

Abstract
Background
Developmental ethanol (EtOH) exposure damages the hippocampus, causing long-lasting alterations in learning and memory. Alterations in glutamatergic synaptic transmission and plasticity may play a role in the mechanism of action of EtOH. This signaling is fundamental for synaptogenesis, which occurs during the third-trimester of human pregnancy (first 12 days of life in rats).
Methods
Acute coronal brain slices were prepared from 7–9 day-old rats. Extracellular and patch-clamp electrophysiological recording techniques were used to characterize the acute effects of EtOH on α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate receptor (AMPAR)- and N-methyl-D-aspartate receptor (NMDAR)-mediated responses and long-term potentiation (LTP) in the CA1 hippocampal region.
Results
EtOH (40 and 80 mM) inhibited AMPAR- and NMDAR-mediated field excitatory postsynaptic potentials (fEPSPs). EtOH (80 mM) also reduced AMPAR-mediated fEPSPs in presence of an inhibitor of Ca2+ permeable AMPARs. The effect of 80 mM EtOH on NMDAR-mediated fEPSPs was significantly greater in presence of Mg2+. EtOH (80 mM) neither affected the paired-pulse ratio of AMPAR-mediated fEPSPs nor the presynaptic volley. The paired-pulse ratio of AMPAR-mediated excitatory postsynaptic currents was not affected either, and the amplitude of these currents was inhibited to a lesser extent than that of fEPSPs. EtOH (80 mM) inhibited LTP of AMPAR-mediated fEPSPs.
Conclusions
Acute EtOH exposure during the third-trimester equivalent of human pregnancy inhibits hippocampal glutamatergic transmission and LTP induction, which could alter synapse refinement and ultimately contribute to the pathophysiology of fetal alcohol spectrum disorder.
Keywords: Ethanol, development, plasticity, glutamate, synaptic
Introduction
Ethanol (EtOH) exposure during development can produce severe and long-lasting deficits in many organs, including the brain (Sokol et al., 2003; Warren and Foudin, 2001). Among the consequences of EtOH-induced brain damage are behavioral disorders, mental retardation, and learning and memory deficits (Warren and Foudin, 2001). Electrophysiological, structural, and behavioral studies performed with human and animal subjects suggest that learning and memory deficits are, in part, a consequence of damage to the hippocampal formation (Berman and Hannigan, 2000; Costa et al., 2000b). Spatial memory tests have demonstrated abnormalities in hippocampal function in children with fetal alcohol syndrome and in animal models of this condition (Hamilton et al., 2003; Johnson and Goodlett, 2002). However, the mechanisms by which EtOH exerts its deleterious effects on the hippocampal formation are not well understood.
Studies from several laboratories suggest that the mechanism of action of EtOH on hippocampal development involves alterations in glutamatergic synaptic transmission (Reviewed in Berman and Hannigan, 2000; Costa et al., 2000b). One study suggested that inhibition of NMDA receptors (NMDARs) induced by acute EtOH administration during the human third trimester-equivalent period of development (i.e., neonatal period in the rat) triggers widespread apoptotic neurodegeneration in many brain regions, including the CA1 hippocampal region (Ikonomidou et al., 2000). However, whether EtOH inhibits NMDAR function in CA1 neurons during this developmental period has not been tested. Studies from our laboratory indicate that NMDARs in CA3 pyramidal neurons in acute brain slices from neonatal rats are relatively insensitive to EtOH; in contrast, AMPA receptor (AMPAR) function and glutamate release were unexpectedly found to be particularly sensitive to EtOH in these neurons (Mameli et al., 2005). In light of these findings with CA3 pyramidal neurons, we hypothesized that acute EtOH exposure inhibits AMPAR function and glutamate release in CA1 pyramidal neurons, without affecting NMDARs.
Since glutamatergic neurotransmission is essential for certain forms of synaptic plasticity, a corollary of this hypothesis is that EtOH exposure should also affect plasticity in developing hippocampal neurons, where this process is thought to play a central role in synaptogenesis (Constantine-Paton and Cline, 1998). Many developing hippocampal Schaffer collateral-CA1 synapses are silent at resting membrane potentials; i.e., they exhibit currents mediated by NMDARs but not AMPARs (Kerchner and Nicoll, 2008; but see Groc et al., 2002). It has been suggested that expression of AMPARs is limited in these immature structures (Fiala et al., 1998; Matsuzaki et al., 2001). Recent evidence indicates that basal activation of NMDARs inhibits AMPAR-mediated responses during the synaptic maturation period (Adesnik et al., 2008; Hall et al., 2007). When NMDARs are activated by strong synchronous activity, they stimulate AMPAR recruitment to the postsynaptic density, contributing to synapse formation (Constantine-Paton and Cline, 1998; Leinekugel, 2003; Citri and Malenka, 2008; Durand et al., 1996; Maletic-Savatic et al., 1999; but see Zito et al., 2009). Excessive spontaneous excitatory activity in cultured cortical neurons was shown to cause premature synaptic stabilization via AMPAR insertion into silent synapses and it was postulated that this could limit the range of connections in the neuronal network (Voigt et al., 2005). Chronic NMDAR blockade during early development in hippocampal organotypic slice cultures demonstrated a decreased threshold for LTP induction and a shift in baseline synaptic efficacy (Savic et al., 2003). Collectively, these studies suggest that if EtOH produced alterations in activity-dependent synaptic plasticity mechanisms, these could have a negative impact on the maturation of developing neuronal networks.
In this study, we used acute brain slices from postnatal day (P) 7–9 rats and electrophysiological techniques to characterize the acute effects of EtOH on AMPAR- and NMDAR-mediated responses in the CA1 region. We then investigated whether EtOH had any presynaptic effects by measuring the paired-pulse ratios of AMPAR-mediated responses and the presynaptic volley. Lastly, we tested the effects of acute EtOH on LTP induced via high-frequency tetanic stimulation.
Experimental Procedures
Tissue preparation and solutions
EtOH (99.8%) molecular biology grade from Sigma (cat #E7023) was used. Unless indicated, all other chemicals were from Sigma (St. Louis, MO) or Tocris Cookson (Bristol, UK). Timed-pregnant Sprague-Dawley rats were provided by Harlan (Indianapolis, IN). Both male and female rat pups (postnatal day (P) 7–9) were anesthetized with 250 mg/kg ketamine and coronal brain slices (300 μm) were prepared using a vibratome, as previously described (Mameli et al., 2005). After a recovery period of 45 min at 35–36 ºC, slices were stored in artificial cerebrospinal fluid (ACSF) for 1–8 hr at room temperature. ACSF contained the following (in mM): 126 NaCl, 2 KCl, 1.25 NaH2PO4, 1 MgSO4, 26 NaHCO3, 2 CaCl2, 10 glucose, and 0.01 gabazine (also known as SR-95531) equilibrated with 95% O2/5% CO2. When indicated, the ACSF contained no MgSO4 and 3 mM CaCl2 (or 0.5 mM MgSO4 and 2.5 mM CaCl2), 50 μM DL-2-amino-5-phosphonovaleric acid (AP5), 10 μM 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide (NBQX), 100 μM 1-naphthylacetyl spermine trihydrochloride (NASPM), 50 μM GYKI-53655, 0.5 μM tetrodotoxin (TTX; Calbiochem, La Jolla, CA) and/or EtOH (20, 40, or 80 mM). Unless indicated, EtOH was bath-applied for 10 min. Animal procedures were approved by the Institutional Animal Care and Use Committee of the University of New Mexico Health Sciences Center and conformed to National Institutes of Health guidelines.
Electrophysiological recordings
Recordings using an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA) were performed in the CA1 stratum radiatum at 32 ºC perfused at 2 ml/min. Recording micropipette glass electrodes had resistances of 2–5 MΩ and were filled with ACSF. AMPAR and NMDAR-mediated fEPSPs were evoked using a concentric bipolar electrode (inner pole 25 μm; outer pole 125 μm; Frederick Haer Company, Bowdoinham, ME) placed in the vicinity of the Schaffer collateral fibers, approximately 200 μm from the recording electrode. Input-output curves were measured at the start of all recordings, and the stimulation intensity was set to produce 40–50% of maximal responses for subsequent experiments. Stimulus duration was 75 μs and stimuli were delivered at 0.033 Hz. For paired-pulse studies, the interpulse interval was 50 ms. LTP was induced via tetanic stimulation (100 Hz train for 1 s; this train was repeated a total of 3 times at 5 s intervals).
Whole-cell patch-clamp recordings were performed as previously described (Mameli et al. 2005). Recording micropipette glass electrodes had resistances of 4–7 MΩ, and were filled with an internal solution containing (in mM): 95 K-gluconate, 40 KCl, 10 HEPES, 2 MgCl2, 3 Na2ATP, 0.4 NaGTP, and 5 QX-314. The pH was 7.2 and the osmolarity was 270 mOsm. Access resistances were between 20–30 MΩ, and recordings were discarded if the access resistance changed more than 20%. The membrane potential was clamped at −65 mV (the liquid junction potential was 11.3; therefore, the corrected voltage was −76.3 mV). For evoked excitatory postsynaptic current recordings (eEPSCs), stimulus duration, intensity, and frequency were the same as for fEPSP recordings. For perforated-patch recordings, we used the same internal solution (without QX-314) containing amphotericin-B (5–10 μg/ml). A stock solution of amphotericin-B (1 mg/mL in dimethylsulfoxide) was prepared every day, sonicated for 10–20 min and maintained under constant vortexing. Amphotericin-B from this stock solution was freshly added to aliquots of internal solution every 1–2 hours. The tips of the glass electrodes were pre-filled with amphotericin-B-free ACSF, and then filled with the amphotericin-B-containing internal solution. A 1–10 GΩ seal was obtained, then the voltage was clamped at −65 mV (the liquid junction potential corrected voltage was −76.3 mV), and the access resistance was monitored for perforation. When access resistances were between 60–80 MΩ, recordings were started and these were discarded if there were sudden changes in access resistance, indicating conversion to the whole-cell configuration. The ACSF contained 10 μM gabazine and 50 μM AP5 for whole-cell and perforated-patch recordings.
