

Abstract
During myogenic differentiation the short mitochondria of myoblasts change into the extensively elongated network observed in myotubes. The functional relevance and the molecular mechanisms driving the formation of this mitochondrial network are unknown. We now show that mitochondrial elongation is required for myogenesis to occur and that this event depends on the cellular generation of nitric oxide (NO). Inhibition of NO synthesis in myogenic precursor cells leads to inhibition of mitochondrial elongation and of myogenic differentiation. This is due to the enhanced activity, translocation and docking of the pro-fission GTPase dynamin-related protein-1 (Drp1) to mitochondria, leading also to a latent mitochondrial dysfunction that increased sensitivity to apoptotic stimuli. These effects of NO inhibition were not observed in myogenic precursor cells containing a dominant-negative form of Drp1. Both NO-dependent repression of Drp1 action and maintenance of mitochondrial integrity and function were mediated through the soluble guanylate cyclase. These data uncover a novel level of regulation of differentiation linking mitochondrial morphology and function to myogenic differentiation.
Keywords: nitric oxide, mitochondrial fission, Drp1, myogenesis
Mitochondria are highly dynamic organelles that continuously and reversibly rearrange their structure through the tightly-regulated processes of fission and fusion of their inner and outer membranes.1,2 Mitochondrial fission is regulated by the GTPase dynamin-related protein-1 (Drp1) that cycles from the cytosol to mitochondria, where it binds to Fis1, an integral outer membrane protein, whereas fusion requires the activity of three large GTPase proteins, mitofusin-1 and 2, and optic atrophy-1 (Opa1).3 Mitochondrial dynamics participate in a cell’s response to fluctuations in oxygen and nutrient availability,4,5 and contribute to regulation of the development and activity of a number of tissues and organs.6-8
Despite the recognized role played by mitochondria in the overall skeletal muscle function, including its repair after damage,9-11 the role of mitochondrial fission and fusion in skeletal muscle bioenergetics and development has not been investigated. Interestingly, during myogenic differentiation the short mitochondria of the myoblasts change into the extensive elongated network observed in the myotubes,12 suggesting that they may have a role in myogenesis.
We report here that inhibition of mitochondrial fission, with the consequent formation of a mitochondrial network, is required for myogenic differentiation and that this is dependent on inhibition of Drp1 function. Furthermore, we report that endogenous nitric oxide (NO) generated by the muscle-specific neuronal NO synthase (NOS) inhibits Drp1-dependent mitochondrial fission, and by this inhibition allows the process of myogenesis to occur. The action of NO was reversible (with a fast onset and offset), physiological, as it was mediated by cyclic GMP (cGMP), and was accompanied by profound changes in the mitochondrial bioenergetics that have a role in myogenesis. Thus NO, by suppressing the activity of Drp1, allows the formation of the mitochondrial network and the changes in cell bioenergetics needed for myogenesis to occur.
Results
NO regulates myogenesis and mitochondrial elongation in differentiating myogenic precursor cells through cGMP
We examined the effects of endogenous generation of NO on myogenic differentiation and on changes in mitochondrial morphology. Myogenic precursor cells, freshly isolated from the muscles of newborn mice, were transiently transfected with a red fluorescent protein targeted to mitochondria (mitoDsRed) and were induced to differentiate.13 Differentiating primary myogenic precursor cells (Figure 1a) expressed progressively the differentiation markers Mef-2A, Myo-D, myogenin and sarcomeric myosin (MyHC) (Figure 1b and Supplementary Figure S1b). In parallel morphological studies, mitochondria that were round in shape in proliferating cells were found to become progressively elongated, forming an extensive branched network at 12 h of differentiation (see Figure 1a and Supplementary Figure S1a for quantification of mitochondrial elongation, and Supplementary Figure S2 for the 3D reconstructions). These changes in mitochondrial morphology were accompanied by changes in the expression of proteins regulating mitochondrial fission and fusion, namely Drp1, mitofusins and Opa1 (Figure 1c and Supplementary Figure S1b). The increased expression of these proteins was not due to mitochondrial biogenesis as expression of other mitochondrial proteins such as cytochrome c and cytochrome c oxidase did not change, neither did we detect an increase in mtDNA (Figure 1c and data not shown). nNOS protein expression in differentiating cells was unchanged whereas NOS activity was increased, indicating that myogenic differentiation triggers NOS activation (Figure 1d). The increased NOS activity was accompanied by increased generation of cGMP, a physiological messenger generated by NO through activation of guanylate cyclase (Figure 1e).14 Both NOS activity and cGMP generation were inhibited by the NOS inhibitor Nω-nitro-l-arginine methylester (l-NAME) (Figure 1d and e). We examined the role of endogenous NO and cGMP generation on myogenic differentiation and mitochondrial morphology. l-NAME and the guanylate cyclase inhibitor H-(1,2,4)-oxadiazolo[4,3-]quinoxalin-1-one (ODQ) inhibited both expression of myogenic markers (Figure 1b) and elongation of mitochondria (Figure 1a and Supplementary Figure S1). Inhibition of NO or cGMP generation did not modify the expression of the mitochondria-shaping proteins or of mtDNA (Figure 1c and data not shown). The neuronal NOS splice variant μ is particularly important in muscle physiology.15 Silencing of the enzyme using a specific siRNA inhibited the expression of myogenic differentiation markers and elongation of mitochondria (Supplementary Figure S3), indicating that NO generation and the consequent elevation of cGMP are required for both mitochondrial elongation and myogenesis.
Figure 1.
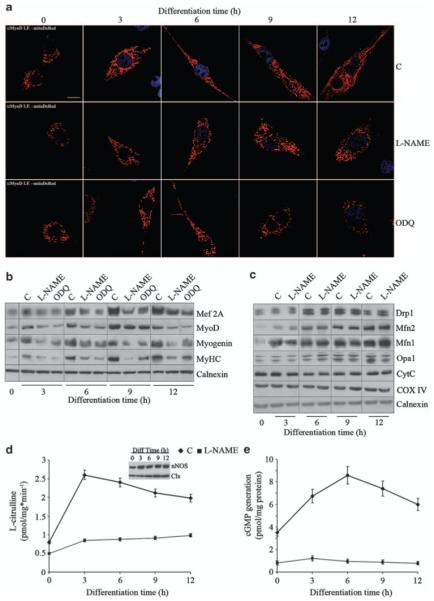
NO regulates myogenesis and mitochondrial network formation through cGMP. Myogenic precursor cells transfected with the red fluorescent mitochondrial protein mitoDsRed were treated with l-NAME, ODQ or vehicle (C) and differentiated by serum withdrawal for up to 12 h. (a) Mitochondrial morphology detected by transient transfection with mitoDsRed; nuclei are stained for Myo-D (blue) to distinguish myogenic cells from possible contaminating cells. Bar: 10 μm. (b) Expression of the differentiation markers Mef-2A, Myo-D, myogenin and sarcomeric myosin (MyHC) as detected by western blot analysis. (c) Expression of the mitochondrial proteins mitofusins (Mfn)-1 and 2, Opa1, Drp1, cytochrome c (CytC) and cytochrome c oxidase subunit-IV (COX-IV). Calnexin (Clx) was used as a loading control in the experiments in panels b and c. (d) NOS activity measured as the conversion of l-[3H]-arginine into l-[3H]-citrulline. The inset shows the levels of expression of nNOS during myogenic differentiation. Clx was used as a loading control. (e) Generation of cGMP. The images are from one of four independent reproducible experiments; the graphs represent the values±S.E.M. (n = 4)
Changes in mitochondrial morphology may be the consequence of toxic cellular effects. In differentiating myoblasts neither l-NAME nor ODQ induced significant changes in the basal rate of apoptosis, as assessed up to 72 h of differentiation by measuring phosphatidylserine exposure to the plasma membrane in 7-amino actinomycin-d (7aad)-excluding cells (Figure 2a) and the number of trypan blue-excluding (viable) cells (Figure 2b). Consistently, we did not observe activation of caspase-3 and 9 (Figure 2c). Likewise, neither ODQ nor l-NAME induced spontaneous apoptosis in proliferating, undifferentiated myoblasts (not shown). These results rule out a toxic action by l-NAME and ODQ.
Figure 2.
