

Abstract
The placenta and its myriad functions are central to successful reproductive outcomes. These functions can be influenced by the environment encountered throughout pregnancy. Such influences can alter the appropriate genetic programming needed to allow for sustained pregnancy and appropriate fetal development. This altered programming may result from epigenetic alterations related to environmental exposures. Epigenetic alterations are now being linked to several important reproductive outcomes, including early pregnancy loss, intrauterine growth restriction, congenital syndromes (eg. Beckwith-Weidemann syndrome), preterm birth and preeclampsia. The diversity of environmental exposures linked to adverse reproductive effects continues to grow. Much attention has focused on the role of endocrine disruptors and other xenobiotics in infertility, but recent work is demonstrating that these chemicals may have adverse effects in pregnancy and development as well. Environmental oxygen is also critical in early pregnancy success. There are clear links between altered oxygen levels and placentation amongst other effects. As research continues to increase our understanding of the molecular processes including epigenetic regulation that influence pregnancy, it will be critical to specifically examine how the environment, broadly defined, may play a role at altering these critical functions.
2. Introduction
Pregnancy is characterized by dramatic changes in metabolism leading to physiologic changes including a 40% increase in cardiac output and the development of insulin resistance. While the mechanisms of these changes are under investigation, it is clear that the placenta plays a crucial role. The placenta is the first complex mammalian organ to develop. It is formed from trophoblast cells that can be identified early during blastocyst formation and prior to implantation. Trophoblast cells are required to attach the embryo to the uterus and invade the maternal vasculature to form the maternal-fetal interface. As development continues, the placenta becomes a robust endocrine organ, producing a spectrum of hormones that are unique to the pregnancy, such as human chorionic gonadotropin, estriol, as well as hormones identical to those produced elsewhere, such as estrogen and progesterone. This hormone production is intrinsically coupled to the physiologic changes of pregnancy and is critical for pregnancy maintenance. Clearly, alterations in placental function can have profound effects on pregnancy. Gene environment interactions have become a major focus of investigation in the post genome era. This review will discuss three common ways that the placental function can be affected by the environment.
3. Endocrine Disruptors
Endocrine disruptors are environmental pollutants that are biologically active in the endocrine system and disrupt endogenous hormonal function. They include a variety of classes of chemicals including pesticides, industrial by-products and chemicals used in manufacturing-particularly plastics. The Centers for Disease Control reported on chemical exposure in over 2000 people in the United States. More than 90% of individuals tested positive for chemicals in their blood; the most common chemicals noted were polybrominated diphenyl ethers and bisphenol A [1].
The endocrine disruptors most commonly associated with reproductive abnormalities are the xenoestrogens such as Bisphenol-A (BPA), polychlorinated biphenyls (PCBs), and antiandrogens such as phthalates. These compounds exhibit weak steroid-like activity and therefore can affect reproductive development along multiple points including the hypothalamus and the gonad.
3.1 Bisphenol-A
BPA is the most ubiquitous of the xenoestrogens. First described in 1891, this chemical was initially developed as a pharmaceutical for estrogen replacement therapy. Today it is widely used as a crosslinking chemical to form plastics. It is used in drinking bottles and epoxy resin that coats the inside of food containers [2]. When substances containing BPA are heated, the chemicals can leach into the food and ingested [3, 4]. During the 2003–2004 NHANES survey, the CDC determined that BPA could be detected in the urine of more than 95% of Americans over the age of six. BPA can be measured in serum, saliva and urine [5–8]. Remarkably, the amniotic fluid contains five times more BPA than corresponding maternal serum [9]. This is likely due to the active transport of BPA across the placenta.
BPA is structurally similar to diethylstilbestrol (DES). Like DES, BPA has been associated with reproductive teratogenicity in animal models [10]. BPA’s toxicity and reproductive disruption has been linked to interference with endogenous estrogens which is mediated through BPA’s binding of the estrogen receptor as well as nuclear-receptor independent activation of key cellular signaling system [11]. Mice and primates exposed to BPA may contain a variety of abnormalities of the breasts, uterus and ovaries [8, 12–14]. The ovaries may contain few or no follicles and may contain ova with a high rate of aneuploidy [15, 16]. BPA can also target the placenta directly [17]. Mouse cytotrophoblast cells cultured in physiologic doses of BPA demonstrate abnormal labyrinthine development and increased rates of apoptosis [18]; in addition, BPA decreases placental aromatase activity leading to lower levels of estrogen production and decreased the amount of estrogen and progesterone receptor expression in the placenta [19, 20]. The affect on birth weight and resorption in animal models has been controversial with some data supporting a causal effect while other suggesting a protective effect [21]. The association with abortion may be due to placental failure or aneuploidy [22].
3.2 Polychlorinated Biphenyls
Recently, a novel mechanism has been described for PCB-induced premature and low birth weight delivery in mice [23]. PCBs cause an increase in amniotic fluid and placental anomalies by inhibiting placental aquaporin 1, a water channel family member. Aquaporin 1 is known to regulate fluid volume and angiogenesis at the maternal-fetal interface [23]. Subsequently, it was shown that PCBs impaired spiral artery remodeling in the mouse utero-placental tissue which could be reversed by interleukin (IL)-10 [23]. Further investigation into the anti-angiogenic effects of PCBs unraveled the involvement of the novel dll4-Notch1-VEGF R2 pathway which could also be reversed by IL-10 [24]. It is thus possible that BPA elicits its effects at the maternal-fetal interface by deregulating aquaporins and angiogenic pathways.
3.3 Phthalates
Phthalates are plasticizers used to increase the flexibility of polyvinyl chloride products. Because they are weakly bound to the plastic, they are readily released in to the environment. Human exposure occurs mainly by ingesting contaminated food and by applying makeup. As noted with BPA, the CDC has detected widespread exposure; however, because there are numerous metabolites, actual exposure levels have been difficult to quantify. Nonetheless, it is clear that women have five-fold greater exposure when compared to age-matched men and children have much larger concentrations per body weight compared to adults [25].
Phthalates are potent reproductive teratogens in male and female animal models. They act principally as antiandrogens and suppress testosterone production [26]. In males, phthalates affect testicular development leading to abnormalities in the seminiferous tubules [27, 28]. In females, phthalates have been associated with uterine abnormalities and an ovarian toxic defect; exposure leads to anovulation with polycystic appearing ovaries [29]. In addition, phthalate exposure decreases the anogenital distance (AGD) in male mice suggesting feminization [30]. AGD is a measure of intrauterine androgen exposure because in male the AGD is twice that of females [31].
There are few studies associating phthalate exposure with reproductive developmental abnormalities in humans. Small studies have correlated phthalate levels with abnormal pubertal development [32, 33]. However, these studies have been criticized because of potential phthalate contamination from diesters in the laboratory equipment. Studies have implicated urinary phthalate metabolites, which are less likely to represent contamination, with sperm semen parameters and sperm DNA damage in men seeking infertility services [34]. The most widely publicized evidence describing an effect of phthalate levels on sexual development is the Swan Study [35]. The investigators measured the AGD in newborn males and demonstrated an indirect correlation with urinary phthalate metabolites. This study has been highly criticized because of the lack of clinical correlation with AGD in humans.
There are no data currently available that suggest that phthalates have a direct effect on implantation or placentation. However, phthalate metabolites are potent PPAR-gamma agonists [36–38]. PPAR-gamma is a critical gene for normal trophoblast differentiation [39–41]. PPAR-gamma null mice show embryonic lethality on day E.10, the time when metabolism switches to the placenta. The placentas of these mice contain poorly developed labyrinthine trophoblasts and thin cardiac muscle. Interestingly, these pathologies could be alleviated based on the trophoblast chimeric models which restore normal PPAR-gamma expression in the trophoblast but not epiblast [39].
