

Introduction
Nature has been recognized as a rich source of therapeutic and medicinal agents for thousands of years. The high activity of these molecules is often attributed to their unique biogenesis, in which evolutionary pressure has selected for secondary biosynthetic pathways capable of producing unique, protein-interacting compounds that confer survival advantages to an organism by facilitating chemical defense, interspecies communication, and adaptation to well defined ecological niches [1,2]. However, despite accounting for close to half of all drug scaffolds discovered in the past 15 years, fewer than 1% of bacterial, 5% of fungal, and 15% of plant species have been systematically investigated for the production of bioactive compounds [3]. This is in part due to the high rediscovery rate of known (and patented) compounds during conventional bioassay guided discovery efforts, which, together with issues of chemical complexity and developments in combinatorial chemistry, helped spur the wholesale elimination of natural product research departments at most pharmaceutical firms over the past decade [4,5].
Rather than leave the vast biosynthetic resources of nature unexplored, enterprising academic groups have filled this gap by continuing natural product isolation and developing new methodologies for the study of natural product biosynthesis [6]. Here we describe recent efforts in which proteomic methods have been applied to link polyketides and nonribosomal peptide natural products to the biosynthetic catalysts responsible for their production using protein mass spectrometry (MS). This approach in effect reverses the drug discovery process by starting from expressed biosynthetic enzymes. The goal of these efforts is to facilitate discovery of new molecules and enzymatic pathways from both sequenced and unsequenced natural product producers. As this represents a relatively nascent field, we focus on providing a brief background before delving into a detailed and in-depth description of two recently reported proteomic approaches to the study of secondary metabolism. Finally, we highlight future opportunities and challenges which must be addressed in order for the full impact of proteomic profiling of natural product biosynthesis to be realized.
Modular Biosynthetic Enzymes
Polyketide and nonribosomal peptide natural products are produced by modular biosynthetic catalysts known as polyketide synthase (PKS) and nonribosomal peptide synthetase (NRPS) enzymes (Figure 1). These enzymes function through activation and condensation of a series of monomer units, acyl-CoAs in the case of PKS and amino acids (both proteinogenic and non-proteinogenic) in the case of NRPS, which are tailored and enzymatically elaborated to produce the structural diversity associated with this class of small molecules. The enzymatic activities responsible for loading and incorporation of each building block is termed a module, and each discrete enzymatic activity within a module (i.e. loading, condensation, methylation) is performed by discrete domains. While the individual details of PKS and NRPS pathways can be highly nuanced and have been superbly reviewed elsewhere [7••,8], the challenges confounding their proteomic identification are common ones: how to efficiently sample this protein population from a complex peptide mixture, and how to increase the dynamic range of detection over which these enzymes may be observed. In this regard it is fortunate that PKS and NRPS enzymes have several distinct features can be used to uniquely distinguish them from the proteomic milieu (Figure 2):
Figure 1.

PKS and NRPS modular biosynthetic pathways. a) Deoxyerythronolide B synthase (DEBS), the type I modular PKS responsible for 6-deoxyerythronolide production. PKS-mediated condensation of propionyl-CoA and methylmalonyl-CoA monomers followed by reduction and macrocyclization produces the final natural product. Three proteins, DEBS1-3 produce the erythromycin aglycone precursor. b) Tyrocidine A synthase, the modular NRPS responsible for tyrocidine production. NRPS-mediated epimerization and condensation of ten amino acids, followed by macrocyclization, affords the final natural product. Modules are indicated with grey bars, and exemplary domains are listed to the right of each synthase.
Figure 2.

Enrichment of modular synthases for mass spectroscopy. Modular synthases may be enriched by size (PrISM) or by activity based labeling (OASIS). Conserved active site chemistries provide a target for tight-binding or irreversible inhibitors that can be used to isolate these enzymes from complex proteomic mixtures. Thioesterase (TE) domains can be targeted by fluorophosphonate-based probes, while posttranslational modification of carrier protein (CP) domains allows their isolation via metabolic delivery of pantetheine analogs. The high molecular weight of type I modular synthases allows PKS and NRPS enzymes to be distinguished by size fractionation methods such as gel-electrophoresis.
1. Posttranslational modification
PKS and NRPS, as well as the functionally related fatty acid synthase (FAS) enzymes all utilize small carrier protein (CP) domains to covalently tether activated monomers and intermediates directly to the enzymes themselves throughout the biosynthetic process [9]. The site of this covalent tethering is at the terminal thiol of the 4′-phosphopantetheine (PPant) prosthetic group, a posttranslational modification that is introduced by a 4′-phosphopantentheinyl transferase (PPTase). PPTase enzymes mediate the transfer of PPant from coenzyme A (CoA) to a conserved serine residue within each CP domain, thereby converting apo-synthase to holo-synthase. Notably, the PPant posttranslational modification has only been observed within PKS, NRPS, FAS and a handful of related enzymes, and can thereby be used to detect these activities through either chemoenzymatic labeling approaches or observation of unique fragmentation during tandem mass spectrometry (described below).