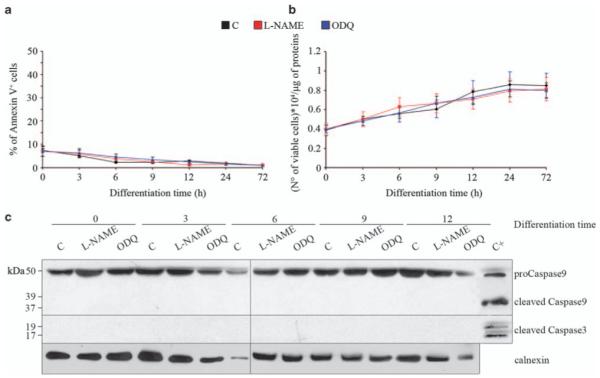
Inhibition of myogenesis and mitochondrial network formation by NO removal are not due to toxic effects. Myogenic precursor cells were treated with l-NAME, ODQ or vehicle (C) and differentiated by serum withdrawal for up to 72 h, during which time the rate of spontaneous apoptosis was measured. (a) Phosphatidylserine exposure to the plasma membrane in 7AAD-excluding cells. (b) The number of trypan blue-excluding cells. (c) Expression of procaspase-9 and its cleaved form, and of cleaved caspase-3. The images are from one of three independent reproducible experiments; the graphs represent the values±S.E.M. (n = 3)
NO/cGMP stimulate myogenesis through inhibition of mitochondrial fission
Myogenic precursor cells were co-transfected with vectors coding for mitoDsRed and cytosolic YFP (pEN1YFP). After induction of differentiation for 6 h we used real-time confocal microscopy and transmission electron microscopy to investigate the effects of l-NAME and ODQ on mitochondrial morphology. Differentiation for this period resulted in the majority of mitochondria being in the elongated form (Figure 3b and c). However, both l-NAME and ODQ induced mitochondrial fragmentation within minutes after their addition, which persisted throughout the time of the analysis (40 min) and was confirmed at the ultrastructural level (see Figure 3a and Supplementary Figure S4 for electron microscopy images, and Supplementary Figure S5 for the films). The NO donor (Z)-1-[2-(2-aminoethyl)-N-(2-ammonioethyl) amino]diazen-1-ium-1,2 diolate (DETA-NO) and the membrane-permeable cGMP mimic 8Br-cGMP reversed the effects of l-NAME and ODQ, respectively, indicating the cGMP dependency of NO’s action (Figure 3b and c). These results indicate that changes in mitochondrial morphology are due to the actions of NO/cGMP.
Figure 3.
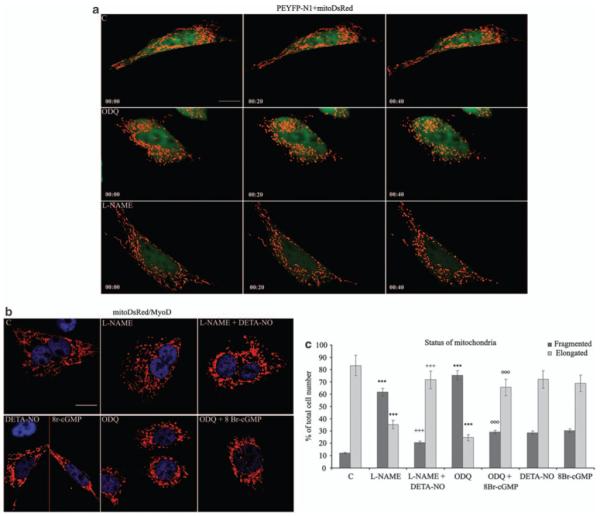
NO and cGMP regulate mitochondrial dynamics. (a) Myogenic precursor cells were co-transfected with vectors coding for mitoDsRed and cytosolic YFP (pEN1YFP) and differentiated. l-NAME, ODQ or vehicle (C) was added after 6 h of differentiation and mitochondrial morphology was examined by time-lapse microscopy at the indicated time points. The images are representative of three independent reproducible experiments. Bar: 10 μm. (b, c) Myogenic precursor cells were transfected with the mitoDsRed-coding vector and differentiated. Six hours later cells were treated with l-NAME, ODQ, DETA-NO, 8Br-cGMP or vehicle (C) in various combinations as indicated, fixed and examined after 50 min. The red and blue colors in panel b show staining of mitoDsRed and an anti-MyoD Ab, respectively. Bar: 10 μm. (c) Fifteen random fields per sample were acquired and the fragmentation index was established using the imagetool 3.0 software. The dimension ranges for fragmented and elongated were 1.20 μm ± 0.2 and >3.50 μm, respectively. The asterisks show the statistical probability versus C, crosses versus l-NAME and circles versus ODQ (P<0.001)
Mitochondrial dynamics depend on the processes of fission and fusion.1,2 We investigated whether NO/cGMP regulates fission, fusion or both processes. To examine mitochondrial fusion, we performed a polyethylene glycol fusion assay on myoblasts differentiated for 6 h. Neither l-NAME nor ODQ changed the rate of mitochondrial fusion, as monitored by measuring the appearance of double mtRFP/mtGFP-positive mitochondria in the heteropolykaryons in the presence of cycloheximide for a further 6 h after polyethylene glycol addition (Figure 4a and b). However, a dominant-negative (K38A) mutant of Drp116 prevented the effects of l-NAME and ODQ on mitochondrial morphology (Figure 4c). This mutant also prevented the slowing of myogenic differentiation induced by l-NAME and ODQ, and enhanced the myogenic process, most likely by inhibiting basal Drp1 function (Figure 4d and Supplementary Figure S6). These results show that mitochondrial elongation by NO is due to reduced fission and that this modulation regulates myogenesis.
Figure 4.
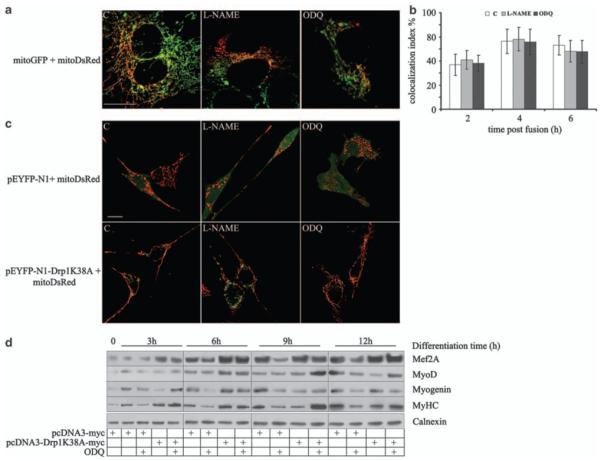
NO and cGMP stimulate myogenesis through inhibition of mitochondrial fission. (a, b) Myogenic precursor cells were transfected with the vector coding for either mitoGFP (green) or mitoDsRed (red), mixed, differentiated for 6 h and exposed for 1 h to l-NAME, ODQ, DETA-NO, 8Br-cGMP or vehicle (C) as indicated. Plasma membrane fusion was induced by addition of PEG 1500 and mitochondrial fusion events were quantified after 2, 4 and 6 h in the heteropolykaryons by measuring the fraction of mitochondria simultaneously positive for both mtGFP and mitoDsRed (colocalization index %; n = 3). Bar: 10 μM. (c) Myogenic precursor cells were transfected with vectors coding for the cytosolic marker pEYFP-N1 (green) or the dominant-negative Drp1, pEYFP-N1-DRPK38A, and differentiated in the presence of l-NAME and ODQ. Mitochondrial morphology was assessed after 6 h. Bar: 10 μM. (d) Expression of the myogenic differentiation markers Mef-2A, MyoD, myogenin and sarcomeric myosin (MyHC) was determined by western blotting in myogenic precursors transfected with either the empty pCDNA3 vector or the dominant-negative Drp1 (pcDNA3-Drp1 K38A) at the indicated time points. The result of one out of three reproducible experiments is shown
NO/cGMP control Drp1 localization and activity
To investigate how NO regulates the function of Drp1 through cGMP we monitored the activity and sub-cellular localization of Drp1 in differentiating myoblasts. Sub-cellular fractionation and immunoblotting for Drp1 showed that mitochondrial Drp1 localization was significantly increased by l-NAME and ODQ (Figure 5a). In addition, ODQ and l-NAME increased the interaction between Drp1 and its mitochondrial receptor Fis1, as shown by co-immunoprecipitation (Figure 5b and data not shown). Finally, l-NAME and ODQ increased the binding of Drp1 to GTP, an assay that measures GTPase activity in intact cells, as shown by pull-down experiments using GTP-conjugated beads (Figure 5c). DETA-NO or 8Br-cGMP reversed all the effects of l-NAME and ODQ. Of note, we did not detect any significant changes in the mitochondrial and cytosolic calcium concentrations upon NOS or guanylate cyclase inhibition, thereby ruling out calcium overload as a trigger for fission under these conditions (not shown). Drp1 is known to be regulated by means of protein kinase-A and C through phosphorylation. We investigated whether NO/cGMP acting through G-kinase induced phosphorylation of Drp1. As shown in Figure 6a myogenic differentiation was accompanied by phosphorylation of Drp1, which was prevented by both l-NAME and ODQ. In addition, DETA-NO and 8Br-cGMP induced the phosphorylation of the protein in a manner that was prevented by the G-kinase inhibitor KT5823 and enhanced by BAY41-2272, which activates guanylate cyclase independently of NO (Figure 6b). These experiments indicate that NO/cGMP phosphorylates Drp1 through G-kinase and inhibits its function during myogenic differentiation.
Figure 5.