3.4 Molecular biology of endocrine disruption
The mechanisms by which endocrine disruptors (EDs) affect cellular activity are unclear. Several researchers have demonstrated that EDs can bind to hormone receptors resulting in altered genomic response(s). However, the binding affinities of the phenol containing xenoestrogens to the classical nuclear hormone receptors are at least 1000-fold lower than that of endogenous estradiol. Therefore, it is more likely that small concentrations of these chemicals found in vivo mediate their activity via non-genomic mechanisms or through different receptors and pathways as seen for certain congeners of PCBs [23]. There have been several reports of xenoestrogens binding to cell membrane bound estrogen receptors and subsequently activating secondary messenger kinases and affecting calcium influx [3, 42, 43]. For example, the estrogen related receptor gamma (ERRg) protein has recently been described as present in the placenta [44]. This receptor has been shown to exhibit a strong affinity for BPA, and thus it may be through binding and activation of this receptor and its downstream signaling that BPA can exert its influence [45–46]. Clearly, more functional and mechanistic studies are needed to more clearly define the modes through which BPA can adverse impact pregnancy.
4. Environmental oxygen
The level of oxygen in the intrauterine environment plays a critical role in pregnancy by affecting embryo development and placentation. Maternal residence at high altitude exerts a profound affect in many pregnant women on pregnancy outcome and growth; the probability of preeclampsia, placental abruption and/or preterm delivery increases at higher altitude [47–51]. There is a profound inverse relationship between altitude and fetal growth. It is estimated that fetal growth decreases by 100g for each 1000m gained [52]. While this results in a five-fold increased probability of small for gestational age delivery, the majority of babies born at high altitude have normal growth. Nonetheless, the risk of fetal morbidity at high altitude is at least two-fold greater than matched pregnancies at lower altitudes [53]. These data, however, need to be interpreted with caution because women at high altitudes may have poorer access to health care services.
4.1. Oxygen tension and development
Early embryo development occurs at low oxygen (Fig. 1). The oocyte is fertilized in the fallopian tube and the embryo undergoes rapid growth as it traverses the tube and enters the uterus on post-conception day 2–3. The embryo remains in the uterus for an additional three days prior to implantation. This intrauterine environment is in continuum with the peritoneal cavity and is, therefore, at an oxygen tension of less than 5% [54]. Several lines of evidence demonstrate that the human embryo remains at low oxygen tension. Hustin and Schaaps injected human pregnant hysterectomy specimens with barium sulfate and demonstrated that there is little vascularization of the first trimester decidua. Further, extravillous trophoblast cells occlude the spiral arteries of the uterus, further separating the intervillous space from the maternal circulation [55]. Several investigators have used Doppler ultrasound to demonstrate that blood flow within the intervillous space is not present before ten weeks of gestation [56–58]. The direct measurement of oxygen content in the first trimester with a small probe proved that, prior to 10 weeks, the intervillous oxygen tension is less than 20mm Hg which is equivalent to less than 3% dissolved oxygen [59].
Figure 1.
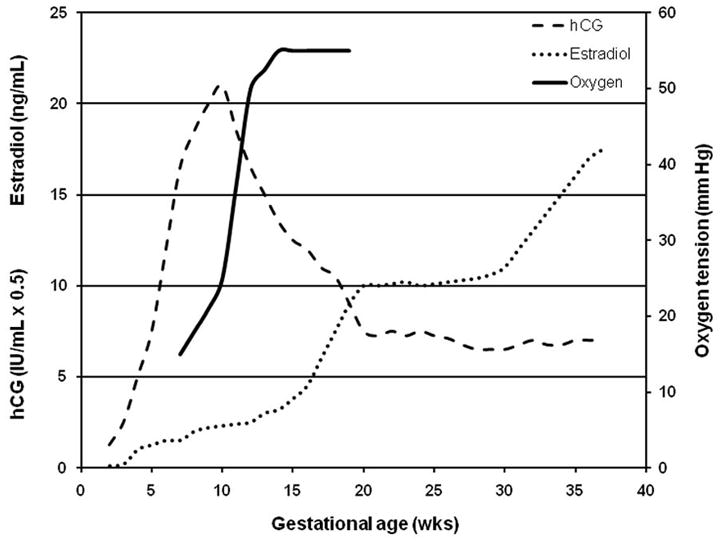
Gestational age-dependent relationship of oxygen tension with fetal development and levels of progesterone and estrogen.
4.2. Oxygen and the placenta
There are few data with in vitro models that have investigated the molecular mechanisms underlying the differentiation pathway of the cytotrophoblast cell. Genbacev and colleagues have suggested that hypoxia promotes extravillous trophoblast proliferation and inhibits differentiation in first trimester placental explant cultures [60, 61]. Similar data were obtained by Caniggia et al [62, 63]. Low oxygen has also been shown to promote extravillous differentiation in a highly purified cytotrophoblast cell culture [64]. This is very relevant data to current practices of culturing embryos for in vitro fertilization. The ideal oxygen environmental conditions for optimal embryo development are under investigation.
Placental hypoxic stress beyond the first trimester is associated with preeclampsia. There are several mechanisms proposed for this phenomenon. In vitro experiments illustrate that severe hypoxia can lead to trophoblast apoptosis, shedding of microparticles and elevated expression of sFlt-1, a critical factor in preeclampsia development [65, 66]. Data further suggests that hypoxia can lead to the inflammatory milieu of preeclampsia. The hypoxia inducible factors (HIFs) promote the release cytokines including IL-10, which plays a critical role in placental function and preeclampsia. HIF also promotes the release of VEGF, TLR and NOS expression [67].
5. Epigenetics
Research in human reproduction, development, fetal programming and many other disciplines is now focusing on the paradigm that gene regulation occurs beyond the DNA sequence and that it is this epigenetic regulation, the mitotically and meiotically heritable control of gene expression not related to DNA sequence that plays a critical role in human health and disease. As epigenetic regulatory mechanisms control gene expression and can be responsive or altered by the environment, including xenoestrogens and environmental oxygen, they may represent the mechanistic basis of gene-environment interactions and their synergistic effects. Figure 2 provides a summary of key epigenetic programming which occurs throughout gestation, and the potential fetopathic exposures whose phenotypic effects may be mediated through alteration of the appropriate epigenetic programming during development.
Figure 2.
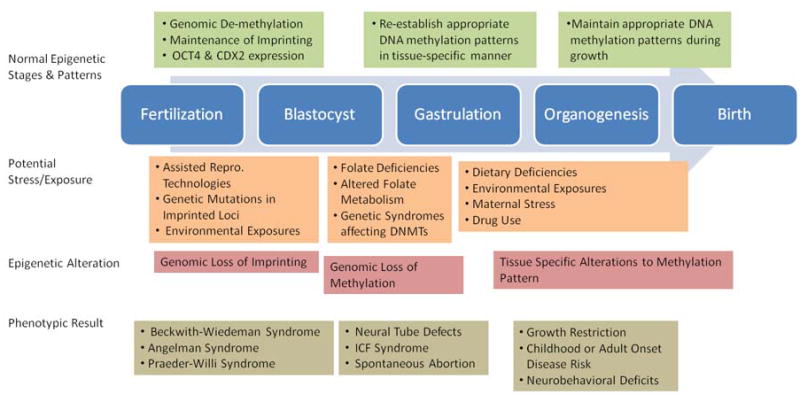
Model of appropriate epigenetic programming throughout gestation and the potential epigenetic effects of environmental factors and their downstream consequences.