2. High molecular weight
PKS and NRPS enzymes utilize a large number of catalytic domains to incorporate and tailor each monomer unit integrated into the final natural product. For example, incorporation and complete reduction of a single acetate unit into a polyketide necessitates an acyltransferase (AT), ketosynthase (KS), ketoreductase (KR), dehydratase (DH), and enoyl reductase (ER) activity, each of which can be ∼30-60 kDa. Therefore type I PKS and NRPS enzymes, in which these domains are all housed on a single polypeptide, are often some of the largest enzymes in the cell, typically in excess of 200 kDa. When more than one module is housed on a single protein the enzymes can be much larger, as exemplified by the cyclosporine synthetase, a single polypeptide chain of 1600 kDa comparable in size to the entire ribosome assembly (2500 kDa).
3. Diverse and well studied active site chemistries
As mentioned above type I PKS and NRPS proteins are enzymologically versatile, utilizing a variety of active site chemistries and cofactors, all housed on a single polypeptide. For example, these include several hydrolase motifs, which are well studied as targets of irreversible inhibitors. Several other activities for which active site inhibitors have been developed owe their importance in primary metabolism and/or as potential antibiotic drug targets [10,11]. Properly derivatized, these scaffolds can provide chemical handles for enrichment and discovery of PKS and NRPS enzymes.
Proteomic Studies of PKS/NRPS Enzymes
Whole Proteome Approaches
Far from being a recent development, PKS and NRPS proteins were commonly isolated and studied directly from natural product producer proteomes in the pre genomic era through the utilization of classical activity based fractionation techniques [12,13]. These approaches were practically abandoned with the advent of modern molecular biology, as recombinant expression of PKS and NRPS enzymes promised some alleviation from the tedium associated with activity based protein fractionation while also affording enzymes of greater purity for in vitro characterization. However, recent years have seen the development of liquid chromatography tandem mass spectrometry (LC-MS/MS) instrumentation and methods whose application affords a new avenue for the routine and automated fractionation and analysis of PKS and NRPS enzymes from proteomic samples. LC-MS/MS proteomic methods are only succinctly introduced here – for an in-depth discussion the reader is directed to reviews of this topic available in this journal and elsewhere [14••,15]. Briefly, LC-MS/MS based proteomics involves breaking cellular proteins down into small peptides, usually through the use of a protease such as trypsin. Liquid chromatography (LC)-based separation is then performed prior to elution into a mass spectrometer, where peptides undergo ionization and are fragmented by tandem mass spectrometry (MS/MS). Search algorithms then match peptide fragmentation spectra to those predicted to be encoded by the genome of the organism, thereby reporting on the identity of proteins present in the cellular proteome [16].
These methods were first applied with the express intent of profiling PKS and NRPS biosynthesis in a study by Muller and coworkers in 2006, in which proteome isolated from late exponential growth phase cultures of the Myxobacteria natural product producer Myxococcus xanthus were interrogated by LC-MS/MS [17]. Offline two-dimensional LC of the soluble proteome followed by MS/MS analysis identified peptides from four PKS/NRPS gene clusters responsible for production of the small molecules myxalamid, DKxanthene, myxochromid, and myxovisescin. Perhaps more intriguingly, peptides from six additional PKS/NRPS gene clusters for which no natural product was known (termed “orphan” gene clusters) were also identified, hinting at the possibilities of using proteomics to guide expression analysis and identification of new natural products. However, no attempt was made to correlate protein expression with natural product production, and the limitations of whole proteome analysis in terms of dynamic range were evidenced by the low overall number of total PKS and NRPS peptides observed. These obstacles spurred the concurrent development of two novel approaches for directed proteomic profiling of PKS and NRPS pathways, termed the OASIS (Orthogonal Active Site Identification System) and PrISM (Proteomic Interrogation of Secondary Metabolism) methods.
The OASIS Method - Profiling PKS/NRPS Through Active Site Enrichment
Proteomic Enrichment Probes for PKS and NRPS Enzymes
To counter the low coverage of PKS/NRPS enzymes observed by whole proteome analysis, OASIS utilizes chemical probes to enrich PKS and NRPS enzymes prior to MS/MS analysis on the basis of i) their use of the PPant posttranslational modification and ii) their unique active site chemistries. The development of these enrichment approaches has been detailed elsewhere but is briefly introduced here to facilitate discussion.
As mentioned above, the 4′-PPant posttranslational modification of CP domains is found in all type I and II PKS and NRPS enzymes. Early studies showed apo-CP domains were capable of accepting fluorophore- and affinity-labeled CoA as PPTase substrates in vitro, resulting in affinity-labeled crypto-CP domains [18•]. However, the application of this technique for proteomic labeling of PKS and NRPS CP domains can be low yielding, due to in vivo modification of these enzymes by endogenous CoA. This lead our group to develop a technique based on co-opting the in vivo CoA biosynthetic pathway with functionalized CoA precursors (Figure 2) [19•,20]. These CoA precursors exhibit uptake and biosynthetic conversion to CoA analogues in vivo by simple feeding and additionally function as substrates for endogenous PPTase enzymes resulting in modified PKS, NRPS, and FAS CP domains. Most useful in this regard have been azido-CoA precursors, whose terminal group functions as a bioorthogonal chemical reporter which can be used for affinity purification or gel-based visualization of labeled CP domains via Cu-catalyzed cycloaddition to a reporter labeled alkyne [21••].