Data Analysis
Data were acquired and analyzed with pClamp 9 (Molecular Devices) and GraphPad Prizm 4.0 (San Diego, CA). The short time interval between the presynaptic volley and fEPSP prevented accurate measurement of the slope in some experiments. Therefore, fEPSP amplitude and area were measured. Recordings with greater inter-electrode separation distance (~400 μm) were attempted to eliminate volley contamination of the fEPSP; however, the amplitude of the fEPSP was substantially reduced, making it difficult to assess the inhibitory effect of EtOH. For each experiment, baseline and washout were defined as the average of 10–20 fEPSPs recorded immediately before EtOH application and the last 5–10 min of washout, respectively. For experimental conditions, 10 fEPSPs immediately preceding the start of the washout or further experimental procedures were averaged for analysis. For LTP studies, the percent change from baseline was measured after ≥30 min from induction and was the average of 20 fEPSPs. Except for the plasticity studies, the effect of acute EtOH exposure was quantified with respect to the average of baseline and washout responses. The paired-pulse ratio was calculated as the ratio of AMPAR-mediated fEPSP2/fEPSP1 (or eEPSC2/eEPSC1). Statistical analyses were performed by unpaired t-test, one sample t-test vs. a theoretical mean of zero or 100%, or one-way ANOVA followed by Bonferroni post hoc test; a p ≤0.05 was considered to be statistically significant. Data are presented as mean ± SEM.
Results
Characterization of input-output relationships for AMPAR- and NMDAR-mediated fEPSPs
To initially characterize AMPAR- and NMDAR-mediated fEPSPs, input-output curves were measured. AMPAR-mediated fEPSPs were recorded in the presence of gabazine (10 μM; GABAA receptor blocker) and AP5 (50 μM; NMDAR blocker) with increasing stimulus intensities (Fig. 1A). NMDAR-mediated fEPSPs were recorded in the presence of gabazine (10 μM), NBQX (10 μM; AMPA/kainate receptor blocker) and in Mg2+-free ACSF (Fig. 1B). The average stimulus intensities that elicited a 50% of maximal stimulation response were 0.67 ± 0.08 mA and 0.41 ± 0.05 mA for AMPAR- and NMDAR-mediated fEPSPs, respectively (Figs. 1C–D). GYKI-53655 (50 μM) blocked AMPAR fEPSPs leaving only the presynaptic volley spike (n=4; 1E), confirming that these were mediated by AMPARs. The non-NMDA antagonist, NBQX (10 μM) produced a similar inhibitory effect (n=6; data not shown). NMDAR-mediated fEPSP responses were inhibited by 50 μM AP5 (n=8; 1F).
Figure 1.

AMPAR- and NMDAR-mediated fEPSP input-output curves in the CA1 hippocampus. Sample traces of (A) AMPAR- and (B) NMDAR-mediated fEPSPs evoked at increasing stimulus intensities via stimulation of the Schaffer collateral fibers. Events were recorded in the stratum radiatum in the presence of gabazine (10 μM) and AP5 (50 μM) for AMPARs, and in the presence of gabazine (10 μM), NBQX (10 μM), and absence of Mg2+ for NMDARs. Pooled data for (C) AMPAR- and (D) NMDAR-mediated fEPSP input-output curves. (E) AMPAR- and (F) NMDAR-mediated fEPSPs were blocked with GYKI-53655 (50 μM) and AP5 (50 μM), respectively.
Acute exposure to EtOH inhibited AMPAR- and NMDAR-mediated fEPSPs
To address the effects of acute bath application of EtOH on ionotropic glutamatergic signaling, we first characterized effects on pharmacologically isolated AMPAR-mediated responses. EtOH (80 mM) reversibly inhibited AMPAR-mediated fEPSPs (Fig. 2A). On average, inhibition was 15.88 ± 1.04% (n= 6; p<0.01 by one-sample t-test vs. zero; Fig 2B and 5A). To determine if Mg2+ concentration affects EtOH mediated inhibition, AMPAR-mediated fEPSPs were recorded in the absence of Mg2+. EtOH (80 mM) reduced AMPAR-mediated fEPSP amplitude by 9.76 ± 3.60% (n= 6; p<0.05 by one-sample t-test vs. zero; Fig. 5A), and this was not different from the inhibition observed in the presence of Mg2+ (not significant (N.S.) by one-way ANOVA followed by Bonferroni post hoc test). We further tested the effects of EtOH (80 mM) in adult rats (P 40) and found that it did not significantly affect AMPAR-mediated fEPSP amplitude or area (n=5; N.S. by one-sample t-test vs. zero; p<0.01 by Bonferroni post hoc test vs P7–9; Fig. 5A and Table 1).
Figure 2.
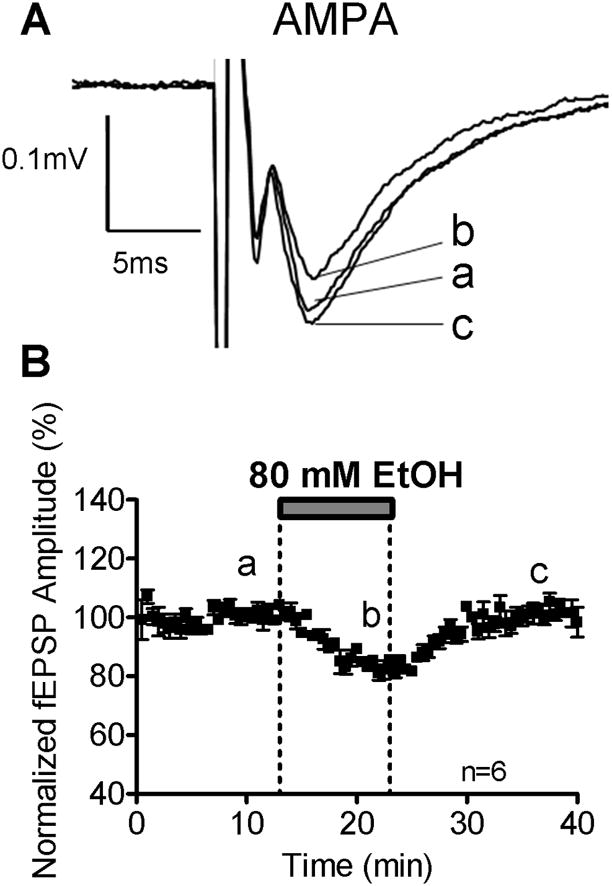
EtOH (80 mM) inhibits AMPAR-mediated fEPSPs. (A) Sample traces of inhibitory effects of EtOH on AMPAR-mediated fEPSP amplitudes recorded in the presence of gabazine (10 μM) and AP5 (50 μM). (B) Time course of the effect of EtOH.
Figure 5.
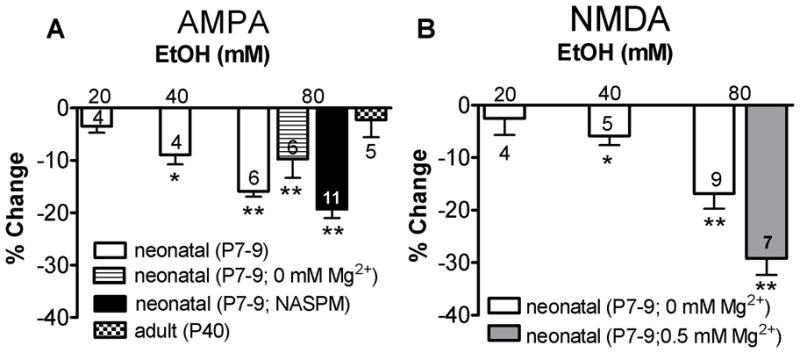
EtOH dose dependently inhibits AMPAR- and NMDAR-mediated fEPSPs. (A) The effects of 20, 40, and 80 mM EtOH are shown as percent inhibition from the average of baseline and washout. AMPAR-mediated fEPSPs were recorded in the presence of gabazine (10 μM) and AP5 (50 μM) in standard ACSF. In addition, AMPAR-mediated fEPSPs were recorded in Mg2+-free ACSF. Also illustrated is the effect of EtOH on AMPAR-mediated fEPSPs in presence of NASPM, as well as the effect of EtOH on AMPAR-mediated fEPSPs in slices from adult rats. (B) Effect of EtOH on NMDAR-mediated fEPSPs recorded in Mg2+-free ACSF, and NMDAR fEPSPs recorded in 0.5 mM Mg2+ ACSF. *p<0.05, and **p<0.01 by one-sample t-test vs. zero. See text for ANOVA results.
Table 1.