Epigenetic regulatory mechanisms include post-translational histone modifications, DNA CpG methylation, imprinting, and small-RNA mediated control. In general, these mechanisms delineate the chromatin structure of a gene and control its transcriptional activity. These processes play key roles in many cellular processes, and are absolutely critical for development, differentiation, and as is becoming increasingly clear, adaptation to the environment throughout life, thereby contributing to disease risk throughout the life course.
5.1. DNA Methylation
The most thoroughly study of the epigenetic mechanisms is that of DNA methylation, specifically methylation of the 5′-carbon of cytosine in the context of CpG dinucleotides [68]. The circumstance of multiple methylated cytosines within a particular CpG island is often associated with total transcriptional silencing of the downstream gene. The presence of the methyl group alone is not sufficient for transcriptional silencing. Instead gene silencing usually requires the recruitment of component proteins related to gene repression and creation/maintenance of a silenced chromatin conformation, complete with the appropriate post-translational modification of histone tails. These methyl groups are added enzymatically through de novo DNA methyltransferases (DNMT3A and DNMT3B) which act to establish DNA methylation patterns in development and possibly later in life in response to environment. In contrast, maintenance methyltransferase (DNMT1) act specifically on hemi-methylated DNA during replication to faithfully recapitulate the pattern through cell division [69].
5.1.1. Reprogramming of methylation during early development
Marks of DNA methylation are thought to remain relatively stable and heritable throughout life, although these marks are entirely reprogrammed during two periods, gametogenesis and pre-implantation embryonic period. This reprogramming, during the pre-implantation period, necessitates a rapid de-methylation of the genome, thought to be accomplished through an active process [70, 71], followed by appropriate, cell and tissue specific methylation of genome. The exact mechanism through which this reprogramming occurs remains a central question in epigenetics, as no demethylation enzymes have been identified which can carry out the rapid demethylation in-vivo [72–74]. This demethylation occurs asynchronously, with the male pronucleus losing its genomic methylation within a few hours after conception through an active process and prior to that of the female, which is thought to occur only passively [75]. The protamine to histone exchange has been posited to explain the asynchronicity [76], and in general chromatin dynamics and changes to histone methylation patterns are tightly linked to this process [77]. Animal models have suggested that this appropriate demethylation can be altered through superovulation, and in-vitro fertilization, and results in developmental failure [78].
Similarly, the reprogramming of the DNA methylation marks and particularly, the appropriate targeting of enzymes responsible for establishing those marks remains a mystery. The de novo methyltransferase DNMT3A along with DNMT3L, a homologous protein which stimulates the activity of DNMT3A and DNMT3B but does not bind DNA [79], are required for appropriate imprinting of primordial germ cells [80]. PiRNAs and their associated Piwi proteins, in addition to their role in transposon control and methylation have also been implicated as specific determinants of DNA methylation in germ cells. The oocyte-derived maintenance methyltransferase DNMT1o plays an important role in preventing epigenetic mosaicism and allowing for appropriate inheritance of imprinted methylation patterns in a developing embryo [81, 82]. DNMT3B is also critical for appropriate genomic methylation re-establishment, and its deficit is linked to ICF syndrome (OMIM:242860) [83]. DNMT3A and DNMT3B have also been implicated as important in mediating trophoblast invasion, as knockdown experiments have demonstrated decreased migration of BeWo cells, possibly related to de-methylation and increased expression of E-cadherin and Plakoglobin [84].
5.1.2. Methylation and cellular differentiation
The earliest markers of differentiation are controlled through epigenetic processes which allow for sustained and heritable expression of genes necessary for tissue development and differentiation. Totipotent stem cells comprising the morula are marked by the expression of OCT4 and CDX2, while the blastocyst is marked by differential expression of these key factors, with the now pluripotent stem cells of the inner cell mass maintaining OCT3/4 expression and downregulated CDX2, and the cells comprising the trophectoderm epigenetically downregulating of OCT4 expression [85] while maintaining CDX2 [86–88]. CDX2 acts through binding of an FGF4-responsive enhancer element in the promoter of BMP4, leading to BMP4 expression, which is critical for trophoblast differentiation from the trophectoderm [89].
Epigenetic regulation is also thought to play a critical role in further differentiating the placenta into regions with known associated functions. Genome-wide scanning of rat placenta tissues identified specific sites of DNA methylation differentiating the placenta’s junctional and labyrinth zones [90]. In addition, differential DNA methylation patterns were observed following differentiation of cultured rat Rcho-1 cells to differentiated giant cells [91].
5.1.3. DNA methylation and poor pregnancy outcome
Links have also been made between fetal DNA methylation and recurrent spontaneous abortion. A study by Park, et al. in a South Korean population, observed that abortuses demonstrated a reduced prevalence of the MTHFR codon 677 CT or TT genotype, which are thought to be related to altered folate metabolism and S-adenosyl methionine availability, although there were no associations observed between these genotypes and methylation of two genes examined [92]. There were no associations observed, though, between MTHFR genotype and measures of embryo quality or spontaneous abortion rates amongst women undergoing IVF procedures, suggesting that appropriate folate supplementation may negate any associations between genotype and pregnancy outcome [93]. A recent study by Pliushch et al. established that 4% of spontaneous abortions and 18% of stillbirths demonstrated hypermethylation of normally imprinted genes and suggested that altered expression patterns of these genes may play a role in pregnancy loss [94].
Examination of circulating nucleic acids, and particularly methylated DNA, in maternal serum is a promising new, non-invasive tool for diagnosis of fetal abnormalities [95]. Muller et al. demonstrated that maternal first-trimester serum derived DNA exhibited a greater prevalence of methylation of the tumor suppressor APC in women who later developed preeclampsia, eclampsia, or HELLP syndrome, compared to healthy pregnant women who did not develop these conditions, and further that this pattern of methylation was similar to that observed in women with advanced breast cancer [96]. Tsui et al examined RASSF1A hypermethylation in third trimester maternal plasma and term placenta tissues, comparing women experiencing preeclampsia and healthy gestational age-matched controls [97]. They found a significantly greater concentration of RASSF1A methylation in the plasma-derived DNA from preeclamptic women, but did not observe any difference in RASSF1A methylation in placental tissues, leaving open the question of the source of this methylated DNA [97]. Yuen et al recently completed a study utilizing an array-based approach to identify CpG loci differentially methylated in placental tissues, and demonstrated that the TUSC3 gene exhibits an increased prevalence of promoter methylation in preeclamptic placentas [98].
5.2. Genomic imprinting
Genomic imprinting refers to the monoallelic expression of a subset of 200 genes in a conserved parent-of-origin fashion orchestrated by the timely placement of epigenetic signals including DNA methylation and histone modification [99]. Based on their key functions in placental and fetal development, the imprinted genes can be classified into three board categories [100]: 1) genes that control the allocation of maternal resources to the fetus; 2) genes that regulate metabolism in the early postnatal period; and 3) prenatal determinants of the metabolism of developing metabolic organs such as the pancreas, muscle, fat cells and the hypothalamus. Alterations to the imprinting status of genes, through both genetic and epigenetic mechanisms, are causally associated with a number of well characterized human syndromes, including, Beckwith-Wiedemann, Angelman, Silver-Russel, and Prader-Willi syndromes [101, 102]. Alterations to imprinting, are thought to be susceptible to environmental influences, with the most publicized example being the loss of imprinting of the IGF2/H19 locus and subsequent increased risk of Beckwith-Wiedemann syndrome associated with the use of assisted reproductive technologies [103–105]. Alterations to imprinting may also be implicated in pregnancy loss, as chorionic villous samples from ART-related pregnancy losses revealed increased level of DNA methylation of the imprinted genes LIT1 and H19 compared to spontaneously conceived pregnancy losses [106].