As an alternative to 4′-PPant-based labeling, hydrolytic PKS/NRPS active sites can be tagged for enrichment employing an approach known as activity based protein profiling (ABPP). ABPP involves the modification of known enzyme inhibitors with fluorescence or affinity agents to allow the purification of active enzymes from complex proteomes [22]. For example the fluorophosphonate (FP) scaffold, a class-wide irreversible inhibitor of serine hydrolases, can been modified with a biotin moiety to facilitate isolation and visualization of this enzyme class (Figure 2). Because the reactivity of ABPP probes depends on the inherent catalytic properties of the enzyme class being profiled, these probes are able to discriminate between active enzymes and their inactive (for example zymogen or inhibitor-bound) states. ABPP thereby provides not only enrichment of low abundance PKS/NRPS enzymes, but also a direct report of their functional activity in a natural product producer proteome. In applying this approach to natural product proteomics, initial studies profiled the reactivity of PKS and NRPS enzymes with fluorescently labeled electrophiles using gel-based assays [23]. Notably, haloacetamide activity based probes showed enhanced reactivity with PKS KS domains, while the aforementioned fluorophosphonates showed strong reactivity with PKS and NRPS TE domains in unfractionated proteomes. In contrast to their intense labeling of TE domains, FP-probes do not label PKS AT domains, demonstrating the ability of this probe to differentiate between two catalytically distinct serine hydrolase active sites. More recent studies have also demonstrated the reactivity of PKS and NRPS TE domains and FAS KS domains with reporter-labeled beta-lactones [24].
Global profiling of PKS and NRPS enzymes of Bacillus subtilis by OASIS
Metabolic labeling provides an avenue for selective labeling of enzymes based on posttranslational modification, but can be stymied by limited pathway incorporation and does not distinguish between primary and secondary biosynthetic enzymes, both of which utilize 4′-phosphopantetheine. Activity based probes provide a handle for selective proteomic enrichment of terminal PKS and NRPS modules, but also shows proteome-wide enrichment of non-PKS/NRPS serine hydrolases. This lead to the exploration of multiple enrichment probes targeted to orthogonal PKS/NRPS active sites to provide selective proteomic detection of PKS/NRPS enzymes, an approach termed OASIS (Figure 3) [25••]. In the initial proof-of-concept study, OASIS CP-probes and ABPP probes were applied to unfractionated whole proteomes of the model natural product producer Bacillus subtilis. Labeled proteins were enriched by biotin-avidin affinity purification, followed by analysis using multidimensional protein identification technology (MudPIT). MudPIT utilizes a two dimensional LC separation, strong cation (SCX) followed by reverse-phase (RP) chromatography, prior to MS/MS analysis to increase resolution and facilitate identification of low abundance peptides in proteomic samples [26]. While primarily a technical improvement, incorporation of MudPIT into the OASIS workflow represented a significant leap over previous, gel-based methods of detection used during development of proteomics probes for PKS/NRPS enzymes, allowing the reactivity of CP-labeling reagents to be profiled for the first time on a global scale. After database search relative protein amounts in each sample were estimated by spectral counting, which sums the number of tandem mass spectrometry spectra assigned to a specific protein to provide a semi-quantitative measure of protein abundance [27].
Figure 3.

Orthogonal Active Site Identification System (OASIS) for proteomic identification and study of PKS and NRPS enzymes. Natural product producer proteomes are enriched with domain-specific probes (RG = protein reactive group), followed by MudPIT MS analysis. The collection of multiple datasets using orthogonal probes allows specific identification of modular biosynthetic enzymes, which contain multiple active sites on a single polypeptide. The application of orthogonal probes can also be used to facilitate study of specific subsets of natural product biosynthetic enzymes.
Analyzing results on the protein level, a CP-probe specifically enriched 12 of the possible 16 PKS and NRPS CP-containing enzymes found in the B. subtilis genome, including hits from all four PKS and NRPS gene clusters. The activity based TE probe proved similarly well-suited to the analysis of modular biosynthetic enzymes, identifying the TE-containing termination modules of 3 out of 4 PKS/NRPS enzymes in both strains of B. subtilis, as well as a type II TE enzyme involved in the editing of misprimed CP domains. Comparing PKS/NRPS enzymes identified in enriched B. subtilis samples to those identified by MudPIT of unfractionated lysates showed that these OASIS probes provide substantial increases in the dynamic range of analysis and allow peptides form PKS and NRPS enzymes to be sampled by MS/MS that were not observed during whole proteome analysis. Spectral counting analysis was also used to compare PKS/NRPS expression in two stains of B. subtilis. While no attempt was made to correlate metabolite profile with enzyme expression, substantial interstrain variations in PKS/NRPS expression were observed which correlated well with gel-based analysis of enzyme abundance [23]. These analyses demonstrated the capabilities of OASIS as a highly sensitive platform for profiling the global expression of PKS and NRPS enzymes directly from natural product producer organisms.
The potential of OASIS as a tool for new PKS/NRPS enzyme discovery was also explored, by analyzing the results of enrichment by CP and TE probes on the peptide level. Cross-referencing peptides specifically enriched in multiple replicate samples by both CP-probes and TE-probes resulted in identification of 39 peptides, eight of which (20%) originated from PKS/NRPS terminal modules. While seemingly modest, this represented a large enrichment relative to the abundance of other enzyme classes found in this enriched peptide pool, and bioinformatic analysis by the error tolerant Basic Local Alignment Search Tool MS-BLAST [28] showed PKS/NRPS peptides could be distinguished by homology from nonspecific background peptides. While not applied in the initial study, it was suggested that such an approach may be similarly applied to identify peptides from unsequenced organisms, and thereby facilitate PCR probe design for the identification of novel PKS/NRPS gene clusters.