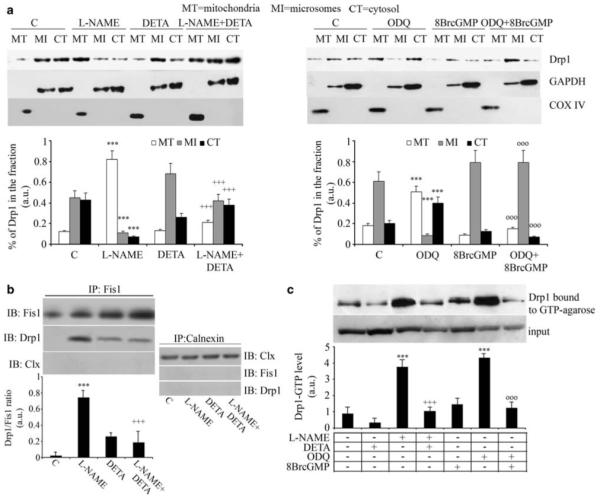
NO and cGMP control the activity and localization of Drp1. Myogenic precursor cells were differentiated for 6 h and treated for 1 h with l-NAME, ODQ, DETA-NO, 8Br-cGMP or vehicle (C), added as indicated. (a) Cells were fractionated and the mitochondrial (MT), microsomal (MI) and cytosolic (CI) fractions were examined for Drp1 content by western blotting, using GAPDH and COX-IV as loading controls for cytosolic and mitochondrial proteins, respectively. The results shown in the images are representative of five reproducible experiments, which are quantified in the corresponding graphs. (b) Co-immunoprecipitation of Drp1 with Fis1 was performed using the an anti-Fis1 Ab for immunoprecipitation (IP). The amount of co-immunoprecipitated Drp1 was determined by immunoblotting (IB) using a specific anti-Drp1 Ab. As control the amount of Fis1 immunoprecipitated was also checked by IB. As a further control for specificity we checked the absence of co-immunoprecipitation of Drp1 with calnexin. (c) Drp1 GTPase activity was measured in pull-down experiments using GTP-conjugated beads. The representative images shown in panels b and c are from four independent reproducible experiments. The graphs below each image report the densitometric values±S.E.M. of the relevant band from the four experiments. In all panels, the asterisks, crosses and circles show statistical probability (P<0.001), calculated versus C, l-NAME and ODQ, respectively
Figure 6.

NO/cGMP triggers G-kinase-dependent phosphorylation of Drp1. (a) Myc-Drp1 was immunoprecipitated from [32P]orthophosphate-labeled proliferating or differentiating myogenic precursor cells. The immunoprecipitates were resolved by SDS-PAGE and 32P-labeled-Myc-Drp1 was visualized by autoradiography. (b) Myc-Drp1 was immunoprecipitated from [32P]orthophosphate-labeled myogenic precursor cells treated with DETA-NO, 8Br cGMP with or without KT5823 and ODQ, respectively, or BAY41-2272. The immunoprecipitates were resolved by SDS-PAGE and 32P-labeled-Myc-Drp1 was visualized by autoradiography
NO/cGMP maintain mitochondrial membrane potential in differentiating myoblasts
We investigated whether changes in mitochondrial morphology and dynamics affected the bioenergetics of differentiating myoblasts, using the F1F0 ATP synthetase inhibitor oligomycin; this is used as a sensitive test of latent mitochondrial dysfunction in intact cells.17 In proliferating myogenic precursor cells and in control differentiating myoblasts (6 h) oligomycin increased the mitochondrial membrane potential, as indicated by an increase in the mitochondrial fluorescence of the potentiometric dye tetramethyl rhodamine methyl ester (TMRM) followed by real-time imaging (Figure 7a); this is as expected for healthy mitochondria.17 By contrast, in l-NAME- or ODQ-treated differentiating myoblasts, addition of oligomycin led to progressive mitochondrial depolarization, which was fully normalized by Drp1 K38A (Figure 7a and data not shown), establishing a link between changes in mitochondrial dynamics and mitochondrial membrane potential. l-NAME and ODQ had no effects on the mitochondrial membrane potential in proliferating cells (not shown). These findings suggest that if NO or cGMP is depleted during myogenesis the fragmented mitochondria that result will show a latent mitochondrial dysfunction.
Figure 7.

The bioenergetic consequences of regulation of fission by NO and cGMP. Myogenic precursor cells were transfected with pEYFP-N1 or pEYFP-N1-Drp1 K38A, differentiated (diff) or allowed to proliferate (pro) for 6 h, and treated as specified. (a) The cells were loaded with the mitochondrial potentiometric dye TMRM and the mitochondrial membrane potential was measured 1 h after treatment with the vehicle (C) or ODQ. The arrows indicate addition of oligomycin (O; 1 μg/ml) and FCCP (4 μM). (b, c) The cells were loaded with luciferin-luciferase and the ATP generated through oxidative phosphorylation (b) or glycolysis (c) was measured after addition of the vehicle or ODQ. (d, e) Respiratory function was measured using a high-sensitivity respirometer. The values shown are for total, oligomycin-resistant, maximal (uncoupled) and residual oxygen consumption (ROC), as well as for the ratio of oligomycin-resistant to maximal (oligomycin resistant/M) and coupled (total minus oligomycin resistant) to maximal (C/M). (f) Activity of the mitochondrial respiratory complexes measured as in panel a assessing ATP generation in cells respiring on pyruvate–malate (P + M) or glutamate–malate (G + M) (complex-I), succinate–rotenone (S + Rot) (complex-II) TMPD–ascorbate–antimycin-A (T + A + AntA) (complex-IV). The panels show values ± S.E.M. (n = 4). The double (P<0.01) and triple (P<0.001) asterisks, and the crosses show statistical probability versus control and ODQ, respectively
NO/cGMP regulate mitochondrial respiration and ATP generation in differentiating myoblasts
We next investigated the effects that the latent dysfunction observed in mitochondria had on the bioenergetic parameters of differentiating myoblasts, in which NO or cGMP generation was inhibited. Total OXPHOS-ATP, measured in the presence of the glycolysis inhibitors fluoride and iodoacetate, was significantly reduced in ODQ- or l-NAME-treated differentiating cells as compared with that in untreated controls. The effects of l-NAME or ODQ were abolished by Drp1 K38A (Figure 7b and data not shown). The amount of ATP generated normally by glycolysis was low in each treatment group, with no significant changes induced by ODQ/l-NAME or Drp1 K38A (Figure 7c).
We analyzed cell respiration (time 6 h) measuring total, oligomycin-resistant (proton leak) and maximal (uncoupled) oxygen consumption in proliferating and differentiating myogenic cells. ODQ and l-NAME did not significantly alter total, oligomycin-resistant or maximal oxygen consumption in proliferating cells (not shown), whereas they reduced all respiratory parameters in the differentiating cells (Figure 7d). Neither l-NAME nor ODQ had any effect on the residual, mitochondrial-independent oxygen consumption (ROC in Figure 7d). To further assess the functional relevance of the changes induced by l-NAME and ODQ on respiration, we measured the ratios of oligomycin-resistant to maximal oxygen consumption and coupled respiration (total minus oligomycin-resistant oxygen consumption) to maximal. These rates give an estimate of whether changes in total respiration can ultimately affect the ability of the mitochondria to use the proton-motive force to synthesize ATP. In ODQ- or l-NAME-treated cells, the ratio of oligomycin-resistant to maximal respiration was reduced, whereas the ratio of coupled respiration to maximal was increased (Figure 7e and data not shown). These data indicate that cells try to compensate for the reduced respiratory capacity induced by impairment of NO or cGMP signaling with enhanced coupling of electron transport with oxidative phosphorylation. All changes induced by l-NAME or ODQ were prevented by Drp1 K38A, indicating that impairment of respiratory function induced by l-NAME or ODQ in differentiating myogenic precursor cells is a consequence of changes in mitochondrial dynamics. The use of selective inhibitors of the electron transport chain showed that l-NAME and ODQ decrease mitochondrially generated ATP regardless of which mitochondrial complex was stimulated (Figure 7f and data not shown), suggesting that cytochrome c oxidase, the terminal component of the respiratory chain, is the likely target. As with the other parameters investigated, mitochondrial complex activities were restored by Drp1 K38A.
NO/cGMP and mitochondrial dynamics: a quality-control check for myogenesis
To investigate the functional role of the induction of fission and the resulting impaired mitochondrial function, we examined cell sensitivity to oxidative stress. In differentiating myoblasts, apoptosis induced by H2O2 was significantly increased in cells in which NO/cGMP signaling was blocked by l-NAME or ODQ (Figure 8). Such increased sensitivity was Drp1-dependent, as shown by its absence in the K38A mutant. No differences in H2O2-induced apoptosis were observed in proliferating cells exposed to ODQ or l-NAME (not shown). These results indicate that in differentiating myoblasts impairment of NO/cGMP signaling and the ensuing changes in mitochondrial function lead to an increased sensitivity to apoptogenic stimuli.
Figure 8.