The Mash2 gene, which is paternally imprinted and in linkage to other critically imprinted genes including Igf2 and H19, is required for further trophoblast development, and expression of the paternal allele is detected in early post-implantation embryos [107], and is required for formation of spongiotrophoblast [108, 109]. A number of other genes involved in critical functions of the placenta and its development have been reported to exhibit imprinting. The alpha-T-catenin gene, necessary for cell-cell adhesion complexes and an important mediator of cell invasion, during the first trimester exhibits preferential expression of the maternal allele in villous cytotrophoblasts and biallelic expression in extravillous trophoblasts, but following the epithelial-mesenchymal transition expression is lost in both villous syncytiotrophoblasts and extravillous trophoblasts, a pattern similar to that observed for p57(KIP2) [110, 111]. The paternally imprinted p57(KIP2) gene has also been utilized in a mouse model of preeclampsia [112]. The imprinted MEST gene is expressed in human villous and invasive cytotrophoblasts and is thought to play a role in angiogenesis [113].
6. Conclusions
As we strive to further understand the mechanisms of reproduction and placentation, we are recognizing the complexity and inefficiency of the processes and their susceptibility to environmental stressors. International health groups are recognizing the increasing importance of environmental toxicants. Despite the dearth of literature on women’s health outcomes, it is clear that many of these toxicants and changing environmental landscape can affect healthy reproduction. Further studies are urgently needed to better identify the molecular basis of these effects, and develop strategies to modify or counteract the environment to improve birth outcomes.
Acknowledgments
The work in our laboratories was supported by grants from the National Institutes of Health, National Center for Research Resources (P20RR018728), and National Institute of Environmental Health Sciences (P42ES013660), and the Rhode Island Foundation.
References
- 1.Center for Disease Control. Fourth National Report on Human Exposure to Environmental Chemicals. National Center for Environmental Health, Division of Laboratory Sciences; Atlanta, GA: 2009. [Google Scholar]
- 2.Dodds EC, Lawson W. Synthetic estrogenic agents without the phenanthrene nucleus [Letter] Nature. 1936;137:996. [Google Scholar]
- 3.Krishnan AV, Stathis P, Permuth SF, Tokes L, Feldman D. Bisphenol-A: an estrogenic substance is released from polycarbonate flasks during autoclaving. Endocrinology. 1993;132(6):2279–86. doi: 10.1210/endo.132.6.8504731. [DOI] [PubMed] [Google Scholar]
- 4.Le HH, Carlson EM, Chua JP, Belcher SM. Bisphenol A is released from polycarbonate drinking bottles and mimics the neurotoxic actions of estrogen in developing cerebellar neurons. Toxicol Lett. 2008;176(2):149–56. doi: 10.1016/j.toxlet.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vandenberg LN, Hauser R, Marcus M, Olea N, Welshons WV. Human exposure to bisphenol A (BPA) Reprod Toxicol. 2007;24(2):139–77. doi: 10.1016/j.reprotox.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 6.Vandenberg LN, Maffini MV, Wadia PR, Sonnenschein C, Rubin BS, Soto AM. Exposure to environmentally relevant doses of the xenoestrogen bisphenol-A alters development of the fetal mouse mammary gland. Endocrinology. 2007;148(1):116–27. doi: 10.1210/en.2006-0561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Calafat AM, Kuklenyik Z, Reidy JA, Caudill SP, Ekong J, Needham LL. Urinary concentrations of bisphenol A and 4-nonylphenol in a human reference population. Environ Health Perspect. 2005;113(4):391–5. doi: 10.1289/ehp.7534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bouskine A, Nebout M, Brucker-Davis F, Benahmed M, Fenichel P. Low doses of bisphenol A promote human seminoma cell proliferation by activating PKA and PKG via a membrane G-protein-coupled estrogen receptor. Environ Health Perspect. 2009;117(7):1053–8. doi: 10.1289/ehp.0800367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ikezuki Y, Tsutsumi O, Takai Y, Kamei Y, Taketani Y. Determination of bisphenol A concentrations in human biological fluids reveals significant early prenatal exposure. Hum Reprod. 2002;17(11):2839–41. doi: 10.1093/humrep/17.11.2839. [DOI] [PubMed] [Google Scholar]
- 10.vom Saal FS, Akingbemi BT, Belcher SM, Birnbaum LS, Crain DA, Eriksen M, Farabollini F, Guillette LJ, Jr, Hauser R, Heindel JJ, Ho SM, Hunt PA, Iguchi T, Jobling S, Kanno J, Keri RA, Knudsen KE, Laufer H, LeBlanc GA, Marcus M, McLachlan JA, Myers JP, Nadal A, Newbold RR, Olea N, Prins GS, Richter CA, Rubin BS, Sonnenschein C, Soto AM, Talsness CE, Vandenbergh JG, Vandenberg LN, Walser-Kuntz DR, Watson CS, Welshons WV, Wetherill Y, Zoeller RT. Chapel Hill bisphenol A expert panel consensus statement, integration of mechanisms, effects in animals and potential to impact human health at current levels of exposure. Reprod Toxicol. 2007;24(2):131–8. doi: 10.1016/j.reprotox.2007.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wetherill YB, Akingbemi BT, Kanno J, McLachlan JA, Nadal A, Sonnenschein C, Watson CS, Zoeller RT, Belcher SM. In vitro molecular mechanisms of bisphenol A action. Reprod Toxicol. 2007;24(2):178–98. doi: 10.1016/j.reprotox.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 12.Maffini MV, Rubin BS, Sonnenschein C, Soto AM. Endocrine disruptors and reproductive health: the case of bisphenol-A. Mol Cell Endocrinol. 2006;254–255:179–86. doi: 10.1016/j.mce.2006.04.033. [DOI] [PubMed] [Google Scholar]
- 13.Markey CM, Luque EH, Munoz De Toro M, Sonnenschein C, Soto AM. In utero exposure to bisphenol A alters the development and tissue organization of the mouse mammary gland. Biol Reprod. 2001;65(4):1215–23. doi: 10.1093/biolreprod/65.4.1215. [DOI] [PubMed] [Google Scholar]
- 14.Munoz-de-Toro M, Markey CM, Wadia PR, Luque EH, Rubin BS, Sonnenschein C, Soto AM. Perinatal exposure to bisphenol-A alters peripubertal mammary gland development in mice. Endocrinology. 2005;146(9):4138–47. doi: 10.1210/en.2005-0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Can A, Semiz O, Cinar O. Bisphenol-A induces cell cycle delay and alters centrosome and spindle microtubular organization in oocytes during meiosis. Mol Hum Reprod. 2005;11(6):389–96. doi: 10.1093/molehr/gah179. [DOI] [PubMed] [Google Scholar]