PrISM - Profiling PKS/NRPS Through High Accuracy Mass Spectrometry
Directly concurrent with the above approach, an alternative method for proteomic profiling of PKS and NRPS enzymes was developed by Kelleher and coworkers known as Proteomic Interrogation of Secondary Metabolism (PrISM –Figure 4) [29••]. PrISM builds on advances in the analysis of PKS and NRPS enzymes by Fourier Transform-Ion Cyclotron Resonance (FT-ICR) mass spectrometry, an excellent review of which can be found in this journal [30••]. FT-ICR MS methods are characterized by their high accuracy and resolution and have been widely applied during in vitro studies of PKS and NRPS enzymes to directly observe mass changes corresponding to 4′-PPant bound intermediates. Such studies have been greatly aided by the highly labile nature of the 4′-PPant posttranslational modification during tandem mass spectrometry, which undergoes neutral or charged ejection to form a low molecular weight PPant ejection fragment to provide a rapid diagnostic for the presence of CP active sites and identity of CP-bound intermediates (Figure 4) [31•,32•]. To explore whether this technology could be extended to profile PKS/NRPS enzymes in proteomic settings, Bumpus et al. first examined proteomic extracts from three systems of increasing complexity: i) Eschericia coli overexpressing a CP-containing NRPS, ii) Bacillus brevis ATCC 9999, producer of the prototypical NRPS product gramicidin, and iii) Streptomyces viridochromogenes DSM 40736, an actinomycete responsible for production of the herbicide phosphothricin. Unfractionated proteomes were subjected to MudPIT-like SCX/reverse phase separation followed by tandem MS analysis using a hybrid FT-ICR ion trap instrument. In each of these studies conventional high accuracy FT-ICR precursor ion scans were followed by a ‘fast’ low resolution FT-ICR scan in which the chromatographic eluent was subjected to source-induced dissociation (resulting in PPant ejection) and selected tandem MS on the 261.1 4′-PPant ejection fragment to confirm the presence of 4′-PPant modified ions [32]. In each of these systems, correlating elution of 4′-PPant ions with highly accurate FT-ICR measurement of precursor ion mass allowed identification of parent ions that corresponded to predicted tryptic NRPS CP-active sites, validating this approach on the proteomic level.
Figure 4.
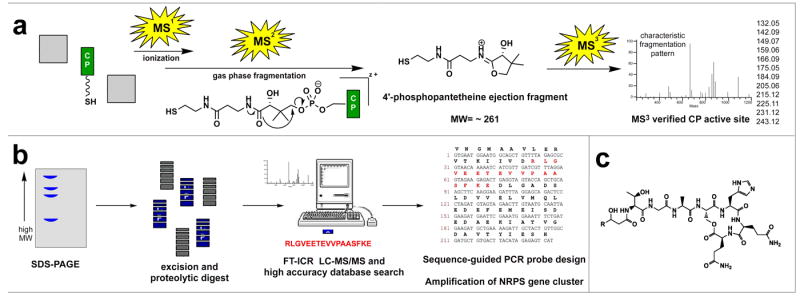
Proteomic Investgation of Secondary Metabolism (PrISM). a) Pseudo-MS3 method for detecting CP active site peptides. Peptides undergo neutral or charged loss of the 4′-PPant modification via pantetheine ejection during tandem MS. The identity of the pantetheine moiety is confirmed by a further round of disassociation (MS3), which yields a characteristic fragmentation pattern. b) PrISM proteomic workflow utilized to identify PKS/NRPS peptides in NK2018. Tryptic digests of gel-fractionated high molecular weight proteins are systematically interrogated by FT-ICR LC-MS/MS. Spectra are searched against the non-redundant protein database (derived from the predicted open reading frames of sequenced organisms). Sequence information is obtained for PKS/NRPS peptides sharing high identity to peptides found in the database, and used to design custom PCR probes and facilitate discovery of biosynthetic gene clusters. c) Characteristic structure of lipoheptapeptides discovered by PrISM. “R” denotes position of the fatty acyl chain.
Not content with proof-of-concept experiments, PrISM was next applied to profile PKS/NRPS production in environmental strains of Bacillus isolated from heat-treated soil samples. Isolated strains were grown in nutrient broth and analyzed by SDS-PAGE for production of high molecular weight proteins characteristic of PKS and NRPS enzymes, leading to identification of a candidate natural product producer strain NK2018. Proteins from NK2018 cell lysate were first separated by off-line SCX chromoatography followed by FT-ICR tandem MS analysis, resulting in detection of a 4′-PPant precursor ion. Upon re-analysis and subjection of this precursor ion to repeated rounds of MS (MSn), a peptide fragmentation spectrum was generated of sufficient quality to allow sequence determination by manual de novo interpretation, resulting in identification of the organism's type II FAS CP active site.