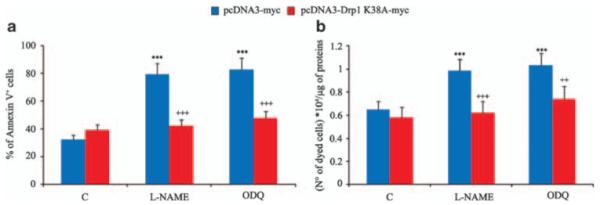
NO/cGMP protect from H2O2-induced apoptosis through inhibition of Drp1. Myogenic precursor cells were transfected with pcDNA3-Drp1-myc or pcDNA3-Drp1 K38A-myc and differentiated for 6 h. The cells were then exposed to H2O2 or vehicle in the presence or absence of l-NAME or ODQ. (a) Phosphatidylserine exposure to the plasma membrane in 7AAD-excluding cells. (b) The number of trypan blue-excluding cells. The panels show values±S.E.M. (n = 4). The asterisks and the crosses show significant differences (P<0.001) from the control of l-NAME or ODQ-treated cells expressing pcDNA-myc, and of the pCDNA3-Drp1 K38A-expressing cells from the cells expressing the empty vector, respectively
Discussion
In this study we show that inhibition of NOS activity prevents the elongation of mitochondria and myogenesis, indicating that NO has a determinant role in these processes by controlling the function and activity of the mitochondrial fission protein Drp1. Indeed expression of the dominant-negative form of Drp1 K38A reversed the effects on both control of mitochondrial dynamics and myogenesis, which occurred due to removal of NO. These effects were specific to differentiating myoblasts as NO had no effect on mitochondrial shape or activity in the proliferating myogenic precursor cells. The effects of NO were dependent on the activation of guanylate cyclase and the generation of cGMP, which is activated by nanomolar concentrations of NO.18 This indicates that the process described takes place under physiological conditions.
NO/cGMP acted selectively on mitochondrial fission, directly controlling Drp1 GTPase activity, translocation to mitochondria and interaction with Fis1. Drp1 activity is known to be tightly regulated through phosphorylation by both cyclin-dependent kinase-1 and the cAMP-dependent protein kinase, and through dephosphorylation mediated by calcineurin.19,20 Our results show that NO phosphorylates Drp1 through G-kinase. This kinase-dependent mechanism of control of Drp1 suggests the need for additional tight levels of regulation of the enzyme, and stresses the importance of this mechanism and the biological relevance of its control by NO. Furthermore, the fact that NO regulates the activity and not the expression of Drp1 points toward a dynamic role of fission control by NO that may be relevant to fine-tuning the process of myogenesis.
In neurons high, cytotoxic concentrations of NO have been reported to promote fission and apoptosis and may contribute to the pathogenesis of Alzheimer’s disease.21,22 This effect is mediated by S-nitrosylation of Drp1,23 that is, a mechanism different from the cGMP/G kinase-dependent phosphorylation we show here and that activates rather than inhibits the enzyme. These findings, although apparently contradictory to the results reported here, are in keeping with the concept that NO is a double-edged messenger that, depending on its concentration and mechanism of action, can have opposing effects on a particular mechanism or target.24,25 Likewise, the role of Drp1 and fission differ depending on the cell type, due to differences in the cell management of mitochondrial morphology and function. Drp1-null embryos fail to undergo developmentally regulated apoptosis during neural tube formation in vivo, whereas embryonic fibroblasts have normal apoptotic responses,26
The specific role of NO in regulating myogenesis physiologically through a mitochondrially linked mechanism raises the question of the biological significance of such control. A possible answer lies in the NO-dependent bioenergetic changes taking places in the mitochondria of differentiating myoblasts and in their pathophysiological consequences that we describe here.
Although the mitochondrial membrane potential was maintained in cells in which fission was taking place as a result of the lack of NO/cGMP, its collapse after treatment with the F1F0 ATP synthetase inhibitor oligomycin and the low rate of ATP generation observed in these conditions showed that mitochondrial respiration is impaired. Such reduced activity was a consequence and not a cause of mitochondrial fission as it was reversed by Drp1 K38A. The functional consequence of such a metabolic defect was evident when apoptosis was induced, so that cells in which fission was taking place were more sensitive to an apoptogenic stimulus. It is possible that mitochondria and NO act as a quality-control check of myogenesis. Indeed activation of NOSs depends crucially on the correct architecture of cells and on intracellular signaling pathways that may be disrupted in damaged cells.18,27
The metabolic changes subsequent to the NO/cGMP-dependent changes in mitochondrial dynamics appear to follow changes in cytochrome c oxidase activity. A recent study highlighted how this enzyme is regulated by cAMP, with important metabolic consequences,22 in a manner analogous to the Drp1-mediated, cGMP-dependent mechanism described here. These observations uncover new and significant mechanisms of regulation of cytochrome c oxidase, with biological consequences that add to the complexity, and recognize the importance of this enzyme for cell pathophysiology.28-30
Previous studies have shown that expression of NOSs is developmentally regulated and that NO acts at various levels in the process of myogenesis, by increasing the secretion of follistatin and follistatin-1-like proteins,31-33 and cooperating with insulin-like growth factor-II.34 In addition, it favors the activation, proliferation and engraftment of myogenic stem cells.13,35,36 We now show that NO controls the early events in myogenic differentiation. It appears therefore that NO presides over several crucial steps of muscle differentiation that are important in repair, and may explain why NO-based therapies appear to work effectively in reducing muscle damage in muscular dystrophy.31,35
The fact that the action of NO is mediated through inhibition of mitochondrial fission and changes in the metabolic status of mitochondria is of great physiological importance in muscles. In particular, NO controls mitochondrial respiration through reversible inhibition of cytochrome c oxidase, acting as an acute sensor for changes in the oxygen concentration.37,38 In addition, it stimulates mitochondrial biogenesis39,40 a key event occurring in muscle hypertrophy and muscle repair that also contributes to the definition of the slow twitch-oxidative versus fast twitch-glycolytic nature of muscle fibers.9,11,15,41 We now describe a third action aimed at regulating mitochondrial dynamics and the resulting changes in bioenergetics. Oxygen sensing and mitochondrial fission are regulated acutely by NO, whereas mitochondrial biogenesis requires sustained changes in NO concentration.38,39 Understanding whether these actions of NO are independent from each other or are causally linked, in particular if the acute NO-regulated effects on mitochondria influence mitochondrial biogenesis, may lead to a better understanding of the role of NO in modulating the mitochondrial behavior, giving new insights into the importance of energy metabolism in the control of muscle development and regeneration. In view of the role of NO and mitochondrial dysfunction in muscle degenerative diseases,35,42-44 these findings will be crucial for designing novel therapeutic approaches to these pathological conditions.
Materials and Methods
Cell culture and treatment
Myogenic precursor cells were prepared as previously described.45 Briefly, muscle fragments were digested with 2% collagenase-II and dispase (Sigma, St. Louis, MO, USA and Gibco-Invitrogen, Carlsbad, CA, USA) for 10 min at 37 °C with gentle agitation. The dispersed cells were then collected, pooled, centrifuged, resuspended in Iscove’s modified Dulbecco’s medium supplemented with 20% fetal bovine serum, 3% chick embryo extract,45 100 U/ml penicillin, 100 μg/ml streptomycin and 50 μg/ml gentamycin, and plated onto matrigel, (BD-Bioscience, San Jose, CA, USA)-coated dishes at a density of 104 cells × cm2. Contamination by non-myogenic cell was reduced by pre-plating the cell suspensions onto plastic dishes where fibroblasts tend to adhere more rapidly. Differentiation was induced by changing the medium to Iscove’s modified Dulbecco’s medium supplemented with 2% horse serum.
Drug treatments were as follows: myogenic precursor cells were differentiated for 6 h and then treated with l-NAME (1 mM), ODQ (1 μM), DETA-NO (30 μM) and 8Br-cGMP (1 mM), alone or in combination, except for time-course differentiation experiments in which drugs were added immediately after changing to differentiation medium.
DNA constructs and cell transfections
Drp1 was purchased from OriGene Technologies (Rockville, MD, USA) and sub-cloned into pcDNA4/myc-His (Invitrogen, Carlsbad, CA, USA) using the following primers containing the sequences for two restriction sites: EcoRV (5′-CGGATATCGTCATGGAGGCGCTAATTCCT-3′) and XhoI (5′-CCTCGAGGTCACCAAAGATGAGTCTCC-3′). PCR conditions, using the Pfu high-fidelity polymerase (Stratagene, La Jolla, CA, USA), were 95 °C for 2 min, then four cycles as follows: 95 °C for 30 s, 50 °C for 30 s, 72 °C for 5 min and then 25 cycles as follows: 95 °C for 30 s, 60 °C for 30 s and 72 °C for 5 min. The 2208-bp fragment thus obtained was sub-cloned into the pcDNA4/myc-Hys (Invitrogen) XhoI-EcoRV-linearized vector, upstream from the Myc tags.