- 16.Hunt PA, Koehler KE, Susiarjo M, Hodges CA, Ilagan A, Voigt RC, Thomas S, Thomas BF, Hassold TJ. Bisphenol a exposure causes meiotic aneuploidy in the female mouse. Curr Biol. 2003;13(7):546–53. doi: 10.1016/s0960-9822(03)00189-1. [DOI] [PubMed] [Google Scholar]
- 17.Benachour N, Aris A. Toxic effects of low doses of Bisphenol-A on human placental cells. Toxicol Appl Pharmacol. 2009;241(3):322–8. doi: 10.1016/j.taap.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 18.Tachibana T, Wakimoto Y, Nakamuta N, Phichitraslip T, Wakitani S, Kusakabe K, Hondo E, Kiso Y. Effects of bisphenol A (BPA) on placentation and survival of the neonates in mice. J Reprod Dev. 2007;53(3):509–14. doi: 10.1262/jrd.18171. [DOI] [PubMed] [Google Scholar]
- 19.Canton RF, Scholten DE, Marsh G, de Jong PC, van den Berg M. Inhibition of human placental aromatase activity by hydroxylated polybrominated diphenyl ethers (OH-PBDEs) Toxicol Appl Pharmacol. 2008;227(1):68–75. doi: 10.1016/j.taap.2007.09.025. [DOI] [PubMed] [Google Scholar]
- 20.Huang H, Leung LK. Bisphenol A downregulates CYP19 transcription in JEG-3 cells. Toxicol Lett. 2009;189(3):248–52. doi: 10.1016/j.toxlet.2009.06.853. [DOI] [PubMed] [Google Scholar]
- 21.Ranjit N, Siefert K, Padmanabhan V. Bisphenol-A and disparities in birth outcomes: a review and directions for future research. J Perinatol. 2010;30(1):2–9. doi: 10.1038/jp.2009.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sugiura-Ogasawara M, Ozaki Y, Sonta S, Makino T, Suzumori K. Exposure to bisphenol A is associated with recurrent miscarriage. Hum Reprod. 2005;20(8):2325–9. doi: 10.1093/humrep/deh888. [DOI] [PubMed] [Google Scholar]
- 23.Tewari N, Kalkunte S, Murray DW, Sharma S. The water channel aquaporin 1 is a novel molecular target of polychlorinated biphenyls for in utero anomalies. J Biol Chem. 2009;284(22):15224–32. doi: 10.1074/jbc.M808892200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sharma S. PCB exposures: Novel in utero targets and adverse effects on Pregnancyy. SRP Annual Meeting; 2009. [Google Scholar]
- 25.Heudorf U, Mersch-Sundermann V, Angerer J. Phthalates: toxicology and exposure. Int J Hyg Environ Health. 2007;210(5):623–34. doi: 10.1016/j.ijheh.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 26.Stroheker T, Cabaton N, Nourdin G, Regnier JF, Lhuguenot JC, Chagnon MC. Evaluation of anti-androgenic activity of di-(2-ethylhexyl)phthalate. Toxicology. 2005;208(1):115–21. doi: 10.1016/j.tox.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 27.Boekelheide K, Kleymenova E, Liu K, Swanson C, Gaido KW. Dose-dependent effects on cell proliferation, seminiferous tubules, and male germ cells in the fetal rat testis following exposure to di(n-butyl) phthalate. Microsc Res Tech. 2009;72(8):629–38. doi: 10.1002/jemt.20684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kleymenova E, Swanson C, Boekelheide K, Gaido KW. Exposure in utero to di(n-butyl) phthalate alters the vimentin cytoskeleton of fetal rat Sertoli cells and disrupts Sertoli cell-gonocyte contact. Biol Reprod. 2005;73(3):482–90. doi: 10.1095/biolreprod.104.037184. [DOI] [PubMed] [Google Scholar]
- 29.Gray LE, Jr, Wilson VS, Stoker T, Lambright C, Furr J, Noriega N, Howdeshell K, Ankley GT, Guillette L. Adverse effects of environmental antiandrogens and androgens on reproductive development in mammals. Int J Androl. 2006;29(1):96–104. doi: 10.1111/j.1365-2605.2005.00636.x. discussion 105–8. [DOI] [PubMed] [Google Scholar]
- 30.Ema M, Miyawaki E, Kawashima K. Reproductive effects of butyl benzyl phthalate in pregnant and pseudopregnant rats. Reprod Toxicol. 1998;12(2):127–32. doi: 10.1016/s0890-6238(97)00127-5. [DOI] [PubMed] [Google Scholar]
- 31.Marty MS, Chapin RE, Parks LG, Thorsrud BA. Development and maturation of the male reproductive system. Birth Defects Res B Dev Reprod Toxicol. 2003;68(2):125–36. doi: 10.1002/bdrb.10015. [DOI] [PubMed] [Google Scholar]
- 32.Colon I, Caro D, Bourdony CJ, Rosario O. Identification of phthalate esters in the serum of young Puerto Rican girls with premature breast development. Environ Health Perspect. 2000;108(9):895–900. doi: 10.1289/ehp.108-2556932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qiao L, Zheng L, Cai D. [Study on the di-n-butyl phthalate and di-2-ethylhexyl phthalate level of girl serum related with precocious puberty in Shanghai] Wei Sheng Yan Jiu. 2007;36(1):93–5. [PubMed] [Google Scholar]
- 34.Hauser R, Meeker JD, Singh NP, Silva MJ, Ryan L, Duty S, Calafat AM. DNA damage in human sperm is related to urinary levels of phthalate monoester and oxidative metabolites. Hum Reprod. 2007;22(3):688–95. doi: 10.1093/humrep/del428. [DOI] [PubMed] [Google Scholar]
- 35.Swan SH, Main KM, Liu F, Stewart SL, Kruse RL, Calafat AM, Mao CS, Redmon JB, Ternand CL, Sullivan S, Teague JL. Decrease in anogenital distance among male infants with prenatal phthalate exposure. Environ Health Perspect. 2005;113(8):1056–61. doi: 10.1289/ehp.8100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hurst CH, Waxman DJ. Activation of PPARalpha and PPARgamma by environmental phthalate monoesters. Toxicol Sci. 2003;74(2):297–308. doi: 10.1093/toxsci/kfg145. [DOI] [PubMed] [Google Scholar]
- 37.Lampen A, Zimnik S, Nau H. Teratogenic phthalate esters and metabolites activate the nuclear receptors PPARs and induce differentiation of F9 cells. Toxicol Appl Pharmacol. 2003;188(1):14–23. doi: 10.1016/s0041-008x(03)00014-0. [DOI] [PubMed] [Google Scholar]
- 38.Maloney EK, Waxman DJ. trans-Activation of PPARalpha and PPARgamma by structurally diverse environmental chemicals. Toxicol Appl Pharmacol. 1999;161(2):209–18. doi: 10.1006/taap.1999.8809. [DOI] [PubMed] [Google Scholar]
- 39.Barak Y, Nelson MC, Ong ES, Jones YZ, Ruiz-Lozano P, Chien KR, Koder A, Evans RM. PPAR gamma is required for placental, cardiac, and adipose tissue development. Mol Cell. 1999;4(4):585–95. doi: 10.1016/s1097-2765(00)80209-9. [DOI] [PubMed] [Google Scholar]