Complementary to the above CP-directed approach, simple size-based fractionation was also utilized for enrichment of NK2018 type I PKS and NRPS enzymes on SDS PAGE gels, followed by tryptic digest of putative high molecular weight PKS/NRPS bands and analysis using MudPIT-like protocols. The presence of PKS/NRPS CP-active sites in these samples was confirmed by observation of authentic PPant ejection ions. Furthermore, search of peptide fragmentation data against the nonredundant protein database resulted in identification of peptides mapping to known and orphan PKS/NPS gene clusters encoded by the recently sequenced Bacillus cereus AH1134. These peptides (along with B. cereus sequence information) were then used to design PCR primers allowing amplification and sequencing of the corresponding PKS/NRPS gene clusters from NK2018. While the amplified NK2018 gene clusters were found to be highly similar (>97% identity) to PKS and NRPS enzymes encoded by B. cereus AH1134, this experiment provide an important proof-of-concept demonstrating how proteomic analyses could be reverse-translated into identification of PKS/NRPS gene clusters. The specific gene clusters themselves were predicted to encode zwittermicin A, a known antibiotic, as well as an orphan natural product of unknown structure. Having already verified expression of the orphan gene cluster by proteomic analyses, NK2018 extracts were analyzed to determine the structure of the encoded compound. A powerful combination of bioinformatics, high accuracy MS, and de novo spectral interpretation allowed discovery of a novel cyclic lipoheptapeptide, providing the first complete characterization of an orphan PKS-NRPS pathway by purely MS-centric methodology.
Comparison of OASIS and PrISM Methods
OASIS and PrISM methods represent innovative and complementary methods in many ways. Foremost among the differences between the two methods is the choice of the MS instrumentation. Tandem MS during OASIS analyses is performed using a conventional linear ion trap for mass analysis, while PrISM utilizes a more recently developed hybrid instrument, in which a FT-ICR and linear ion trap mass analyzer are applied in parallel to allow for highly accurate determination of precursor ion mass (FT-ICR) followed by unit resolution of tandem MS data. The advantages of conventional linear ion trap instruments are their relative accessibility (for example, in proteomics core facilities) and comparatively fast cycle times, which maximize the sampling of enriched peptides during shotgun proteomics. However, hybrid FT-ICR instruments determine the mass of precursor ions during MS1 with much higher resolution (≫ 50,000 for FT-ICR compared to ∼ 1000 for linear ion trap) and mass accuracy (<10 ppm compared to 50-200 ppm), allowing for more stringent search parameters to be employed during database analysis. This latter advantage proved valuable during PrISM analysis of the unsequenced NK2018 proteome, as the high mass accuracy of the FT-ICR instrument allowed database search to be performed utilizing a precursor ion search tolerance of +/- 0.01 Da compared to the more standard +/- 1.5 Da employed during search of ion trap data. By immediately eliminating a large proportion of database peptides from comparison based on precursor ion mass, FT-ICR instruments greatly reduce the computational cost of database searches. This lowers the computational cost of searching extremely large databases (e.g. the entire nonredundant protein database), and thus can aid PKS/NRPS peptide identification in unsequenced organisms. In contrast, the more efficient peptide sampling provided by conventional ion trap instruments may prove preferable for proteomic studies aimed at strain optimization in sequenced organisms.
These comparisons also highlight advantages which may be gained by merging aspects of the two approaches. For example, the increased dynamic range provided by OASIS probes may prove especially useful in offsetting the reduced sampling efficiency of FT-ICR instruments. Similarly, size-based fractionation limits PrISM to the analysis of high molecular weight type I PKS/NRPS proteins. The disadvantages of this approach are evident when considering the drastic effect a single unanticipated post-lysis proteolytic event can have on the migration of a type I PKS/NRPS megasynthase on SDS-PAGE, potentially shifting the molecular weight of a 150 kDa species to 75 Da. OASIS probes are resilient to such degradation and have the potential to detect PKS/NRPS proteins of diverse architechtures. However, the development of OASIS active site probes is time and labor intensive and requires moderate synthetic expertise. Therefore the continued collaboration of organic chemistry and systems biology will be of utmost importance to the development of new methods.
Future directions and challenges
De Novo Sequencing of PKS/NRPS Peptides from Unsequenced Organisms
In speculating on the potential of proteomics to facilitate discovery of novel natural product biosynthetic pathways from unsequenced organisms, it is important to note that neither OASIS nor PrISM approaches have yet demonstrated true utility in de novo identification of PKS/NRPS peptides from such entities. This is because both methods utilize database searching, in which tandem MS spectra are not interpreted but rather matched to a finite database of predicted tryptic peptides [16]. Database searching biases proteomic discovery towards PKS/NRPS enzymes homologous to those found in sequenced organisms, as evidenced by PriSM's discovery of two gene clusters nearly identical to PKS/NRPS found in B. cereus.