Summary of Effects of EtOH on Synaptic Event Area
| Experiment | Event | % change | SEM | One sample t-test vs. zero |
|---|---|---|---|---|
| AMPA | ||||
| 20 mM EtOH | fEPSP | −3.77 | 1.70 | N.S. |
| 40 mM EtOH | fEPSP | −8.61 | 0.83 | p<0.01 |
| 80 mM EtOH | fEPSP | −16.10 | 1.30 | p<0.01 |
| 80 mM EtOH (0 Mg2+) | fEPSP | −15.37 | 4.12 | p<0.05 |
| 80 mM EtOH (NASPM) | fEPSP | −21.38 | 1.57 | p<0.01 |
| 80 mM EtOH (P40 Animal)a | fEPSP | −3.17 | 3.22 | N.S. |
| 80 mM EtOH Perforated-Patch | eEPSC | −9.59 | 1.20 | p<0.01 |
| 80 mM EtOH Whole-cell | eEPSC | −3.97 | 9.72 | N.S. |
| AMPA (Pulse 2 / Pulse 1) | ||||
| 80 mM Control | fEPSP | −2.89 | 3.18 | N.S. |
| 80 mM EtOHb | fEPSP | −5.63 | 2.39 | p<0.05 |
| 80 mM EtOH (Perforated-Patch) | eEPSC | −3.28 | 2.30 | N.S. |
| 80 mM EtOH (Whole-Cell) | eEPSC | 0.10 | 5.20 | N.S. |
| NMDA | ||||
| 20 mM EtOH (0 Mg2+) | fEPSP | −6.15 | 0.72 | p<0.01 |
| 40 mM EtOH (0 Mg2+) | fEPSP | −23.26 | 10.80 | N.S. |
| 80 mM EtOH (0 Mg2+) | fEPSP | −21.55 | 2.34 | p<0.01 |
| 80 mM EtOH (0.5 Mg2+)c | fEPSP | −34.40 | 2.49 | p<0.01 |
| LTP | ||||
| Control | fEPSP | 13.20 | 4.51 | p<0.01 |
| 40 mM EtOHb | fEPSP | 9.30 | 2.49 | p<0.01 |
| 40 mM EtOH (washout)d | fEPSP | 13.50 | 5.39 | p<0.05 |
| 80 mM EtOH | fEPSP | 2.40 | 3.28 | N.S. |
| 80 mM EtOH (washout)e | fEPSP | 18.50 | 6.00 | p<0.05 |
All Other recordings were performed in slices from P7–9 rat pups
Not significantly different from control (by unpaired t-test)
Significantly different from 80 mM EtOH 0 Mg2+ (by unpaired t-test)
Not significantly different than LTP in the presence of 40 mM EtOH (by paired t-test)
Significantly different than LTP in the presence of 80 mM EtOH (by paired t-test)
To determine if Ca2+ influx through Ca2+-permeable AMPARs alters EtOH sensitivity, we blocked these receptors with the synthetic polyamine NASPM (100 μM) and tested the effects of EtOH on the remaining responses (i.e. mediated by Ca2+ impermeable AMPARs). NASPM inhibited the fEPSP amplitude by 18.46 ± 2.85% (n=11; p<0.01 by one-sample t-test vs. zero; Fig. 3). In presence of NASPM, EtOH rapidly and reversibly inhibited fEPSP amplitude by 19.32 ± 1.66% (n=11; p<0.01 by one-sample t-test vs. zero; Fig. 3 and 5A).
Figure 3.

EtOH sensitivity of AMPAR-mediated fEPSPs is not affected by inhibition of Ca2+ permeable AMPARs. (A) Sample traces demonstrating the effects of NASPM (100 μM) on AMPAR-mediated fEPSPs (10 μM gabazine and 50 μM AP5). Also shown is the EtOH (80 mM)-induced reversibly inhibition of fEPSPs in the presence of NASPM. (B) Time course of the effect of NASPM and EtOH.
NMDAR-mediated fEPSPs were recorded in Mg2+-free ACSF, as described above. Acute EtOH (80 mM) application reversibly inhibited NMDAR-mediated fEPSPs (Fig. 4A). The percent inhibition from the average of baseline and washout was 16.81 ± 2.91% (n=9; p<0.01 by one sample t-test vs. zero; Fig. 4C and 5B). Because Mg2+ has been shown to modulate NMDAR sensitivity to EtOH, we measured its effect on NMDAR-mediated fEPSPs in ACSF containing 0.5 mM Mg2+ and 2.5 mM Ca2+ (Fig. 4B) Under these conditions, we found that 80 mM EtOH inhibited the NMDAR-mediated fEPSPs by 29.18 ± 3.16% (n=7; p<0.01 by one-sample t-test vs. zero; Fig. 4D and 5B). This effect was significantly larger when compared to NMDAR-mediated fEPSPs recorded in Mg2+-free ACSF (p<0.05 by one-way ANOVA followed by Bonferroni post hoc test; Fig. 5B).
Figure 4.

EtOH inhibits NMDAR-mediated fEPSPs. Sample traces of inhibitory effects of 80 mM EtOH on the amplitude of NMDAR-mediated fEPSPs recorded in the presence of gabazine (10 μM), NBQX (10μM), in Mg2+-free ACSF (A) and 0.5 mM Mg2+ ACSF (B). Time courses of the effects of EtOH in Mg2+-free ACSF (C) and 0.5 mM Mg2+ ACSF (D).
Fig. 5 summarizes these effects of 80 mM EtOH on fEPSP amplitude, and also shows the effects of lower EtOH concentrations. Small but significant effects on AMPAR- and NMDAR-mediated fEPSP amplitudes were detected with 40 mM EtOH; however, no significant effects were observed with 20 mM EtOH. Dose dependent effects of EtOH revealed significant differences between 80 mM vs. 20 mM and 40 mM EtOH for both AMPAR- and NMDAR-mediated fEPSPs (p<0.05; by one-way ANOVA followed by Bonferroni post hoc test).
Table 1 summarizes the effects of EtOH on field, whole-cell and perforated patch fEPSP or eEPSC recordings analyzed for event area. In virtually all cases, these results were similar to those obtained with amplitude analysis, with the exception that NMDAR-mediated fEPSP area was significantly inhibited by 20 mM EtOH (n=4; p<0.05 by one-sample t-test vs. zero) but not by 40 mM EtOH exposure (n=5; N.S. by one-sample t-test vs. zero; Table 1) in ACSF lacking Mg2+.
Acute EtOH does not affect glutamate release or the presynaptic volley
To assess the effects of EtOH on glutamate release, paired-pulse plasticity of AMPAR-mediated fEPSPs was assessed (Fig. 6). Recordings were performed in the presence of gabazine (10 μM) and AP5 (50 μM). The percent change in the paired-pulse ratio of control recordings (−2.9 ± 11.6%; relative to the average of the baseline and washout periods in the same slice) was not significantly different from the percent change in the paired-pulse ratio (4.33 ± 1.71%) recordings obtained in presence of 80 mM EtOH (n=7–10; N.S. by unpaired t-test). Paired-pulse ratios of AMPAR-mediated eEPSCs were also recorded under whole-cell patch-clamp and perforated-patch conditions. EtOH (80 mM) did not significantly affect the paired-pulse ratios of AMPAR-mediated eEPSCs (whole cell = 0.1 ± 5.2 % data not shown; perforated-patch = −3.28 ± 2.3%; Fig 6E; N.S. by one sample t-test v. zero; n=4 for each configuration).
Figure 6.

Paired-pulse ratios of AMPAR-mediated fEPSPs and eEPSCs were not affected by acute EtOH (80 mM) exposure. Recordings were performed in the presence of gabazine (10 μM) and AP5 (50 μM). The interpulse interval was 50 ms. (A) Sample fEPSP traces from control (i.e. no EtOH) and (B) EtOH groups. (C) Time course of the fEPSP paired-pulse ratio for these groups. (D) Sample traces of perforated-patch eEPSC paired-pulse recordings. (E) Summary of the EtOH-induced percent change of eEPSC1 amplitude and paired-pulse ratio. *p<0.01 by one sample t-test vs. 100%.
In the whole-cell configuration, 80 mM EtOH did not significantly inhibit eEPSC1 amplitude (−11.97 ± 7.90% from the average of baseline and washout by one sample t-test v. zero; n=4, data not shown). In the perforated-patch configuration, 80 mM EtOH had a small but significant inhibitory effect on eEPSC1 amplitude (−4.84 ± 1.3% from the average of baseline and washout by one sample t-test v. zero; n=4, Fig. 6D–E) and area (Table 1). The percent inhibition of eEPSC1 amplitude (but not area) was significantly smaller than that observed in the AMPAR-mediated fEPSP recordings (p<0.01 by unpaired t-test). In perforated-patch experiments, EtOH did not significantly affect the holding current (4.96 ± 6.45% of control), membrane time constant (13.2 ± 8.57% of control) and membrane resistance (4.9 ± 2.95% of control) from the average of baseline and washout (n=4; N.S. by one sample t-test v. zero; membrane time constant and membrane resistance were measured from responses to a 10 mV hyperpolarizing step of 10 ms duration). Similar results were found for the holding current (6.7 ± 8.9 % of control), membrane time constant (13.57 ± 6.69% of control) and membrane resistance (4.38 ± 2.36% of control) in whole-cell recordings (n=4; N.S. by one sample t-test v. zero).
To assess the effect of EtOH on presynaptic excitability, measurements of the presynaptic volley were performed (Fig. 7). Recordings were performed in the presence of gabazine (10 μM), NBQX (10 μM), and AP5 (50 μM). EtOH (80 mM) did not significantly affect the presynaptic volley (21.9 ± 10.10%; n=7; N.S. by one-sample t-test vs. zero). As expected, the presynaptic volley was blocked by the Na+ channel antagonist, TTX (0.5 μM). After TTX application, a residual stimulus artifact could be observed, which was subtracted to obtain the isolated presynaptic volley response.
Figure 7.