- 40.Schaiff WT, Carlson MG, Smith SD, Levy R, Nelson DM, Sadovsky Y. Peroxisome proliferator-activated receptor-gamma modulates differentiation of human trophoblast in a ligand-specific manner. J Clin Endocrinol Metab. 2000;85(10):3874–81. doi: 10.1210/jcem.85.10.6885. [DOI] [PubMed] [Google Scholar]
- 41.Shalom-Barak T, Nicholas JM, Wang Y, Zhang X, Ong ES, Young TH, Gendler SJ, Evans RM, Barak Y. Peroxisome proliferator-activated receptor gamma controls Muc1 transcription in trophoblasts. Mol Cell Biol. 2004;24(24):10661–9. doi: 10.1128/MCB.24.24.10661-10669.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Olea N, Pulgar R, Perez P, Olea-Serrano F, Rivas A, Novillo-Fertrell A, Pedraza V, Soto AM, Sonnenschein C. Estrogenicity of resin-based composites and sealants used in dentistry. Environ Health Perspect. 1996;104(3):298–305. doi: 10.1289/ehp.96104298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Derfoul A, Lin FJ, Awumey EM, Kolodzeski T, Hall DJ, Tuan RS. Estrogenic endocrine disruptive components interfere with calcium handling and differentiation of human trophoblast cells. J Cell Biochem. 2003;89(4):755–70. doi: 10.1002/jcb.10558. [DOI] [PubMed] [Google Scholar]
- 44.Matsushima A, Kakuta Y, Teramoto T, Koshiba T, Liu X, Okada H, Tokunaga T, Kawabata S, Kimura M, Shimohigashi Y. Structural evidence for endocrine disruptor bisphenol A binding to human nuclear receptor ERR gamma. J Biochem. 2007;142(4):517–24. doi: 10.1093/jb/mvm158. [DOI] [PubMed] [Google Scholar]
- 45.Matsushima A, Teramoto T, Okada H, Liu X, Tokunaga T, Kakuta Y, Shimohigashi Y. ERRgamma tethers strongly bisphenol A and 4-alpha-cumylphenol in an induced-fit manner. Biochem Biophys Res Commun. 2008;373(3):408–13. doi: 10.1016/j.bbrc.2008.06.050. [DOI] [PubMed] [Google Scholar]
- 46.Takeda Y, Liu X, Sumiyoshi M, Matsushima A, Shimohigashi M, Shimohigashi Y. Placenta expressing the greatest quantity of bisphenol A receptor ERR{gamma} among the human reproductive tissues: Predominant expression of type-1 ERRgamma isoform. J Biochem. 2009;146(1):113–22. doi: 10.1093/jb/mvp049. [DOI] [PubMed] [Google Scholar]
- 47.Khong TY, De Wolf F, Robertson WB, Brosens I. Inadequate maternal vascular response to placentation in pregnancies complicated by pre-eclampsia and by small-for-gestational age infants. Br J Obstet Gynaecol. 1986;93(10):1049–59. doi: 10.1111/j.1471-0528.1986.tb07830.x. [DOI] [PubMed] [Google Scholar]
- 48.Khong TY, Liddell HS, Robertson WB. Defective haemochorial placentation as a cause of miscarriage: a preliminary study. Br J Obstet Gynaecol. 1987;94(7):649–55. doi: 10.1111/j.1471-0528.1987.tb03169.x. [DOI] [PubMed] [Google Scholar]
- 49.Dommisse J, Tiltman AJ. Placental bed biopsies in placental abruption. Br J Obstet Gynaecol. 1992;99(8):651–4. doi: 10.1111/j.1471-0528.1992.tb13848.x. [DOI] [PubMed] [Google Scholar]
- 50.Pringle KG, Kind KL, Sferruzzi-Perri AN, Thompson JG, Roberts CT. Beyond oxygen, complex regulation and activity of hypoxia inducible factors in pregnancy. Hum Reprod Update. 2009 doi: 10.1093/humupd/dmp046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kingdom JC, Kaufmann P. Oxygen and placental villous development: origins of fetal hypoxia. Placenta. 1997;18(8):613–21. doi: 10.1016/s0143-4004(97)90000-x. discussion 623–6. [DOI] [PubMed] [Google Scholar]
- 52.Jensen GM, Moore LG. The effect of high altitude and other risk factors on birthweight: independent or interactive effects? Am J Public Health. 1997;87(6):1003–7. doi: 10.2105/ajph.87.6.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moore LG, Niermeyer S, Zamudio S. Human adaptation to high altitude: regional and life-cycle perspectives. Am J Phys Anthropol. 1998;(Suppl 27):25–64. doi: 10.1002/(sici)1096-8644(1998)107:27+<25::aid-ajpa3>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 54.Spokane RB, Clark LC, Jr, Bhargava HK, Burden MK, Davis SL. An implanted peritoneal oxygen tonometer that can be calibrated in situ. ASAIO Trans. 1990;36(3):M719–22. [PubMed] [Google Scholar]
- 55.Hustin J, Schaaps JP. Echographic [corrected] and anatomic studies of the maternotrophoblastic border during the first trimester of pregnancy. Am J Obstet Gynecol. 1987;157(1):162–8. doi: 10.1016/s0002-9378(87)80371-x. [DOI] [PubMed] [Google Scholar]
- 56.Jaffe R, Woods JR., Jr Color Doppler imaging and in vivo assessment of the anatomy and physiology of the early uteroplacental circulation. Fertil Steril. 1993;60(2):293–7. doi: 10.1016/s0015-0282(16)56100-7. [DOI] [PubMed] [Google Scholar]
- 57.Jauniaux E, Jurkovic D, Campbell S, Kurjak A, Hustin J. Investigation of placental circulations by color Doppler ultrasonography. Am J Obstet Gynecol. 1991;164(2):486–8. doi: 10.1016/s0002-9378(11)80005-0. [DOI] [PubMed] [Google Scholar]
- 58.Jurkovic D, Jauniaux E, Kurjak A, Hustin J, Campbell S, Nicolaides KH. Transvaginal color Doppler assessment of the uteroplacental circulation in early pregnancy. Obstet Gynecol. 1991;77(3):365–9. [PubMed] [Google Scholar]
- 59.Jauniaux E, Watson AL, Hempstock J, Bao YP, Skepper JN, Burton GJ. Onset of maternal arterial blood flow and placental oxidative stress. A possible factor in human early pregnancy failure. Am J Pathol. 2000;157(6):2111–22. doi: 10.1016/S0002-9440(10)64849-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Genbacev O, Joslin R, Damsky CH, Polliotti BM, Fisher SJ. Hypoxia alters early gestation human cytotrophoblast differentiation/invasion in vitro and models the placental defects that occur in preeclampsia. J Clin Invest. 1996;97(2):540–50. doi: 10.1172/JCI118447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Genbacev O, Zhou Y, Ludlow JW, Fisher SJ. Regulation of human placental development by oxygen tension. Science. 1997;277(5332):1669–72. doi: 10.1126/science.277.5332.1669. [DOI] [PubMed] [Google Scholar]
- 62.Caniggia I, Winter JL. Adriana and Luisa Castellucci Award lecture 2001. Hypoxia inducible factor-1: oxygen regulation of trophoblast differentiation in normal and pre-eclamptic pregnancies--a review. Placenta. 2002;23(Suppl A):S47–57. doi: 10.1053/plac.2002.0815. [DOI] [PubMed] [Google Scholar]
- 63.De Marco CS, Caniggia I. Mechanisms of oxygen sensing in human trophoblast cells. Placenta. 2002;23(Suppl A):S58–68. doi: 10.1053/plac.2002.0809. [DOI] [PubMed] [Google Scholar]
