PubMed] [Google Scholar]
- Wu M, Jung L, Cooper AB, Fleet C, Chen L, Breault L, Clark K, Cai Z, Vincent S, Bottega S, Shen Q, Richardson A, Bosenburg M, Naber SP, DePinho RA, Kuperwasser C, Robinson MO. Dissecting genetic requirements of human breast tumorigenesis in a tissue transgenic model of human breast cancer in mice. Proc Natl Acad Sci U S A. 2009;106:7022–7027. doi: 10.1073/pnas.0811785106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Q, Hu YF, Zhong H, Nye AC, Belmont AS, Li R. BRCA1-induced large-scale chromatin unfolding and allele-specific effects of cancer-predisposing mutations. J Cell Biol. 2001;155:911–921. doi: 10.1083/jcb.200108049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Malhotra GK, Lele SM, Lele MS, West WW, Eudy JD, Band H, Band V. Telomerase-immortalized human mammary stem/progenitor cells with ability to self-renew and differentiate. Proc Natl Acad Sci U S A. 2010 Aug 10;107(32):14146–51. doi: 10.1073/pnas.1009030107. Epub 2010 Jul 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.