Acute EtOH (80 mM) application did not affect the presynaptic volley amplitude. Recordings were performed in the presence of gabazine (10 μM), NBQX (10 μM), and AP5 (50 μM). (A) Sample traces and time course (B) are shown. Presynaptic volleys were blocked by TTX (0.5 μM). The residual signal corresponds to a portion of the stimulus artifact.
EtOH acutely inhibited LTP induction
To determine the effect of acute EtOH exposure on the induction of LTP, a high-frequency stimulation protocol was used. Experiments were performed in the presence of gabazine (10 μM) in standard ACSF. LTP induction was blocked by AP5 (50 μM) plus MK-801 (50 μM), demonstrating that it was NMDAR-dependent (10.2 ± 14.92% change in fEPSP amplitude; n=9; N.S. by one-sample t-test vs. zero; data not shown). In the absence of EtOH, high-frequency stimulation increased the amplitude of the AMPAR-mediated fEPSP by 25.3 ± 7.33% from baseline (n=6; p<0.05; by one-sample t-test vs. zero; Fig. 8A). For EtOH studies, the LTP induction protocol was delivered in the middle of a 10 min EtOH application. EtOH 40 mM (Fig. 8B) did not significantly inhibit the induction of LTP; the fEPSP amplitude increase in the presence of EtOH (40 mM) was 22.5 ± 4.18% (p<0.01; n=7 by one-sample t-test vs. zero). The LTP induction protocol was repeated in the same slice in the absence of EtOH, and it induced an increase of 29.7 ± 8.11% from baseline, which was not significantly different than the increase in the presence of 40 mM EtOH (N.S. by paired t-test).
Figure 8.

Acute EtOH (80 mM, but not 40 mM) inhibited the induction of LTP in the CA1 hippocampus of P7–9 animals. (A) LTP induction in control slices. The LTP induction protocol (represented by the dashed lines) consisted of 3 one second 100 Hz trains (trains were separated by 5s). Recordings were obtained in the presence of gabazine (10 μM). Effect of 40 mM (B) and 80 mM (C) EtOH on LTP. The first LTP induction protocol was delivered in the presence of EtOH, and repeated in its absence. (D) Effects of continuous 80 mM EtOH exposure on LTP induction; also shown are control recordings obtained from slices in which LTP was not induced. Note different time scales in x-axis (A–D).
For 80 mM EtOH (Fig. 8C), the percent change in AMPAR-mediated fEPSP amplitude was 6.0 ± 5.39% (n=8; N.S. by one-sample t-test vs. zero); the LTP induction protocol was then repeated in the same slice in the absence of EtOH, resulting in a 24.8 ± 8.43% increase in the amplitude of the fEPSP from baseline (p<0.05 by one-sample t-test vs. zero).
We also measured the effect of longer exposure to 80 mM EtOH on LTP. A stable baseline of AMPAR-mediated fEPSPs was recorded and EtOH was bath–applied (Fig. 8D), causing a 14.01 ± 3.94% reduction (at t = 13–15 min) in the fEPSP amplitude (n=6; p<0.05 by one sample t-test from baseline). This effect was followed by gradual run-down of the responses, which was observed independently of whether or not high-frequency stimulation was delivered (Fig. 8D). High-frequency stimulation failed to potentiate the fEPSP under these conditions (Fig. 8D).
Table 1 summarizes the effects of EtOH on LTP of fEPSP area. In all cases, the effects of EtOH were similar to its effects on fEPSP amplitude.
Discussion
To the best of our knowledge, this is the first characterization of the acute effects of EtOH on glutamatergic transmission and plasticity in the rat CA1 hippocampal region during the third trimester-equivalent period of development. Two effects of EtOH were demonstrated: 1) inhibition of both AMPAR- and NMDAR-mediated responses and 2) inhibition of NMDAR-dependent LTP. These findings add to growing evidence indicating that alterations in excitatory amino acid-mediated neurotransmission play a role in the pathophysiology of fetal alcohol spectrum disorder (Berman and Hannigan, 2000; Costa et al., 2000b; Kimura et al., 2000; Olney, 2004; Thomas and Riley, 1998; Valenzuela et al., 2007).
Acute EtOH exposure inhibits AMPAR-mediated fEPSPs
Acute inhibitory effects of EtOH have been observed on AMPAR-mediated responses evoked by exogenous agonist application in developing and mature neurons in different brain regions (Carta, et al, 2003; Costa, et al, 2000; Hsiao, and Frye, 2003; Lovinger, et al, 1989; Lu, and Yeh, 1999; Martin, et al, 1995; Moykkynen, et al, 2003; Nie, et al, 1994; Valenzuela, et al, 1998). EtOH has also been shown to acutely inhibit the function of non-NMDA glutamate receptors in central amygdala synapses from adult rats (Zhu et al., 2007; Roberto et al., 2004). In contrast to these studies, AMPAR function has been shown to be relatively insensitive to acute EtOH exposure in CA3 pyramidal and dentate granule neurons from juvenile or adult rats (Weiner et al., 1999; Ariwodola et al., 2003). Similar findings have been obtained in the CA1 hippocampal subfield, where Lovinger et al. (1990) were the first to study the effect of EtOH on synaptic AMPAR function using the acute slice preparation; these investigators reported that 50 mM EtOH exposure did not significantly affect non-NMDAR-mediated fEPSPs and that 100 mM EtOH reduced the amplitude of these events by only ~10%. Similarly, Randall et al. (1995) found that non-NMDAR-mediated synaptic responses were not affected by 22 mM EtOH at Schaffer collateral-to-CA1 synapses from 130–160 g rats, and Carta et al. (2003) demonstrated that 80 mM EtOH did not inhibit AMPAR-mediated EPSCs in CA1 pyramidal neurons from P21–40 rats. Durand et al. (1981) also demonstrated that EtOH (100 mM) had little effect on the amplitude of fEPSPs that were, in part, mediated by AMPARs in the CA1 hippocampal region of adult rats. A 30 min application of 100 mM EtOH inhibited non-NMDAR-mediated fEPSPs by ~20% in the CA1 hippocampus of C56Bl/6 mice (Gordey et al., 2001). In addition, EtOH (25–75 mM) did not affect the amplitude of AMPAR-mediated miniature EPSCs in CA1 pyramidal neurons of the rat hippocampus from P12–20 rats (Hendricson et al., 2003).
In contrast to the findings of the studies described above, we found that acute EtOH exposure inhibits non-NMDAR-mediated fEPSPs in the CA1 region of neonatal (but not adult) rats, where EtOH had a small but significant effect at a concentration as low as 40 mM. Under our recording conditions, non-NMDAR-mediated fEPSPs were abolished by the selective AMPAR antagonist, GYKI-53655, indicating that kainate receptors do not contribute to the generation of these events. It should be noted, however, that the effects of EtOH on AMPAR function were either undetectable or less robust under somatic voltage-clamp conditions. In the whole-cell configuration, 80 mM EtOH had no significant effect on eEPSC amplitude, and, in the perforated-patch configuration, it had a smaller action than in dendritic field recordings. These findings suggest that: 1) intracellular signal transduction pathways that are disrupted during whole-cell recording are required for EtOH’s action, 2) EtOH may preferentially act on dendritic AMPARs that are not sampled under somatic patch-clamp recordings (Williams, and Mitchell, 2008), and/or 3) EtOH may affect non-AMPAR-mediated components of the fEPSP–for instance, those mediated by leak channels or capacitive currents. This possibility is unlikely given that EtOH did not affect the holding current, membrane time constant or membrane resistance.
Previous studies have suggested that AMPAR subunit composition and postsynaptic proteins involved in AMPAR function are developmentally regulated in the hippocampus (CA1), which may explain the increased EtOH sensitivity of AMPARs at P7–9 (Elias et al., 2008; Zhu et al., 2000; Fukaya et al., 2006; Rouach et al., 2005; Morimoto-Tomita et al., 2009; Sager et al., 2009). In addition, other factors, such as neurosteroids, play a role in the effects of EtOH in developing CA1 pyramidal neurons. Studies from our laboratory previously showed that EtOH strengthens AMPAR-mediated synaptic transmission in CA1 pyramidal neurons from P3–4 (but not P6–10 rats) under conditions of minimal stimulation; this effect was mediated by an endogenous pregnenolone sulfate-like neurosteroid that acts at the presynaptic level (Mameli et al., 2005; Mameli and Valenzuela, 2006; reviewed in Valenzuela et al., 2007). Thus, EtOH can either potentiate or inhibit CA1 AMPAR-mediated synaptic transmission during the early or late portions of the third trimester-equivalent period of rat development. We previously detected relatively potent inhibitory effects of EtOH on AMPAR-mediated synaptic transmission in CA3 pyramidal neurons of the developing hippocampus (Mameli et al., 2005), suggesting that immature CA1 and CA3 AMPARs may share some of the properties that confer EtOH sensitivity and this should be explored in the future. However, synaptic AMPARs are not sensitive to EtOH in all developing brain regions and under all experimental conditions, as shown by the voltage-clamp studies presented here and our recently reported results with neocortical layer II and III pyramidal neurons from P7–9 rats (Sanderson et al., 2009).