- 64.Robins JC, Heizer A, Hardiman A, Hubert M, Handwerger S. Oxygen tension directs the differentiation pathway of human cytotrophoblast cells. Placenta. 2007;28(11–12):1141–6. doi: 10.1016/j.placenta.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 65.Nevo O, Soleymanlou N, Wu Y, Xu J, Kingdom J, Many A, Zamudio S, Caniggia I. Increased expression of sFlt-1 in in vivo and in vitro models of human placental hypoxia is mediated by HIF-1. Am J Physiol Regul Integr Comp Physiol. 2006;291(4):R1085–93. doi: 10.1152/ajpregu.00794.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Redman CW, Sargent IL. Placental debris, oxidative stress and pre-eclampsia. Placenta. 2000;21(7):597–602. doi: 10.1053/plac.2000.0560. [DOI] [PubMed] [Google Scholar]
- 67.Nizet V, Johnson RS. Interdependence of hypoxic and innate immune responses. Nat Rev Immunol. 2009;9(9):609–17. doi: 10.1038/nri2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bird A. Perceptions of epigenetics. Nature. 2007;447(7143):396–8. doi: 10.1038/nature05913. [DOI] [PubMed] [Google Scholar]
- 69.Chen T, Li E. Establishment and maintenance of DNA methylation patterns in mammals. Curr Top Microbiol Immunol. 2006;301:179–201. doi: 10.1007/3-540-31390-7_6. [DOI] [PubMed] [Google Scholar]
- 70.Hajkova P, Erhardt S, Lane N, Haaf T, El-Maarri O, Reik W, Walter J, Surani MA. Epigenetic reprogramming in mouse primordial germ cells. Mech Dev. 2002;117(1–2):15–23. doi: 10.1016/s0925-4773(02)00181-8. [DOI] [PubMed] [Google Scholar]
- 71.Lee J, Inoue K, Ono R, Ogonuki N, Kohda T, Kaneko-Ishino T, Ogura A, Ishino F. Erasing genomic imprinting memory in mouse clone embryos produced from day 11.5 primordial germ cells. Development. 2002;129(8):1807–17. doi: 10.1242/dev.129.8.1807. [DOI] [PubMed] [Google Scholar]
- 72.Weiss A, Keshet I, Razin A, Cedar H. DNA demethylation in vitro: involvement of RNA. Cell. 1996;86(5):709–18. doi: 10.1016/s0092-8674(00)80146-4. [DOI] [PubMed] [Google Scholar]
- 73.Bhattacharya SK, Ramchandani S, Cervoni N, Szyf M. A mammalian protein with specific demethylase activity for mCpG DNA. Nature. 1999;397(6720):579–83. doi: 10.1038/17533. [DOI] [PubMed] [Google Scholar]
- 74.Zhu B, Benjamin D, Zheng Y, Angliker H, Thiry S, Siegmann M, Jost JP. Overexpression of 5-methylcytosine DNA glycosylase in human embryonic kidney cells EcR293 demethylates the promoter of a hormone-regulated reporter gene. Proc Natl Acad Sci U S A. 2001;98(9):5031–6. doi: 10.1073/pnas.091097298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yamagata K. Capturing epigenetic dynamics during pre-implantation development using live cell imaging. J Biochem. 2008;143(3):279–86. doi: 10.1093/jb/mvn001. [DOI] [PubMed] [Google Scholar]
- 76.Polanski Z, Motosugi N, Tsurumi C, Hiiragi T, Hoffmann S. Hypomethylation of paternal DNA in the late mouse zygote is not essential for development. Int J Dev Biol. 2008;52(2–3):295–8. doi: 10.1387/ijdb.072347zp. [DOI] [PubMed] [Google Scholar]
- 77.Hajkova P, Ancelin K, Waldmann T, Lacoste N, Lange UC, Cesari F, Lee C, Almouzni G, Schneider R, Surani MA. Chromatin dynamics during epigenetic reprogramming in the mouse germ line. Nature. 2008;452(7189):877–81. doi: 10.1038/nature06714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shi W, Haaf T. Aberrant methylation patterns at the two-cell stage as an indicator of early developmental failure. Mol Reprod Dev. 2002;63(3):329–34. doi: 10.1002/mrd.90016. [DOI] [PubMed] [Google Scholar]
- 79.Hata K, Okano M, Lei H, Li E. Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Development. 2002;129(8):1983–93. doi: 10.1242/dev.129.8.1983. [DOI] [PubMed] [Google Scholar]
- 80.Kaneda M, Okano M, Hata K, Sado T, Tsujimoto N, Li E, Sasaki H. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature. 2004;429(6994):900–3. doi: 10.1038/nature02633. [DOI] [PubMed] [Google Scholar]
- 81.Cirio MC, Martel J, Mann M, Toppings M, Bartolomei M, Trasler J, Chaillet JR. DNA methyltransferase 1o functions during preimplantation development to preclude a profound level of epigenetic variation. Dev Biol. 2008;324(1):139–50. doi: 10.1016/j.ydbio.2008.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hirasawa R, Chiba H, Kaneda M, Tajima S, Li E, Jaenisch R, Sasaki H. Maternal and zygotic Dnmt1 are necessary and sufficient for the maintenance of DNA methylation imprints during preimplantation development. Genes Dev. 2008;22(12):1607–16. doi: 10.1101/gad.1667008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ehrlich M. The ICF syndrome, a DNA methyltransferase 3B deficiency and immunodeficiency disease. Clin Immunol. 2003;109(1):17–28. doi: 10.1016/s1521-6616(03)00201-8. [DOI] [PubMed] [Google Scholar]
- 84.Rahnama F, Shafiei F, Gluckman PD, Mitchell MD, Lobie PE. Epigenetic regulation of human trophoblastic cell migration and invasion. Endocrinology. 2006;147(11):5275–83. doi: 10.1210/en.2006-0288. [DOI] [PubMed] [Google Scholar]
- 85.Hattori N, Nishino K, Ko YG, Ohgane J, Tanaka S, Shiota K. Epigenetic control of mouse Oct-4 gene expression in embryonic stem cells and trophoblast stem cells. J Biol Chem. 2004;279(17):17063–9. doi: 10.1074/jbc.M309002200. [DOI] [PubMed] [Google Scholar]
- 86.Jedrusik A, Parfitt DE, Guo G, Skamagki M, Grabarek JB, Johnson MH, Robson P, Zernicka-Goetz M. Role of Cdx2 and cell polarity in cell allocation and specification of trophectoderm and inner cell mass in the mouse embryo. Genes Dev. 2008;22(19):2692–706. doi: 10.1101/gad.486108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Johnson MH, McConnell JM. Lineage allocation and cell polarity during mouse embryogenesis. Semin Cell Dev Biol. 2004;15(5):583–97. doi: 10.1016/j.semcdb.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 88.Avilion AA, Nicolis SK, Pevny LH, Perez L, Vivian N, Lovell-Badge R. Multipotent cell lineages in early mouse development depend on SOX2 function. Genes Dev. 2003;17(1):126–40. doi: 10.1101/gad.224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Murohashi M, Nakamura T, Tanaka S, Ichise T, Yoshida N, Yamamoto T, Shibuya M, Schlessinger J, Gotoh N. An FGF4-FRS2alpha-Cdx2 axis in trophoblast stem cells induces Bmp4 to regulate proper growth of early mouse embryos. Stem Cells. 2010;28(1):113–21. doi: 10.1002/stem.247. [DOI] [PubMed] [Google Scholar]