However, despite the limitations imposed by database searching, continuing experimental and bioinformatics innovations offer a number of potential routes forwards. One is through the further development of automated de novo sequencing algorithms for analysis of MS/MS data [33]. While de novo sequencing has a high error rate, incorporation of BLAST homology searching can lead to the identification of novel peptides [34]. Importantly, such approaches could be aided by the application of OASIS probes with validated specificities (whose known reactivity lends credence to homology based identifications) as well as through the application of high accuracy mass spectrometry (PrISM) [35]. Another potential approach to identification of novel PKS/NRPS enzymes is through detection and manual de novo sequencing of 4′-PPant peptides, as demonstrated during PrISm identification of the NK2018 FAS ACP active site. This method circumvents homology-based identification entirely by relying on experimental observation of 4′-PPant ejection to assign PKS/NRPS/FAS biogenesis. In addition to fragmentation-based approaches, recent work has focused on development of bioinformatic methods to detecting distinct PPant signatures from MS/MS spectra through the use of support vector machines [36]. Integrating bioinformatic and experimental methods for 4′-PPant peptide identification promises to further strengthen approaches to proteomic identification of PKS/NRPS enzymes from novel systems.
Development of New OASIS Probes
The OASIS technique has shown the ability to be improved for sensitivity by two means: 1) increase the number of MudPIT runs collected, and 2) increasing the number of orthogonal domains selected for probe labeling and pull-down. Since an average MudPIT run requires 12 hours of instrument time, option #1 offers limited scalability. Therefore, the most effective means of improving sensitivity is to develop new OASIS probes for modular synthases. The prospect exists to develop probes specific for many of the existent domains within modular synthases (Figure 1), offering significant sensitivity advances with each additional probe [37,38]. In addition to these canonical PKS/NRPS activities, another opportunity lies in the development of probes designed to capture specific enzyme activities predicted to be associated with a given biosynthetic pathways. For example, the biosynthetic pathways for dinoflagellate polyether polyketides are currently unknown but are predicted to utilize enzyme catalyzed Favorskii-like chemistry during polyether biosynthesis [39]. Could specific probes be designed to target these enzymatic steps? Here probe design is limited only by the synthetic chemist's ingenuity.
Conclusions
The proteomic analysis of natural product biosynthesis is a rapidly evolving field epitomized by the complimentary OASIS and PrISM methods, which combine novel enrichment tools developed through years of mechanistic and enzymological characterization with advanced mass spectroscopic data collection and analysis. These technological improvements will enable and accelerate new discoveries in the natural products arena, ranging from drug discovery to pathway engineering.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
Papers of particular interest for further insights into the topic of PKS and NRPS proteomics have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Danishefsky S. On the potential of natural products in the discovery of pharma leads: a case for reassessment. Nat Prod Rep. 27:1114–1116. doi: 10.1039/c003211p. [DOI] [PubMed] [Google Scholar]
- 2.Clardy J, Fischbach MA, Currie CR. The natural history of antibiotics. Curr Biol. 2009;19:R437–441. doi: 10.1016/j.cub.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harvey AL. Natural products in drug discovery. Drug Discov Today. 2008;13:894–901. doi: 10.1016/j.drudis.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 4.Li JW, Vederas JC. Drug discovery and natural products: end of an era or an endless frontier? Science. 2009;325:161–165. doi: 10.1126/science.1168243. [DOI] [PubMed] [Google Scholar]
- 5.Baltz RH. Marcel Faber Roundtable: is our antibiotic pipeline unproductive because of starvation, constipation or lack of inspiration? J Ind Microbiol Biotechnol. 2006;33:507–513. doi: 10.1007/s10295-005-0077-9. [DOI] [PubMed] [Google Scholar]
- 6.Hopwood DA. Developments in the study of natural products biosynthesis. Preface. Methods Enzymol. 2009;458:xix–xxi. doi: 10.1016/S0076-6879(09)58027-5. [DOI] [PubMed] [Google Scholar]
- 7••.Fischbach MA, Walsh CT. Assembly-line enzymology for polyketide and nonribosomal Peptide antibiotics: logic, machinery, and mechanisms. Chem Rev. 2006;106:3468–3496. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]; An excellent primer on the modular biosynthetic paradigm and canonical mechanisms utilized by PKS and NRPS enzymes.
- 8.Sieber SA, Marahiel MA. Molecular mechanisms underlying nonribosomal peptide synthesis: approaches to new antibiotics. Chem Rev. 2005;105:715–738. doi: 10.1021/cr0301191. [DOI] [PubMed] [Google Scholar]
- 9.Mercer AC, Burkart MD. The ubiquitous carrier protein--a window to metabolite biosynthesis. Nat Prod Rep. 2007;24:750–773. doi: 10.1039/b603921a. [DOI] [PubMed] [Google Scholar]
- 10.Zhang YM, White SW, Rock CO. Inhibiting bacterial fatty acid synthesis. J Biol Chem. 2006;281:17541–17544. doi: 10.1074/jbc.R600004200. [DOI] [PubMed] [Google Scholar]
- 11.Foster RJ, Poulose AJ, Bonsall RF, Kolattukudy PE. Measurement of distance between the active serine of the thioesterase domain and the pantetheine thiol of fatty acid synthase by fluorescence resonance energy transfer. J Biol Chem. 1985;260:2826–2831. [PubMed] [Google Scholar]
- 12.Roskoski R, Jr, Gevers W, Kleinkauf H, Lipmann F. Tyrocidine biosynthesis by three complementary fractions from Bacillus brevis (ATCC 8185) Biochemistry. 1970;9:4839–4845. doi: 10.1021/bi00827a002. [DOI] [PubMed] [Google Scholar]
- 13.Gevers W, Kleinkauf H, Lipmann F. The activation of amino acids for biosynthesis of gramicidin S. Proc Natl Acad Sci U S A. 1968;60:269–276. doi: 10.1073/pnas.60.1.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14••.Domon B, Aebersold R. Mass spectrometry and protein analysis. Science. 2006;312:212–217. doi: 10.1126/science.1124619. [DOI] [PubMed] [Google Scholar]; A well-written and accessible account of developments from the past decade in the area of MS-driven proteomics studies, with a comprehensive description of common methods and instrumentation utilized during LC-MS/MS shotgun proteomics analyses.