Ca2+ influx through AMPARs is not required for modulation by EtOH
A previous study with recombinant AMPARs expressed in Xenopus oocytes found that EtOH inhibited Ca2+-permeable (i.e. containing GluR1 + GluR3 subunits) and Ca2+-impermeable (containing GluR2 + GluR3 subunits) receptors to a similar extent. However, for GluR1 + GluR3 receptors, greater EtOH inhibition was observed when the only permeant ion was Ca2+ (Dildy-Mayfield and Harris, 1995). These findings suggest that increased intracellular Ca2+ levels modulate AMPAR sensitivity to EtOH. Consequently, we tested the effect of EtOH on synaptic AMPARs in the presence of an antagonist of Ca2+-permeable AMPARs. The functional expression ratio of Ca2+-permeable to -impermeable AMPARs was approximately 1/5, consistent with a previous report (Pickard et al., 2000). In the presence of NASPM, an inhibitor of Ca2+-permeable AMPARs, EtOH reduced the fEPSP amplitude to a similar extent as controls. These results suggest that EtOH does not preferentially inhibit Ca2+-permeable AMPARs in developing CA1 hippocampal synapses, and Ca2+ influx through Ca2+ -permeable AMPARs does not affect the sensitivity to EtOH. However, we were unable to directly test the effects of EtOH on Ca2+-permeable AMPARs due to the lack of availability of pharmacological agents to isolate these receptors. Studies using molecular biological approaches and/or genetically modified mice could be used to further address this issue.
Acute EtOH exposure inhibits NMDAR-mediated fEPSPs
We found that acute EtOH inhibits NMDAR-mediated fEPSPs in the CA1 region of neonatal rats; small but significant effects were detected at EtOH concentrations of 40 mM (~5%) and 80 mM (~15%). Our findings are in general agreement with those of Gordey et al. (2001) who showed that 100 mM EtOH inhibits NMDAR-mediated fEPSPs by ~20% in the CA1 region of slices from P4–7 C56Bl/6 mice. Other slice electrophysiological studies performed in our laboratory have investigated the modulation of NMDARs by EtOH in pyramidal neurons from another hippocampal subfield; i.e., the CA3 region. In CA3 pyramidal neurons from P5 rats, currents evoked by exogenous application of NMDA were significantly inhibited (~10%) by 75 mM EtOH, but were unaffected by lower EtOH concentrations (Mameli et al., 2005). In slices from older rats (P9–10), 50 mM EtOH significantly inhibited (~8%) exogenous NMDA-evoked currents in these neurons (Mameli et al., 2005). Although these results are in general agreement with the findings of the present study, it must be kept in mind that currents evoked by exogenous NMDA are likely mediated by synaptic and extrasynaptic NMDARs. We examined the effect of EtOH on NMDAR currents triggered by synaptic glutamate release in CA3 pyramidal neurons and found that these are inhibited (up to 40%) by 50 mM EtOH in slices from P3–10 rats; however, this effect is likely a consequence of an EtOH-induced decrease of glutamate release as discussed in more detail below (Mameli et al., 2005). In the present study, we did not detect an effect of EtOH on glutamate release in the CA1 region, but did find an effect on NMDAR-mediated fEPSPs. These findings indicate that the mechanism by which EtOH modulates NMDARs in the neonatal hippocampus is regionally specific; i.e., it predominantly inhibits NMDAR-mediated responses in the CA1 and CA3 region via postsynaptic and presynaptic mechanisms, respectively.
The magnitude of inhibition of synaptic NMDAR-mediated responses in neonatal CA1 pyramidal neurons reported here is slightly lower than that observed in some studies with acute slices from juvenile or adult animals. For instance, Lovinger et al. (1990) demonstrated that 25–100 mM EtOH inhibits NMDAR-mediated fEPSPs by ~20–50% in slices from adult rats. Swartzwelder et al. (1995) reported that CA1 NMDAR-mediated fEPSPs were inhibited by ~30% and 40% in P20–25 rat slices acutely exposed to 30 and 100 mM EtOH, respectively. Similarly, 80–100 mM EtOH was shown to inhibit CA1 NMDAR-mediated fEPSPs by ~50% in slices from P21–26 C57BL/6J mice (Suvarna et al., 2005; Yaka et al., 2003) and by ~20–30% in slices from adult +/fynZ mice (Miyakawa et al., 1997). The relative insensitivity to EtOH of NMDARs in neonatal CA1 pyramidal neurons could be attributed to expression of NR2D-containing NMDARs, which have been shown be less sensitive to EtOH inhibition (Chu et al., 1995). However, NMDARs containing the NR2D subunit may not be present in the CA1 hippocampus from rats older than P6 (Mameli et al., 2005). Therefore, differences in the subunit composition, phosphorylation state or association with other proteins between NMDARs in neurons from neonatal vs. more mature animals could be responsible for age-dependent differences in EtOH sensitivity in the CA1 region (Elias et al., 2008; Yasuda et al., 2003). Another reason could be related to Mg2+ concentration in the ACSF. Morrisett et al. (1991) reported that EtOH inhibits NMDAR-mediated fEPSPs in slices from adult rats with an EC50 of ~50 mM in the presence of 1 mM Mg2+ and an EC50 of 100 mM in its absence. Jin et al. (2008) also observed enhanced inhibition of recombinant NMDARs in the presence of Mg2+. In agreement with these findings, we found that 80 mM EtOH had a significantly greater inhibitory effect on NMDAR-mediated fEPSPs when recorded in the presence of 0.5 mM Mg2+ than under 0 mM Mg2+ conditions.
Lack of Presynaptic Effects of EtOH
We show here that EtOH acutely reduces the amplitude of both AMPAR- and NMDAR-mediated fEPSPs in neonatal CA1 pyramidal neurons. However, it did not affect the paired-pulse ratio of AMPAR-mediated fEPSPs or eEPSCs under whole-cell patch-clamp and perforated-patch clamp conditions. Since alterations in paired-pulse plasticity are an indicator of changes in transmitter release (Zucker and Regehr, 2002), these findings suggests that EtOH does not affect glutamate release at Schaffer collateral-to-CA1 pyramidal neuron synapses from P7–9 rats. As mentioned above, EtOH induced a neurosteroid-dependent increase in glutamate release at these synapses in slices from younger rats (P3–4) (Mameli et al., 2005; Mameli and Valenzuela, 2006; Reviewed in Valenzuela et al., 2007). Therefore, acute EtOH exposure exerts developmentally-regulated actions on glutamate release in this hippocampal region. A similar observation was made in the CA3 hippocampal region where EtOH acutely inhibits glutamate release in slices from rats younger than P10; this effect is mediated by inhibition of presynaptic N-type Ca2+ channels (Mameli et al., 2005). Previous studies have shown that EtOH affects glutamate release in several brain regions, including the CA1 hippocampal region from more mature rats (reviewed in Siggins et al., 2005). In cultured hippocampal CA1 neurons, EtOH decreased mEPSC frequency without affecting amplitude, suggesting that it inhibits glutamate release; this effect was shown to be mediated by endocannabinoid release triggered by an elevation in postsynaptic Ca2+ levels (Basavarajappa et al., 2008). EtOH inhibited KCl-induced vesicular FM1–43 destaining in the CA1 stratum radiatum of P21–28 rats, and this effect was occluded by antagonists of N-type and P/Q-type Ca2+ channels; EtOH also reduced frequency of AMPAR-mediated mEPSCs in the presence, but not in the absence of KCl (Maldve et al., 2004). Moreover, EtOH decreased both the frequency of asynchronous NMDAR-mediated mEPSCs and the paired-pulse ratio of NMDAR-mediated EPSCs in CA1 pyramidal neurons (Hendricson et al., 2004). Future experiments should determine whether the lack of a presynaptic effect of EtOH in the neonatal CA1 region is, in part, a consequence of a unique pattern of voltage-gated Ca2+ channel expression in glutamatergic axonal terminals.
Acute EtOH Exposure Inhibits NMDAR-dependent LTP
We found that the induction of LTP was suppressed by application of 80 mM EtOH. This was not observed at a concentration of 40 mM. These findings are in agreement with the literature and indicate that exposure to relatively high EtOH concentrations is required to inhibit LTP in the CA1 hippocampal region (Izumi et al., 2007; Schummers et al., 1997; Sinclair and Lo, 1986; Sugiura et al., 1995; Tokuda et al., 2007; Zhang and Morrisett, 1993; Zhang et al., 2005). One paper showed that LTP was virtually abolished by 60 mM EtOH in slices from P15–25 rats, but not P70–100 rats (Swartzwelder et al., 1995). However, other studies have reported more potent effects of EtOH on LTP in the rat CA1 hippocampal region than those reported here. This could be a consequence of differences in experimental conditions (i.e., slice preparation protocol, recording temperature, LTP induction paradigm, and age of rats) (Randall et al., 1995; Blitzer et al., 1990; Pyapali et al., 1999; Fujii et al., 2008). For instance, in some but not all cases, LTP was induced by theta burst stimulation, and in other studies there were variances in the number of pulses in the LTP induction train (Pyapali et al., 1999; Fujii et al., 2008). Future studies should address the effect of EtOH on neonatal LTP evoked by different patterns of stimulation.
The mechanism by which EtOH blocks LTP in the developing CA1 hippocampus may involve inhibition of NMDARs (Blitzer et al., 1990) because the dose-dependency of EtOH’s effect on NMDAR-mediated fEPSPs matches that of its effect on LTP. Since our studies were performed in presence of gabazine, we did not investigate the contribution of an EtOH-induced increase in GABAA receptor-mediated transmission. However, future studies should examine a potential participation of GABAA receptors in the mechanism of action of EtOH. Studies suggest that EtOH could indirectly inhibit NMDAR-mediated responses and LTP by increasing GABAergic transmission (Schummers et al., 1997; Schummers and Browning, 2001). GABAergic neurosteroids were recently shown to mediate the effects of EtOH on LTP (Izumi et al., 2007). During the third-trimester equivalent, GABAA receptors will likely have a dual excitatory and inhibitory action and may actually facilitate LTP induction under some conditions (Caillard et al., 1999). In addition, the inhibitory actions of EtOH on LTP were occluded by an angiotensin 1 receptor blocker, and this receptor may also be involved in the mechanism by which EtOH affects LTP during the neonatal period of rat development (Wayner et al., 1993). Inhibition of AMPARs could also contribute to LTP blockade because these receptors are needed for removal of Mg2+ block from NMDARs (reviewed in Kerchner and Nicoll, 2008). LTP during this neonatal period is not dependent on CaMKII as in more mature animals, but requires protein kinase A (Yasuda et al., 2003). EtOH may also interfere with these pathways uniquely involved in developmental LTP (Newton and Messing, 2006).