Drp1 K38A was generated through site-directed mutagenesis of pcDNA4-Myc-Drp1 by using a QuikChange II Site-directed Mutagenesis kit (Stratagene, La Jolla, CA, USA). The endogenous Drp1 stop codon was removed using the following primers: 5′-GGAGACTCATCTTTGGTCCTCGAGTCTAGAGGGCCCG-3′ (sense) and 5′-CGGGCCCTCTAGACTCGAGGACCAAAGATGAGTCTCC-3′ (antisense). The second mutagenesis step to obtain Drp1 K38A was performed using the following primers: 5′-GGGAACGCAGAGCAGCGGAGCGAGCTCAGTG-3′ (sense) and 5′-CACTGAGCTCGCTCCGCTGCTCTGCGTTCCC-3′ (antisense). The constructs were sequence-verified using a Big Dye Terminator Sequencing kit (version 3.1; Applied Biosystems, Foster City, CA, USA) run on an Applied Biosystems ABI 3100 Avant Genetic Analyzer.
To obtain the constructs pEYFP-N1-Drp1 and pEYFP-N1-Drp1K38A, the original OriGene construct was digested with NotI and the DrpI fragment was sub-cloned in NotI-linearized pEYFP-N1 (Invitrogen) downstream from YFP. Site-specific mutagenesis was performed first to remove a stop codon in the EYFP sequence using the following primers: 5′-CTCGGCATGGACGAGCTGTACAAGAGCAAGGCCGCGAATTC-3′ (sense) and 5′-GAATTCGCGGCCTTGCTCTTGTACAGCTCGTCCATGCCGAG-3′ (antisense). Drp1 K38A was finally obtained with a second mutagenesis step as described above. The final constructs were verified by sequencing, as described above.
Cell transfections were performed with the FuGENE HD reagent (Roche Diagnostics, Rotkreuz, Switzerland) according to the manufacturer’s instructions. The cells were used 24 h after transfection in the various experimental settings described.
To silence nNOS we used the ‘ON-TARGETplus SMARTpool siRNA’ system developed by Thermo Scientific Dharmacon Laboratories (Lafayette, CO, USA) in which silencing is obtained by transfection of 40 nM of a pool of four different siRNAs. The nNOS-silencing siRNAs used in the pool were 5′-AGAUCGACAGCUUUGGUGG-3′; 5′-AAUGACGUUGGGUUGGAUC-3′; 5′-UUGGAUAAGGCCGCUCUGC-3′; 5′-GAUUCCUGUGUCUUUCAUC-3′. Transfections were performed using the INTERFERIN polyplus transfection reagent from Euroclone (Euroclone Siziano (PV)).
Immunoblotting
The extracted proteins (50 μg) were separated by 4–12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (NuPAGE; Invitrogen) and transferred onto nitrocellulose membranes (Perkin Elmer, Waltham, MA, USA) as described.46 The membranes were probed using the following antibodies (Abs): anti-calnexin (1 : 5000; Genetex Inc, San Antonio, TX, USA); anti-Mfn2 (1 : 200; Abnova, Taipei, Taiwan); anti-Mfn1 (1 : 200, Abnova); anti Drp1 (1 : 1000; BD Transduction Laboratories); anti-COX-IV(oxidative phosphorylation Complex-IV subunit-IV, 1 : 400; Ibridoma Bank Iowa City, Iowa); anti-Myc (1 : 1000; Cell Signaling, Boston, MA, USA); anti-Myo-D (1 : 500; Ibridoma Bank Iowa City, Iowa); anti-sarcomeric myosin MyHC (1 : 10; Developmental Studies Hybridoma Bank); anti myogenin (1 : 10; Developmental Studies Hybridoma Bank); anti Mef2A (1 : 500; Santa Cruz Biotechnology); anti-GAPDH (AbD; Serotec, Oxford, UK) and anti-Opa1 (1 : 1000; BD Transduction Laboratories).
Assay of NOS activity
The time course of NOS activity was assayed in intact cells by measuring the conversion of l-[3H]-arginine into l-[3H]-citrulline.46 In brief, the reaction was performed using 145 mM NaCl, 5 mM KCl, 1 mM MgSO4, 10 mM glucose, 1 mM CaCl2 and 10 mM HEPES (pH 7.4). A mix of 10 μCi/ml l-[3H]-arginine and 10 μM cold arginine was added at various time points and the reaction was stopped after 5 min by washing with ice-cold phosphate-buffered saline (PBS) supplemented with 5 mM l-arginine and 4 mM EDTA. A 0.5-ml volume of 100% cold ethanol was added to the dishes and left to evaporate before a final addition of 20 mM HEPES (pH 6.0). l-NAME-treated cells were run in parallel as a control for specificity. l-[3H]-citrulline was separated from l-[3H]-arginine by DOWEX 50X8-400 chromatography. The l-[3H]-citrulline formed was normalized to protein content, which was evaluated by the bicinchoninic acid procedure (BCA; Perbio, Bezons, France).
Measurement of cGMP generation
At each time point myogenic precursor cell cultures were incubated for 30 min at 37 °C either in growth medium or in differentiation medium with 0.5 mM of the phosphodiesterase inhibitor 3-isobutyl-1-methylxanthine, supplemented with 1 mM l-NAME, or a vehicle.46 The reaction was terminated by rapid removal of the medium and washing with ice-cold PBS. The cells were lysed by addition of ice-cold trichloroacetic acid (final concentration: 6%). After ether extraction, cGMP levels were measured using a radioimmunoassay kit and normalized to protein content.
Confocal and time-lapse microscopy
For confocal z-axis stacks of the mitochondrial network, coverslips were placed on the stage of an UltraVIEW ERS Spinning-Disk Confocal microscope equipped with a stage incubator (OkoLab, Quarto, ITALY) and an EM-CCD Hamamatsu (Hamamatsu City, Shizuoka, Japan) C9100, allowing us to follow highly dynamic processes. Cells expressing mtDsRed47 were excited using the 514-nm line of an HeNe laser with exposure times of 100 ms using a × 63 immersion objective. Stacks of 20 images, separated by 1 μm along the z-axis, were acquired for steady-state and time-resolved 3D imaging. The total acquisition time for each stack was 1.1 s for the 20 planes to minimize reconstruction artifacts caused by mitochondrial movement. Reconstruction of the stacks was performed using the Volocity software.
For confocal imaging of fixed cells, 16-mm-diameter round coverslips were placed on the stage of a Leica TCS SP2 Laser-Scanning Confocal microscope equipped with an electronically controlled and freely definable Acousto-Optical Beam Splitter. Images were acquired with ×63 magnification oil-immersion lenses at 1024 × 1024 pixel resolution. The cells were excited with the 543-nm line for mtDsRed and the emitted light was collected with 570 long-pass filters. Morphometric analyses were performed using imagetool 3.0 (University of Texas Health Science Center, San Antonio, USA). Images of cells expressing mitoGFP or mitoDsRed47 were thresholded by using the automatic threshold function. For each identified object, the major-axis length and the roundness index were calculated. Cells were scored with elongated mitochondria when >50% of the objects in the image (i.e., mitochondria) had a major axis longer than 5 mm and a roundness index below 0.5 (maximum value is 1).
Serial images (z-stacks) of mitoDsRed-expressing myogenic cells were acquired with a Leica TCS SP2 Laser-Scanning Confocal microscope and collected with a z-step of 0.5 μm. Image analysis and processing was performed with the public domain software MBF_ImageJ for Microscopy (http://www.macbiophotonics.ca/index.htm). Single-channel surface-rendered images were processed using ImageJ running the VolumeJ plugin48 as described by Collins et al.49
Electron transmission microscopy
After the various treatments, the myogenic precursor cell pellets were prepared for electron microscopy, as described by Nisoli et al.39 In brief, thin sections were obtained using a MT-X ultramicrotome (RMC, Tucson, AZ, USA), stained with lead citrate and examined using a transmission electron microscope (Philips CM10; Eindhoven, The Netherlands). A minimum of 50 cells from each treatment were assessed.
Apoptotic cell death analysis
For cell death experiments, cells were plated in six-well dishes, exposed to the treatments indicated in the section ‘Cell culture and treatment’ and treated in the presence or absence of 30 μM H2O2 for 4 h at 37 °C. The cells were detached and stained with tetramethyl rhodamine isothiocyanate–annexin-V (1 : 20) and (1 : 5) (BD Biosciences) according to the kit manufacturer’s instructions and analyzed by flow cytometry using a FACStar Plus (Becton Dickinson, Franklin Lakes, NJ, USA) as described by Sciorati et al.13 Cells staining positive for annexin-V and excluding 7AAD were considered apoptotic. Cell death was also assessed by the trypan blue exclusion assay of cell viability and by detecting activated caspase-3 and caspase-9 with specific Abs (Cell Signaling-New England Biolabs, Ipswich, MA, USA).