- 90.Ohgane J, Aikawa J, Ogura A, Hattori N, Ogawa T, Shiota K. Analysis of CpG islands of trophoblast giant cells by restriction landmark genomic scanning. Dev Genet. 1998;22(2):132–40. doi: 10.1002/(SICI)1520-6408(1998)22:2<132::AID-DVG3>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 91.Ohgane J, Hattori N, Oda M, Tanaka S, Shiota K. Differentiation of trophoblast lineage is associated with DNA methylation and demethylation. Biochem Biophys Res Commun. 2002;290(2):701–6. doi: 10.1006/bbrc.2001.6258. [DOI] [PubMed] [Google Scholar]
- 92.Park HM, Shin SJ, Choi DH, Oh D, Lee S, Kim NK. Association between folate metabolism-related gene polymorphisms and methylation of p16(INK4A) and hMLH1 genes in spontaneously aborted embryos with normal chromosomal integrity. Fertil Steril. 2008;90(5):1605–10. doi: 10.1016/j.fertnstert.2007.09.046. [DOI] [PubMed] [Google Scholar]
- 93.Dobson AT, Davis RM, Rosen MP, Shen S, Rinaudo PF, Chan J, Cedars MI. Methylenetetrahydrofolate reductase C677T and A1298C variants do not affect ongoing pregnancy rates following IVF. Hum Reprod. 2007;22(2):450–6. doi: 10.1093/humrep/del396. [DOI] [PubMed] [Google Scholar]
- 94.Pliushch G, Schneider E, Weise D, El Hajj N, Tresch A, Seidmann L, Coerdt W, Muller AM, Zechner U, Haaf T. Extreme methylation values of imprinted genes in human abortions and stillbirths. Am J Pathol. 2010;176(3):1084–90. doi: 10.2353/ajpath.2010.090764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Oudejans CB. Noncoding RNA and DNA as biomarkers, toward an epigenetic fetal barcode for use in maternal plasma. Clin Chem. 2008;54(3):456–7. doi: 10.1373/clinchem.2007.100123. [DOI] [PubMed] [Google Scholar]
- 96.Muller HM, Ivarsson L, Schrocksnadel H, Fiegl H, Widschwendter A, Goebel G, Kilga-Nogler S, Philadelphy H, Gutter W, Marth C, Widschwendter M. DNA methylation changes in sera of women in early pregnancy are similar to those in advanced breast cancer patients. Clin Chem. 2004;50(6):1065–8. doi: 10.1373/clinchem.2003.030387. [DOI] [PubMed] [Google Scholar]
- 97.Tsui DW, Chan KC, Chim SS, Chan LW, Leung TY, Lau TK, Lo YM, Chiu RW. Quantitative aberrations of hypermethylated RASSF1A gene sequences in maternal plasma in pre-eclampsia. Prenat Diagn. 2007;27(13):1212–8. doi: 10.1002/pd.1897. [DOI] [PubMed] [Google Scholar]
- 98.Yuen RK, Avila L, Penaherrera MS, von Dadelszen P, Lefebvre L, Kobor MS, Robinson WP. Human placental-specific epipolymorphism and its association with adverse pregnancy outcomes. PLoS One. 2009;4(10):e7389. doi: 10.1371/journal.pone.0007389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tycko B, Efstratiadis A. Genomic imprinting: piece of cake. Nature. 2002;417(6892):913–4. doi: 10.1038/417913a. [DOI] [PubMed] [Google Scholar]
- 100.Charalambous M, da Rocha ST, Ferguson-Smith AC. Genomic imprinting, growth control and the allocation of nutritional resources: consequences for postnatal life. Curr Opin Endocrinol Diabetes Obes. 2007;14(1):3–12. doi: 10.1097/MED.0b013e328013daa2. [DOI] [PubMed] [Google Scholar]
- 101.Eggermann T. Silver-Russell and Beckwith-Wiedemann syndromes: opposite (epi)mutations in 11p15 result in opposite clinical pictures. Horm Res. 2009;71(Suppl 2):30–5. doi: 10.1159/000192433. [DOI] [PubMed] [Google Scholar]
- 102.Gurrieri F, Accadia M. Genetic imprinting: the paradigm of Prader-Willi and Angelman syndromes. Endocr Dev. 2009;14:20–8. doi: 10.1159/000207473. [DOI] [PubMed] [Google Scholar]
- 103.Owen CM, Segars JH., Jr Imprinting disorders and assisted reproductive technology. Semin Reprod Med. 2009;27(5):417–28. doi: 10.1055/s-0029-1237430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chang AS, Moley KH, Wangler M, Feinberg AP, Debaun MR. Association between Beckwith-Wiedemann syndrome and assisted reproductive technology: a case series of 19 patients. Fertil Steril. 2005;83(2):349–54. doi: 10.1016/j.fertnstert.2004.07.964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.DeBaun MR, Niemitz EL, Feinberg AP. Association of in vitro fertilization with Beckwith-Wiedemann syndrome and epigenetic alterations of LIT1 and H19. Am J Hum Genet. 2003;72(1):156–60. doi: 10.1086/346031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zechner U, Pliushch G, Schneider E, El Hajj N, Tresch A, Shufaro Y, Seidmann L, Coerdt W, Muller AM, Haaf T. Quantitative methylation analysis of developmentally important genes in human pregnancy losses after ART and spontaneous conception. Mol Hum Reprod. 2009 doi: 10.1093/molehr/gap107. [DOI] [PubMed] [Google Scholar]
- 107.Guillemot F, Caspary T, Tilghman SM, Copeland NG, Gilbert DJ, Jenkins NA, Anderson DJ, Joyner AL, Rossant J, Nagy A. Genomic imprinting of Mash2, a mouse gene required for trophoblast development. Nat Genet. 1995;9(3):235–42. doi: 10.1038/ng0395-235. [DOI] [PubMed] [Google Scholar]
- 108.Guillemot F, Nagy A, Auerbach A, Rossant J, Joyner AL. Essential role of Mash-2 in extraembryonic development. Nature. 1994;371(6495):333–6. doi: 10.1038/371333a0. [DOI] [PubMed] [Google Scholar]
- 109.Tanaka M, Gertsenstein M, Rossant J, Nagy A. Mash2 acts cell autonomously in mouse spongiotrophoblast development. Dev Biol. 1997;190(1):55–65. doi: 10.1006/dbio.1997.8685. [DOI] [PubMed] [Google Scholar]
- 110.Fukunaga M. Immunohistochemical characterization of p57(KIP2) expression in early hydatidiform moles. Hum Pathol. 2002;33(12):1188–92. doi: 10.1053/hupa.2002.129421. [DOI] [PubMed] [Google Scholar]
- 111.Fisher RA, Hodges MD, Rees HC, Sebire NJ, Seckl MJ, Newlands ES, Genest DR, Castrillon DH. The maternally transcribed gene p57(KIP2) (CDNK1C) is abnormally expressed in both androgenetic and biparental complete hydatidiform moles. Hum Mol Genet. 2002;11(26):3267–72. doi: 10.1093/hmg/11.26.3267. [DOI] [PubMed] [Google Scholar]
- 112.Kanayama N, Takahashi K, Matsuura T, Sugimura M, Kobayashi T, Moniwa N, Tomita M, Nakayama K. Deficiency in p57Kip2 expression induces preeclampsia-like symptoms in mice. Mol Hum Reprod. 2002;8(12):1129–35. doi: 10.1093/molehr/8.12.1129. [DOI] [PubMed] [Google Scholar]
- 113.Mayer W, Hemberger M, Frank HG, Grummer R, Winterhager E, Kaufmann P, Fundele R. Expression of the imprinted genes MEST/Mest in human and murine placenta suggests a role in angiogenesis. Dev Dyn. 2000;217(1):1–10. doi: 10.1002/(SICI)1097-0177(200001)217:1<1::AID-DVDY1>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]