A limitation of our study is that the EtOH concentration increased in the recording chamber over a short period of time (2–3 min). Human consumption of alcoholic beverages does not cause such a sudden increase in brain EtOH levels, even during binge drinking. Tokuda et al., (2007) showed that a gradual increase in EtOH concentration induces the emergence of an NMDAR- and Ca2+ channel-independent form of synaptic plasticity that depends on Ca2+ release from internal stores. Future studies should examine if EtOH produces a similar effect in developing neurons. Moreover, to further assess the effects of gradual increases in EtOH concentrations, the effects of EtOH exposure on developmental LTP in vivo should also be explored in the future.
Implications of Findings
Plasticity of glutamatergic transmission may contribute to the maturation of developing neuronal networks, for example, by stabilizing newly formed synapses (Constantine-Paton and Cline, 1998; Leinekugel, 2003; Zhu et al., 2000; Molnar et al., 2002). Alterations in these processes likely contribute to the long-lasting effects of EtOH on the CA1 hippocampus of rodents exposed to EtOH during the third trimester-equivalent of human pregnancy. For instance, abnormal circuit formation secondary to LTP blockade in the CA1 region could explain the deficits in input/output curves detected in this hippocampal subfield in P45–60 rats that were exposed to EtOH vapor at P4–6 (Bellinger et al., 1999). EtOH inhibition of NMDAR-mediated responses could trigger apoptosis in the hippocampal CA1 region (Ikonomidou et al., 2000), although this mechanism may not apply to other regions such as layer II/III of neocortex or CA3 pyramidal neurons (Mameli et al., 2005; Sanderson et al., 2009). Alternatively, EtOH-induced alterations in glutamatergic transmission could contribute to the neurogenesis or neuronal proliferation deficits that may be responsible for decreased pyramidal neuronal numbers in the CA1 hippocampal region of rats exposed to EtOH during the neonatal period (Livy et al., 2003; Miller, 1995; Martel et al., 2009). In addition, EtOH-induced NMDAR inhibition during the neonatal period could contribute to the compensatory upregulation of these receptors that has been detected during EtOH withdrawal (Thomas et al., 2004). It can be concluded that therapeutic interventions that prevent the EtOH-induced inhibition of glutamatergic transmission and plasticity in the CA1 region during the third trimester of pregnancy could mitigate the learning and memory alterations associated with fetal alcohol spectrum disorder.
Acknowledgments
This work was supported by NIH grants RO1-AA15614, T32-AA14127, F30-AA017813-01, and the UNM-SOM M.D./Ph.D program. We thank Drs. L. D. Partridge and K. K. Caldwell for critically reading the manuscript.
References
- Adesnik H, Li G, During MJ, Pleasure SJ, Nicoll RA. NMDA receptors inhibit synapse unsilencing during brain development. Proc Natl Acad Sci U S A. 2008;105:5597–5602. doi: 10.1073/pnas.0800946105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariwodola OJ, Crowder TL, Grant KA, Daunais JB, Friedman DP, Weiner JL. Ethanol modulation of excitatory and inhibitory synaptic transmission in rat and monkey dentate granule neurons. Alcohol Clin Exp Res. 2003;27:1632–1639. doi: 10.1097/01.ALC.0000089956.43262.17. [DOI] [PubMed] [Google Scholar]
- Basavarajappa BS, Ninan I, Arancio O. Acute ethanol suppresses glutamatergic neurotransmission through endocannabinoids in hippocampal neurons. J Neurochem. 2008;107:1001–1013. doi: 10.1111/j.1471-4159.2008.05685.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellinger FP, Bedi KS, Wilson P, Wilce PA. Ethanol exposure during the third trimester equivalent results in long-lasting decreased synaptic efficacy but not plasticity in the CA1 region of the rat hippocampus. Synapse. 1999;31:51–58. doi: 10.1002/(SICI)1098-2396(199901)31:1<51::AID-SYN7>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Berman RF, Hannigan JH. Effects of prenatal alcohol exposure on the hippocampus: spatial behavior, electrophysiology, and neuroanatomy. Hippocampus. 2000;10:94–110. doi: 10.1002/(SICI)1098-1063(2000)10:1<94::AID-HIPO11>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Blitzer RD, Gil O, Landau EM. Long-term potentiation in rat hippocampus is inhibited by low concentrations of ethanol. Brain Res. 1990;537:203–208. doi: 10.1016/0006-8993(90)90359-j. [DOI] [PubMed] [Google Scholar]
- Caillard O, Ben-Ari Y, Gaiarsa JL. Mechanisms of induction and expression of long-term depression at GABAergic synapses in the neonatal rat hippocampus. J Neurosci. 1999;19:7568–7577. doi: 10.1523/JNEUROSCI.19-17-07568.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carta M, Ariwodola OJ, Weiner JL, Valenzuela CF. Alcohol potently inhibits the kainate receptor-dependent excitatory drive of hippocampal interneurons. Proc Natl Acad Sci U S A. 2003;100:6813–6818. doi: 10.1073/pnas.1137276100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu B, Anantharam V, Treistman SN. Ethanol inhibition of recombinant heteromeric NMDA channels in the presence and absence of modulators. J Neurochem. 1995;65:140–148. doi: 10.1046/j.1471-4159.1995.65010140.x. [DOI] [PubMed] [Google Scholar]
- Citri A, Malenka RC. Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology. 2008;33:18–41. doi: 10.1038/sj.npp.1301559. [DOI] [PubMed] [Google Scholar]
- Constantine-Paton M, Cline HT. LTP and activity-dependent synaptogenesis: the more alike they are, the more different they become. Curr Opin Neurobiol. 1998;8:139–148. doi: 10.1016/s0959-4388(98)80017-2. [DOI] [PubMed] [Google Scholar]
- Costa ET, Olivera DS, Meyer DA, Ferreira VM, Soto EE, Frausto S, Savage DD, Browning MD, Valenzuela CF. Fetal alcohol exposure alters neurosteroid modulation of hippocampal N-methyl-D-aspartate receptors. J Biol Chem. 2000a;275:38268–38274. doi: 10.1074/jbc.M004136200. [DOI] [PubMed] [Google Scholar]
- Costa ET, Savage DD, Valenzuela CF. A review of the effects of prenatal or early postnatal ethanol exposure on brain ligand-gated ion channels. Alcohol Clin Exp Res. 2000b;24:706–715. [PubMed] [Google Scholar]
- Dildy-Mayfield JE, Harris RA. Ethanol inhibits kainate responses of glutamate receptors expressed in Xenopus oocytes: role of calcium and protein kinase C. J Neurosci. 1995;15:3162–3171. doi: 10.1523/JNEUROSCI.15-04-03162.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand D, Corrigall WA, Kujtan P, Carlen PL. Effect of low concentrations of ethanol on CA1 hippocampal neurons in vitro. Can J Physiol Pharmacol. 1981;59:979–984. doi: 10.1139/y81-149. [DOI] [PubMed] [Google Scholar]
- Durand GM, Kovalchuk Y, Konnerth A. Long-term potentiation and functional synapse induction in developing hippocampus. Nature. 1996;381:71–75. doi: 10.1038/381071a0. [DOI] [PubMed] [Google Scholar]
- Elias GM, Elias LA, Apostolides PF, Kriegstein AR, Nicoll RA. Differential trafficking of AMPA and NMDA receptors by SAP102 and PSD-95 underlies synapse development. Proc Natl Acad Sci U S A. 2008;105:20953–20958. doi: 10.1073/pnas.0811025106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiala JC, Feinberg M, Popov V, Harris KM. Synaptogenesis via dendritic filopodia in developing hippocampal area CA1. J Neurosci. 1998;18:8900–8911. doi: 10.1523/JNEUROSCI.18-21-08900.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii S, Yamazaki Y, Sugihara T, Wakabayashi I. Acute and chronic ethanol exposure differentially affect induction of hippocampal LTP. Brain Res. 2008;1211:13–21. doi: 10.1016/j.brainres.2008.02.052. [DOI] [PubMed] [Google Scholar]
- Fukaya M, Tsujita M, Yamazaki M, Kushiya E, Abe M, Akashi K, Natsume R, Kano M, Kamiya H, Watanabe M, Sakimura K. Abundant distribution of TARP gamma-8 in synaptic and extrasynaptic surface of hippocampal neurons and its major role in AMPA receptor expression on spines and dendrites. Eur J Neurosci. 2006;24:2177–2190. doi: 10.1111/j.1460-9568.2006.05081.x. [DOI] [PubMed] [Google Scholar]
- Gordey M, Mekmanee L, Mody I. Altered effects of ethanol in NR2A(DeltaC/DeltaC) mice expressing C-terminally truncated NR2A subunit of NMDA receptor. Neuroscience. 2001;105:987–997. doi: 10.1016/s0306-4522(01)00234-2. [DOI] [PubMed] [Google Scholar]