PEG fusion assay
Fusion experiments were performed as described by Cipolat et al.47 In brief, 5 × 105 mouse myogenic precursor cells were transfected either with mitoGFP or with mitoDsRed alone. After 24 h, cells labeled with different fluorescent proteins were co-plated at a 1 : 1 ratio onto 13-mm-diameter round coverslips. Differentiation was induced for 6 h after which the cells were treated with l-NAME or ODQ for 1 h; fusion was induced by a 60-s treatment with a 50% (wt/vol) solution of polyethylene glycol (PEG) 1500 in PBS (Sigma), followed by extensive washes in DMEM supplemented with 10% fetal calf serum. To inhibit de novo synthesis of fluorescent proteins, 30 min before PEG treatment cells were incubated with the protein synthesis inhibitor cycloheximide (20 μg/ml, Sigma), which was subsequently added in all solutions and tissue culture media until cells were fixed for 30 min with ice-cold 3.7% (vol/vol) formaldehyde in PBS. After two washes with PBS, the coverslips were mounted on slides using the Anti-Fade Reagent (Molecular Probes). For imaging of polykaryons, fixed cells on 13-mm-diameter round coverslips were placed on the stage of a Bio-Rad (Hercules, CA, USA) MRC 1024 Laser Scanning Confocal microscope. The cells were excited with the 488-nm laser line for yellow fluorescent protein and the 568-nm line for mitoDsRed, and the emitted light was collected with 515/30 band-pass and 570 long-pass filters, respectively. The intensity of double-labeling in the polykaryons was quantified in three different experiments using the ‘Colocalizer’ plugins of ImageJ software.
Sub-cellular fractionation
Myogenic precursor cells were scraped in 400 μl of homogenization medium (0.3 M sucrose, 10 mM MES K+, 1mM K2EGTA, 1 mM Mg2SO4, protease inhibitors (pH 7.4)) and homogenized with 25 strokes of a ball-bearing homogenizer on ice before centrifugation at 1000 × g for 10 min at 4 °C. The resulting low-speed pellet containing nuclei and unbroken cells was discarded. The post-nuclear supernatant (PNS) was further centrifuged at 10 000 × g for 30 min at 4 °C and the resulting pellet contained both heavy and light mitochondrial fractions. The pellet was washed twice at 10 000 × g for 30 min at 4 °C. The post-mitochondrial supernatant was centrifuged at 55 000 r.p.m. for 2 h with TLA-100 to obtain a microsomal pellet and a cytosolic fraction. Equal amounts of mitochondria, microsomes and cytosolic fraction were separated on 12% polyacrylamide gel, blotted onto a nitrocellulose membrane (Perkin Elmer) and probed with anti-Drp1 anti-COX-IV, anti GAPDH Abs.50
Detection of the interaction between Fis1 and Drp1 by co-immunoprecipitation
To test for a Drp1–Fis1 interaction in intact cells, in view of the reversible nature of such an interaction, myogenic precursor cells were subjected to chemical cross-linking. The cleavable, homo-bifunctional cross-linker dithiobis (succinimidylpropionate) (DSP; Pierce, Rockford, IL, USA) was diluted to a final concentration of 1.0 mM in PBS and added to the cultured cells. After incubation for 1 h at room temperature, cross-linking was stopped by addition of Tris HCl (pH 7.7) to a final concentration of 20 mM. The cells were lysed with a lysis buffer containing 20 mM Tris HCl, 150 mM NaCl, 10 mM EGTA, 10% glycerol and 1% Triton X-100 (pH 7.4) and immunoprecipitation was performed using anti-Fis1 Abs and protein-A–Sepharose (GE Healthcare, Chalfont, St Giles, UK) for 16 h at 4 °C. The samples were washed three times in lysis buffer and boiled in SDS sample buffer containing 5% 2-mercaptoethanol to cleave the cross-linking. The samples were run on 15% polyacrylamide gels and subjected to western blot analysis using anti-Drp1, anti-Fis1 Abs (Enzo Life Science, Farmingdale, NY, USA). The specificity of the interaction between the two proteins and of the binding to the beads was verified in a control immunoprecipitation experiment using an anti-calnexin Ab (Genetex Inc.), followed by immunoblotting with anti Drp1 or Fis1 Abs.
Assay for GTPase activity
Cells were suspended in 400 μl of homogenization buffer containing 0.22 M mannitol, 0.07 M sucrose, the protease inhibitor cocktail and 10 mM HEPES-KOH (pH 7.4) and homogenized with 10 strokes using a syringe equipped with a 27-gauge needle. The PNS was recovered by centrifugation at 1500 r.p.m. for 5 min and 300 μl were diluted in a reaction buffer (homogenization buffer containing 50 mM Tris-HCl, 150 mM NaCl, 1 mM MgOAc2, 1 mM dithiothreitol, the protease inhibitor cocktail and 1% digitonin (pH 7.5)). The cleared lysate was incubated with 30 μl GTP-agarose beads (Sigma) at 30 °C for 1 h. The beads were washed three times in the same buffer. The proteins were resolved by SDS-PAGE and analyzed by immunoblotting using anti-Drp1 Abs.51
Measurement of Ca2+ concentrations
Cells grown on 13-mm-diameter round glass coverslips at 50% confluence were co-transfected with cytosolic or mitochondrial aequorin.52 Aequorin reconstitution, measurement and calibration were performed in a Krebs Ringer HEPES buffer containing 1 mM Ca2+ (pH 7.4). All measurements were performed in this buffer.52
Measurement of mitochondrial membrane potential
Cells were seeded onto 24-mm-diameter round glass coverslips and the mitochondrial membrane potential was measured on the basis of the accumulation of TMRM (Molecular Probes Invitrogen) as described by Angelin et al.17 Cells were rinsed once and then incubated in serum-free Hank’s buffered salt solution supplemented with 1.6 μM cyclosporine-H to minimize variability in the extent of mitochondrial loading using potentiometric probes, and loaded with 10 nM TMRM for 30 min. Oligomycin at 1 μg/ml was added during time lapse as indicated in the figures and, at the end of each experiment, mitochondria were fully depolarized by addition of 4 μM of the protonophore carbonyl cyanide p-trifluoromethoxy phenylhydrazone (FCCP). Cellular fluorescence images were acquired using the epifluorescence-based IN Cell Analyzer 1000 microscope system (GE Healthcare) equipped with a high-resolution CCD camera, fast laser-based confocal auto-focus and optical Z-sectioning with Opti-Grid. Images were analyzed using the IN Cell Investigator Software for automated analysis of cellular images. Sequential digital images were acquired every 1 min and the average fluorescence intensity of all relevant regions was recorded and stored for subsequent analysis.17
Measurement of ATP formation
ATP concentration was determined using the luciferinluciferase method.53 Briefly, cells were resuspended in buffer-A (150 mM KCl, 25 mM Tris-HCl, 2 mM EDTA, 0.1% BSA, 10 mM potassium phosphate and 0.1 mM MgCl (pH 7.4)) and incubated for 1 min with 50 μg/ml digitonin at room temperature with gentle agitation. They were then washed by adding 1 ml of buffer-A and pelletted at 1000 × g. The cell pellets were resuspended in 160 μl of buffer-A and plated in 96 wells in triplicate. The samples were treated with 30 μl of a mix containing 1 mM malate, 1 mM pyruvate, 0.1 mM ADP (Sigma), 10 μl buffer-B (containing 0.8 mM luciferin and 20 mg/ml luciferase in 0.5 M Tris-acetate (pH 7.75)) and either 1 μg/ml oligomycin or 100 μM iodoacetate or 4 mM fluoride to assess OXPHOS ATP and glycolytic ATP, respectively. ATP was measured using a GloMax luminometer (Promega, Madison, WI, USA).
Tests for respiratory chain defects were essentially performed as described by Robinson.54 In brief, digitonin-permeabilized cells were incubated at 37 °C for 15 min in a buffer containing 0.25 M sucrose, 20 mM MOPS, 1 mM EDTA, 5 mM inorganic phosphate, 0.1% fatty acid-free BSA and 1 mM ADP (pH 7.4), and supplemented with the following specific substrates and inhibitors of the respiratory chain complexes: 1 mM pyruvate and 5 mM malate, or 5 mM glutamate and 5 mM malate (complex-I); 1 μM rotenone and 10 mM succinate (complex-II); 10 μg/ml antimycin-A, 0.1 mM N,N,N’-N’-tetramethyl-p-phenylenediamine and 2 mM ascorbate (complex-IV).
Analysis of phosphorylation of Drp1
Myogenic precursor cells were transfected with pcDNA3-Drp1-myc and its control empty vector. After 24 h, the myogenic precursor cells were differentiated for 6 h, incubated in serum-free medium and labeled with 0.5 mCi/ml [32P]orthophosphate (8500–9120 Ci/mmol; PerkinElmer Life Science) for 3.5 h in the presence of 100 μM 3-isobutyl-1-methylxanthine. To determine the role of NO directly, cells were incubated in serum-free medium containing 0.5 mCi/ml [32P]orthophosphate for 3 h and treated with 30 μM DETA-NO, 1 μM KT5823, 1 mM 8Br cGMP, 1 μM ODQ and 2 μM BAY41-2272 for 1.5 h, in the presence of 100 μM 3-isobutyl-1-methylxanthine. The cells were lysed in a radio-immunoprecipitation buffer supplemented with a protease inhibitor mixture (Sigma) and a phosphatase inhibitor mixture (Sigma). Myc-Drp1 was immunoprecipitated using an anti-Myc monoclonal Ab (9E10) immobilized on agarose beads (Invitrogen). The immunoprecipitates were resolved by SDS-PAGE and 32P-labeled Drp1 was visualized by autoradiography using Amersham Hyperfilm MP (GE Healthcare).