- 15.Han X, Aslanian A, Yates JR., 3rd Mass spectrometry for proteomics. Curr Opin Chem Biol. 2008;12:483–490. doi: 10.1016/j.cbpa.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nesvizhskii AI, Vitek O, Aebersold R. Analysis and validation of proteomic data generated by tandem mass spectrometry. Nat Methods. 2007;4:787–797. doi: 10.1038/nmeth1088. [DOI] [PubMed] [Google Scholar]
- 17.Schley C, Altmeyer MO, Swart R, Muller R, Huber CG. Proteome analysis of Myxococcus xanthus by off-line two-dimensional chromatographic separation using monolithic poly-(styrene-divinylbenzene) columns combined with ion-trap tandem mass spectrometry. J Proteome Res. 2006;5:2760–2768. doi: 10.1021/pr0602489. [DOI] [PubMed] [Google Scholar]
- 18•.La Clair JJ, Foley TL, Schegg TR, Regan CM, Burkart MD. Manipulation of carrier proteins in antibiotic biosynthesis. Chem Biol. 2004;11:195–201. doi: 10.1016/j.chembiol.2004.02.010. [DOI] [PubMed] [Google Scholar]; An initial foray into the chemoenzymatic labeling and detection of PKS and NRPS peptides in complex proteomic mixtures through specific labeling of the 4′-PPant posttranslational modification. The promise as well as the limitations of this approach lead to the development of in vivo labeling approaches detailed in the next two references.
- 19•.Clarke KM, Mercer AC, La Clair JJ, Burkart MD. In vivo reporter labeling of proteins via metabolic delivery of coenzyme A analogues. J Am Chem Soc. 2005;127:11234–11235. doi: 10.1021/ja052911k. [DOI] [PubMed] [Google Scholar]; Details the initial steps towards an in vivo approach to labeling PKS and NRPS carrier protein domains by metabolic transformation of fluorescent CoA precursors into CoA analogues in Escherichia coli.
- 20.Meier JL, Mercer AC, Rivera H, Jr, Burkart MD. Synthesis and evaluation of bioorthogonal pantetheine analogues for in vivo protein modification. J Am Chem Soc. 2006;128:12174–12184. doi: 10.1021/ja063217n. [DOI] [PubMed] [Google Scholar]
- 21••.Mercer AC, Meier JL, Torpey JW, Burkart MD. In vivo modification of native carrier protein domains. Chembiochem. 2009;10:1091–1100. doi: 10.1002/cbic.200800838. [DOI] [PMC free article] [PubMed] [Google Scholar]; This report details the first application of bioorthogonal CoA precursors to the labeling of fatty acyl CP domains in genetically unmodified bacteria. This phenomena is demonstrated in a number of bacteria as well as a human cancer cell line. Most notably, this study demonstrated that metabolic labeling, followed by SDS-PAGE and MALDI TOF-TOF analysis of peptides extracted from a fluorescently labeled gel band could be used to provide sequence information and ultimately allowed facilitate cloning of the primary FAS ACP domain of an unsequenced bacterium. While demonstrated in the context of a relatively high abundance bacterial fatty acyl ACP, this study constituted an important proof-of-concept for proteomics driven discovery of natural product gene clusters.
- 22.Evans MJ, Cravatt BF. Mechanism-based profiling of enzyme families. Chem Rev. 2006;106:3279–3301. doi: 10.1021/cr050288g. [DOI] [PubMed] [Google Scholar]
- 23.Meier JL, Mercer AC, Burkart MD. Fluorescent profiling of modular biosynthetic enzymes by complementary metabolic and activity based probes. J Am Chem Soc. 2008;130:5443–5445. doi: 10.1021/ja711263w. [DOI] [PubMed] [Google Scholar]
- 24.Bottcher T, Sieber SA. Beta-lactones as privileged structures for the active-site labeling of versatile bacterial enzyme classes. Angew Chem Int Ed Engl. 2008;47:4600–4603. doi: 10.1002/anie.200705768. [DOI] [PubMed] [Google Scholar]
- 25••.Meier JL, Niessen S, Hoover HS, Foley TL, Cravatt BF, Burkart MD. An orthogonal active site identification system (OASIS) for proteomic profiling of natural product biosynthesis. ACS Chem Biol. 2009;4:948–957. doi: 10.1021/cb9002128. [DOI] [PMC free article] [PubMed] [Google Scholar]; The first comprehensive application of metabolic labeling and small molecule affinity tags to profile the natural product production capacities of a bacterium (B. subtilis) on a global scale. Detailed extensively in the main text, this method and PrISM (see below) represent the current state of the art methods for proteomic profiling of PKS and NRPS biosynthesis.