- Groc L, Gustafsson B, Hanse E. Spontaneous unitary synaptic activity in CA1 pyramidal neurons during early postnatal development: constant contribution of AMPA and NMDA receptors. J Neurosci. 2002;22:5552–5562. doi: 10.1523/JNEUROSCI.22-13-05552.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall BJ, Ripley B, Ghosh A. NR2B signaling regulates the development of synaptic AMPA receptor current. J Neurosci. 2007;27:13446–13456. doi: 10.1523/JNEUROSCI.3793-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton DA, Kodituwakku P, Sutherland RJ, Savage DD. Children with Fetal Alcohol Syndrome are impaired at place learning but not cued-navigation in a virtual Morris water task. Behav Brain Res. 2003;143:85–94. doi: 10.1016/s0166-4328(03)00028-7. [DOI] [PubMed] [Google Scholar]
- Hendricson AW, Sibbald JR, Morrisett RA. Ethanol alters the frequency, amplitude, and decay kinetics of Sr2+-supported, asynchronous NMDAR mEPSCs in rat hippocampal slices. J Neurophysiol. 2004;91:2568–2577. doi: 10.1152/jn.00997.2003. [DOI] [PubMed] [Google Scholar]
- Hendricson AW, Thomas MP, Lippmann MJ, Morrisett RA. Suppression of L-type voltage-gated calcium channel-dependent synaptic plasticity by ethanol: analysis of miniature synaptic currents and dendritic calcium transients. J Pharmacol Exp Ther. 2003;307:550–558. doi: 10.1124/jpet.103.055137. [DOI] [PubMed] [Google Scholar]
- Hsiao SH, Frye GD. AMPA receptors on developing medial septum/diagonal band neurons are sensitive to early postnatal binge-like ethanol exposure. Brain Res Dev Brain Res. 2003;142:89–99. doi: 10.1016/s0165-3806(03)00034-8. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Bittigau P, Ishimaru MJ, Wozniak DF, Koch C, Genz K, Price MT, Stefovska V, Horster F, Tenkova T, Dikranian K, Olney JW. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science. 2000;287:1056–1060. doi: 10.1126/science.287.5455.1056. [DOI] [PubMed] [Google Scholar]
- Izumi Y, Murayama K, Tokuda K, Krishnan K, Covey DF, Zorumski CF. GABAergic neurosteroids mediate the effects of ethanol on long-term potentiation in rat hippocampal slices. Eur J Neurosci. 2007;26:1881–1888. doi: 10.1111/j.1460-9568.2007.05809.x. [DOI] [PubMed] [Google Scholar]
- Jin C, Smothers CT, Woodward JJ. Enhanced ethanol inhibition of recombinant N-methyl-D-aspartate receptors by magnesium: role of NR3A subunits. Alcohol Clin Exp Res. 2008;32:1059–1066. doi: 10.1111/j.1530-0277.2008.00667.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston MV. Clinical disorders of brain plasticity. Brain Dev. 2004;26:73–80. doi: 10.1016/S0387-7604(03)00102-5. [DOI] [PubMed] [Google Scholar]
- Kerchner GA, Nicoll RA. Silent synapses and the emergence of a postsynaptic mechanism for LTP. Nat Rev Neurosci. 2008;9:813–825. doi: 10.1038/nrn2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura KA, Reynolds JN, Brien JF. Ethanol neurobehavioral teratogenesis and the role of the hippocampal glutamate-N-methyl-D-aspartate receptor-nitric oxide synthase system. Neurotoxicol Teratol. 2000;22:607–616. doi: 10.1016/s0892-0362(00)00089-1. [DOI] [PubMed] [Google Scholar]
- Leinekugel X. Developmental patterns and plasticities: the hippocampal model. J Physiol Paris. 2003;97:27–37. doi: 10.1016/j.jphysparis.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Livy DJ, Miller EK, Maier SE, West JR. Fetal alcohol exposure and temporal vulnerability: effects of binge-like alcohol exposure on the developing rat hippocampus. Neurotoxicol Teratol. 2003;25:447–458. doi: 10.1016/s0892-0362(03)00030-8. [DOI] [PubMed] [Google Scholar]
- Lovinger DM, White G, Weight FF. NMDA receptor-mediated synaptic excitation selectively inhibited by ethanol in hippocampal slice from adult rat. J Neurosci. 1990;10:1372–1379. doi: 10.1523/JNEUROSCI.10-04-01372.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovinger DM, White G, Weight FF. Ethanol inhibits NMDA-activated ion current in hippocampal neurons. Science. 1989;243:1721–1724. doi: 10.1126/science.2467382. [DOI] [PubMed] [Google Scholar]
- Lu SM, Yeh HH. Ethanol modulates AMPA-induced current responses of primary somatosensory cortical neurons. Neurochem Int. 1999;35:175–183. doi: 10.1016/s0197-0186(99)00059-5. [DOI] [PubMed] [Google Scholar]
- Maldve RE, Chen X, Zhang TA, Morrisett RA. Ethanol selectively inhibits enhanced vesicular release at excitatory synapses: real-time visualization in intact hippocampal slices. Alcohol Clin Exp Res. 2004;28:143–152. doi: 10.1097/01.ALC.0000106304.39174.AD. [DOI] [PubMed] [Google Scholar]
- Maletic-Savatic M, Malinow R, Svoboda K. Rapid dendritic morphogenesis in CA1 hippocampal dendrites induced by synaptic activity. Science. 1999;283:1923–1927. doi: 10.1126/science.283.5409.1923. [DOI] [PubMed] [Google Scholar]
- Mameli M, Carta M, Partridge LD, Valenzuela CF. Neurosteroid-induced plasticity of immature synapses via retrograde modulation of presynaptic NMDA receptors. J Neurosci. 2005;25:2285–2294. doi: 10.1523/JNEUROSCI.3877-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mameli M, Valenzuela CF. Alcohol increases efficacy of immature synapses in a neurosteroid-dependent manner. Eur J Neurosci. 2006;23:835–839. doi: 10.1111/j.1460-9568.2006.04597.x. [DOI] [PubMed] [Google Scholar]
- Mameli M, Zamudio PA, Carta M, Valenzuela CF. Developmentally regulated actions of alcohol on hippocampal glutamatergic transmission. J Neurosci. 2005;25:8027–8036. doi: 10.1523/JNEUROSCI.2434-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martel MA, Wyllie DJ, Hardingham GE. In developing hippocampal neurons, NR2B-containing N-methyl-D-aspartate receptors (NMDARs) can mediate signaling to neuronal survival and synaptic potentiation, as well as neuronal death. Neuroscience. 2009;158:334–343. doi: 10.1016/j.neuroscience.2008.01.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin D, Tayyeb MI, Swartzwelder HS. Ethanol inhibition of AMPA and kainate receptor-mediated depolarizations of hippocampal area CA1. Alcohol Clin Exp Res. 1995;19:1312–1316. doi: 10.1111/j.1530-0277.1995.tb01617.x. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, Ellis-Davies GC, Nemoto T, Miyashita Y, Iino M, Kasai H. Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat Neurosci. 2001;4:1086–1092. doi: 10.1038/nn736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MW. Generation of neurons in the rat dentate gyrus and hippocampus: effects of prenatal and postnatal treatment with ethanol. Alcohol Clin Exp Res. 1995;19:1500–1509. doi: 10.1111/j.1530-0277.1995.tb01014.x. [DOI] [PubMed] [Google Scholar]
- Miyakawa T, Yagi T, Kitazawa H, Yasuda M, Kawai N, Tsuboi K, Niki H. Fyn-kinase as a determinant of ethanol sensitivity: relation to NMDA-receptor function. Science. 1997;278:698–701. doi: 10.1126/science.278.5338.698. [DOI] [PubMed] [Google Scholar]
- Molnar E, Pickard L, Duckworth JK. Developmental changes in ionotropic glutamate receptors: lessons from hippocampal synapses. Neuroscientist. 2002;8:143–153. doi: 10.1177/107385840200800210. [DOI] [PubMed] [Google Scholar]
- Morimoto-Tomita M, Zhang W, Straub C, Cho CH, Kim KS, Howe JR, Tomita S. Autoinactivation of neuronal AMPA receptors via glutamate-regulated TARP interaction. Neuron. 2009;61:101–112. doi: 10.1016/j.neuron.2008.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrisett RA, Martin D, Oetting TA, Lewis DV, Wilson WA, Swartzwelder HS. Ethanol and magnesium ions inhibit N-methyl-D-aspartate-mediated synaptic potentials in an interactive manner. Neuropharmacology. 1991;30:1173–1178. doi: 10.1016/0028-3908(91)90162-5. [DOI] [PubMed] [Google Scholar]
- Moykkynen T, Korpi ER, Lovinger DM. Ethanol inhibits alpha-amino-3-hydyroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor function in central nervous system neurons by stabilizing desensitization. J Pharmacol Exp Ther. 2003;306:546–555. doi: 10.1124/jpet.103.050666. [DOI] [PubMed] [Google Scholar]
- Newton PM, Messing RO. Intracellular signaling pathways that regulate behavioral responses to ethanol. Pharmacol Ther. 2006;109:227–237. doi: 10.1016/j.pharmthera.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Nie Z, Madamba SG, Siggins GR. Ethanol inhibits glutamatergic neurotransmission in nucleus accumbens neurons by multiple mechanisms. J Pharmacol Exp Ther. 1994;271:1566–1573. [PubMed] [Google Scholar]