High-resolution respirometry
The respiration rate of the myogenic precursor cells transfected with pEYFP-N1 Drp1 K38A or the empty vector, differentiated for 6 h and treated with ODQ for 1 h, was measured using two-channel titration injection respirometers (Oroboros, Instruments Oroboros, Innsbruck, Austria)55 at 37 °C in a buffer containing 0.5 mM EGTA, 3 mM MgCl2 60 mM K-lactobionate, 20 mM taurine, 10 mM KH2PO4, 20 mM HEPES, 110 mM sucrose and 1 g/l bovine serum albumin. Before each experiment the medium was equilibrated with air in the oxygraph chambers at 37 °C until a stable signal was obtained for oxygen calibration. The medium was then replaced by the aerated cell suspensions previously counted and the chambers were closed at a volume of 2 ml by insertion of gas-tight stoppers. The cell suspension was stirred continuously in the respirometer chamber at 460–600 r.p.m. the DatLab software (Oroboros Instruments) was used for data acquisition (1 s time intervals) and analysis, which includes online calculation of the time derivative of oxygen concentration and correction for instrumental background oxygen flux.55 The respiratory effect of cytochrome c was used as a test for plasma membrane integrity and mitochondrial function.55 After steady-state respiratory flux recording, titration was performed using oligomycin (1 μg/ml) followed by uncoupling of oxidative phosphorylation by stepwise titration of FCCP up to optimum concentrations in the range of 2.5 to 4 μM. Finally, respiration was inhibited by sequential addition of 0.5 μM rotenone and 2.5 μM antimycin-A. All data were normalized to citrate synthase activity.
Statistical analysis
The results are expressed as means±S.E.M. Statistical analysis was performed using a two-tailed t-test for unpaired variables. The asterisks and crosses in the figures refer to statistical probabilities, measured under the various experimental conditions as detailed in the figure legends. Statistical probability values of <0.05 were considered significant.
Acknowledgements
We thank Annie Higgs (London) for critically reading the paper, Erich Gnaiger (Innsbruck) for help with respirometry. This work was supported by Telethon Italia (GP007006, to EC), European Community 7th framework programme (OPTISTEM and ENDOSTEM large cooperative programmes, to EC and GC), Cariplo (to EC, MTB and SB), Associazione Italiana Ricerca sul Cancro (AIRC, to EC) Fondazione Romeo ed Enrica Invernizzi (to EC), Association Française contre Les Myopathies (AFM13478, to EC) and Italian Ministry of University (PRIN 2007, to EC). LS is a Senior Telethon Scientist of the Dulbecco-Telethon Institute and an EMBO Young Investigator. SC was a recipient of a fellowship from AIRC. CDeP, SF and SP performed the majority of the experiments and data analysis, and participated in project planning; SC and SP contributed to the bioenergetics experiments; CP prepared the various molecular biology tools; AD contributed to time-lapse microscopy; RA performed electron microscopy; MTB, GC, TP and L.S. performed some of the experiments and data analysis, and participated in project planning; LS, SM, SB and EC conceived the study, participated in project planning and wrote the paper.
Abbreviations
- NO
nitric oxide
- Drp1
dynamin-related protein-1
- Opa1
optic atrophy-1
- NOS
NO synthase
- cGMP
cyclic GMP
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Supplementary Information accompanies the paper on Cell Death and Differentiation website (http://www.nature.com/cdd)
References
- 1.Detmer SA, Chan DC. Functions and dysfunctions of mitochondrial dynamics. Nat Rev Mol Cell Biol. 2007;8:870–879. doi: 10.1038/nrm2275. [DOI] [PubMed] [Google Scholar]
- 2.Dimmer KS, Scorrano L. (De)constructing mitochondria: what for? Physiology (Bethesda) 2006;21:233–241. doi: 10.1152/physiol.00010.2006. [DOI] [PubMed] [Google Scholar]
- 3.Santel A, Frank S. Shaping mitochondria: the complex posttranslational regulation of the mitochondrial fission protein DRP1. IUBMB Life. 2008;60:448–455. doi: 10.1002/iub.71. [DOI] [PubMed] [Google Scholar]
- 4.Skulachev VP. Mitochondrial filaments and clusters as intracellular power-transmitting cables. Trends Biochem Sci. 2001;26:23–29. doi: 10.1016/s0968-0004(00)01735-7. [DOI] [PubMed] [Google Scholar]
- 5.Westermann B. Molecular machinery of mitochondrial fusion and fission. J Biol Chem. 2008;283:13501–13505. doi: 10.1074/jbc.R800011200. [DOI] [PubMed] [Google Scholar]
- 6.Campello S, Lacalle RA, Bettella M, Manes S, Scorrano L, Viola A. Orchestration of lymphocyte chemotaxis by mitochondrial dynamics. J Exp Med. 2006;203:2879–2886. doi: 10.1084/jem.20061877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160:189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Finkel T, Hwang PM. The Krebs cycle meets the cell cycle: mitochondria and the G1–S transition. Proc Natl Acad Sci USA. 2009;106:11825–11826. doi: 10.1073/pnas.0906430106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Puigserver P, Spiegelman BM. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr Rev. 2003;24:78–90. doi: 10.1210/er.2002-0012. [DOI] [PubMed] [Google Scholar]
- 10.Wenz T, Diaz F, Spiegelman BM, Moraes CT. Activation of the PPAR/PGC-1alpha pathway prevents a bioenergetic deficit and effectively improves a mitochondrial myopathy phenotype. Cell Metab. 2008;8:249–256. doi: 10.1016/j.cmet.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11.Hood DA, Irrcher I, Ljubicic V, Joseph AM. Coordination of metabolic plasticity in skeletal muscle. J Exp Biol. 2006;209(Part 12):2265–2275. doi: 10.1242/jeb.02182. [DOI] [PubMed] [Google Scholar]
- 12.Bach D, Pich S, Soriano FX, Vega N, Baumgartner B, Oriola J, et al. Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. J Biol Chem. 2003;278:17190–17197. doi: 10.1074/jbc.M212754200. [DOI] [PubMed] [Google Scholar]
- 13.Sciorati C, Galvez BG, Brunelli S, Tagliafico E, Ferrari S, Cossu G, et al. Ex vivo treatment with nitric oxide increases mesoangioblast therapeutic efficacy in muscular dystrophy. J Cell Sci. 2006;119(Part 24):5114–5123. doi: 10.1242/jcs.03300. [DOI] [PubMed] [Google Scholar]
- 14.Garthwaite J, Southam E, Boulton CL, Nielsen EB, Schmidt K, Mayer B. Potent and selective inhibition of nitric oxide-sensitive guanylyl cyclase by 1H-[1,2,4]oxadiazolo[4,3-a] quinoxalin-1-one. Mol Pharmacol. 1995;48:184–188. [PubMed] [Google Scholar]
- 15.Stamler JS, Meissner G. Physiology of nitric oxide in skeletal muscle. Physiol Rev. 2001;81:209–237. doi: 10.1152/physrev.2001.81.1.209. [DOI] [PubMed] [Google Scholar]
- 16.van der Bliek AM, Redelmeier TE, Damke H, Tisdale EJ, Meyerowitz EM, Schmid SL. Mutations in human dynamin block an intermediate stage in coated vesicle formation. J Cell Biol. 1993;122:553–563. doi: 10.1083/jcb.122.3.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Angelin A, Tiepolo T, Sabatelli P, Grumati P, Bergamin N, Golfieri C, et al. Mitochondrial dysfunction in the pathogenesis of Ullrich congenital muscular dystrophy and prospective therapy with cyclosporins. Proc Natl Acad Sci USA. 2007;104:991–996. doi: 10.1073/pnas.0610270104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- 19.Carlucci A, Lignitto L, Feliciello A. Control of mitochondria dynamics and oxidative metabolism by cAMP, AKAPs and the proteasome. Trends Cell Biol. 2008;18:604–613. doi: 10.1016/j.tcb.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 20.Cereghetti GM, Stangherlin A, Martins de Brito O, Chang CR, Blackstone C, Bernardi P, et al. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci USA. 2008;105:15803–15808. doi: 10.1073/pnas.0808249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barsoum MJ, Yuan H, Gerencser AA, Liot G, Kushnareva Y, Graber S, et al. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 2006;25:3900–3911. doi: 10.1038/sj.emboj.7601253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cho DH, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z, et al. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science. 2009;324:102–105. doi: 10.1126/science.1171091. 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Acin-Perez R, Salazar E, Kamenetsky M, Buck J, Levin LR, Manfredi G. Cyclic AMP produced inside mitochondria regulates oxidative phosphorylation. Cell Metab. 2009;9:265–276. doi: 10.1016/j.cmet.2009.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thippeswamy T, McKay JS, Quinn JP, Morris R. Nitric oxide, a biological double-faced janus – is this good or bad? Histol Histopathol. 2006;21:445–458. doi: 10.14670/HH-21.445. [DOI] [PubMed] [Google Scholar]