- 26.Wolters DA, Washburn MP, Yates JR., 3rd An automated multidimensional protein identification technology for shotgun proteomics. Anal Chem. 2001;73:5683–5690. doi: 10.1021/ac010617e. [DOI] [PubMed] [Google Scholar]
- 27.Liu H, Sadygov RG, Yates JR., 3rd A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal Chem. 2004;76:4193–4201. doi: 10.1021/ac0498563. [DOI] [PubMed] [Google Scholar]
- 28.Shevchenko A, Sunyaev S, Loboda A, Bork P, Ens W, Standing KG. Charting the proteomes of organisms with unsequenced genomes by MALDI-quadrupole time-of-flight mass spectrometry and BLAST homology searching. Anal Chem. 2001;73:1917–1926. doi: 10.1021/ac0013709. [DOI] [PubMed] [Google Scholar]
- 29••.Bumpus SB, Evans BS, Thomas PM, Ntai I, Kelleher NL. A proteomics approach to discovering natural products and their biosynthetic pathways. Nat Biotechnol. 2009;27:951–956. doi: 10.1038/nbt.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]; A pioneering study in which high accuracy FT-ICR MS methods are extended to the proteomic profiling of PKS/NRPS biosynthesis in model as well as unsequenced organisms. Importantly, this resulted in the identification of a novel natural product from an unsequenced environmental isolate. Further detailed in the main text, this method and OASIS (see above) represent the current state of the art methods for proteomic profiling of PKS and NRPS biosynthesis.
- 30••.Bumpus SB, Kelleher NL. Accessing natural product biosynthetic processes by mass spectrometry. Curr Opin Chem Biol. 2008;12:475–482. doi: 10.1016/j.cbpa.2008.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]; An excellent introduction to recent “bottom-up” LC-MS/MS studies of reconstituted PKS and NRPS biosynthetic enzymes using FT-ICR instruments. These reports provide much of the technical and functional groundwork for development of the PrISM method.
- 31•.Dorrestein PC, Bumpus SB, Calderone CT, Garneau-Tsodikova S, Aron ZD, Straight PD, Kolter R, Walsh CT, Kelleher NL. Facile detection of acyl and peptidyl intermediates on thiotemplate carrier domains via phosphopantetheinyl elimination reactions during tandem mass spectrometry. Biochemistry. 2006;45:12756–12766. doi: 10.1021/bi061169d. [DOI] [PMC free article] [PubMed] [Google Scholar]; A comprehensive investigation into the utility of identifying 4′-PPant bound biosynthetic intermediates from reconstituted PKS and NRPS enzymes by direct observation of modified PPant ejection fragments during tandem mass spectrometry, this study reinvigorated approaches toward the examination of the PPant posttranslational modification by MS methods.
- 32•.Meluzzi D, Zheng WH, Hensler M, Nizet V, Dorrestein PC. Top-down mass spectrometry on low-resolution instruments: characterization of phosphopantetheinylated carrier domains in polyketide and non-ribosomal biosynthetic pathways. Bioorg Med Chem Lett. 2008;18:3107–3111. doi: 10.1016/j.bmcl.2007.10.104. [DOI] [PMC free article] [PubMed] [Google Scholar]; Details the development of an MS assay for identifying CP active sites based on the unique MS3 fragmentation signature of the 4′-PPant modification. Compatible with low-resolution “benchtop”instruments, the fragmentation pattern identified was utilized for confirmation of 4′-PPant peptides in the PrISM proteomic workflow.
- 33.Frank A, Pevzner P. PepNovo: de novo peptide sequencing via probabilistic network modeling. Anal Chem. 2005;77:964–973. doi: 10.1021/ac048788h. [DOI] [PubMed] [Google Scholar]
- 34.Waridel P, Frank A, Thomas H, Surendranath V, Sunyaev S, Pevzner P, Shevchenko A. Sequence similarity-driven proteomics in organisms with unknown genomes by LC-MS/MS and automated de novo sequencing. Proteomics. 2007;7:2318–2329. doi: 10.1002/pmic.200700003. [DOI] [PubMed] [Google Scholar]
- 35.Frank AM, Savitski MM, Nielsen ML, Zubarev RA, Pevzner PA. De novo peptide sequencing and identification with precision mass spectrometry. J Proteome Res. 2007;6:114–123. doi: 10.1021/pr060271u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Patel AD, Bafna V, Dorrestein PD. Manuscript in preparation. 2010 [Google Scholar]
- 37.Finking R, Neumuller A, Solsbacher J, Konz D, Kretzschmar G, Schweitzer M, Krumm T, Marahiel MA. Aminoacyl adenylate substrate analogues for the inhibition of adenylation domains of nonribosomal peptide synthetases. Chembiochem. 2003;4:903–906. doi: 10.1002/cbic.200300666. [DOI] [PubMed] [Google Scholar]
- 38.Meier JL, Haushalter RW, Burkart MD. A mechanism based protein crosslinker for acyl carrier protein dehydratases. Bioorg Med Chem Lett. 20:4936–4939. doi: 10.1016/j.bmcl.2010.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wright JLC, Hu T, McLachlan JL, Needham J, Walter JA. Biosynthesis of DTX-4: Confirmation of a Polyketide Pathway, Proof of a Baeyer-Villiger Oxidation Step, and Evidence for an Unusual Carbon Deletion Process. Journal of the American Chemical Socierty. 1996;118:8757–8758. [Google Scholar]