- Olney JW. Fetal alcohol syndrome at the cellular level. Addict Biol. 2004;9:137–49. doi: 10.1080/13556210410001717006. discussion 151. [DOI] [PubMed] [Google Scholar]
- Pickard L, Noel J, Henley JM, Collingridge GL, Molnar E. Developmental changes in synaptic AMPA and NMDA receptor distribution and AMPA receptor subunit composition in living hippocampal neurons. J Neurosci. 2000;20:7922–7931. doi: 10.1523/JNEUROSCI.20-21-07922.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyapali GK, Turner DA, Wilson WA, Swartzwelder HS. Age and dose-dependent effects of ethanol on the induction of hippocampal long-term potentiation. Alcohol. 1999;19:107–111. doi: 10.1016/s0741-8329(99)00021-x. [DOI] [PubMed] [Google Scholar]
- Randall RD, Lee SY, Meyer JH, Wittenberg GF, Gruol DL. Acute alcohol blocks neurosteroid modulation of synaptic transmission and long-term potentiation in the rat hippocampal slice. Brain Res. 1995;701:238–248. doi: 10.1016/0006-8993(95)01007-9. [DOI] [PubMed] [Google Scholar]
- Roberto M, Schweitzer P, Madamba SG, Stouffer DG, Parsons LH, Siggins GR. Acute and chronic ethanol alter glutamatergic transmission in rat central amygdala: an in vitro and in vivo analysis. J Neurosci. 2004;24:1594–1603. doi: 10.1523/JNEUROSCI.5077-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouach N, Byrd K, Petralia RS, Elias GM, Adesnik H, Tomita S, Karimzadegan S, Kealey C, Bredt DS, Nicoll RA. TARP gamma-8 controls hippocampal AMPA receptor number, distribution and synaptic plasticity. Nat Neurosci. 2005;8:1525–1533. doi: 10.1038/nn1551. [DOI] [PubMed] [Google Scholar]
- Sager C, Tapken D, Kott S, Hollmann M. Functional modulation of AMPA receptors by transmembrane AMPA receptor regulatory proteins. Neuroscience. 2009;158:45–54. doi: 10.1016/j.neuroscience.2007.12.046. [DOI] [PubMed] [Google Scholar]
- Sanderson JL, Partridge LD, Valenzuela CF. Modulation of GABAergic and glutamatergic transmission by ethanol in the developing neocortex: An in vitro test of the excessive inhibition hypothesis of fetal alcohol spectrum disorder. Neuropharmacology. 2009;56:541–555. doi: 10.1016/j.neuropharm.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savic N, Luthi A, Gahwiler BH, McKinney RA. N-methyl-D-aspartate receptor blockade during development lowers long-term potentiation threshold without affecting dynamic range of CA3-CA1 synapses. Proc Natl Acad Sci U S A. 2003;100:5503–5508. doi: 10.1073/pnas.0831035100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schummers J, Bentz S, Browning MD. Ethanol's inhibition of LTP may not be mediated solely via direct effects on the NMDA receptor. Alcohol Clin Exp Res. 1997;21:404–408. doi: 10.1111/j.1530-0277.1997.tb03783.x. [DOI] [PubMed] [Google Scholar]
- Schummers J, Browning MD. Evidence for a role for GABA(A) and NMDA receptors in ethanol inhibition of long-term potentiation. Brain Res Mol Brain Res. 2001;94:9–14. doi: 10.1016/s0169-328x(01)00161-9. [DOI] [PubMed] [Google Scholar]
- Siggins GR, Roberto M, Nie Z. The tipsy terminal: presynaptic effects of ethanol. Pharmacol Ther. 2005;107:80–98. doi: 10.1016/j.pharmthera.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Sinclair JG, Lo GF. Ethanol blocks tetanic and calcium-induced long-term potentiation in the hippocampal slice. Gen Pharmacol. 1986;17:231–233. doi: 10.1016/0306-3623(86)90144-8. [DOI] [PubMed] [Google Scholar]
- Sokol RJ, Delaney-Black V, Nordstrom B. Fetal alcohol spectrum disorder. JAMA. 2003;290:2996–2999. doi: 10.1001/jama.290.22.2996. [DOI] [PubMed] [Google Scholar]
- Sugiura M, Shoyama Y, Saito H, Abe K. The effects of ethanol and crocin on the induction of long-term potentiation in the CA1 region of rat hippocampal slices. Jpn J Pharmacol. 1995;67:395–397. doi: 10.1254/jjp.67.395. [DOI] [PubMed] [Google Scholar]
- Suvarna N, Borgland SL, Wang J, Phamluong K, Auberson YP, Bonci A, Ron D. Ethanol alters trafficking and functional N-methyl-D-aspartate receptor NR2 subunit ratio via H-Ras. J Biol Chem. 2005;280:31450–31459. doi: 10.1074/jbc.M504120200. [DOI] [PubMed] [Google Scholar]
- Swartzwelder HS, Wilson WA, Tayyeb MI. Differential sensitivity of NMDA receptor-mediated synaptic potentials to ethanol in immature versus mature hippocampus. Alcohol Clin Exp Res. 1995;19:320–323. doi: 10.1111/j.1530-0277.1995.tb01509.x. [DOI] [PubMed] [Google Scholar]
- Thomas JD, Garcia GG, Dominguez HD, Riley EP. Administration of eliprodil during ethanol withdrawal in the neonatal rat attenuates ethanol-induced learning deficits. Psychopharmacology (Berl) 2004;175:189–195. doi: 10.1007/s00213-004-1806-x. [DOI] [PubMed] [Google Scholar]
- Thomas JD, Riley EP. Fetal alcohol syndrome: does alcohol withdrawal play a role? Alcohol Health Res World. 1998;22:47–53. [PMC free article] [PubMed] [Google Scholar]
- Tokuda K, Zorumski CF, Izumi Y. Modulation of hippocampal long-term potentiation by slow increases in ethanol concentration. Neuroscience. 2007;146:340–349. doi: 10.1016/j.neuroscience.2007.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzuela CF, Bhave S, Hoffman P, Harris RA. Acute effects of ethanol on pharmacologically isolated kainate receptors in cerebellar granule neurons: comparison with NMDA and AMPA receptors. J Neurochem. 1998;71:1777–1780. doi: 10.1046/j.1471-4159.1998.71041777.x. [DOI] [PubMed] [Google Scholar]
- Valenzuela CF, Partridge LD, Mameli M, Meyer DA. Modulation of glutamatergic transmission by sulfated steroids: Role in fetal alcohol spectrum disorder. Brain Res Rev. 2007 doi: 10.1016/j.brainresrev.2007.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voigt T, Opitz T, de Lima AD. Activation of early silent synapses by spontaneous synchronous network activity limits the range of neocortical connections. J Neurosci. 2005;25:4605–4615. doi: 10.1523/JNEUROSCI.3803-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren KR, Foudin LL. Alcohol-related birth defects--the past, present, and future. Alcohol Res Health. 2001;25:153–158. [PMC free article] [PubMed] [Google Scholar]
- Wayner MJ, Armstrong DL, Polan-Curtain JL, Denny JB. Ethanol and diazepam inhibition of hippocampal LTP is mediated by angiotensin II and AT1 receptors. Peptides. 1993;14:441–444. doi: 10.1016/0196-9781(93)90129-5. [DOI] [PubMed] [Google Scholar]
- Weiner JL, Dunwiddie TV, Valenzuela CF. Ethanol inhibition of synaptically evoked kainate responses in rat hippocampal CA3 pyramidal neurons. Mol Pharmacol. 1999;56:85–90. doi: 10.1124/mol.56.1.85. [DOI] [PubMed] [Google Scholar]
- Williams SR, Mitchell SJ. Direct measurement of somatic voltage clamp errors in central neurons. Nat Neurosci. 2008;11:790–798. doi: 10.1038/nn.2137. [DOI] [PubMed] [Google Scholar]
- Yaka R, Phamluong K, Ron D. Scaffolding of Fyn kinase to the NMDA receptor determines brain region sensitivity to ethanol. J Neurosci. 2003;23:3623–3632. doi: 10.1523/JNEUROSCI.23-09-03623.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda H, Barth AL, Stellwagen D, Malenka RC. A developmental switch in the signaling cascades for LTP induction. Nat Neurosci. 2003;6:15–16. doi: 10.1038/nn985. [DOI] [PubMed] [Google Scholar]
- Zhang G, Morrisett RA. Ethanol inhibits tetraethylammonium chloride-induced synaptic plasticity in area CA1 of rat hippocampus. Neurosci Lett. 1993;156:27–30. doi: 10.1016/0304-3940(93)90431-j. [DOI] [PubMed] [Google Scholar]
- Zhang TA, Hendricson AW, Wilkemeyer MF, Lippmann MJ, Charness ME, Morrisett RA. Synergistic effects of the peptide fragment D-NAPVSIPQ on ethanol inhibition of synaptic plasticity and NMDA receptors in rat hippocampus. Neuroscience. 2005;134:583–593. doi: 10.1016/j.neuroscience.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Zhu JJ, Esteban JA, Hayashi Y, Malinow R. Postnatal synaptic potentiation: delivery of GluR4-containing AMPA receptors by spontaneous activity. Nat Neurosci. 2000;3:1098–1106. doi: 10.1038/80614. [DOI] [PubMed] [Google Scholar]
- Zhu W, Bie B, Pan ZZ. Involvement of non-NMDA glutamate receptors in central amygdala in synaptic actions of ethanol and ethanol-induced reward behavior. J Neurosci. 2007;27:289–298. doi: 10.1523/JNEUROSCI.3912-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zito K, Scheuss V, Knott G, Hill T, Svoboda K. Rapid functional maturation of nascent dendritic spines. Neuron. 2009;61:247–258. doi: 10.1016/j.neuron.2008.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]