- 25.Moncada S, Bolanos JP. Nitric oxide, cell bioenergetics and neurodegeneration. J Neurochem. 2006;97:1676–1689. doi: 10.1111/j.1471-4159.2006.03988.x. [DOI] [PubMed] [Google Scholar]
- 26.Wakabayashi J, Zhang Z, Wakabayashi N, Tamura Y, Fukaya M, Kensler TW, et al. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J Cell Biol. 2009;186:805–816. doi: 10.1083/jcb.200903065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brenman JE, Chao DS, Xia H, Aldape K, Bredt DS. Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell. 1995;82:743–752. doi: 10.1016/0092-8674(95)90471-9. [DOI] [PubMed] [Google Scholar]
- 28.Erusalimsky JD, Moncada S. Nitric oxide and mitochondrial signaling: from physiology to pathophysiology. Arterioscler Thromb Vasc Biol. 2007;27:2524–2531. doi: 10.1161/ATVBAHA.107.151167. [DOI] [PubMed] [Google Scholar]
- 29.Brunelli S, Rovere-Querini P, Sciorati C, Manfredi AA, Clementi E. Nitric oxide: emerging concepts about its use in cell-based therapies. Expert Opin Investig Drugs. 2007;16:33–43. doi: 10.1517/13543784.16.1.33. [DOI] [PubMed] [Google Scholar]
- 30.Herrmann PC, Herrmann EC. Oxygen metabolism and a potential role for cytochrome c oxidase in the Warburg effect. J Bioenerg Biomembr. 2007;39:247–250. doi: 10.1007/s10863-007-9084-z. [DOI] [PubMed] [Google Scholar]
- 31.Colussi C, Mozzetta C, Gurtner A, Illi B, Rosati J, Straino S, et al. HDAC2 blockade by nitric oxide and histone deacetylase inhibitors reveals a common target in Duchenne muscular dystrophy treatment. Proc Natl Acad Sci USA. 2008;105:19183–19187. doi: 10.1073/pnas.0805514105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pisconti A, Brunelli S, Di Padova M, De Palma C, Deponti D, Baesso S, et al. Follistatin induction by nitric oxide through cyclic GMP: a tightly regulated signaling pathway that controls myoblast fusion. J Cell Biol. 2006;172:233–244. doi: 10.1083/jcb.200507083. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33.Ouchi N, Oshima Y, Ohashi K, Higuchi A, Ikegami C, Izumiya Y, et al. Follistatin-like 1, a secreted muscle protein, promotes endothelial cell function and revascularization in ischemic tissue through a nitric-oxide synthase-dependent mechanism. J Biol Chem. 2008;283:32802–32811. doi: 10.1074/jbc.M803440200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaliman P, Canicio J, Testar X, Palacin M, Zorzano A. Insulin-like growth factor-II, phosphatidylinositol 3-kinase, nuclear factor-kappaB and inducible nitric-oxide synthase define a common myogenic signaling pathway. J Biol Chem. 1999;274:17437–17444. doi: 10.1074/jbc.274.25.17437. [DOI] [PubMed] [Google Scholar]
- 35.Brunelli S, Sciorati C, D’Antona G, Innocenzi A, Covarello D, Galvez BG, et al. Nitric oxide release combined with nonsteroidal antiinflammatory activity prevents muscular dystrophy pathology and enhances stem cell therapy. Proc Natl Acad Sci USA. 2007;104:264–269. doi: 10.1073/pnas.0608277104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anderson JE. A role for nitric oxide in muscle repair: nitric oxide-mediated activation of muscle satellite cells. Mol Biol Cell. 2000;11:1859–1874. doi: 10.1091/mbc.11.5.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moncada S, Erusalimsky JD. Does nitric oxide modulate mitochondrial energy generation and apoptosis? Nat Rev Mol Cell Biol. 2002;3:214–220. doi: 10.1038/nrm762. [DOI] [PubMed] [Google Scholar]
- 38.Clementi E, Brown GC, Foxwell N, Moncada S. On the mechanism by which vascular endothelial cells regulate their oxygen consumption. Proc Natl Acad Sci USA. 1999;96:1559–1562. doi: 10.1073/pnas.96.4.1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, et al. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science. 2003;299:896–899. doi: 10.1126/science.1079368. [DOI] [PubMed] [Google Scholar]
- 40.Nisoli E, Falcone S, Tonello C, Cozzi V, Palomba L, Fiorani M, et al. Mitochondrial biogenesis by NO yields functionally active mitochondria in mammals. Proc Natl Acad Sci USA. 2004;101:16507–16512. doi: 10.1073/pnas.0405432101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O, et al. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797–801. doi: 10.1038/nature00904. [DOI] [PubMed] [Google Scholar]
- 42.Figueiredo PA, Mota MP, Appell HJ, Duarte JA. The role of mitochondria in aging of skeletal muscle. Biogerontology. 2008;9:67–84. doi: 10.1007/s10522-007-9121-7. [DOI] [PubMed] [Google Scholar]
- 43.Wehling M, Spencer MJ, Tidball JG. A nitric oxide synthase transgene ameliorates muscular dystrophy in mdx mice. J Cell Biol. 2001;155:123–131. doi: 10.1083/jcb.200105110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wells GD, Noseworthy MD, Hamilton J, Tarnopolski M, Tein I. Skeletal muscle metabolic dysfunction in obesity and metabolic syndrome. Can J Neurol Sci. 2008;35:31–40. doi: 10.1017/s0317167100007538. [DOI] [PubMed] [Google Scholar]
- 45.Cossu G, Eusebi F, Grassi F, Wanke E. Acetylcholine receptor channels are present in undifferentiated satellite cells but not in embryonic myoblasts in culture. Dev Biol. 1987;123:43–50. doi: 10.1016/0012-1606(87)90425-8. [DOI] [PubMed] [Google Scholar]
- 46.Bulotta S, Barsacchi R, Rotiroti D, Borgese N, Clementi E. Activation of the endothelial nitric-oxide synthase by tumor necrosis factor-alpha. A novel feedback mechanism regulating cell death. J Biol Chem. 2001;276:6529–6536. doi: 10.1074/jbc.M006535200. [DOI] [PubMed] [Google Scholar]
- 47.Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci USA. 2004;101:15927–15932. doi: 10.1073/pnas.0407043101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abramoff MD, Viergever MA. Computation and visualization of three-dimensional soft tissue motion in the orbit. IEEE Trans Med Imaging. 2002;21:296–304. doi: 10.1109/TMI.2002.1000254. [DOI] [PubMed] [Google Scholar]
- 49.Collins TJ, Berridge MJ, Lipp P, Bootman MD. Mitochondria are morphologically and functionally heterogeneous within cells. EMBO J. 2002;21:1616–1627. doi: 10.1093/emboj/21.7.1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Varadi A, Johnson-Cadwell LI, Cirulli V, Yoon Y, Allan VJ, Rutter GA. Cytoplasmic dynein regulates the subcellular distribution of mitochondria by controlling the recruitment of the fission factor dynamin-related protein-1. J Cell Sci. 2004;117(Part 19):4389–4400. doi: 10.1242/jcs.01299. [DOI] [PubMed] [Google Scholar]
- 51.Ishihara N, Eura Y, Mihara K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J Cell Sci. 2004;117(Part 26):6535–6546. doi: 10.1242/jcs.01565. [DOI] [PubMed] [Google Scholar]
- 52.Brini M, De Giorgi F, Murgia M, Marsault R, Massimino ML, Cantini M, et al. Subcellular analysis of Ca2+ homeostasis in primary cultures of skeletal muscle myotubes. Mol Biol Cell. 1997;8:129–143. doi: 10.1091/mbc.8.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Manfredi G, Yang L, Gajewski CD, Mattiazzi M. Measurements of ATP in mammalian cells. Methods. 2002;26:317–326. doi: 10.1016/S1046-2023(02)00037-3. [DOI] [PubMed] [Google Scholar]
- 54.Robinson BH. Use of fibroblast and lymphoblast cultures for detection of respiratory chain defects. Methods Enzymol. 1996;264:454–464. doi: 10.1016/s0076-6879(96)64041-5. [DOI] [PubMed] [Google Scholar]
- 55.Gnaiger E. Bioenergetics at low oxygen: dependence of respiration and phosphorylation on oxygen and adenosine diphosphate supply. Respir Physiol. 2001;128:277–297. doi: 10.1016/s0034-5687(01)00307-3. [DOI] [PubMed] [Google Scholar]